

Immune Microenvironment and Immunotherapies for Diffuse Intrinsic Pontine Glioma

Abstract
:Simple Summary
Abstract
1. Introduction
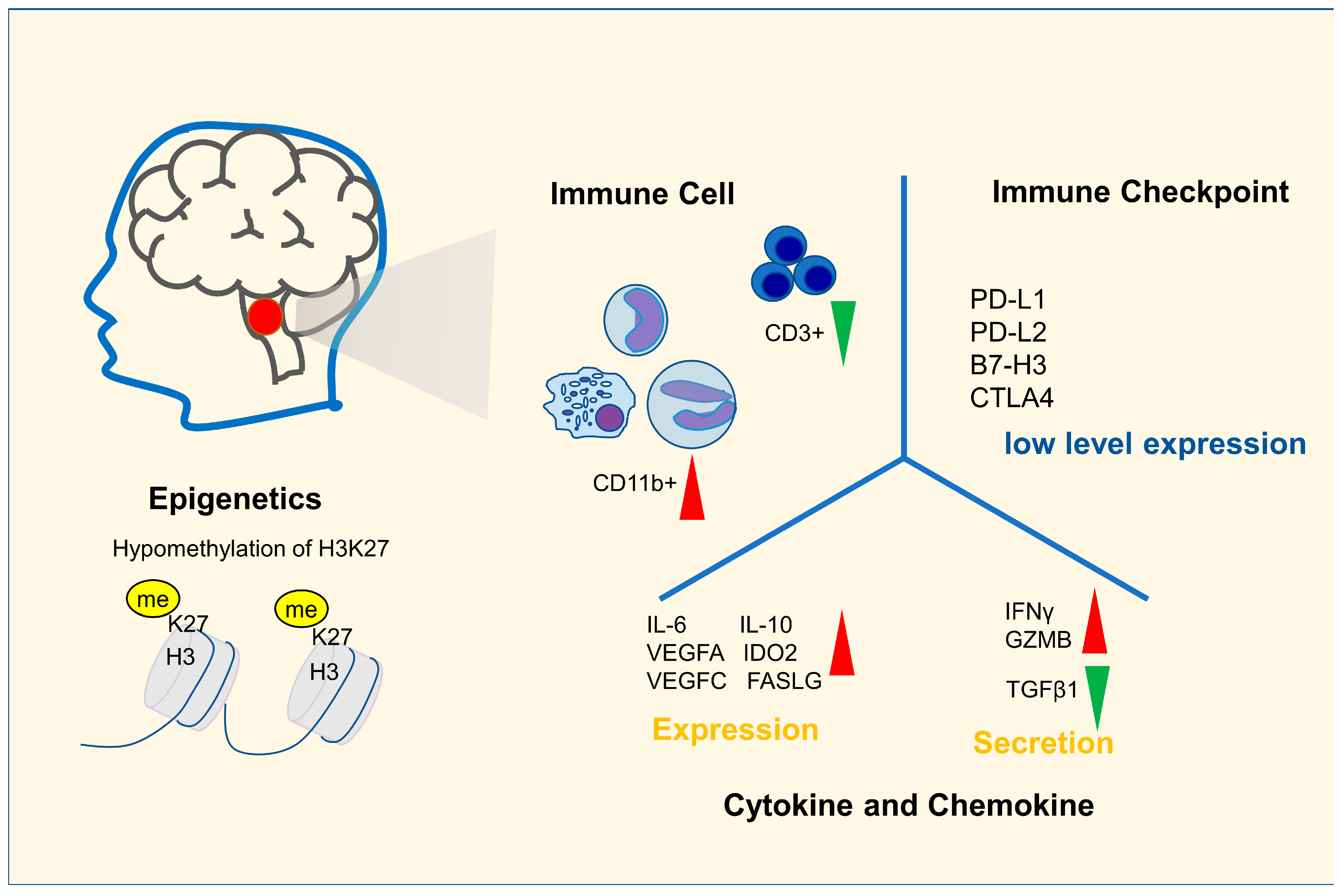
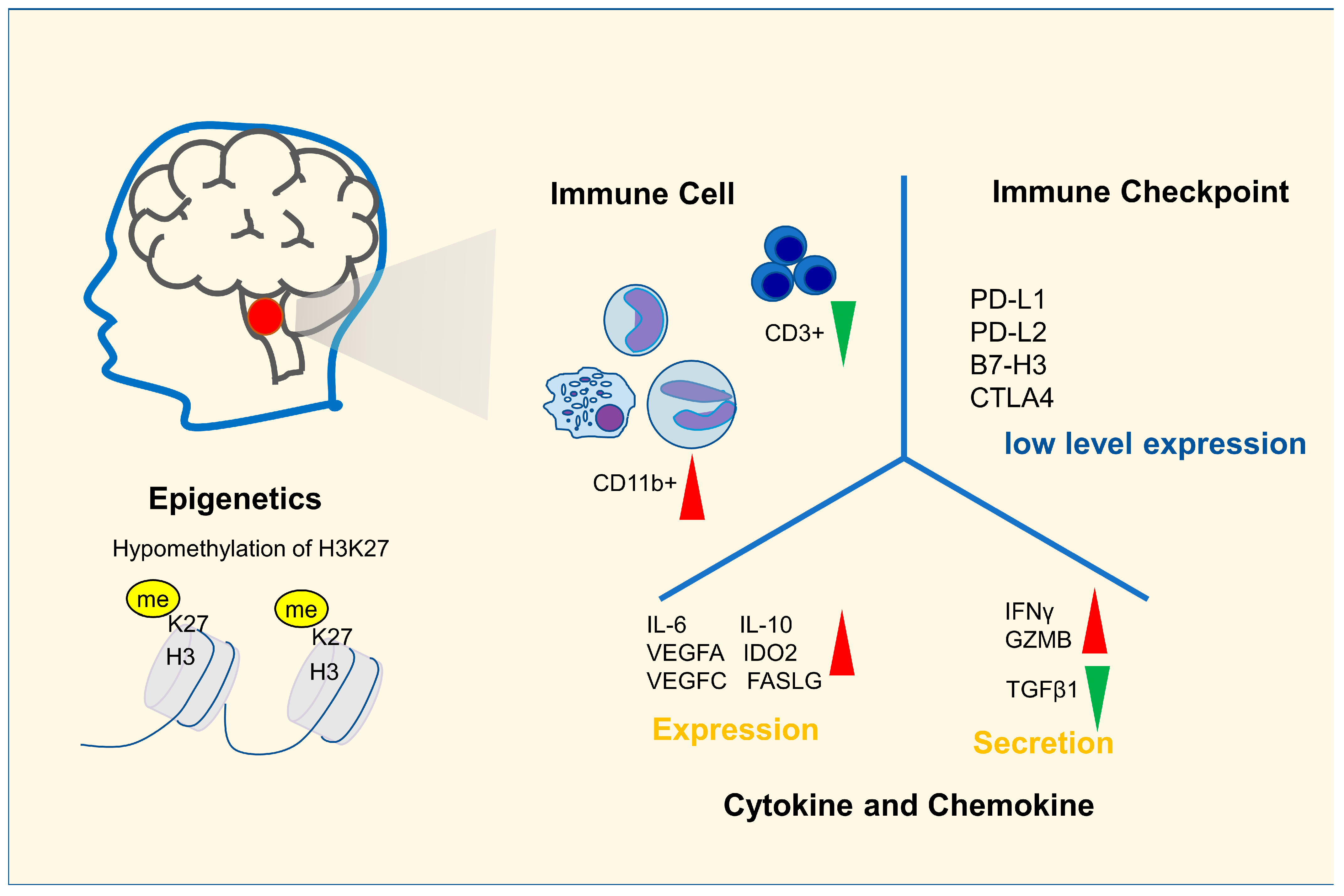
2. Immune Characteristics of DIPG
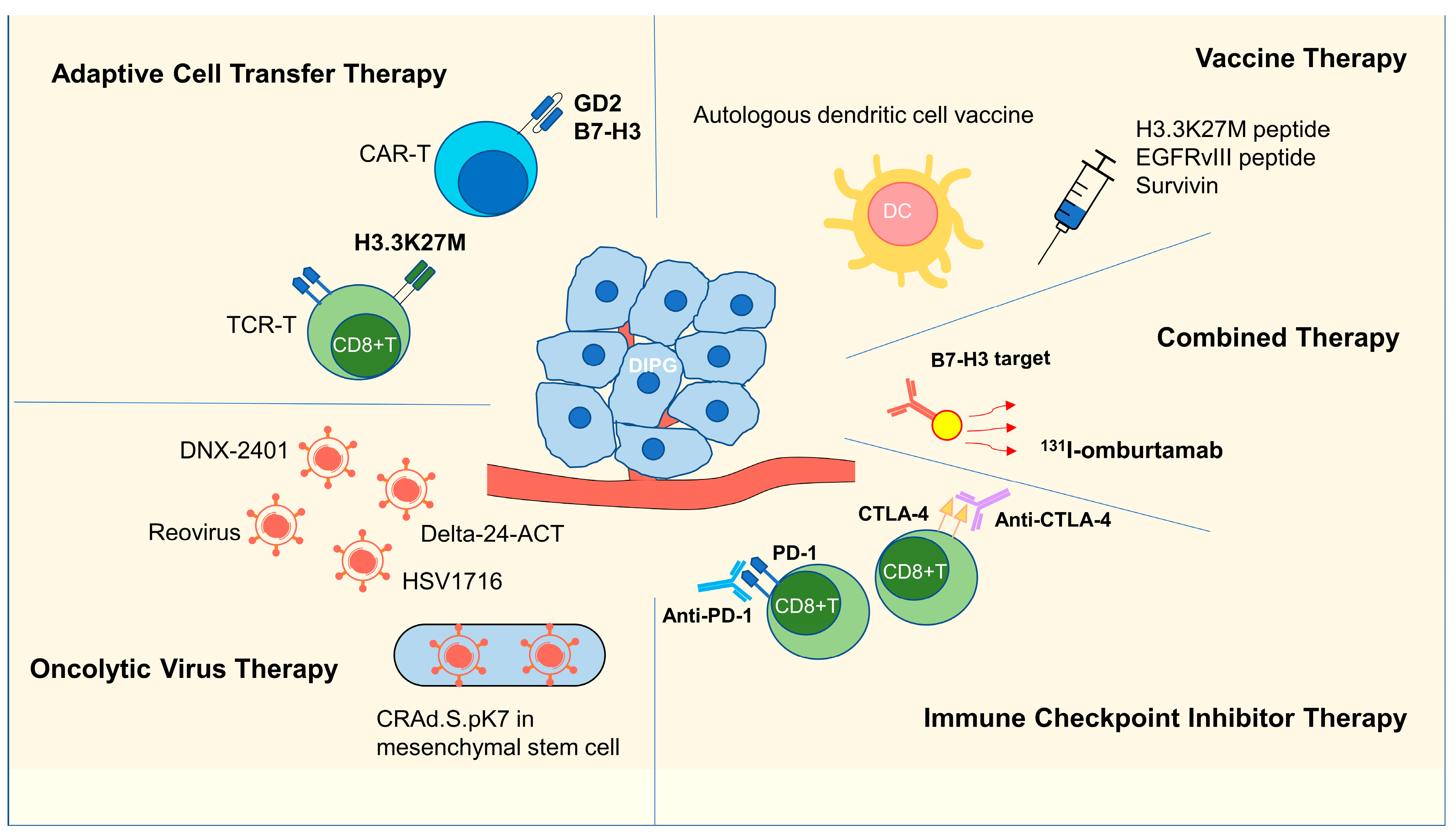
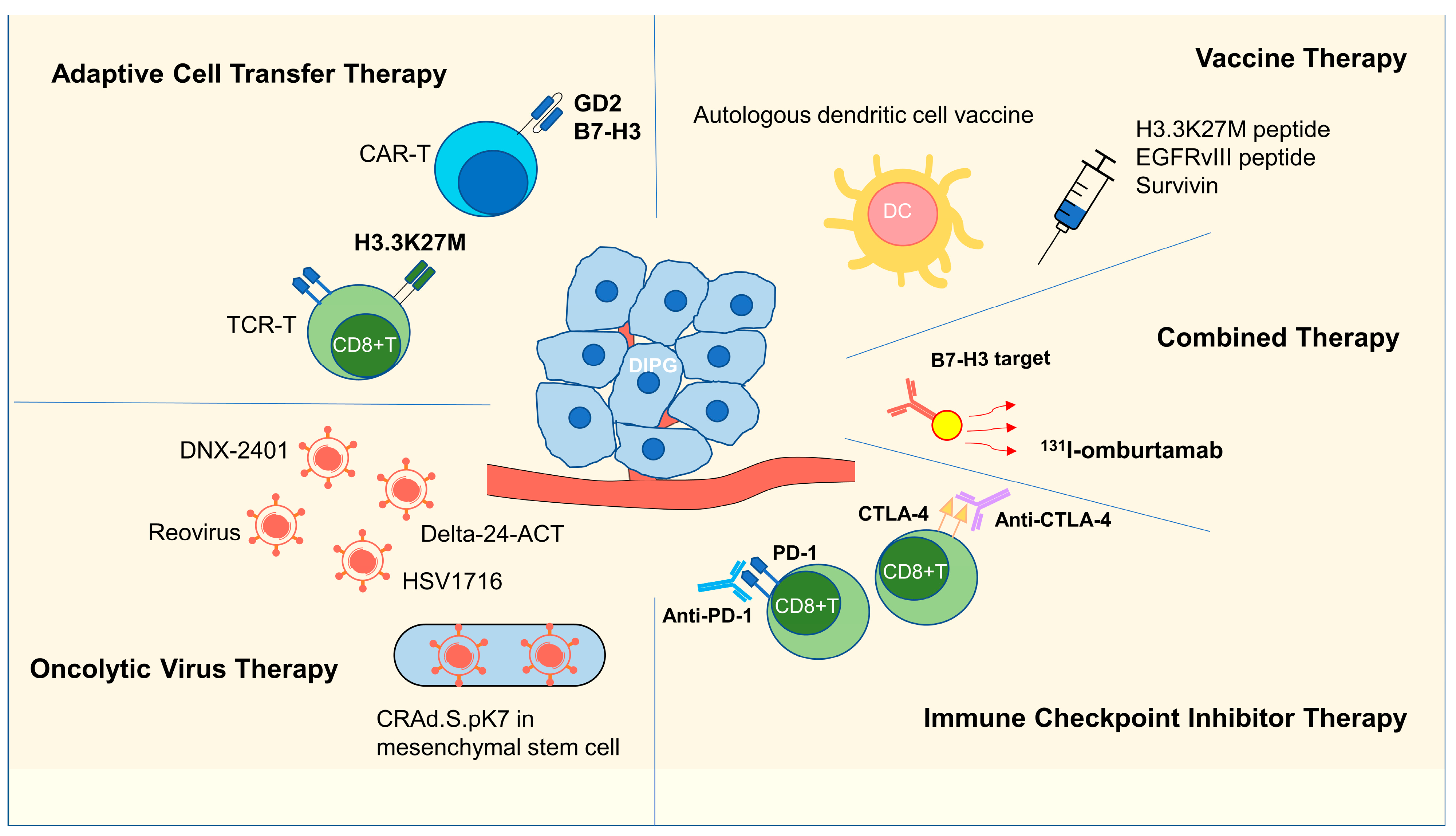
3. Immunotherapy Research in DIPG
3.1. Adoptive Cell Transfer Therapy
3.1.1. CAR-T Therapy
3.1.2. TCR-T Therapy
3.2. Vaccine Therapy
3.2.1. H3K27M Peptide Vaccine
3.2.2. EGFRvIII and Survivin Vaccines
3.2.3. DC Vaccines
3.3. Oncolytic Virus Therapy
3.4. ICI Therapy
3.5. Combination Therapy
3.6. Summary of Immunotherapy Research on DIPG
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ostrom, Q.T.; Fahmideh, M.A.; Cote, D.J.; Muskens, I.S.; Schraw, J.M.; Scheurer, M.E.; Bondy, M.L. Risk factors for childhood and adult primary brain tumors. Neuro Oncol. 2019, 21, 1357–1375. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Luther, N.; Ibrahim, G.M.; Hawkins, C.; Vibhakar, R.; Handler, M.H.; Souweidane, M.M. B7-H3, a potential therapeutic target, is expressed in diffuse intrinsic pontine glioma. J. Neurooncol. 2013, 111, 257–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimm, S.A.; Chamberlain, M.C. Brainstem glioma: A review. Curr. Neurol. Neurosci. Rep. 2013, 13, 346. [Google Scholar] [CrossRef] [PubMed]
- Warren, K.E. Diffuse intrinsic pontine glioma: Poised for progress. Front. Oncol. 2012, 2, 205. [Google Scholar] [CrossRef] [Green Version]
- Perrone, M.G.; Ruggiero, A.; Centonze, A.; Carrieri, A.; Ferorelli, S.; Scilimati, A. Diffuse Intrinsic Pontine Glioma (DIPG): Breakthrough and Clinical Perspective. Curr. Med. Chem. 2021, 28, 3287–3317. [Google Scholar] [CrossRef]
- Chan, K.-M.; Fang, D.; Gan, H.; Hashizume, R.; Yu, C.; Schroeder, M.; Gupta, N.; Mueller, S.; James, C.D.; Jenkins, R.; et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013, 27, 985–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persson, M.L.; Douglas, A.M.; Alvaro, F.; Faridi, P.; Larsen, M.R.; Alonso, M.M.; Vitanza, N.A.; Dun, M.D. The intrinsic and microenvironmental features of diffuse midline glioma; implications for the development of effective immunotherapeutic treatment strategies. Neuro Oncol. 2022, 24, 1408–1422. [Google Scholar] [CrossRef]
- Yu, J.-R.; LeRoy, G.; Bready, D.; Frenster, J.D.; Saldaña-Meyer, R.; Jin, Y.; Descostes, N.; Stafford, J.M.; Placantonakis, D.G.; Reinberg, D. The H3K36me2 writer-reader dependency in H3K27M-DIPG. Sci. Adv. 2021, 7, eabg7444. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.-Y.; Jones, D.T.W.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.-A.K.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- WHO Classification of Tumours Editorial Board (Ed.) Central Nervous System Tumours WHO Classification of Tumours; IARC Press: Lyon, France, 2021. [Google Scholar]
- Wiese, M.; Hamdan, F.H.; Kubiak, K.; Diederichs, C.; Gielen, G.H.; Nussbaumer, G.; Carcaboso, A.M.; Hulleman, E.; Johnsen, S.A.; Kramm, C.M. Combined treatment with CBP and BET inhibitors reverses inadvertent activation of detrimental super enhancer programs in DIPG cells. Cell Death Dis. 2020, 11, 673. [Google Scholar] [CrossRef]
