
Preferential Expansion of HPV16 E1-Specific T Cells from Healthy Donors’ PBMCs after Ex Vivo Immunization with an E1E2E6E7 Fusion Antigen
 , , , , , ,
, , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Antigen Design and Adenoviral Vectors
2.2. Peripheral Blood Mononuclear Cells (PBMCs) Isolation and Cryopreservation
2.3. Monocyte Isolation and Differentiation to Dendritic Cells (DCs)
2.4. Transduction of Mature DCs
2.5. Cancer Cell Lines
2.6. PBMC Stimulation and HPV16-Specific T-Cell Expansion
2.7. Sorting of Interferon Gamma-Positive Cells
2.8. Rapid Expansion Protocol (REP)
2.9. Surface and Intracellular Cytokine Staining (ICS) and Flow Cytometry
2.10. Chromium-51 (51Cr)-Release Assay
3. Results
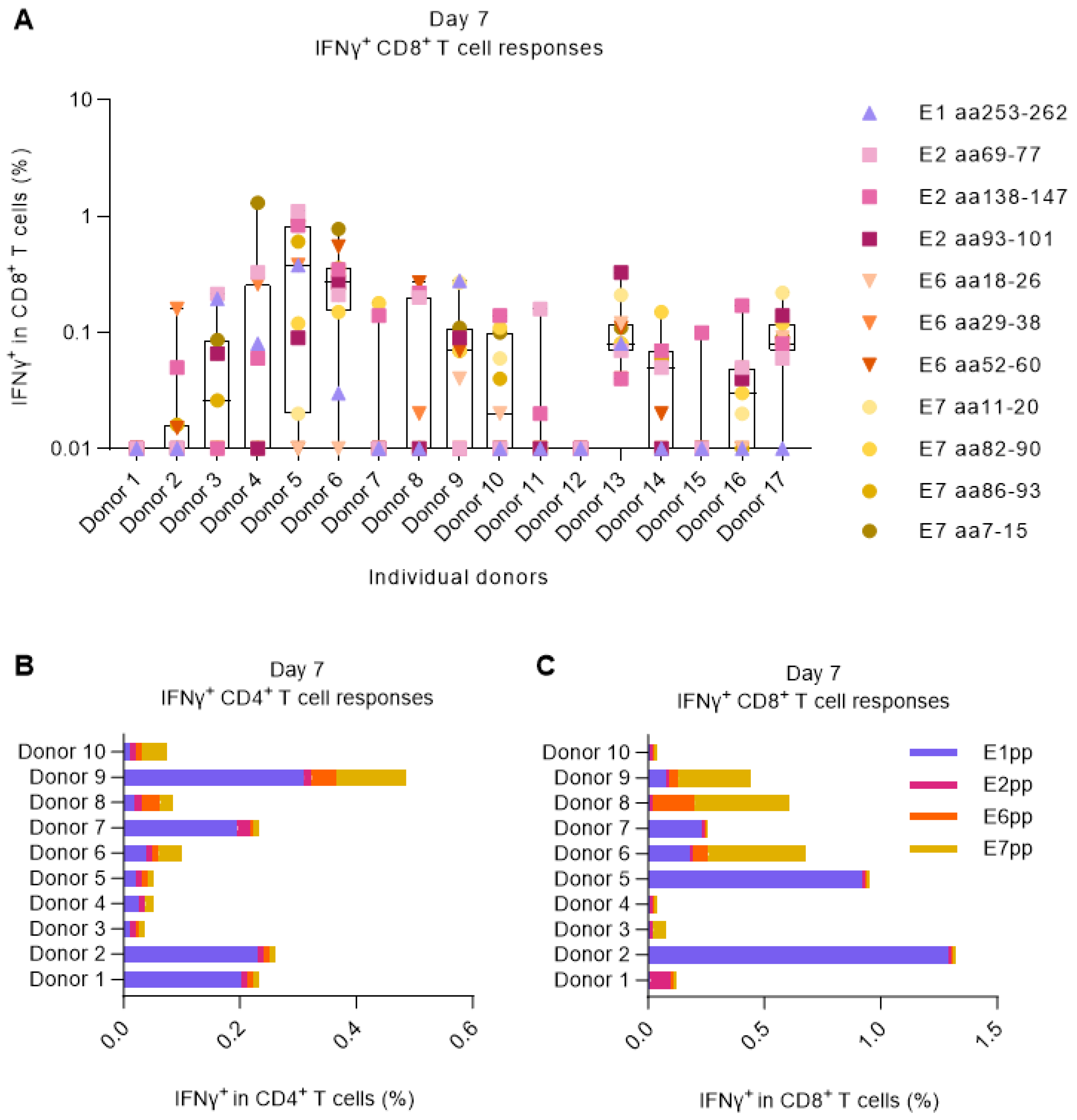
3.1. Screening for HPV16-Responding Donors
