
Clinical, Histopathological and Molecular Spectrum of Cutaneous Lesions in Myelodysplastic Syndrome and Myeloproliferative Neoplasms (MDS/MPN): An Integrative Review



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Myelodysplastic/Myeloproliferative Related Dermatosis with Indolent Clinical Outcomes
2.1. Neutrophilic Dermatoses
2.1.1. Sweet Syndrome
Histiocytoid SS (H-SS)
Other Morphological Variants of the Sweet Syndrome
2.1.2. Vexas Syndrome
2.1.3. Myelodysplasia Cutis
2.2. Granulomatous Dermatoses
2.3. Mature Plasmacytoid Dendritic Cell Dermatoses
2.4. Other Myelodysplastic/Myeloproliferative Dermatosis
3. Cutaneous Processes Related to Either MDS/MNP and Aggressive Clinical Behavior
3.1. Leukemia Cutis/Myeloid Leukemia Cutis
3.1.1. Granulocytic Sarcoma
3.1.2. Aleukemic LC
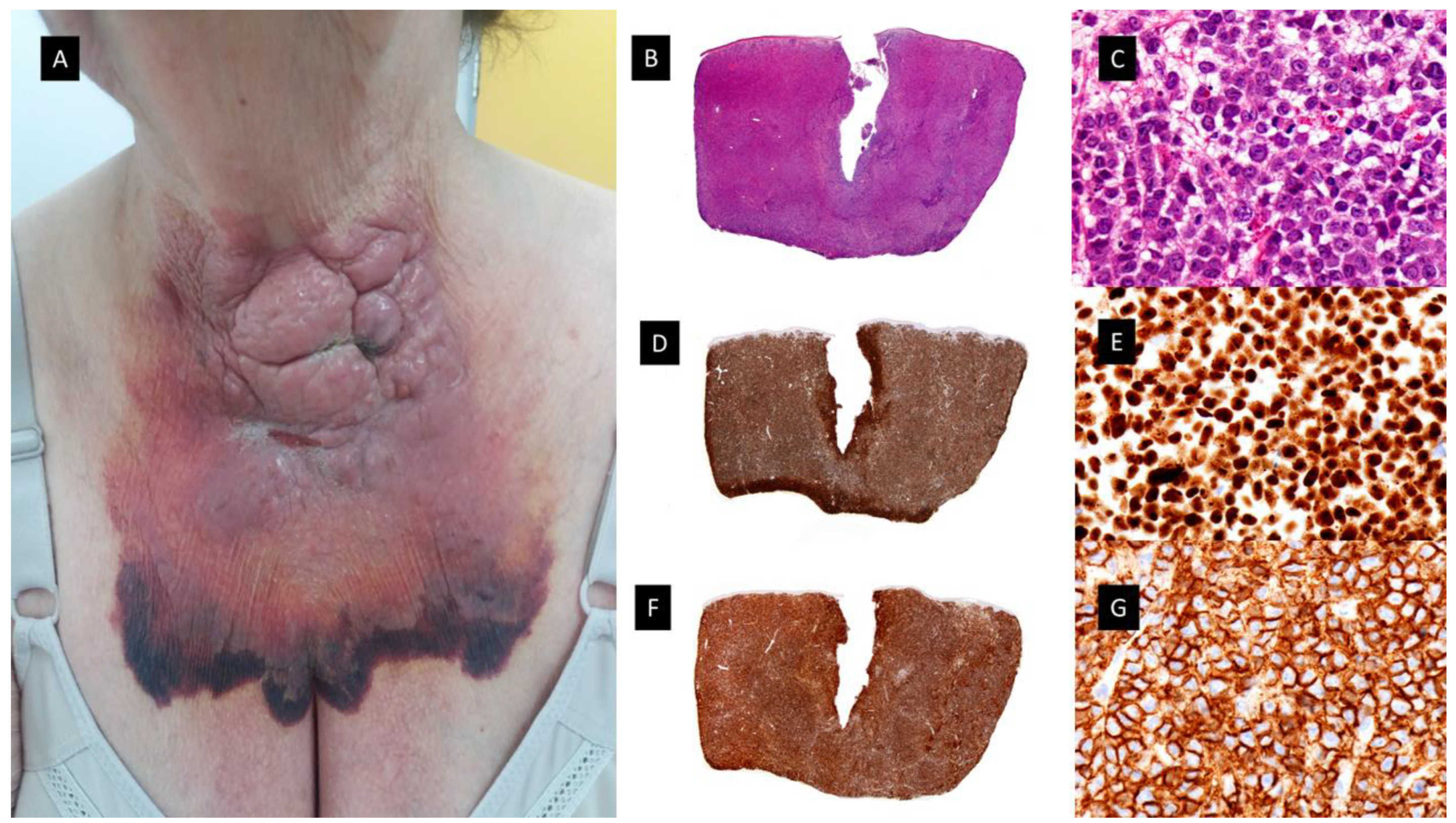
3.2. Blastic Plasmacytoid Dendritic Cell Tumor (see Figure 5)
3.3. Histiocytoid Disorders/Histiocytosis
3.3.1. Histiocytic Sarcoma
3.3.2. Other Histiocytosis
4. Other Cutaneous Disorders in MDS/MPN Patients
4.1. Cutaneous Manifestations Secundary to Microcirculation Abnormalities
4.2. Other Inflammatory/Autoimmune Diseases
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef] [PubMed]
- Orazi, A.; Germing, U. The myelodysplastic/myeloproliferative neoplasms: Myeloproliferative diseases with dysplastic features. Leukemia 2008, 22, 1308–1319. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.; Fenchel, K.; Back, W.; Schulz, A.; Sachse, M.M. Leukemia cutis—Epidemiology, clinical presentation, and differential diagnoses. J. Dtsch. Dermatol. Ges. 2012, 10, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Passet, M.; Lepelletier, C.; Vignon-Pennamen, M.D.; Chasset, F.; Hirsch, P.; Battistella, M.; Duriez, P.; Sicre de Fontbrune, F.; Boissel, N.; Legrand, O.; et al. Next-Generation Sequencing in Myeloid Neoplasm-Associated Sweet’s Syndrome Demonstrates Clonal Relation between Malignant Cells and Skin-Infiltrating Neutrophils. J. Invest. Dermatol. 2020, 140, 1873–1876.e1875. [Google Scholar] [CrossRef] [PubMed]
- Martin de Frémont, G.; Hirsch, P.; Gimenez de Mestral, S.; Moguelet, P.; Ditchi, Y.; Emile, J.F.; Senet, P.; Georgin-Lavialle, S.; Hanslik, T.; Maurier, F.; et al. Myeloid Clonal Infiltrate Identified With Next-Generation Sequencing in Skin Lesions Associated With Myelodysplastic Syndromes and Chronic Myelomonocytic Leukemia: A Case Series. Front. Immunol. 2021, 12, 715053. [Google Scholar] [CrossRef] [PubMed]
- Whittington, C.P.; Ross, C.W.; Ramirez, J.A.; Lowe, L.; Brown, N.; Hristov, A.C. Myelodysplasia Cutis. Arch. Pathol. Lab. Med. 2023. [Google Scholar] [CrossRef]
- Weiss, E.H.; Ko, C.J.; Leung, T.H.; Micheletti, R.G.; Mostaghimi, A.; Ramachandran, S.M.; Rosenbach, M.; Nelson, C.A. Neutrophilic Dermatoses: A Clinical Update. Curr. Dermatol. Rep. 2022, 11, 89–102. [Google Scholar] [CrossRef]
- Sweet, R.D. An Acute Febrile Neutrophilic Dermatosis. Br. J. Dermatol. 1964, 76, 349–356. [Google Scholar] [CrossRef]
- Ferea, C.R.; Mihai, S.N.; Balan, G.; Badescu, M.C.; Tutunaru, D.; Tatu, A.L. Sweet Syndrome Associated with Myelodysplastic Syndrome-A Review of a Multidisciplinary Approach. Life 2023, 13, 809. [Google Scholar] [CrossRef]
