
Evaluation of the Aggressive-Variant Prostate Cancer Molecular Signature in Clinical Laboratory Improvement Amendments (CLIA) Environments

 , , ,
, , ,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design and Sample Collection
2.2. Immunohistochemistry: AVPCmIHC
2.3. Next-Generation Sequencing of FFPE Tumor DNA: AVPCmstDNA
2.4. Next-Generation Sequencing of Circulating Tumor DNA: AVPCmctDNA
2.5. Outcome Variables and Statistical Analysis
3. Results
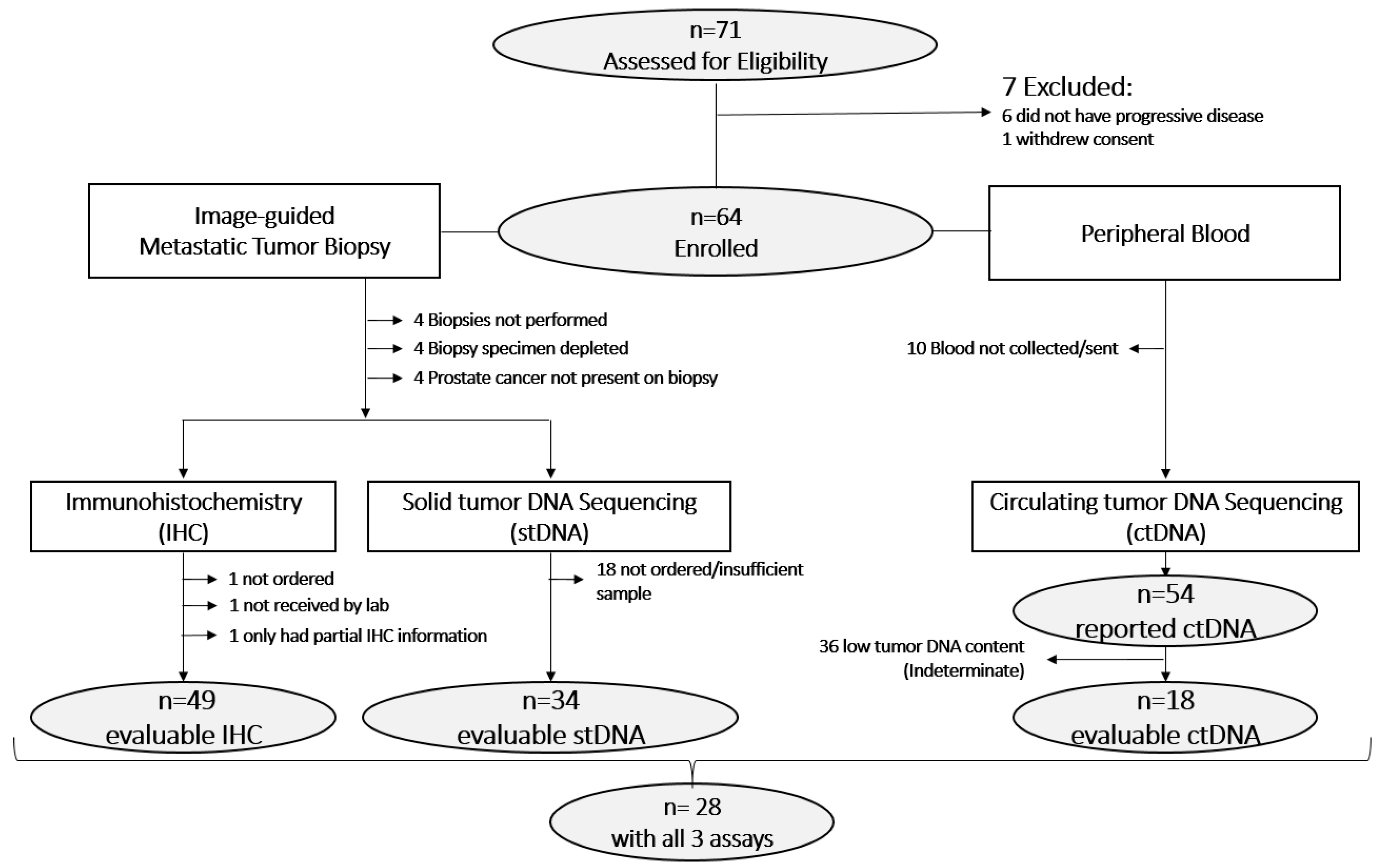
3.1. Sample Evaluability
3.2. Immunohistochemistry Results and Inter-Reader Variability
3.3. Next-Generation Sequencing of stDNA
3.4. Next-Generation Sequencing of ctDNA
3.5. Inter-Assay Agreements
3.6. Assay Turnaround Times
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hussain, M.; Tangen, C.M.; Higano, C.; Schelhammer, P.F.; Faulkner, J.; Crawford, E.D.; Wilding, G.; Akdas, A.; Small, E.J.; Donnelly, B.; et al. Absolute prostate-specific antigen value after androgen deprivation is a strong independent predictor of survival in new metastatic prostate cancer: Data from Southwest Oncology Group Trial 9346 (INT-0162). J. Clin. Oncol. 2006, 24, 3984–3990. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Beltran, H. Clinical and Biological Features of Neuroendocrine Prostate Cancer. Curr. Oncol. Rep. 2021, 23, 15. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.T.; Yao, Y.H.; Li, B.G.; Tang, Y.; Chang, J.W.; Zhang, J. Neuroendocrine Prostate Cancer (NEPC) progressing from conventional prostatic adenocarcinoma: Factors associated with time to development of NEPC and survival from NEPC diagnosis-a systematic review and pooled analysis. J. Clin. Oncol. 2014, 32, 3383–3390. [Google Scholar] [CrossRef] [PubMed]
- Uo, T.; Sprenger, C.C.; Plymate, S.R. Androgen Receptor Signaling and Metabolic and Cellular Plasticity During Progression to Castration Resistant Prostate Cancer. Front. Oncol. 2020, 10, 580617. [Google Scholar] [CrossRef]
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Tzelepi, V.; Zhang, J.; Lu, J.F.; Kleb, B.; Wu, G.; Wan, X.; Hoang, A.; Efstathiou, E.; Sircar, K.; Navone, N.M.; et al. Modeling a lethal prostate cancer variant with small-cell carcinoma features. Clin. Cancer Res. 2012, 18, 666–677. [Google Scholar] [CrossRef]