3.2. HPV16 Peptide-Primed PBMCs Can Specifically Recognize HPV16+ Ca Ski Cancer Cells
3.3. Verification of the Human Ex Vivo Model of Immunization for Boosting of HPV16-Specific CD8+ T Cells
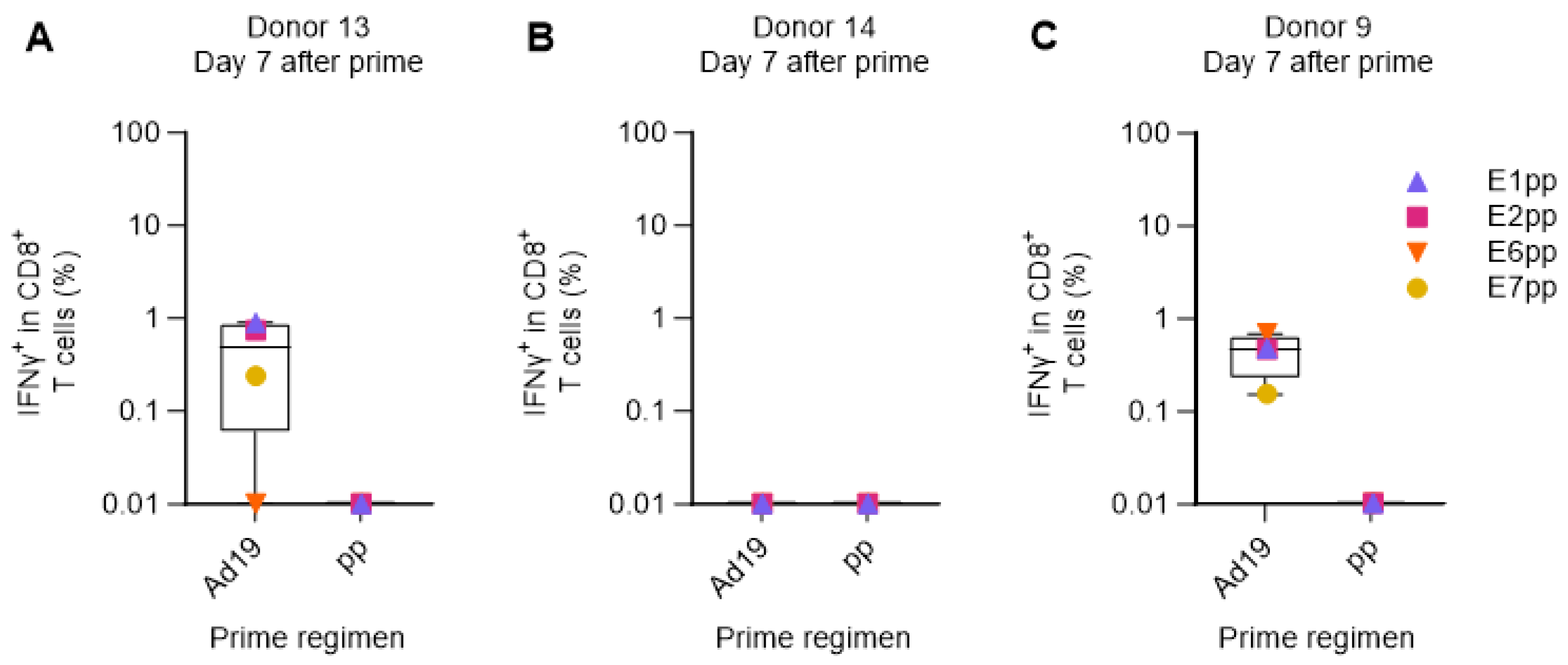
3.4. Verification of the Human Ex Vivo Immunization Model for the Priming of HPV16-Specific CD8+ T Cells
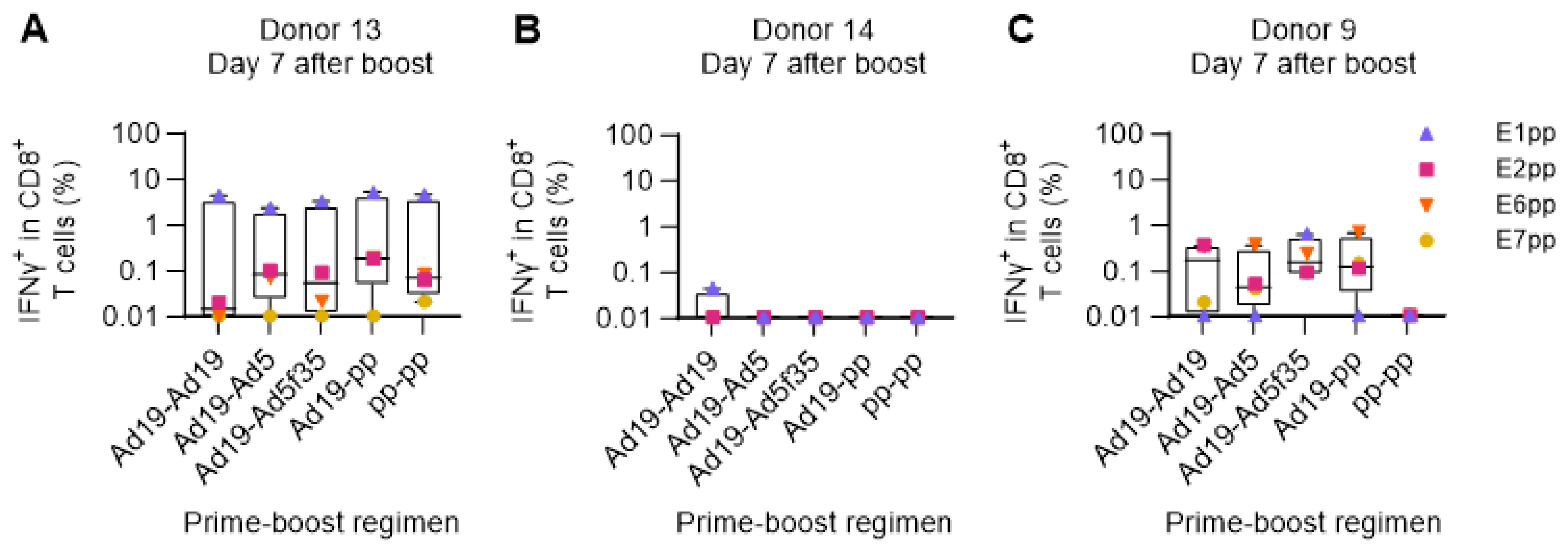
3.5. Use of the Ex Vivo Human Immunization Model for the Comparison of Different Prime-Boost Strategies
3.6. Only Vaccine-Expanded/Sorted HPV16-E1-Specific T Cells Are Functional Regarding Killing of Cervical Carcinoma Cancer Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walboomers, J.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.; Peto, J.; Meijer, C.J.; Muñoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- Zamani, M.; Grønhøj, C.; Jensen, D.H.; Carlander, A.F.; Agander, T.; Kiss, K.; Olsen, C.; Baandrup, L.; Nielsen, F.C.; Andersen, E.; et al. The current epidemic of HPV-associated oropharyngeal cancer: An 18-year Danish population-based study with 2169 patients. Eur. J. Cancer 2020, 134, 52–59. [Google Scholar] [CrossRef]
- Steinau, M.; Steinau, M.; Saraiya, M.; Goodman, M.T.; Peters, E.S.; Watson, M.; Cleveland, J.L.; Lynch, C.F.; Wilkinson, E.J.; Hernandez, B.Y.; et al. Human papillomavirus prevalence in oropharyngeal cancer before vaccine introduction, United States. Emerg. Infect. Dis. 2014, 20, 822–828. [Google Scholar] [CrossRef]
- National Cancer Institute at the National Institutes of Health. HPV and Cancer. 2023. Available online: https://www.cancer.gov/about-cancer/causes-prevention/risk/infectious-agents/hpv-and-cancer (accessed on 12 December 2023).