- Khuong-Quang, D.-A.; Buczkowicz, P.; Rakopoulos, P.; Liu, X.-Y.; Fontebasso, A.M.; Bouffet, E.; Bartels, U.; Albrecht, S.; Schwartzentruber, J.; Letourneau, L.; et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012, 124, 439–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell. Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M.; et al. Integrated Molecular Meta-Analysis of 1000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 32, 520–537.e525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, C.P.; Wang, R.; Figueroa, M.; Zhang, S.; Wang, L.; Chandra, J. Computational immune infiltration analysis of pediatric high-grade gliomas (pHGGs) reveals differences in immunosuppression and prognosis by tumor location Computational and Systems. Oncology 2021, 1, e1016. [Google Scholar] [CrossRef]
- Lieberman, N.A.P.; DeGolier, K.; Kovar, H.M.; Davis, A.; Hoglund, V.; Stevens, J.; Winter, C.; Deutsch, G.; Furlan, S.N.; Vitanza, N.A.; et al. Characterization of the immune microenvironment of diffuse intrinsic pontine glioma: Implications for development of immunotherapy. Neuro Oncol. 2019, 21, 83–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, G.L.; Nagaraja, S.; Filbin, M.G.; Suvà, M.L.; Vogel, H.; Monje, M. Non-inflammatory tumor microenvironment of diffuse intrinsic pontine glioma. Acta Neuropathol. Commun. 2018, 6, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bockmayr, M.; Klauschen, F.; Maire, C.L.; Rutkowski, S.; Westphal, M.; Lamszus, K.; Schüller, U.; Mohme, M. Immunologic Profiling of Mutational and Transcriptional Subgroups in Pediatric and Adult High-Grade Gliomas. Cancer Immunol. Res. 2019, 7, 1401–1411. [Google Scholar] [CrossRef]
- Zhu, X.; Lazow, M.A.; Schafer, A.; Bartlett, A.; Kumar, S.S.; Mishra, D.K.; Dexheimer, P.; DeWire, M.; Fuller, C.; Leach, J.L.; et al. A pilot radiogenomic study of DIPG reveals distinct subgroups with unique clinical trajectories and therapeutic targets. Acta Neuropathol. Commun. 2021, 9, 14. [Google Scholar] [CrossRef]
- Chheda, Z.S.; Kohanbash, G.; Okada, K.; Jahan, N.; Sidney, J.; Pecoraro, M.; Yang, X.; Carrera, D.A.; Downey, K.M.; Shrivastav, S.; et al. Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J. Exp. Med. 2018, 215, 141–157. [Google Scholar] [CrossRef] [Green Version]
- Brudno, J.N.; Kochenderfer, J.N. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev 2019, 34, 45–55. [Google Scholar] [CrossRef]
- Mount, C.W.; Majzner, R.G.; Sundaresh, S.; Arnold, E.P.; Kadapakkam, M.; Haile, S.; Labanieh, L.; Hulleman, E.; Woo, P.J.; Rietberg, S.P.; et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M diffuse midline gliomas. Nat. Med. 2018, 24, 572–579. [Google Scholar] [CrossRef] [PubMed]
- de Billy, E.; Pellegrino, M.; Orlando, D.; Pericoli, G.; Ferretti, R.; Businaro, P.; Ajmone-Cat, M.A.; Rossi, S.; Petrilli, L.L.; Maestro, N.; et al. Dual IGF1R/IR inhibitors in combination with GD2-CAR T-cells display a potent anti-tumor activity in diffuse midline glioma H3K27M-mutant. Neuro Oncol. 2021, 24, 1150–1163. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Ramakrishna, S.; Yeom, K.W.; Patel, S.; Chinnasamy, H.; Schultz, L.M.; Richards, R.M.; Jiang, L.; Barsan, V.; Mancusi, R.; et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature 2022, 603, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Franson, A.; Koschmann, C. Enhancing GD2 CAR-T Therapy with IGF1R Blockade: Are DIPG CAR-Ts ready for combinatorial therapy? Neuro Oncol. 2022, 24. [Google Scholar] [CrossRef] [Green Version]
- Majzner, R.G.; Ramakrishna, S.; Mochizuki, A.; Patel, S.; Chinnasamy, H.; Yeom, K.; Schultz, L.; Richards, R.; Campen, C.; Reschke, A.; et al. Abstract CT031: GD2 CAR T cells mediate clinical activity and manageable toxicity in children and young adults with DIPG and H3K27M-mutated diffuse midline gliomas. Cancer Res. 2021, 81, CT031. [Google Scholar] [CrossRef]