- Cohen, P.R. Sweet’s syndrome—A comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J. Rare Dis. 2007, 2, 34. [Google Scholar] [CrossRef] [PubMed]
- Requena, L.; Kutzner, H.; Palmedo, G.; Pascual, M.; Fernández-Herrera, J.; Fraga, J.; García-Díez, A.; Yus, E.S. Histiocytoid Sweet syndrome: A dermal infiltration of immature neutrophilic granulocytes. Arch. Dermatol. 2005, 141, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Alegría-Landa, V.; Rodríguez-Pinilla, S.M.; Santos-Briz, A.; Rodríguez-Peralto, J.L.; Alegre, V.; Cerroni, L.; Kutzner, H.; Requena, L. Clinicopathologic, Immunohistochemical, and Molecular Features of Histiocytoid Sweet Syndrome. JAMA Dermatol. 2017, 153, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Vignon-Pennamen, M.D.; Juillard, C.; Rybojad, M.; Wallach, D.; Daniel, M.T.; Morel, P.; Verola, O.; Janin, A. Chronic recurrent lymphocytic Sweet syndrome as a predictive marker of myelodysplasia: A report of 9 cases. Arch. Dermatol. 2006, 142, 1170–1176. [Google Scholar] [CrossRef] [PubMed]
- Ghoufi, L.; Ortonne, N.; Ingen-Housz-Oro, S.; Barhoumi, W.; Begon, E.; Haioun, C.; Pautas, C.; Beckerich, F.; Robin, C.; Wolkenstein, P.; et al. Histiocytoid Sweet Syndrome Is More Frequently Associated with Myelodysplastic Syndromes Than the Classical Neutrophilic Variant: A Comparative Series of 62 Patients. Medicine 2016, 95, e3033. [Google Scholar] [CrossRef] [PubMed]
- Delaleu, J.; Kim, R.; Zhao, L.P.; de Masson, A.; Vignon-Pennamen, M.D.; Cassius, C.; Ram-Wolff, C.; Bagot, M.; Clappier, E.; Adès, L.; et al. Clinical, pathological, and molecular features of myelodysplasia cutis. Blood 2022, 139, 1251–1253. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, A.; Yanagisawa, H.; Tsunemi, Y.; Kusano, T.; Arai, E.; Tsuchida, T.; Nakamura, K. Normolipemic xanthomatized Sweet’s syndrome: A variant of Sweet’s syndrome with myelodysplastic syndrome. J. Dermatol. 2021, 48, 695–698. [Google Scholar] [CrossRef]
- Sutra-Loubet, C.; Carlotti, A.; Guillemette, J.; Wallach, D. Neutrophilic panniculitis. J. Am. Acad. Dermatol. 2004, 50, 280–285. [Google Scholar] [CrossRef]
- Jagdeo, J.; Campbell, R.; Long, T.; Muglia, J.; Telang, G.; Robinson-Bostom, L. Sweet’s syndrome—Like neutrophilic lobular panniculitis associated with all-trans-retinoic acid chemotherapy in a patient with acute promyelocytic leukemia. J. Am. Acad. Dermatol. 2007, 56, 690–693. [Google Scholar] [CrossRef]
- Chan, M.P.; Duncan, L.M.; Nazarian, R.M. Subcutaneous Sweet syndrome in the setting of myeloid disorders: A case series and review of the literature. J. Am. Acad. Dermatol. 2013, 68, 1006–1015. [Google Scholar] [CrossRef]