- McCormack, P.L. Quadrivalent human papillomavirus (types 6, 11, 16, 18) recombinant vaccine (gardasil(®)): A review of its use in the prevention of premalignant anogenital lesions, cervical and anal cancers, and genital warts. Drugs 2014, 74, 1253–1283. [Google Scholar] [CrossRef]
- Mirabello, L.; Clarke, M.A.; Nelson, C.W.; Dean, M.; Wentzensen, N.; Yeager, M.; Cullen, M.; Boland, J.F.; Workshop, N.H.; Schiffman, M.; et al. The Intersection of HPV Epidemiology, Genomics and Mechanistic Studies of HPV-Mediated Carcinogenesis. Viruses 2018, 10, 80. [Google Scholar] [CrossRef]
- Global Cancer Observatory. Estimated Number of New Cases in 2020, Worldwide, Both Sexes, All Ages (excl. NMSC). Available online: https://gco.iarc.fr/today/online-analysis-table?v=2020&mode=cancer&mode_population=continents&population=900&populations=900&key=asr&sex=0&cancer=39&type=0&statistic=5&prevalence=0&population_group=0&ages_group%5B%5D=0&ages_group%5B%5D=17&group_cancer=1&include_nmsc=0&include_nmsc_other=1 (accessed on 2 October 2023).
- Crosbie, E.J.; Einstein, M.H.; Franceschi, S.; Kitchener, H.C. Human papillomavirus and cervical cancer. Lancet 2013, 382, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Sriplung, H.; Singkham, P.; Iamsirithaworn, S.; Jiraphongsa, C.; Bilheem, S. Success of a cervical cancer screening program: Trends in incidence in songkhla, southern Thailand, 1989–2010, and prediction of future incidences to 2030. Asian Pac. J. Cancer Prev. 2014, 15, 10003–10008. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.K.; Engels, E.A.; Pfeiffer, R.M.; Hernandez, B.Y.; Xiao, W.; Kim, E.; Jiang, B.; Goodman, M.T.; Sibug-Saber, M.; Cozen, W.; et al. Human Papillomavirus and Rising Oropharyngeal Cancer Incidence in the United States. J. Clin. Oncol. 2011, 29, 4294–4301. [Google Scholar] [CrossRef] [PubMed]
- Kreimer, A.R.; Shiels, M.S.; Fakhry, C.; Johansson, M.; Pawlita, M.; Brennan, P.; Hildesheim, A.; Waterboer, T. Screening for human papillomavirus-driven oropharyngeal cancer: Considerations for feasibility and strategies for research. Cancer 2018, 124, 1859–1866. [Google Scholar] [CrossRef]
- von Witzleben, A.; Wang, C.; Laban, S.; Savelyeva, N.; Ottensmeier, C.H. HNSCC: Tumour Antigens and Their Targeting by Immunotherapy. Cells 2020, 9, 2103. [Google Scholar] [CrossRef] [PubMed]
- Boilesen, D.R.; Nielsen, K.N.; Holst, P.J. Novel antigenic targets of hpv therapeutic vaccines. Vaccines 2021, 9, 1262. [Google Scholar] [CrossRef]
- Hancock, G.; Hellner, K.; Dorrell, L. Therapeutic HPV vaccines. Best Pract. Res. Clin. Obstet. Gynaecol. 2018, 47, 59–72. [Google Scholar] [CrossRef]
- Feltkamp, M.C.; Smits, H.L.; Vierboom, M.P.M.; Minnaar, R.P.; De Jongh, B.M.; Drijfhout, J.W.; Ter Schegget, J.; Melief, C.J.M.; Kast, W.M. Vaccination with cytotoxic T lymphocyte epitope-containing peptide protects against a tumor induced by human papillomavirus type 16-transformed cells. Eur. J. Immunol. 1993, 23, 2242–2249. [Google Scholar] [CrossRef] [PubMed]
- Tombak, E.M.; Männik, A.; Burk, R.D.; Le Grand, R.; Ustav, E.; Ustav, M. The molecular biology and HPV drug responsiveness of cynomolgus macaque papillomaviruses support their use in the development of a relevant in vivo model for antiviral drug testing. PLoS ONE 2019, 14, e0211235. [Google Scholar] [CrossRef] [PubMed]
