
CAR-T Cell Therapy and the Gut Microbiota


Abstract
Simple Summary
Abstract
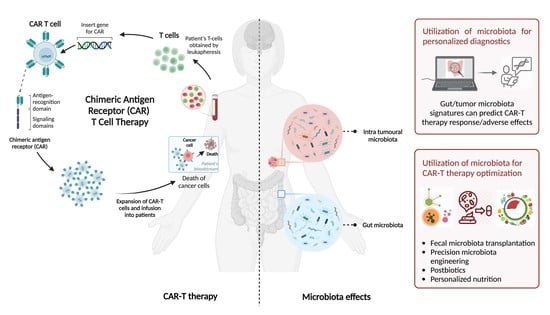
{kind=link}
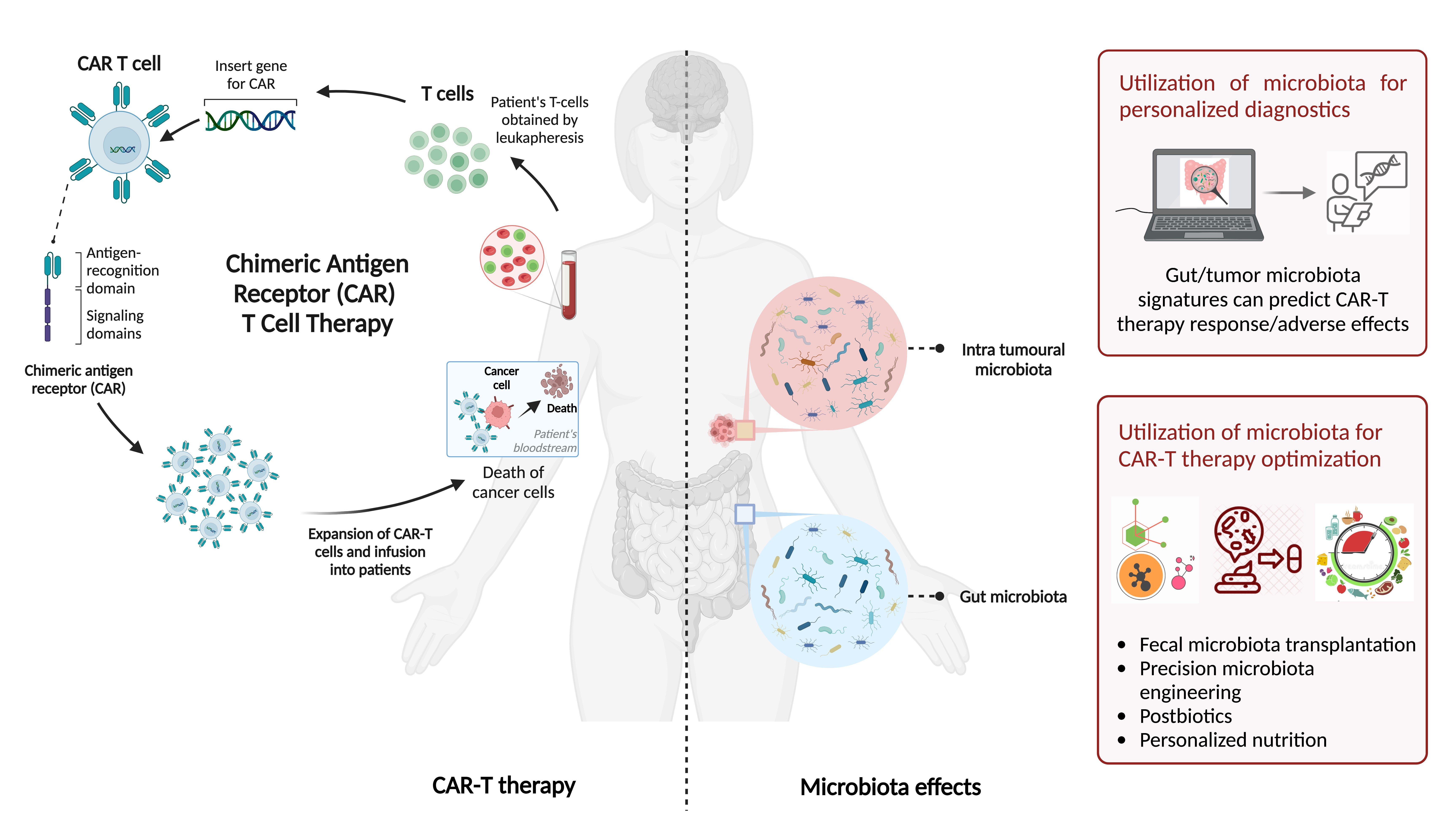
{kind=link}
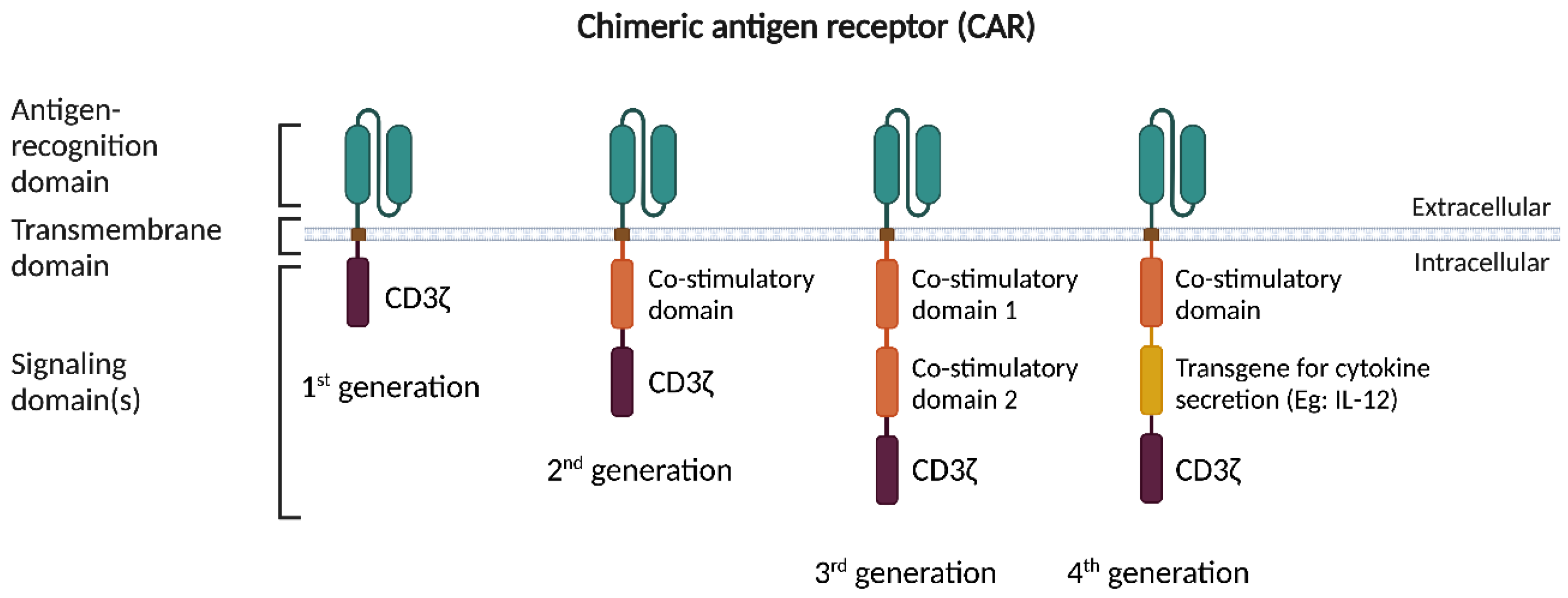
{kind=link}
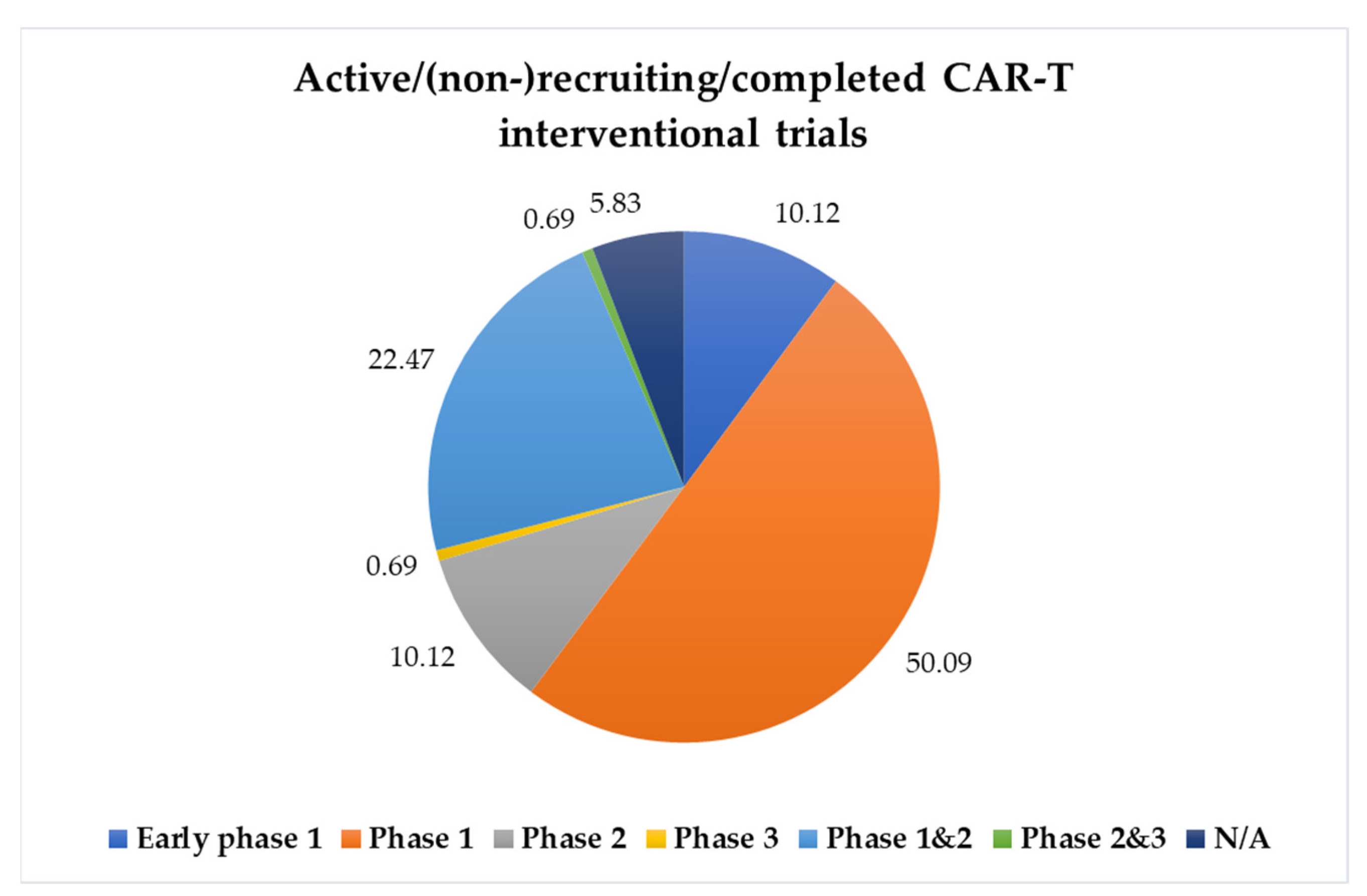
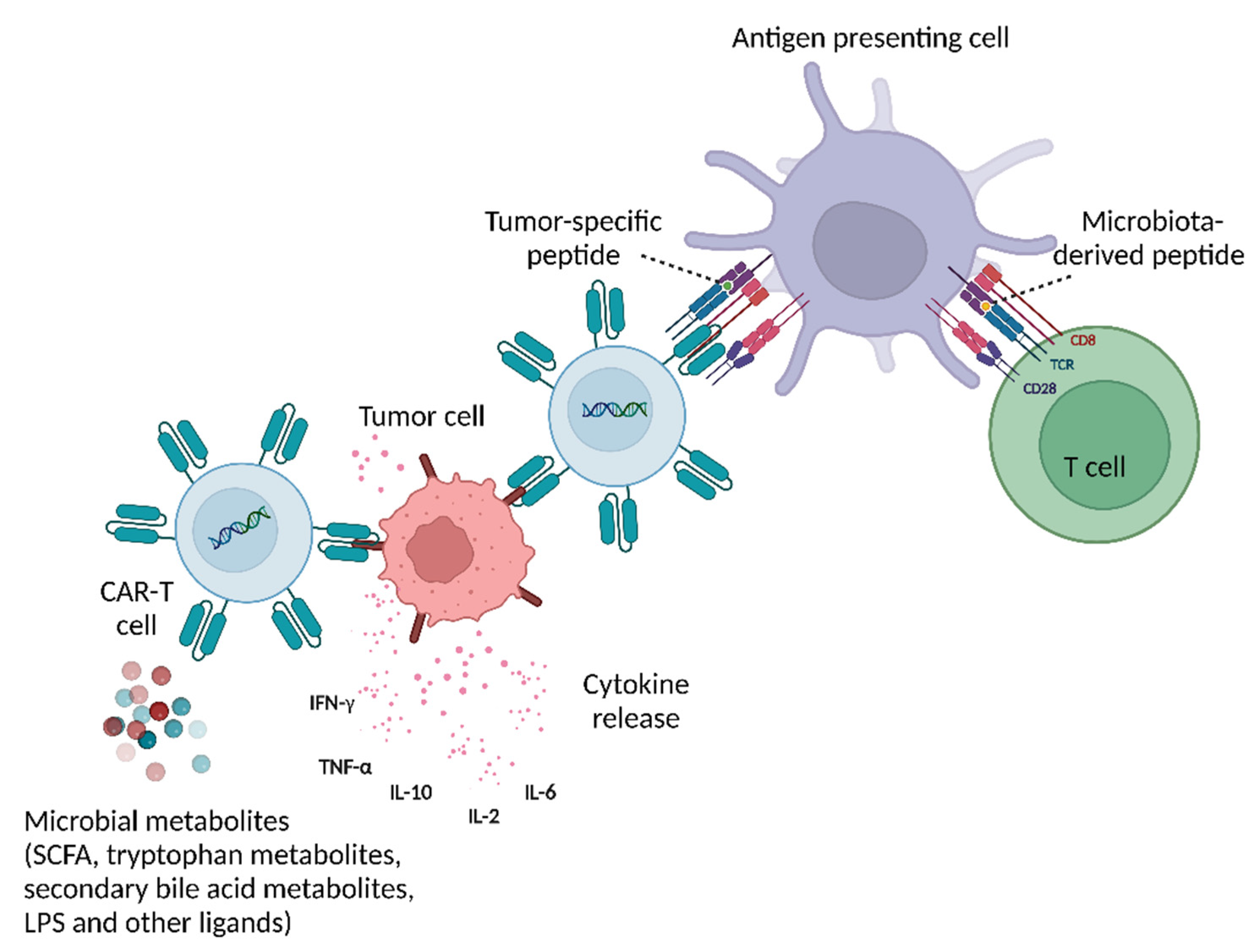{kind=link}
1. Introduction
2. CAR-T Cell Therapy
3. Toxicity Associated with CAR-T Therapy
4. Microbiota Involvement in CAR-T Response and Toxicity
5. Therapeutic Microbiota-Mediated Modulation of CAR-T Efficacy
6. Limitations and Challenges
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AhR | aryl hydrocarbon receptors |
| ALL | acute lymphoblastic leukemia |
| allo HCT | allogeneic hematopoietic cell transplantation |
| CAR | Chimeric antigen receptor |