- Kramer, K.; Kushner, B.H.; Modak, S.; Pandit-Taskar, N.; Smith-Jones, P.; Zanzonico, P.; Humm, J.L.; Xu, H.; Wolden, S.L.; Souweidane, M.M.; et al. Compartmental intrathecal radioimmunotherapy: Results for treatment for metastatic CNS neuroblastoma. J. Neurooncol. 2010, 97, 409–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, T.; Bunse, L.; Pusch, S.; Sahm, F.; Wiestler, B.; Quandt, J.; Menn, O.; Osswald, M.; Oezen, I.; Ott, M.; et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 2014, 512, 324–327. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Thorne, A.H.; Zanca, C.; Furnari, F. Epidermal growth factor receptor targeting and challenges in glioblastoma. Neuro Oncol. 2016, 18, 914–918. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar]
- Buczkowicz, P.; Hoeman, C.; Rakopoulos, P.; Pajovic, S.; Letourneau, L.; Dzamba, M.; Morrison, A.; Lewis, P.; Bouffet, E.; Bartels, U.; et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat. Genet. 2014, 46, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.A.; Wood, M.D.; Tihan, T.; Bollen, A.W.; Gupta, N.; Phillips, J.J.J.; Perry, A. Diffuse Midline Gliomas with Histone H3-K27M Mutation: A Series of 47 Cases Assessing the Spectrum of Morphologic Variation and Associated Genetic Alterations. Brain Pathol. 2016, 26, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Ochs, K.; Ott, M.; Bunse, T.; Sahm, F.; Bunse, L.; Deumelandt, K.; Sonner, J.K.; Keil, M.; von Deimling, A.; Wick, W.; et al. K27M-mutant histone-3 as a novel target for glioma immunotherapy. Oncoimmunology 2017, 6, e1328340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, S.; Taitt, J.M.; Villanueva-Meyer, J.E.; Bonner, E.R.; Nejo, T.; Lulla, R.R.; Goldman, S.; Banerjee, A.; Chi, S.N.; Whipple, N.S.; et al. Mass cytometry detects H3.3K27M-specific vaccine responses in diffuse midline glioma. J. Clin. Invest. 2020, 130, 6325–6337. [Google Scholar] [CrossRef] [PubMed]
- Aldape, K.D.; Ballman, K.; Furth, A.; Buckner, J.C.; Giannini, C.; Burger, P.C.; Scheithauer, B.W.; Jenkins, R.B.; James, C.D. Immunohistochemical detection of EGFRvIII in high malignancy grade astrocytomas and evaluation of prognostic significance. J. Neuropathol. Exp. Neurol. 2004, 63, 700–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Mitra, S.S.; Monje, M.; Henrich, K.N.; Bangs, C.D.; Nitta, R.T.; Wong, A.J. Expression of epidermal growth factor variant III (EGFRvIII) in pediatric diffuse intrinsic pontine gliomas. J. Neurooncol. 2012, 108, 395–402. [Google Scholar] [CrossRef]
- Dunn-Pirio, A.M.; Vlahovic, G. Immunotherapy approaches in the treatment of malignant brain tumors. Cancer 2017, 123, 734–750. [Google Scholar] [CrossRef]
- Sampson, J.H.; Archer, G.E.; Mitchell, D.A.; Heimberger, A.B.; Herndon, J.E.; Lally-Goss, D.; McGehee-Norman, S.; Paolino, A.; Reardon, D.A.; Friedman, A.H.; et al. An epidermal growth factor receptor variant III-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol. Cancer Ther. 2009, 8, 2773–2779. [Google Scholar] [CrossRef] [Green Version]
- Schuster, J.; Lai, R.K.; Recht, L.D.; Reardon, D.A.; Paleologos, N.A.; Groves, M.D.; Mrugala, M.M.; Jensen, R.; Baehring, J.M.; Sloan, A.; et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: The ACT III study. Neuro Oncol. 2015, 17, 854–861. [Google Scholar] [CrossRef] [Green Version]
- Altieri, D.C. Validating survivin as a cancer therapeutic target. Nat. Rev. Cancer 2003, 3, 46–54. [Google Scholar] [CrossRef]