- Pauken, K.E.; Wherry, E.J. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015, 36, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, C.S.; Kissick, H.T.; Patel, M.R.; Cardenas, M.A.; Prokhnevska, N.; Obeng, R.C.; Nasti, T.H.; Griffith, C.C.; Im, S.J.; Wang, X.; et al. Functional HPV-specific PD-1+ stem-like CD8 T cells in head and neck cancer. Nature 2021, 597, 279–284. [Google Scholar] [CrossRef]
- Peng, X.; Woodhouse, I.; Hancock, G.; Parker, R.; Marx, K.; Müller, J.; Salatino, S.; Partridge, T.; Nicastri, A.; Liao, H.; et al. Novel canonical and non-canonical viral antigens extend current targets for immunotherapy of HPV-driven cervical cancer. iScience 2023, 26, 106101. [Google Scholar] [CrossRef] [PubMed]
- McInnis, C.; Bhatia, S.; Vijaykumar, B.; Tian, Q.; Sun, Y.; Leistritz-Edwards, D.; Quinn, C.T.; Uppaluri, R.; Egloff, A.M.; Srinivasan, L.; et al. Identification of HPV16 E1 and E2-specific T cells in the oropharyngeal cancer tumor microenvironment. J. Immunother. Cancer 2023, 11, e006721. [Google Scholar] [CrossRef]
- Boilesen, D.R.; Neckermann, P.; Willert, T.; Müller, M.D.; Schrödel, S.; Pertl, C.; Thirion, C.; Asbach, B.; Wagner, R.; Holst, P.J. Efficacy and synergy with cisplatin of an adenovirus vectored therapeutic E1E2E6E7 vaccine against HPV genome positive C3 cancers in mice. Cancer Immunol. Res. 2022, 11, 261–275. [Google Scholar] [CrossRef]
- Kiener, R.; Fleischmann, M.; Schwegler, C.; Ruzsics, Z.; Thirion, C.; Schrödel, S.; Asbach, B.; Wagner, R. Vaccine vectors based on Adenovirus 19a/64 exhibit broad cellular tropism and potently restimulate HCMV-specific T cell responses ex vivo. Sci. Rep. 2018, 8, 1474. [Google Scholar] [CrossRef]
- Ragonnaud, E.; Schroedel, S.; Mariya, S.; Iskandriati, D.; Pamungkas, J.; Fougeroux, C.; Daradoumis, J.; Thomsen, A.R.; Neukirch, L.; Ruzsics, Z.; et al. Replication deficient human adenovirus vector serotype 19a/64: Immunogenicity in mice and female cynomolgus macaques. Vaccine 2018, 36, 6212–6222. [Google Scholar] [CrossRef]
- Ruzsics, Z.; Lemnitzer, F.; Thirion, C. Engineering adenovirus genome by bacterial artificial chromosome (BAC) technology. Methods Mol. Biol. 2014, 1089, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Flickinger, J.C., Jr.; Singh, J.; Carlson, R.; Leong, E.; Baybutt, T.R.; Barton, J.; Caparosa, E.; Pattison, A.; Rappaport, J.A.; Roh, J.; et al. Chimeric Ad5.F35 vector evades anti-adenovirus serotype 5 neutralization opposing GUCY2C-targeted antitumor immunity. J. Immunother. Cancer 2020, 8, e001046. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.; Ljungberg, J.; Richter, J.; Kiefer, T.; Magnusson, M.; Lieber, A.; Widegren, B.; Karlsson, S.; Fan, X. Development of an adenoviral vector system with adenovirus serotype 35 tropism; efficient transient gene transfer into primary malignant hematopoietic cells. J. Gene Med. 2004, 6, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Chirmule, N.; Propert, K.; Magosin, S.; Qian, Y.; Qian, R.; Wilson, J. Immune responses to adenovirus and adeno-associated virus in humans. Gene Ther. 1999, 6, 1574–1583. [Google Scholar] [CrossRef]
- Fausther-Bovendo, H.; Kobinger, G.P. Pre-existing immunity against Ad vectors: Humoral, cellular, and innate response, what’s important? Hum. Vaccin. Immunother. 2014, 10, 2875–2884. [Google Scholar] [CrossRef]
- Barouch, D.H.; Kik, S.V.; Weverling, G.J.; Dilan, R.; King, S.L.; Maxfield, L.F.; Clark, S.; Ng’ang’a, D.; Brandariz, K.L.; Abbink, P.; et al. International seroepidemiology of adenovirus serotypes 5, 26, 35, and 48 in pediatric and adult populations. Vaccine 2011, 29, 5203–5209. [Google Scholar] [CrossRef] [PubMed]