- Srisuttiyakorn, C.; Reeve, J.; Reddy, S.; Imaeda, S.; Lazova, R. Subcutaneous histiocytoid Sweet’s syndrome in a patient with myelodysplastic syndrome and acute myeloblastic leukemia. J. Cutan. Pathol. 2014, 41, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cornejo, K.M.; Rork, J.; Rothman, K.; Deng, A. Subcutaneous Histiocytoid Sweet Syndrome in a Patient With Relapsed Acute Myeloblastic Leukemia. Am. J. Dermatopathol. 2018, 40, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Zhang, Q.; Chen, M. Subcutaneous histiocytoid Sweet’s syndrome in a patient associated with myelodysplastic syndrome-refractory anemia. J. Dermatol. 2012, 39, 99–101. [Google Scholar] [CrossRef]
- Beck, D.B.; Ferrada, M.A.; Sikora, K.A.; Ombrello, A.K.; Collins, J.C.; Pei, W.; Balanda, N.; Ross, D.L.; Ospina Cardona, D.; Wu, Z.; et al. Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease. N. Engl. J. Med. 2020, 383, 2628–2638. [Google Scholar] [CrossRef] [PubMed]
- Beck, D.B.; Bodian, D.L.; Shah, V.; Mirshahi, U.L.; Kim, J.; Ding, Y.; Magaziner, S.J.; Strande, N.T.; Cantor, A.; Haley, J.S.; et al. Estimated Prevalence and Clinical Manifestations of UBA1 Variants Associated with VEXAS Syndrome in a Clinical Population. JAMA 2023, 329, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.N.; Young, N.S. Clonality in context: Hematopoietic clones in their marrow environment. Blood 2017, 130, 2363–2372. [Google Scholar] [CrossRef] [PubMed]
- Haines, P.; Pullarkat, S.; Said, J. VEXAS syndrome: Vacuoles in myeloid, erythroid, and lymphoid lineages. Int. J. Lab. Hematol. 2023. [Google Scholar] [CrossRef]
- Djerbi, N.; Zimmermann, K.; Balabanov, S.; Manz, M.G.; Becker, M.O.; Roncador, M. Intra-patient competition of VEXAS syndrome and CML clones. Blood Adv. 2023, 7, 6815–6818. [Google Scholar] [CrossRef]
- Nguyen, J.K.; Routledge, D.; van Der Weyden, C.; Blombery, P.; Angel, C.M.; Johnson, D.; Goh, M.S.; Lee, A. VEXAS syndrome: A dermatological perspective. Australas. J. Dermatol. 2022, 63, 488–492. [Google Scholar] [CrossRef]
- Osio, A.; Battistella, M.; Feugeas, J.P.; Cuccuini, W.; Noguera, M.E.; Petrella, T.; Raffoux, E.; Janin, A.; Pennamen, V. Myelodysplasia Cutis Versus Leukemia Cutis. J. Invest. Dermatol. 2015, 135, 2321–2324. [Google Scholar] [CrossRef]
- Sujobert, P.; Cuccuini, W.; Vignon-Pennamen, D.; Martin-Garcia, N.; Albertini, A.F.; Uzunov, M.; Redjoul, R.; Dombret, H.; Raffoux, E. Evidence of differentiation in myeloid malignancies associated neutrophilic dermatosis: A fluorescent in situ hybridization study of 14 patients. J. Invest. Dermatol. 2013, 133, 1111–1114. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, G.J.; Duve, R.S.; Ackerman, A.B. Interstitial granulomatous dermatitis with cutaneous cords and arthritis: Linear subcutaneous bands in rheumatoid arthritis revisited. Dermatopathol. Pract. Concept. 1995, 1, 3–6. [Google Scholar]
- Chu, P.; Connolly, M.K.; LeBoit, P.E. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch. Dermatol. 1994, 130, 1278–1283. [Google Scholar] [CrossRef] [PubMed]