- Fenstermaker, R.A.; Ciesielski, M.J. Challenges in the development of a survivin vaccine (SurVaxM) for malignant glioma. Exp. Rev. Vaccines 2014, 13, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Adida, C.; Crotty, P.L.; McGrath, J.; Berrebi, D.; Diebold, J.; Altieri, D.C. Developmentally regulated expression of the novel cancer anti-apoptosis gene survivin in human and mouse differentiation. Am. J. Pathol. 1998, 152, 43–49. [Google Scholar]
- Chastkofsky, M.I.; Pituch, K.C.; Katagi, H.; Zannikou, M.; Ilut, L.; Xiao, T.; Han, Y.; Sonabend, A.M.; Curiel, D.T.; Bonner, E.R.; et al. Mesenchymal Stem Cells Successfully Deliver Oncolytic Virotherapy to Diffuse Intrinsic Pontine Glioma. Clin. Cancer Res. 2021, 27, 1766–1777. [Google Scholar] [CrossRef] [PubMed]
- Heimberger, A.B.; Crotty, L.E.; Archer, G.E.; McLendon, R.E.; Friedman, A.; Dranoff, G.; Bigner, D.D.; Sampson, J.H. Bone marrow-derived dendritic cells pulsed with tumor homogenate induce immunity against syngeneic intracerebral glioma. J. Neuroimmunol. 2000, 103, 16–25. [Google Scholar] [CrossRef]
- Weller, M.; Roth, P.; Preusser, M.; Wick, W.; Reardon, D.A.; Platten, M.; Sampson, J.H. Vaccine-based immunotherapeutic approaches to gliomas and beyond. Nat. Rev. Neurol. 2017, 13, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Grauer, O.M.; Sutmuller, R.P.M.; van Maren, W.; Jacobs, J.F.M.; Bennink, E.; Toonen, L.W.J.; Nierkens, S.; Adema, G.J. Elimination of regulatory T cells is essential for an effective vaccination with tumor lysate-pulsed dendritic cells in a murine glioma model. Int. J. Cancer 2008, 122, 1794–1802. [Google Scholar] [CrossRef]
- Fedorova, L.; Mudry, P.; Pilatova, K.; Selingerova, I.; Merhautova, J.; Rehak, Z.; Valik, D.; Hlavackova, E.; Cerna, D.; Faberova, L.; et al. Assessment of Immune Response Following Dendritic Cell-Based Immunotherapy in Pediatric Patients with Relapsing Sarcoma. Front. Oncol. 2019, 9, 1169. [Google Scholar] [CrossRef]
- Garg, A.D.; Vandenberk, L.; Koks, C.; Verschuere, T.; Boon, L.; Van Gool, S.W.; Agostinis, P. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci. Transl. Med. 2016, 8, 328ra327. [Google Scholar] [CrossRef]
- Benitez-Ribas, D.; Cabezón, R.; Flórez-Grau, G.; Molero, M.C.; Puerta, P.; Guillen, A.; Paco, S.; Carcaboso, A.M.; Santa-Maria Lopez, V.; Cruz, O.; et al. Immune Response Generated with the Administration of Autologous Dendritic Cells Pulsed with an Allogenic Tumoral Cell-Lines Lysate in Patients with Newly Diagnosed Diffuse Intrinsic Pontine Glioma. Front. Oncol. 2018, 8, 127. [Google Scholar] [CrossRef] [Green Version]
- Harrington, K.; Freeman, D.J.; Kelly, B.; Harper, J.; Soria, J.-C. Optimizing oncolytic virotherapy in cancer treatment. Nat. Rev. Drug Discov. 2019, 18, 689–706. [Google Scholar] [CrossRef]
- Grigg, C.; Blake, Z.; Gartrell, R.; Sacher, A.; Taback, B.; Saenger, Y. Talimogene laherparepvec (T-Vec) for the treatment of melanoma and other cancers. Semin. Oncol. 2016, 43, 638–646. [Google Scholar] [CrossRef]
- Gállego Pérez-Larraya, J.; Garcia-Moure, M.; Labiano, S.; Patiño-García, A.; Dobbs, J.; Gonzalez-Huarriz, M.; Zalacain, M.; Marrodan, L.; Martinez-Velez, N.; Puigdelloses, M.; et al. Oncolytic DNX-2401 Virus for Pediatric Diffuse Intrinsic Pontine Glioma. New Engl. J. Med. 2022, 386, 2471–2481. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Vélez, N.; Garcia-Moure, M.; Marigil, M.; González-Huarriz, M.; Puigdelloses, M.; Gallego Pérez-Larraya, J.; Zalacaín, M.; Marrodán, L.; Varela-Guruceaga, M.; Laspidea, V.; et al. The oncolytic virus Delta-24-RGD elicits an antitumor effect in pediatric glioma and DIPG mouse models. Nat. Commun. 2019, 10, 2235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Yung, W.K.A.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J. Clin Oncol. 2018, 36, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Tejada, S.; Alonso, M.; Patiño, A.; Fueyo, J.; Gomez-Manzano, C.; Diez-Valle, R. Phase I Trial of DNX-2401 for Diffuse Intrinsic Pontine Glioma Newly Diagnosed in Pediatric Patients. Neurosurgery 2018, 83, 1050–1056. [Google Scholar] [CrossRef] [PubMed]