- Mennechet, F.J.D.; Paris, O.; Ouoba, A.R.; Arenas, S.S.; Sirima, S.B.; Dzomo, G.R.T.; Diarra, A.; Traore, I.T.; Kania, D.; Eichholz, K.; et al. A review of 65 years of human adenovirus seroprevalence. Expert Rev. Vaccines 2019, 18, 597–613. [Google Scholar] [CrossRef]
- Neckermann, P.; Boilesen, D.R.; Willert, T.; Pertl, C.; Schrödel, S.; Thirion, C.; Asbach, B.; Holst, P.J.; Wagner, R. Design and Immunological Validation of Macaca fascicularis Papillomavirus Type 3 Based Vaccine Candidates in Outbred Mice: Basis for Future Testing of a Therapeutic Papillomavirus Vaccine in NHPs. Front. Immunol. 2021, 12, 761214. [Google Scholar] [CrossRef]
- Raab, D.; Graf, M.; Notka, F.; Schödl, T.; Wagner, R. The GeneOptimizer Algorithm: Using a sliding window approach to cope with the vast sequence space in multiparameter DNA sequence optimization. Syst. Synth. Biol. 2010, 4, 215–225. [Google Scholar] [CrossRef]
- Olofsson, G.H.; Idorn, M.; Simões, A.M.C.; Aehnlich, P.; Skadborg, S.K.; Noessner, E.; Debets, R.; Moser, B.; Met, M.; Straten, P.T. Vγ9Vδ2 T Cells Concurrently Kill Cancer Cells and Cross-Present Tumor Antigens. Front. Immunol. 2021, 12, 645131. [Google Scholar] [CrossRef]
- Esposito, I.; Cicconi, P.; D’alise, A.M.; Brown, A.; Esposito, M.; Swadling, L.; Holst, P.J.; Bassi, M.R.; Stornaiuolo, M.; Mori, F.; et al. MHC class II invariant chain-adjuvanted viral vectored vaccines enhances T cell responses in humans. Sci. Transl. Med. 2020, 12, 1–36. [Google Scholar] [CrossRef]
- Goldstein, N.; Bockstal, V.; Bart, S.; Luhn, K.; Robinson, C.; Gaddah, A.; Callendret, B.; Douoguih, M. Safety and Immunogenicity of Heterologous and Homologous 2-Dose Regimens of Adenovirus Serotype 26- and Modified Vaccinia Ankara-Vectored Ebola Vaccines: A Randomized, Controlled Phase 1 Study. J. Infect. Dis. 2022, 226, 595–607. [Google Scholar] [CrossRef] [PubMed]
- Ewer, K.; Rampling, T.; Venkatraman, N.; Bowyer, G.; Wright, D.; Lambe, T.; Imoukhuede, E.B.; Payne, R.; Fehling, S.K.; Strecker, T.; et al. A Monovalent Chimpanzee Adenovirus Ebola Vaccine Boosted with MVA. N. Engl. J. Med. 2016, 374, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.L.; Bråve, A.; Scarlatti, G.; Manrique, A.; Buonaguro, L. Progress towards development of an HIV vaccine: Report of the AIDS Vaccine 2009 Conference. Lancet. Infect. Dis. 2010, 10, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Trimble, C.; Wu, L.; Pardoll, D.; Roden, R.; Hung, C.-F.; Wu, T.-C. HLA-DQB1*02-restricted HPV-16 E7 peptide-specific CD4+ T-cell immune responses correlate with regression of HPV-16-associated high-grade squamous intraepithelial lesions. Clin. Cancer Res. 2007, 13, 2479–2487. [Google Scholar] [CrossRef]
- Stevanović, S.; Pasetto, A.; Helman, S.R.; Gartner, J.J.; Prickett, T.D.; Howie, B.; Robins, H.S.; Robbins, P.F.; Klebanoff, C.A.; Rosenberg, S.A.; et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science 2017, 356, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.; Ulrich, P.; Wilson, E.; Parikh, F.; Narang, P.; Yang, S.; Read, A.K.; Kim-Schulze, S.; Park, J.G.; Posner, M.; et al. Human Papilloma Virus Specific Immunogenicity and Dysfunction of CD8+ T Cells in Head and Neck Cancer. Cancer Res. 2018, 78, 6159–6170. [Google Scholar] [CrossRef]