- Rosenbach, M.; English, J.C., 3rd. Reactive Granulomatous Dermatitis: A Review of Palisaded Neutrophilic and Granulomatous Dermatitis, Interstitial Granulomatous Dermatitis, Interstitial Granulomatous Drug Reaction, and a Proposed Reclassification. Dermatol. Clin. 2015, 33, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Yoneta, K.; Fujimoto, N.; Teramura, K.; Takayama, S.; Tanaka, T. Disseminated granulomatous skin lesions associated with myelodysplastic syndrome treated successfully with tranilast: A case report and review of the literature. Eur. J. Dermatol. 2016, 26, 398–400. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Torres, L.; Santonja, C.; Chamizo, C.; García-Gil, M.F.; Lezcano, V.; Arranz-Sánchez, D.M.; Rodríguez-Pinilla, S.M.; Azaceta, G.; García-García, M. Granulomatous Dermatitis Heralding Myelodisplastic/Myeloproliferative Neoplasms. Neoplastic or Reactive Cells? A Study of 2 Cases. Am. J. Dermatopathol. 2022, 44, 456–460. [Google Scholar] [CrossRef] [PubMed]
- Federmann, B.; Bonzheim, I.; Yazdi, A.S.; Schmidt, J.; Fend, F.; Metzler, G. Generalized palisaded neutrophilic and granulomatous dermatitis-a cutaneous manifestation of chronic myelomonocytic leukemia? A clinical, histopathological, and molecular study of 3 cases. Hum. Pathol. 2017, 64, 198–206. [Google Scholar] [CrossRef]
- Kyriakou, A.; Patsatsi, A.; Papadopoulos, V.; Kioumi, A.; Efstratiou, I.; Lazaridou, E. A case of palisaded neutrophilic granulomatous dermatitis with subsequent development of chronic myelomonocytic leukemia. Clin. Case Rep. 2019, 7, 695–698. [Google Scholar] [CrossRef]
- Enescu, C.D.; Patel, A.; Friedman, B.J. Unique Recognizable Histopathologic Variant of Palisaded Neutrophilic and Granulomatous Dermatitis that Is Associated With SRSF2-Mutated Chronic Myelomonocytic Leukemia: Case Report and Review of the Literature. Am. J. Dermatopathol. 2022, 44, e33–e36. [Google Scholar] [CrossRef]
- Tzankov, A.; Hebeda, K.; Kremer, M.; Leguit, R.; Orazi, A.; van der Walt, J.; Gianelli, U. Plasmacytoid dendritic cell proliferations and neoplasms involving the bone marrow: Summary of the workshop cases submitted to the 18th Meeting of the European Association for Haematopathology (EAHP) organized by the European Bone Marrow Working Group, Basel 2016. Ann. Hematol. 2017, 96, 765–777. [Google Scholar]
- Wang, P.; Feng, Y.; Deng, X.; Liu, S.; Qiang, X.; Gou, Y.; Li, J.; Yang, W.; Peng, X.; Zhang, X. Tumor-forming plasmacytoid dendritic cells in acute myelocytic leukemia: A report of three cases and literature review. Int. J. Clin. Exp. Pathol. 2017, 10, 7285–7291. [Google Scholar] [PubMed]
- Facchetti, F.; Cigognetti, M.; Fisogni, S.; Rossi, G.; Lonardi, S.; Vermi, W. Neoplasms derived from plasmacytoid dendritic cells. Mod. Pathol. 2016, 29, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Orazi, A.; Chiu, R.; O’Malley, D.P.; Czader, M.; Allen, S.L.; An, C.; Vance, G.H. Chronic myelomonocytic leukemia: The role of bone marrow biopsy immunohistology. Mod. Pathol. 2006, 19, 1536–1545. [Google Scholar] [CrossRef] [PubMed]
- Lucas, N.; Duchmann, M.; Rameau, P.; Noël, F.; Michea, P.; Saada, V.; Kosmider, O.; Pierron, G.; Fernandez-Zapico, M.E.; Howard, M.T.; et al. Biology and prognostic impact of clonal plasmacytoid dendritic cells in chronic myelomonocytic leukemia. Leukemia 2019, 33, 2466–2480. [Google Scholar] [CrossRef] [PubMed]