- Aggarwal, R.; Huang, J.; Alumkal, J.J.; Zhang, L.; Feng, F.Y.; Thomas, G.V.; Weinstein, A.S.; Friedl, V.; Zhang, C.; Witte, O.N.; et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J. Clin. Oncol. 2018, 36, 2492–2503. [Google Scholar] [CrossRef]
- Zou, M.; Toivanen, R.; Mitrofanova, A.; Floch, N.; Hayati, S.; Sun, Y.; Le Magnen, C.; Chester, D.; Mostaghel, E.A.; Califano, A.; et al. Transdifferentiation as a Mechanism of Treatment Resistance in a Mouse Model of Castration-Resistant Prostate Cancer. Cancer Discov. 2017, 7, 736–749. [Google Scholar] [CrossRef]
- Nadal, R.; Schweizer, M.; Kryvenko, O.N.; Epstein, J.I.; Eisenberger, M.A. Small cell carcinoma of the prostate. Nat. Rev. Urol. 2014, 11, 213–219. [Google Scholar] [CrossRef]
- Spetsieris, N.; Boukovala, M.; Patsakis, G.; Alafis, I.; Efstathiou, E. Neuroendocrine and Aggressive-Variant Prostate Cancer. Cancers 2020, 12, 3792. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, A.M.; Shen, L.; Tapia, E.L.; Lu, J.F.; Chen, H.C.; Zhang, J.; Wu, G.; Wang, X.; Troncoso, P.; Corn, P.; et al. Combined Tumor Suppressor Defects Characterize Clinically Defined Aggressive Variant Prostate Cancers. Clin. Cancer Res. 2016, 22, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.C.; Wongvipat, J.; Ku, S.Y.; Gao, D.; Cao, Z.; et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbe, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Corn, P.G.; Heath, E.I.; Zurita, A.; Ramesh, N.; Xiao, L.; Sei, E.; Li-Ning-Tapia, E.; Tu, S.M.; Subudhi, S.K.; Wang, J.; et al. Cabazitaxel plus carboplatin for the treatment of men with metastatic castration-resistant prostate cancers: A randomised, open-label, phase 1-2 trial. Lancet Oncol. 2019, 20, 1432–1443. [Google Scholar] [CrossRef]
- Guedes, L.B.; Almutairi, F.; Haffner, M.C.; Rajoria, G.; Liu, Z.; Klimek, S.; Zoino, R.; Yousefi, K.; Sharma, R.; De Marzo, A.M.; et al. Analytic, Preanalytic, and Clinical Validation of p53 IHC for Detection of TP53 missense mutation in prostate cancer. Clin. Cancer Res. 2017, 23, 4693–4703. [Google Scholar] [CrossRef]
- Kato, S.; Han, S.Y.; Liu, W.; Otsuka, K.; Shibata, H.; Kanamaru, R.; Ishioka, C. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 8424–8429. [Google Scholar] [CrossRef]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef]