- Ma, M.; Feng, Y.; Fan, P.; Yao, X.; Peng, Y.; Dong, T.; Wang, R. Human papilloma virus E1-specific T cell immune response is associated with the prognosis of cervical cancer patients with squamous cell carcinoma. Infect. Agent. Cancer 2018, 13, 35. [Google Scholar] [CrossRef]
- Dillon, S.; Sasagawa, T.; Crawford, A.; Prestidge, J.; Inder, M.K.; Jerram, J.; Mercer, A.A.; Hibma, M. Resolution of cervical dysplasia is associated with T-cell proliferative responses to human papillomavirus type 16 E2. J. Gen. Virol. 2007, 88 Pt 3, 803–813. [Google Scholar] [CrossRef]
- Woo, Y.L.; Hende, M.v.D.; Sterling, J.C.; Coleman, N.; Crawford, R.A.; Kwappenberg, K.M.; Stanley, M.A.; van der Burg, S.H. A prospective study on the natural course of low-grade squamous intraepithelial lesions and the presence of HPV16 E2-, E6- and E7-specific T-cell responses. Int. J. Cancer 2010, 126, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Evnouchidou, I.; van Endert, P. Peptide trimming by endoplasmic reticulum aminopeptidases: Role of MHC class I binding and ERAP dimerization. Hum. Immunol. 2019, 80, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, M.; Holst, P.J.; Bukh, J.; Thomsen, A.R.; Christensen, J.P. Enhanced and sustained CD8+ T cell responses with an adenoviral vector-based hepatitis C virus vaccine encoding NS3 linked to the MHC class II chaperone protein invariant chain. J. Immunol. 2011, 186, 2355–2364. [Google Scholar] [CrossRef]
- Sorensen, M.R.; Holst, P.J.; Pircher, H.; Christensen, J.P.; Thomsen, A.R. Vaccination with an adenoviral vector encoding the tumor antigen directly linked to invariant chain induces potent CD4+ T-cell-independent CD8+ T-cell-mediated tumor control. Eur. J. Immunol. 2009, 39, 2725–2736. [Google Scholar] [CrossRef]
- Holst, P.J.; Sorensen, M.R.; Jensen, C.M.M.; Orskov, C.; Thomsen, A.R.; Christensen, J.P. MHC class II-associated invariant chain linkage of antigen dramatically improves cell-mediated immunity induced by adenovirus vaccines. J. Immunol. 2008, 180, 3339–3346. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daradoumis, J.; Müller, M.D.; Neckermann, P.; Asbach, B.; Schrödel, S.; Thirion, C.; Wagner, R.; thor Straten, P.; Holst, P.J.; Boilesen, D. Preferential Expansion of HPV16 E1-Specific T Cells from Healthy Donors’ PBMCs after Ex Vivo Immunization with an E1E2E6E7 Fusion Antigen. Cancers 2023, 15, 5863. https://doi.org/10.3390/cancers15245863
Daradoumis J, Müller MD, Neckermann P, Asbach B, Schrödel S, Thirion C, Wagner R, thor Straten P, Holst PJ, Boilesen D. Preferential Expansion of HPV16 E1-Specific T Cells from Healthy Donors’ PBMCs after Ex Vivo Immunization with an E1E2E6E7 Fusion Antigen. Cancers. 2023; 15(24):5863. https://doi.org/10.3390/cancers15245863
Chicago/Turabian StyleDaradoumis, Joana, Mikkel Dons Müller, Patrick Neckermann, Benedikt Asbach, Silke Schrödel, Christian Thirion, Ralf Wagner, Per thor Straten, Peter Johannes Holst, and Ditte Boilesen. 2023. "Preferential Expansion of HPV16 E1-Specific T Cells from Healthy Donors’ PBMCs after Ex Vivo Immunization with an E1E2E6E7 Fusion Antigen" Cancers 15, no. 24: 5863. https://doi.org/10.3390/cancers15245863
APA StyleDaradoumis, J., Müller, M. D., Neckermann, P., Asbach, B., Schrödel, S., Thirion, C., Wagner, R., thor Straten, P., Holst, P. J., & Boilesen, D. (2023). Preferential Expansion of HPV16 E1-Specific T Cells from Healthy Donors’ PBMCs after Ex Vivo Immunization with an E1E2E6E7 Fusion Antigen. Cancers, 15(24), 5863. https://doi.org/10.3390/cancers15245863