- Vitte, F.; Fabiani, B.; Bénet, C.; Dalac, S.; Balme, B.; Delattre, C.; Vergier, B.; Beylot-Barry, M.; Vignon-Pennamen, D.; Ortonne, N.; et al. Specific skin lesions in chronic myelomonocytic leukemia: A spectrum of myelomonocytic and dendritic cell proliferations: A study of 42 cases. Am. J. Surg. Pathol. 2012, 36, 1302–1316. [Google Scholar] [CrossRef] [PubMed]
- Machan, S.; Alonso-Dominguez, J.M.; Sánchez García, F.J.; Nieves Salgado, R.; Soto, C.; Castro, Y.; Pajares, R.; Manso, R.; Santonja, C.; Serrano Del Castillo, C.; et al. Plasmacytoid Dendritic Cell Dermatosis Associated to Myeloproliferative/Myelodysplastic Neoplasms. Am. J. Surg. Pathol. 2022, 46, 1623–1632. [Google Scholar] [CrossRef] [PubMed]
- Dargent, J.L.; Henne, S.; Pranger, D.; Balzarini, P.; Sartenaer, D.; Bulliard, G.; Rack, K.; Facchetti, F. Tumor-forming plasmacytoid dendritic cells associated with myeloid neoplasms. Report of a peculiar case with histopathologic features masquerading as lupus erythematosus. J. Cutan. Pathol. 2016, 43, 280–286. [Google Scholar] [CrossRef]
- Jian, J.; Qiao, Y.; Li, Y.; Guo, Y.; Ma, H.; Liu, B. Mutations in chronic myelomonocytic leukemia and their prognostic relevance. Clin. Transl. Oncol. 2021, 23, 1731–1742. [Google Scholar] [CrossRef]
- Wang, N.; Wang, F.; Shan, N.; Sui, X.; Xu, H. IDH1 Mutation Is an Independent Inferior Prognostic Indicator for Patients with Myelodysplastic Syndromes. Acta Haematol. 2017, 138, 143–151. [Google Scholar] [CrossRef]
- Brazão, C.; Mancha, D.; Antunes-Duarte, S.; Kempf, W.; Soares-de-Almeida, L. Chilblain-Like Eruption Unveiling Cutaneous Aleukemic Relapse of Acute Myeloid Leukemia. Am. J. Dermatopathol. 2023, 45, 847–851. [Google Scholar] [CrossRef]
- Yazawa, H.; Saga, K.; Omori, F.; Jimbow, K.; Sasagawa, Y. The chilblain-like eruption as a diagnostic clue to the blast crisis of chronic myelocytic leukemia. J. Am. Acad. Dermatol. 2004, 50 (Suppl. S2), S42–S44. [Google Scholar] [CrossRef] [PubMed]
- Affleck, A.G.; Ravenscroft, J.C.; Leach, I.H. Chilblain-like leukemia cutis. Pediatr. Dermatol. 2007, 24, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Faria, C.; Tzankov, A. Progression in Myeloid Neoplasms: Beyond the Myeloblast. Pathobiology 2023, 1–21. [Google Scholar] [CrossRef]
- Bénet, C.; Gomez, A.; Aguilar, C.; Delattre, C.; Vergier, B.; Beylot-Barry, M.; Fraitag, S.; Carlotti, A.; Dechelotte, P.; Hospital, V.; et al. Histologic and immunohistologic characterization of skin localization of myeloid disorders: A study of 173 cases. Am. J. Clin. Pathol. 2011, 135, 278–290. [Google Scholar] [CrossRef]
- Cho-Vega, J.H.; Medeiros, L.J.; Prieto, V.G.; Vega, F. Leukemia cutis. Am. J. Clin. Pathol. 2008, 129, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Kaddu, S.; Zenahlik, P.; Beham-Schmid, C.; Kerl, H.; Cerroni, L. Specific cutaneous infiltrates in patients with myelogenous leukemia: A clinicopathologic study of 26 patients with assessment of diagnostic criteria. J. Am. Acad. Dermatol. 1999, 40 Pt 1, 966–978. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.A.; Bennett, J.M.; Liu, J.J.; Komrokji, R.S.; Lancet, J.E.; Naghashpour, M.; Messina, J.L.; List, A.F.; Moscinski, L.C.; Zhang, L. Cutaneous manifestations in CMML: Indication of disease acceleration or transformation to AML and review of the literature. Leuk. Res. 2012, 36, 72–80. [Google Scholar] [CrossRef]