- Tan, H.L.; Sood, A.; Rahimi, H.A.; Wang, W.; Gupta, N.; Hicks, J.; Mosier, S.; Gocke, C.D.; Epstein, J.I.; Netto, G.J.; et al. Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin. Cancer Res. 2014, 20, 890–903. [Google Scholar] [CrossRef]
- Lotan, T.L.; Heumann, A.; Rico, S.D.; Hicks, J.; Lecksell, K.; Koop, C.; Sauter, G.; Schlomm, T.; Simon, R. PTEN loss detection in prostate cancer: Comparison of PTEN immunohistochemistry and PTEN FISH in a large retrospective prostatectomy cohort. Oncotarget 2017, 8, 65566–65576. [Google Scholar] [CrossRef]
- Navone, N.M.; Troncoso, P.; Pisters, L.L.; Goodrow, T.L.; Palmer, J.L.; Nichols, W.W.; von Eschenbach, A.C.; Conti, C.J. p53 protein accumulation and gene mutation in the progression of human prostate carcinoma. J. Natl. Cancer Inst. 1993, 85, 1657–1669. [Google Scholar] [CrossRef] [PubMed]
- Eastham, J.A.; Stapleton, A.M.; Gousse, A.E.; Timme, T.L.; Yang, G.; Slawin, K.M.; Wheeler, T.M.; Scardino, P.T.; Thompson, T.C. Association of p53 mutations with metastatic prostate cancer. Clin. Cancer Res. 1995, 1, 1111–1118. [Google Scholar]
- Soundararajan, R.; Viscuse, P.; Pilie, P.; Liu, J.; Logotheti, S.; Laberiano Fernández, C.; Lorenzini, D.; Hoang, A.; Lu, W.; Soto, L.M.S.; et al. Genotype-to-Phenotype Associations in the Aggressive Variant Prostate Cancer Molecular Profile (AVPC-m) Components. Cancers 2022, 14, 3233. [Google Scholar] [CrossRef] [PubMed]
- Kuo, A.J.; Paulson, V.A.; Hempelmann, J.A.; Beightol, M.; Todhunter, S.; Colbert, B.G.; Salipante, S.J.; Konnick, E.Q.; Pritchard, C.C.; Lockwood, C.M. Validation and implementation of a modular targeted capture assay for the detection of clinically significant molecular oncology alterations. Pract. Lab. Med. 2020, 19, e00153. [Google Scholar] [CrossRef]
- Pritchard, C.C.; Salipante, S.J.; Koehler, K.; Smith, C.; Scroggins, S.; Wood, B.; Wu, D.; Lee, M.K.; Dintzis, S.; Adey, A.; et al. Validation and implementation of targeted capture and sequencing for the detection of actionable mutation, copy number variation, and gene rearrangement in clinical cancer specimens. J. Mol. Diagn. 2014, 16, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Bujang, M.A.; Baharum, N. Guidelines of the minimum sample size requirements for Kappa agreement test. Epidemiol. Biostat. Public Health 2017, 14, e12267-1. [Google Scholar] [CrossRef]