- Tejada, S.; Díez-Valle, R.; Domínguez, P.D.; Patiño-García, A.; González-Huarriz, M.; Fueyo, J.; Gomez-Manzano, C.; Idoate, M.A.; Peterkin, J.; Alonso, M.M. DNX-2401, an Oncolytic Virus, for the Treatment of Newly Diagnosed Diffuse Intrinsic Pontine Gliomas: A Case Report. Front. Oncol. 2018, 8, 61. [Google Scholar] [CrossRef] [Green Version]
- Kicielinski, K.P.; Chiocca, E.A.; Yu, J.S.; Gill, G.M.; Coffey, M.; Markert, J.M. Phase 1 clinical trial of intratumoral reovirus infusion for the treatment of recurrent malignant gliomas in adults. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 1056–1062. [Google Scholar] [CrossRef] [Green Version]
- Schuelke, M.R.; Wongthida, P.; Thompson, J.; Kottke, T.; Driscoll, C.B.; Huff, A.L.; Shim, K.G.; Coffey, M.; Pulido, J.; Evgin, L.; et al. Diverse immunotherapies can effectively treat syngeneic brainstem tumors in the absence of overt toxicity. J. Immunother Cancer 2019, 7, 188. [Google Scholar] [CrossRef] [Green Version]
- Laspidea, V.; Puigdelloses, M.; Labiano, S.; Marrodán, L.; Garcia-Moure, M.; Zalacain, M.; Gonzalez-Huarriz, M.; Martínez-Vélez, N.; Ausejo-Mauleon, I.; de la Nava, D.; et al. Exploiting 4-1BB immune checkpoint to enhance the efficacy of oncolytic virotherapy for diffuse intrinsic pontine gliomas. JCI Insight 2022, 7, e154812. [Google Scholar] [CrossRef]
- Cockle, J.V.; Brüning-Richardson, A.; Scott, K.J.; Thompson, J.; Kottke, T.; Morrison, E.; Ismail, A.; Carcaboso, A.M.; Rose, A.; Selby, P.; et al. Oncolytic Herpes Simplex Virus Inhibits Pediatric Brain Tumor Migration and Invasion. Mol. Ther. Oncolytics 2017, 5, 75–86. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.U.; Rolle, C.E.; Tyler, M.A.; Han, Y.; Sengupta, S.; Wainwright, D.A.; Balyasnikova, I.V.; Ulasov, I.V.; Lesniak, M.S. Bone marrow mesenchymal stem cells loaded with an oncolytic adenovirus suppress the anti-adenoviral immune response in the cotton rat model. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 1846–1856. [Google Scholar] [CrossRef] [PubMed]
- Morshed, R.A.; Gutova, M.; Juliano, J.; Barish, M.E.; Hawkins-Daarud, A.; Oganesyan, D.; Vazgen, K.; Yang, T.; Annala, A.; Ahmed, A.U.; et al. Analysis of glioblastoma tumor coverage by oncolytic virus-loaded neural stem cells using MRI-based tracking and histological reconstruction. Cancer Gene. Ther. 2015, 22, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [Green Version]
- Vranic, S.; Cyprian, F.S.; Gatalica, Z.; Palazzo, J. PD-L1 status in breast cancer: Current view and perspectives. Semin. Cancer Biol. 2021, 72, 146–154. [Google Scholar] [CrossRef]
- Yaghoubi, N.; Soltani, A.; Ghazvini, K.; Hassanian, S.M.; Hashemy, S.I. PD-1/PD-L1 blockade as a novel treatment for colorectal cancer. Biomed Pharm. 2019, 110, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Bagcchi, S. Pembrolizumab for treatment of refractory melanoma. Lancet. Oncol. 2014, 15, e419. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Ciuleanu, T.-E.; Cobo, M.; Schenker, M.; Zurawski, B.; Menezes, J.; Richardet, E.; Bennouna, J.; Felip, E.; Juan-Vidal, O.; et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): An international, randomised, open-label, phase 3 trial. Lancet. Oncol. 2021, 22, 198–211. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. New Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Motzer, R.J.; Rini, B.I.; McDermott, D.F.; Arén Frontera, O.; Hammers, H.J.; Carducci, M.A.; Salman, P.; Escudier, B.; Beuselinck, B.; Amin, A.; et al. Nivolumab plus ipilimumab versus sunitinib in first-line treatment for advanced renal cell carcinoma: Extended follow-up of efficacy and safety results from a randomised, controlled, phase 3 trial. Lancet. Oncol. 2019, 20, 1370–1385. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. New Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Janjigian, Y.Y.; Shitara, K.; Moehler, M.; Garrido, M.; Salman, P.; Shen, L.; Wyrwicz, L.; Yamaguchi, K.; Skoczylas, T.; Campos Bragagnoli, A.; et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): A randomised, open-label, phase 3 trial. Lancet 2021, 398, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.A.; Forsyth, P.A.; Algazi, A.; Hamid, O.; Hodi, F.S.; Moschos, S.J.; Khushalani, N.I.; Lewis, K.; Lao, C.D.; Postow, M.A.; et al. Combined Nivolumab and Ipilimumab in Melanoma Metastatic to the Brain. New Engl. J. Med. 2018, 379, 722–730. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Long-Term Outcomes with Nivolumab Plus Ipilimumab or Nivolumab Alone Versus Ipilimumab in Patients with Advanced Melanoma. J. Clin. Oncol. 2022, 40, 127–137. [Google Scholar] [CrossRef]
- Reardon, D.A.; Kaley, T.J.; Dietrich, J.; Clarke, J.L.; Dunn, G.; Lim, M.; Cloughesy, T.F.; Gan, H.K.; Park, A.J.; Schwarzenberger, P.; et al. Phase II study to evaluate safety and efficacy of MEDI4736 (durvalumab) + radiotherapy in patients with newly diagnosed unmethylated MGMT glioblastoma (new unmeth GBM). J. Clin. Oncol. 2019, 37, 2032. [Google Scholar] [CrossRef]
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro Oncol. 2020, 22, 1073–1113. [Google Scholar] [CrossRef]
- Nayak, L.; Standifer, N.; Dietrich, J.; Clarke, J.L.; Dunn, G.P.; Lim, M.; Cloughesy, T.; Gan, H.K.; Flagg, E.; George, E.; et al. Circulating Immune Cell and Outcome Analysis from the Phase II Study of PD-L1 Blockade with Durvalumab for Newly Diagnosed and Recurrent Glioblastoma. Clin. Cancer Res. 2022, 28, 2567–2578. [Google Scholar] [CrossRef]
- Lee, E.Q. Immune checkpoint inhibitors in GBM. J. Neuro Oncol. 2021, 155, 1–11. [Google Scholar] [CrossRef]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients with Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef]
- Cacciotti, C.; Choi, J.; Alexandrescu, S.; Zimmerman, M.A.; Cooney, T.M.; Chordas, C.; Clymer, J.; Chi, S.; Yeo, K.K. Immune checkpoint inhibition for pediatric patients with recurrent/refractory CNS tumors: A single institution experience. J. Neuro Oncol. 2020, 149, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Kline, C.; Liu, S.J.; Duriseti, S.; Banerjee, A.; Nicolaides, T.; Raber, S.; Gupta, N.; Haas-Kogan, D.; Braunstein, S.; Mueller, S. Reirradiation and PD-1 inhibition with nivolumab for the treatment of recurrent diffuse intrinsic pontine glioma: A single-institution experience. J. Neuro Oncol. 2018, 140, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Hwang, E.; Onar, A.; Young-Poussaint, T.; Mitchell, D.; Kilburn, L.; Margol, A.; Gilheeny, S.; Lin, T.; Dunkel, I.; Fouladi, M. Immu-09. outcome of patients with recurrent diffuse intrinsic pontine glioma (DIPG) treated with pembrolizumab (ANTI-PD-1): A pediatric brain tumor consortium study (PBTC045). Neuro Oncol. 2018, 20, i100. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Classification | NCT Number | Title | Status | Interventions | Age (Years) | Phases | Enrollment | Combination with Other Therapy |