- Ohno, S.; Yokoo, T.; Ohta, M.; Yamamoto, M.; Danno, K.; Hamato, N.; Tomii, K.; Ohno, Y.; Kobashi, Y. Aleukemic leukemia cutis. J. Am. Acad. Dermatol. 1990, 22 Pt 2, 374–377. [Google Scholar] [CrossRef]
- Heskel, N.S.; White, C.R.; Fryberger, S.; Neerhout, R.C.; Spraker, M.; Hanifin, J.M. Aleukemic leukemia cutis: Juvenile chronic granulocytic leukemia presenting with figurate cutaneous lesions. J. Am. Acad. Dermatol. 1983, 9, 423–427. [Google Scholar] [CrossRef]
- Gil-Mateo, M.P.; Miquel, F.J.; Piris, M.A.; Sánchez, M.; Martin-Aragonés, G. Aleukemic "leukemia cutis" of monocytic lineage. J. Am. Acad. Dermatol. 1997, 36 Pt 2, 837–840. [Google Scholar] [CrossRef]
- Aboutalebi, A.; Korman, J.B.; Sohani, A.R.; Hasserjian, R.P.; Louissaint, A., Jr.; Le, L.; Kraft, S.; Duncan, L.M.; Nazarian, R.M. Aleukemic cutaneous myeloid sarcoma. J. Cutan. Pathol. 2013, 40, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Iliadis, A.; Koletsa, T.; Georgiou, E.; Patsatsi, A.; Sotiriadis, D.; Kostopoulos, I. Bilateral Aleukemic Myeloid Sarcoma of the Eyelids With Indolent Course. Am. J. Dermatopathol. 2016, 38, 312–314. [Google Scholar] [CrossRef] [PubMed]
- Dijkman, R.; van Doorn, R.; Szuhai, K.; Willemze, R.; Vermeer, M.H.; Tensen, C.P. Gene-expression profiling and array-based CGH classify CD4+CD56+ hematodermic neoplasm and cutaneous myelomonocytic leukemia as distinct disease entities. Blood 2007, 109, 1720–1727. [Google Scholar] [CrossRef] [PubMed]
- Khanlari, M.; Yin, C.C.; Takahashi, K.; Lachowiez, C.; Tang, G.; Loghavi, S.; Bah, I.; Wang, W.; Konoplev, S.; Medeiros, L.J.; et al. Bone marrow clonal hematopoiesis is highly prevalent in blastic plasmacytoid dendritic cell neoplasm and frequently sharing a clonal origin in elderly patients. Leukemia 2022, 36, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Hiramoto, N.; Mori, T.; Yamashita, D.; Tai, Y.; Yamamoto, R.; Nishikubo, M.; Maruoka, H.; Sakamoto, K.; Takeuchi, K.; et al. Coincidence of cutaneous blastic plasmacytoid dendritic cell neoplasm and myelodysplastic syndrome derived from clonal hematopoiesis. Blood Cancer J. 2023, 13, 119. [Google Scholar] [CrossRef] [PubMed]
- Emile, J.F.; Abla, O.; Fraitag, S.; Horne, A.; Haroche, J.; Donadieu, J.; Requena-Caballero, L.; Jordan, M.B.; Abdel-Wahab, O.; Allen, C.E.; et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood 2016, 127, 2672–2681. [Google Scholar] [CrossRef] [PubMed]
- Ansari, J.; Naqash, A.R.; Munker, R.; El-Osta, H.; Master, S.; Cotelingam, J.D.; Griffiths, E.; Greer, A.H.; Yin, H.; Peddi, P.; et al. Histiocytic sarcoma as a secondary malignancy: Pathobiology, diagnosis, and treatment. Eur. J. Haematol. 2016, 97, 9–16. [Google Scholar] [CrossRef]
- Zhao, J.; Niu, X.; Wang, Z.; Lu, H.; Lin, X.; Lu, Q. Histiocytic sarcoma combined with acute monocytic leukemia: A case report. Diagn. Pathol. 2015, 10, 110. [Google Scholar] [CrossRef]