- Khalaf, D.; Annala, M.; Beja, K.; Vandekerkhove, G.; Zulfiqar, M.; Finch, D.L.; Oja, C.D.; Vergidis, J.; Gleave, M.; Wyatt, A.W.; et al. Circulating tumor DNA (ctDNA) and correlations with clinical prognostic factors in patients with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2017, 35, 186. [Google Scholar] [CrossRef]
- Herberts, C.; Annala, M.; Sipola, J.; Ng, S.W.S.; Chen, X.E.; Nurminen, A.; Korhonen, O.V.; Munzur, A.D.; Beja, K.; Schönlau, E.; et al. Deep whole-genome ctDNA chronology of treatment-resistant prostate cancer. Nature 2022, 608, 199–208. [Google Scholar] [CrossRef]
- Milbury, C.A.; Creeden, J.; Yip, W.K.; Smith, D.L.; Pattani, V.; Maxwell, K.; Sawchyn, B.; Gjoerup, O.; Meng, W.; Skoletsky, J.; et al. Clinical and analytical validation of FoundationOne®CDx, a comprehensive genomic profiling assay for solid tumors. PLoS ONE 2022, 17, e0264138. [Google Scholar] [CrossRef]
- Beaubier, N.; Tell, R.; Lau, D.; Parsons, J.R.; Bush, S.; Perera, J.; Sorrells, S.; Baker, T.; Chang, A.; Michuda, J.; et al. Clinical validation of the tempus xT next-generation targeted oncology sequencing assay. Oncotarget 2019, 10, 2384–2396. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| All Patients | IHC Evaluable * | stDNA Evaluable | ctDNA Evaluable | ctDNA Sufficient ** | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient Characteristics | N | (%) | N | (%) | N | (%) | N | (%) | N | (%) | |
| All | 64 | (100%) | 49 | (100%) | 34 | (100%) | 54 | (100%) | 19 | (100%) | |
| Age—median (min, max) | 66.5 | (48, 86) | 66 | (48, 86) | 66 | (48, 86) | 67 | (48, 86) | 65 | (48, 75) | |
| Biopsy Year | 2020 | 12 | (19%) | 11 | (22%) | 10 | (29%) | 10 | (19%) | 5 | (26%) |
| 2021 | 47 | (73%) | 36 | (73%) | 23 | (68%) | 41 | (76%) | 12 | (63%) | |
| 2022 | 2 | (3%) | 2 | (4%) | 1 | (3%) | 2 | (4%) | 1 | (5%) | |
| None | 3 | (5%) | 0 | (0%) | 0 | (0%) | 1 | (2%) | 1 | (5%) | |
| Biopsy Site | Not Applicable | 3 | (5%) | 0 | (0%) | 0 | (0%) | 1 | (2%) | 1 | (5%) |
| Adrenal | 1 | (2%) | 1 | (2%) | 0 | (0%) | 1 | (2%) | 0 | (0%) | |
| Bladder | 1 | (2%) | 0 | (0%) | 0 | (0%) | 1 | (2%) | 0 | (0%) | |
| Bone | 19 | (30%) | 15 | (31%) | 10 | (29%) | 15 | (28%) | 2 | (11%) | |