|---|---|---|---|---|---|---|---|---|
| Adoptive Transfer Cell Therapy | NCT04196413 | GD2 CAR T Cells in Diffuse Intrinsic Pontine Gliomas (DIPG) and Spinal Diffuse Midline Glioma (DMG) | Recruiting | GD2 CAR T cells + Fludarabine +Cyclophosphamide | 2–30 | Phase 1 | 54 | Combination with Chemotherapy |
| NCT04099797 | C7R-GD2.CAR T Cells for Patients with GD2-expressing Brain Tumors (GAIL-B) | Recruiting | (C7R)-GD2.CART cells + Cyclophosphamide+ Fludarabine | 1–21 | Phase 1 | 34 | Combination with Chemotherapy | |
| NCT05298995 | GD2-CAR T Cells for Pediatric Brain Tumors | Not yet recruiting | GD2-CART01 (iC9-GD2-CAR T cells) | 0.5–30 | Phase 1 | 54 | No | |
| NCT04185038 | Study of B7-H3-Specific CAR T-Cell Locoregional Immunotherapy for Diffuse Intrinsic Pontine Glioma/Diffuse Midline Glioma and Recurrent or Refractory Pediatric Central Nervous System Tumors | Recruiting | SCRI-CARB7H3(s); B7H3-specific chimeric antigen receptor (CAR) T cells | 1–26 | Phase 1 | 90 | No | |
| Vaccine | NCT02960230 | H3.3K27M Peptide Vaccine With Nivolumab for Children With Newly Diagnosed DIPG and Other Gliomas | Active, not recruiting | K27M peptide+ Nivolumab | 3–21 | Phase 1|Phase 2 | 50 | Combination with an Anti-PD-1 mAb |
| NCT04749641 | Neoantigen Vaccine Therapy Against H3.3-K27M Diffuse Intrinsic Pontine Glioma | Recruiting | Histone H3.3-K27M Neoantigen Vaccine | 5- | Phase 1 | 30 | No | |
| NCT04808245 | A MultIceNTER Phase I Peptide VaCcine Trial for the Treatment of H3-Mutated Gliomas | Not yet recruiting | Tecentriq +H3K27M peptide vaccine+ Imiquimod (5%) | 18- | Phase 1 | 15 | Combination with an Anti-PD-L1 mAb | |
| NCT01058850 | Phase I Rindopepimut After Conventional Radiation in Children w/Diffuse Intrinsic Pontine Gliomas | Terminated | Rindopepimut | 3–18 | Phase 1 | 3 | No | |
| NCT04978727 | A Pilot Study of SurVaxM in Children Progressive or Relapsed Medulloblastoma, High Grade Glioma, Ependymoma and Newly Diagnosed Diffuse Intrinsic Pontine Glioma | Recruiting | SurVaxM | 1–21 | Phase 1 | 35 | Part of patients combination with radiotherapy | |
| NCT04943848 | rHSC-DIPGVax Plus Checkpoint Blockade for the Treatment of Newly Diagnosed DIPG and DMG | Recruiting | rHSC-DIPGVax+ Balstilimab+ Zalifrelimab | 1–18 | Phase 1 | 36 | Combination an Anti-PD-1 mAb and an Anti-CTLA4 mAb | |
| NCT02750891 | A Study of DSP-7888 in Pediatric Patients with Relapsed or Refractory High-grade Gliomas | Completed | DSP-7888 | 0–19 | Phase 1|Phase 2 | 18 | No | |
| NCT02840123 | Safety Study of DIPG Treatment with Autologous Dendritic Cells Pulsed with Lysates from Allogenic Tumor Lines | Unknown status | Autologous dendritic cells | 3–18 | Phase 1 | 10 | No | |
| NCT03914768 | Immune Modulatory DC Vaccine Against Brain Tumor | Enrolling by invitation | Immunomodulatory DC vaccine to target DIPG and GBM | 1–75 | Phase 1 | 10 | No | |
| NCT04911621 | Adjuvant Dendritic Cell Immunotherapy for Pediatric Patients With High-grade Glioma or Diffuse Intrinsic Pontine Glioma | Recruiting | Dendritic cell vaccination + temozolomide-based chemoradiation+- conventional next-line treatment | 1–17 | Phase 1|Phase 2 | 10 | Combination with Temozolomide-based Chemoradiation | |
| NCT03396575 | Brain Stem Gliomas Treated with Adoptive Cellular Therapy During Focal Radiotherapy Recovery Alone or with Dose-intensified Temozolomide (Phase I) | Recruiting | TTRNA-DC vaccines with GM-CSF/TTRNA-xALT/cyclophosphamide + fludarabine lymphodepletive conditioning/dose-intensified TMZ/Td vaccine/autologous hematopoietic stem cells (HSCs) | 3–30 | Phase 1 | 21 | Combination with GM-CSF or Chemotherapy or Autologous Hematopoietic Stem Cells | |