- Mori, M.; Matsushita, A.; Takiuchi, Y.; Arima, H.; Nagano, S.; Shimoji, S.; Kimura, T.; Inoue, D.; Tabata, S.; Yanagita, S.; et al. Histiocytic sarcoma and underlying chronic myelomonocytic leukemia: A proposal for the developmental classification of histiocytic sarcoma. Int. J. Hematol. 2010, 92, 168–173. [Google Scholar] [CrossRef]
- Pérez-Sáenz, M.A.; Rodriguez-Pinilla, S.M.; Salgado, R.N.; Carvajal, N.; Serrano, C.; Soto, C.; Serrano, J.; Atance, M.; López-Lorenzo, J.L.; Requena, L.; et al. Three monocytic neoplasms in a single patient. Leuk. Lymphoma 2020, 61, 2523–2526. [Google Scholar] [CrossRef]
- Papo, M.; Diamond, E.L.; Cohen-Aubart, F.; Emile, J.F.; Roos-Weil, D.; Gupta, N.; Durham, B.H.; Ozkaya, N.; Dogan, A.; Ulaner, G.A.; et al. High prevalence of myeloid neoplasms in adults with non-Langerhans cell histiocytosis. Blood 2017, 130, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Kemps, P.G.; Hebeda, K.M.; Pals, S.T.; Verdijk, R.M.; Lam, K.H.; Bruggink, A.H.; de Lil, H.S.; Ruiterkamp, B.; de Heer, K.; van Laar, J.A.; et al. Spectrum of histiocytic neoplasms associated with diverse haematological malignancies bearing the same oncogenic mutation. J. Pathol. Clin. Res. 2021, 7, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Miralles, E.S.; Escribano, L.; Bellas, C.; Núñez, M.; Ledo, A. Cutaneous xanthomatous tumours as an expression of chronic myelomonocytic leukaemia? Clin. Exp. Dermatol. 1996, 21, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, P.; Chasset, F.; Moguelet, P.; Abisror, N.; Itzykson, R.; Bouaziz, J.D.; Hirsch, P.; Barbaud, A.; Haroche, J.; Mekinian, A.; et al. Erdheim-Chester disease associated with chronic myelomonocytic leukemia harboring the same clonal mutation. Haematologica 2019, 104, e530–e533. [Google Scholar] [CrossRef] [PubMed]
- Fiegl, A.; Dirnhofer, S.; Juskevicius, D.; Zagrapan, B.; Dertinger, S.; Bösl, A.; Milos, S.; Brunner, J.; Bertolini, F.; Offner, F.A. Testicular Rosai-Dorfman disease clonally related to CMML—Case report and literature review. Pathol. Res. Pract. 2023, 247, 154548. [Google Scholar] [CrossRef] [PubMed]
- Kiavash, K.; Malone, J.C. Langerhans Cell Histiocytosis Associated With Underlying Hematolymphoid Disorders in Adults: Report of 2 Cases and Review of the Literature. Am. J. Dermatopathol. 2018, 40, 588–593. [Google Scholar] [CrossRef]
- Billings, S.D.; Hans, C.P.; Schapiro, B.L.; Martin, R.W., 3rd; Fivenson, D.; Fruland, J.E.; Moores, W.B.; Cotton, J. Langerhans cell histiocytosis associated with myelodysplastic syndrome in adults. J. Cutan. Pathol. 2006, 33, 171–174. [Google Scholar] [CrossRef]
- Durham, B.H.; Roos-Weil, D.; Baillou, C.; Cohen-Aubart, F.; Yoshimi, A.; Miyara, M.; Papo, M.; Hélias-Rodzewicz, Z.; Terrones, N.; Ozkaya, N.; et al. Functional evidence for derivation of systemic histiocytic neoplasms from hematopoietic stem/progenitor cells. Blood 2017, 130, 176–180. [Google Scholar] [CrossRef]
- Milne, P.; Bigley, V.; Bacon, C.M.; Néel, A.; McGovern, N.; Bomken, S.; Haniffa, M.; Diamond, E.L.; Durham, B.H.; Visser, J.; et al. Hematopoietic origin of Langerhans cell histiocytosis and Erdheim-Chester disease in adults. Blood 2017, 130, 167–175. [Google Scholar] [CrossRef]