| Bone marrow | 1 | (2%) | 0 | (0%) | 0 | (0%) | 1 | (2%) | 0 | (0%) | |
| LN | 23 | (36%) | 21 | (43%) | 15 | (44%) | 20 | (37%) | 6 | (32%) | |
| Liver | 11 | (17%) | 8 | (16%) | 8 | (24%) | 11 | (20%) | 9 | (47%) | |
| Lung | 3 | (5%) | 2 | (4%) | 0 | (0%) | 3 | (6%) | 1 | (5%) | |
| Prostate | 2 | (3%) | 2 | (4%) | 1 | (3%) | 1 | (2%) | 0 | (0%) | |
| IHC Evaluable | No | 15 | (23%) | 0 | (0%) | 2 | (6%) | 12 | (22%) | 4 | (21%) |
| Yes | 49 | (77%) | 49 | (100%) | 32 | (94%) | 42 | (78%) | 15 | (79%) | |
| stDNA Evaluable | No | 30 | (47%) | 17 | (35%) | 0 | (0%) | 26 | (48%) | 8 | (42%) |
| Yes | 34 | (53%) | 32 | (65%) | 34 | (100%) | 28 | (52%) | 11 | (58%) | |
| ctDNA Evaluable | No | 10 | (16%) | 7 | (14%) | 6 | (18%) | 0 | (0%) | 0 | (0%) |
| Yes | 54 | (84%) | 42 | (86%) | 28 | (82%) | 54 | (100%) | 19 | (100%) | |
| Tp53 Abnormal | Rev2, n | Kappa (95% CI) | ||
| No | Yes | |||
| Rev1, n | No | 21 | 5 | 0.76 (0.58, 0.94) |
| Yes | 1 | 23 | ||
| RB1 abnormal | Rev2, n | |||
| No | Yes | |||
| Rev1, n | No | 36 | 2 | 0.58 (0.31, 0.86) |
| Yes | 5 | 7 | ||
| PTEN abnormal | Rev2, n | |||
| No | Yes | |||
| Rev1, n | No | 31 | 1 | 0.91 (0.79, 1.00) |
| Yes | 1 | 16 | ||
| AVPCmIHC | Rev2, n | |||
| No | Yes | |||
| Rev1, n | No | 32 | 4 | 0.66 (0.43, 0.89) |
| Yes | 3 | 11 | ||
| A | ||||
|---|---|---|---|---|
| AVPCmIHC vs. AVPCmctDNA | ||||
| AVPCmctDNA− | AVPCmctDNA+ | total | Kappa (95% CI) | |
| AVPCmIHC− | 29 | 2 | 31 | 0.35 (0.03, 0.67) |
| AVPCmIHC+ | 7 | 4 | 11 | |
| total | 36 | 6 | 42 | |
| AVPCmIHC vs. AVPCmstDNA | ||||
| AVPCmstDNA− | AVPCmstDNA+ | total | Kappa (95% CI) | |
| AVPCmIHC− | 21 | 1 | 22 | 0.07 (−0.19, 0.33) |
| AVPCmIHC+ | 9 | 1 | 10 | |
| total | 30 | 2 | 32 | |
| AVPCmstDNA vs. AVPCmctDNA | ||||
| AVPCmctDNA− | AVPCmctDNA+ | total | Kappa (95% CI) | |
| AVPCmstDNA− | 22 | 4 | 26 | 0.20 (−0.24, 0.65) |
| AVPCmstDNA+ | 1 | 1 | 2 | |
| total | 23 | 5 | 28 | |
| AVPCmstDNA adjusted vs. AVPCmIHC | ||||
| AVPCstDNA adjusted− | AVPCstDNA adjusted+ | total | Kappa (95% CI) | |
| AVPCIHC− | 14 | 8 | 22 | 0.30 (−0.02, 0.61) |
| AVPCIHC+ | 3 | 7 | 10 | |
| Total | 17 | 15 | 32 | |
| AVPCmstDNA adjusted vs. AVPCmctDNA | ||||
| AVPCmctDNA− | AVPCmctDNA+ | total | Kappa (95% CI) | |