| NCT02750891 | A Study of DSP-7888 in Pediatric Patients with Relapsed or Refractory High-grade Gliomas | Completed | DSP-7888 | 0–19 | Phase 1|Phase 2 | 18 | No | |
| Oncolytic Virus Therapy | NCT03330197 | A Study of Ad-RTS-hIL-12 + Veledimex in Pediatric Subjects with Brain Tumors Including DIPG | Terminated | Ad-RTS-hIL-12+ Oral Veledimex | 0–21 | Phase 1|Phase 2 | 6 | No |
| NCT03178032 | Oncolytic Adenovirus, DNX-2401, for Naive Diffuse Intrinsic Pontine Gliomas | Active, not recruiting | DNX-2401 | 1–18 | Phase 1 | 12 | No | |
| NCT02444546 | Wild-Type Reovirus in Combination with Sargramostim in Treating Younger Patients With High-Grade Relapsed or Refractory Brain Tumors | Active, not recruiting | Sargramostim+ Wild-type Reovirus | 10–21 | Phase 1 | 6 | Combination with Sargramostim | |
| NCT04758533 | Clinical Trial to Assess the Safety and Efficacy of AloCELYVIR with Newly Diagnosed Diffuse Intrinsic Pontine Glioma (DIPG) in Combination with Radiotherapy or Medulloblastoma in Monotherapy | Recruiting | AloCELYVIR | 1–21 | Phase 1|Phase 2 | 12 | No | |
| NCT05096481 | PEP-CMV Vaccine Targeting CMV Antigen to Treat Newly Diagnosed Pediatric HGG and DIPG and Recurrent Medulloblastoma | Not yet recruiting | PEP-CMV+ Temozolomide+ Tetanus Diphtheria Vaccine | 3–25 | Phase 2 | 120 | Combination with Temozolomide | |
| Immune Checkpoint Inhibitor Therapy | NCT01952769 | Anti PD1 Antibody in Diffuse Intrinsic Pontine Glioma | Unknown status | MDV9300 | 3–21 | Phase 1|Phase 2 | 50 | No |
| NCT02359565 | Pembrolizumab in Treating Younger Patients With Recurrent, Progressive, or Refractory High-grade Gliomas, Diffuse Intrinsic Pontine Gliomas, Hypermutated Brain Tumors, Ependymoma or Medulloblastoma | Recruiting | Pembrolizumab | 1–29 | Phase 1 | 110 | No | |
| NCT03690869 | REGN2810 in Pediatric Patients with Relapsed, Refractory Solid, or Central Nervous System (CNS) Tumors and Safety and Efficacy of REGN2810 in Combination With Radiotherapy in Pediatric Patients With Newly Diagnosed or Recurrent Glioma | Recruiting | Cemiplimab | 0–25 | Phase 1|Phase 2 | 130 | No | |
| Other | NCT05063357 | 131I-omburtamab Delivered by Convection-Enhanced Delivery in Patients with Diffuse Intrinsic Pontine Glioma | Not yet recruiting | 131I-Omburtamab | 3–21 | Phase 1 | 36 | No |
| NCT01502917 | Convection-Enhanced Delivery of 124I-Omburtamab for Patients with Non-Progressive Diffuse Pontine Gliomas Previously Treated with External Beam Radiation Therapy | Completed | Radioactive iodine-labeled monoclonal antibody omburtamab+ external beam radiotherapy | 2–21 | Phase 1 | 50 | Radiotherapy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Zhao, C.; Li, S.; Wang, J.; Zhang, H. Immune Microenvironment and Immunotherapies for Diffuse Intrinsic Pontine Glioma. Cancers 2023, 15, 602. https://doi.org/10.3390/cancers15030602
Chen Y, Zhao C, Li S, Wang J, Zhang H. Immune Microenvironment and Immunotherapies for Diffuse Intrinsic Pontine Glioma. Cancers. 2023; 15(3):602. https://doi.org/10.3390/cancers15030602
Chicago/Turabian StyleChen, Yujia, Chao Zhao, Shenglun Li, Jun Wang, and Hongwei Zhang. 2023. "Immune Microenvironment and Immunotherapies for Diffuse Intrinsic Pontine Glioma" Cancers 15, no. 3: 602. https://doi.org/10.3390/cancers15030602
APA StyleChen, Y., Zhao, C., Li, S., Wang, J., & Zhang, H. (2023). Immune Microenvironment and Immunotherapies for Diffuse Intrinsic Pontine Glioma. Cancers, 15(3), 602. https://doi.org/10.3390/cancers15030602