- Marks, R.; Lim, C.C.; Borrie, P.F. A perniotic syndrome with monocytosis and neutropenia--a possible association with a preleukaemic state. Br. J. Dermatol. 1969, 81, 327–332. [Google Scholar] [CrossRef]
- Kelly, J.W.; Dowling, J.P. Pernio. A possible association with chronic myelomonocytic leukemia. Arch. Dermatol. 1985, 121, 1048–1052. [Google Scholar] [CrossRef] [PubMed]
- Dreno, B.; Gandon, P.; Bureau, B.; Milpied, N.; Barrière, H. Skin lesions from hypersensitivity to cold during chronic myelomonocytic leukaemia. Br. J. Dermatol. 1986, 115, 607–609. [Google Scholar] [CrossRef] [PubMed]
- Nazzaro, G.; Genovese, G.; Marzano, A.V. Idiopathic chilblains in myelomonocytic leukemia: Not a simple association. Int. J. Dermatol. 2018, 57, 596–598. [Google Scholar] [CrossRef] [PubMed]
- Pielasinski, U.; Haro, R.; Santonja, C.; Kutzner, H.; Requena, L. Essential thrombocythemia presenting as localized livedo reticularis. Am. J. Dermatopathol. 2013, 35, e22–e25. [Google Scholar] [CrossRef] [PubMed]
- Lapeña Casado, A.; Santonja, C.; García-García, M.; Olave-Rubio, M.T.; Lorda-Espés, M.; Fink-Puches, R.; Cerroni, L.; Prieto-Torres, L. Essential thrombocythemia manifesting as livedoid and purpuric skin lesions: Report of two cases and literature review. J. Cutan. Pathol. 2023, 50, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Saif, M.W.; Hopkins, J.L.; Gore, S.D. Autoimmune phenomena in patients with myelodysplastic syndromes and chronic myelomonocytic leukemia. Leuk. Lymphoma 2002, 43, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Horiuchi, M.; Ueda, H.; Hagihara, K.; Kanashima, H.; Nakao, T.; Hirata, C.; Inoue, T.; Yamane, T. Cutaneous extramedullary hematopoiesis associated with myelodysplastic/myeloproliferative neoplasm, unclassifiable. Rinsho Ketsueki 2015, 56, 911–914. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prieto-Torres, L.; Requena, L.; Rodríguez-Pinilla, S.M. Clinical, Histopathological and Molecular Spectrum of Cutaneous Lesions in Myelodysplastic Syndrome and Myeloproliferative Neoplasms (MDS/MPN): An Integrative Review. Cancers 2023, 15, 5888. https://doi.org/10.3390/cancers15245888
Prieto-Torres L, Requena L, Rodríguez-Pinilla SM. Clinical, Histopathological and Molecular Spectrum of Cutaneous Lesions in Myelodysplastic Syndrome and Myeloproliferative Neoplasms (MDS/MPN): An Integrative Review. Cancers. 2023; 15(24):5888. https://doi.org/10.3390/cancers15245888
Chicago/Turabian StylePrieto-Torres, Lucía, Luis Requena, and Socorro Maria Rodríguez-Pinilla. 2023. "Clinical, Histopathological and Molecular Spectrum of Cutaneous Lesions in Myelodysplastic Syndrome and Myeloproliferative Neoplasms (MDS/MPN): An Integrative Review" Cancers 15, no. 24: 5888. https://doi.org/10.3390/cancers15245888
APA StylePrieto-Torres, L., Requena, L., & Rodríguez-Pinilla, S. M. (2023). Clinical, Histopathological and Molecular Spectrum of Cutaneous Lesions in Myelodysplastic Syndrome and Myeloproliferative Neoplasms (MDS/MPN): An Integrative Review. Cancers, 15(24), 5888. https://doi.org/10.3390/cancers15245888