| AVPCstDNA adjusted− | 13 | 0 | 13 | 0.32 (0.07, 0.57) |
| AVPCstDNA adjusted+ | 10 | 5 | 15 | |
| total | 23 | 5 | 28 | |
| B | ||||
| AVPCmIHC vs. AVPCmctDNA | ||||
| AVPCmctDNA− | AVPCmctDNA+ | total | Kappa (95% CI) | |
| AVPCmIHC− | 8 | 2 | 10 | 0.57 (0.15, 1.00) |
| AVPCmIHC+ | 1 | 4 | 5 | |
| total | 9 | 6 | 15 | |
| AVPCmIHC vs. AVPCmstDNA | ||||
| AVPCmstDNA− | AVPCmstDNA+ | total | Kappa (95% CI) | |
| AVPCmIHC− | 6 | 0 | 6 | 0.21 (−0.17, 0.59) |
| AVPCmIHC+ | 4 | 1 | 5 | |
| total | 10 | 1 | 11 | |
| AVPCmstDNA vs. AVPCmctDNA | ||||
| AVPCmctDNA− | AVPCmctDNA+ | total | Kappa (95% CI) | |
| AVPCmstDNA− | 6 | 4 | 10 | 0.21 (−0.17, 0.59) |
| AVPCmstDNA+ | 0 | 1 | 1 | |
| total | 6 | 5 | 11 | |
| AVPCmIHC vs. AVPCmstDNA adjusted | ||||
| AVPCstDNA adjusted− | AVPCstDNA adjusted+ | total | Kappa (95% CI) | |
| AVPCIHC− | 3 | 3 | 6 | 0.29 (−0.23, 0.81) |
| AVPCIHC+ | 1 | 4 | 5 | |
| total | 4 | 7 | 11 | |
| AVPCmstDNA adjusted vs. AVPCmctDNA | ||||
| AVPCmctDNA− | AVPCmctDNA+ | total | Kappa (95% CI) | |
| AVPCstDNA adjusted− | 4 | 0 | 4 | 0.65 (−0.23, 1.00) |
| AVPCstDNA adjusted+ | 2 | 5 | 7 | |
| total | 6 | 5 | 11 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viscuse, P.V.; Slack-Tidwell, R.S.; Zhang, M.; Rohra, P.; Zhu, K.; San Lucas, F.A.; Konnick, E.; Pilie, P.G.; Siddiqui, B.; Logothetis, C.J.; et al. Evaluation of the Aggressive-Variant Prostate Cancer Molecular Signature in Clinical Laboratory Improvement Amendments (CLIA) Environments. Cancers 2023, 15, 5843. https://doi.org/10.3390/cancers15245843
Viscuse PV, Slack-Tidwell RS, Zhang M, Rohra P, Zhu K, San Lucas FA, Konnick E, Pilie PG, Siddiqui B, Logothetis CJ, et al. Evaluation of the Aggressive-Variant Prostate Cancer Molecular Signature in Clinical Laboratory Improvement Amendments (CLIA) Environments. Cancers. 2023; 15(24):5843. https://doi.org/10.3390/cancers15245843
Chicago/Turabian StyleViscuse, Paul V., Rebecca S. Slack-Tidwell, Miao Zhang, Prih Rohra, Keyi Zhu, F. Anthony San Lucas, Eric Konnick, Patrick G. Pilie, Bilal Siddiqui, Christopher J. Logothetis, and et al. 2023. "Evaluation of the Aggressive-Variant Prostate Cancer Molecular Signature in Clinical Laboratory Improvement Amendments (CLIA) Environments" Cancers 15, no. 24: 5843. https://doi.org/10.3390/cancers15245843