| CD3ζ | cluster of differentiation 3 zeta-chain |
| CEA | carcinoembryonic antigen |
| CNS | central nervous system |
| CRS | cytokine release syndrome |
| CTLA-4 | cytotoxic T lymphocyte-associated protein 4 |
| DLBCL | diffuse large B cell lymphoma |
| EGFR | epidermal growth factor receptor |
| EMA | European Medicines Agency |
| ERBB2 | Erb-B2 receptor tyrosine kinase 2 |
| FAP | fibroblast activation protein |
| FDA | U.S. Food and Drug Administration |
| FMT | fecal microbiota transplantation |
| G2 | diganglioside |
| GVDH | graft-versus-host disease |
| Her2 | human epidermal growth factor receptor 2 |
| ICANS | immune effector cell-associated neurotoxicity syndrome |
| IFN-γ | interferon gamma |
| IL | interleukin |
| IL-13Rα | interleukin 13 receptor α |
| L1CAM | L1 cell adhesion molecule |
| LEfSe | linear discriminant analysis effect size |
| LPS | lipopolysaccharide |
| MM | multiple myeloma |
| MUC1 | mucin 1 |
| NHL | non-Hodgkin lymphoma |
| PD-1 | programmed cell death protein 1 |
| PSMA | prostate-specific membrane antigen |
| ROR1 | receptor tyrosine kinase-like orphan 1 |
| SCFA | short chain fatty acids |
| scFv | single-chain variable fragment |
| TCR | T cell receptors |
| TNF-α | tumor necrosis factor alpha |
| TRUCK | T cells redirected for universal cytokine-mediated killing |
References
- Kenderian, S.S.; Porter, D.L.; Gill, S. Chimeric Antigen Receptor T Cells and Hematopoietic Cell Transplantation: How Not to Put the CART Before the Horse. Biol. Blood Marrow Transpl. 2017, 23, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-H.; Kim, C.H. Evolution of Chimeric Antigen Receptor (CAR) T Cell Therapy: Current Status and Future Perspectives. Arch. Pharm. Res. 2019, 42, 607–616. [Google Scholar] [CrossRef]
- Subklewe, M.; von Bergwelt-Baildon, M.; Humpe, A. Chimeric Antigen Receptor T Cells: A Race to Revolutionize Cancer Therapy. Transfus. Med. Hemother. Off. Organ Dtsch. Ges. Transfus. Immunhamatol. 2019, 46, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of Immunoglobulin-T-Cell Receptor Chimeric Molecules as Functional Receptors with Antibody-Type Specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, Y.; Asakura, Y.; Utsunomiya, N.; Nakanishi, M.; Arata, Y.; Itoh, S.; Nagase, F.; Kurosawa, Y. Expression of Chimeric Receptor Composed of Immunoglobulin-Derived V Regions and T-Cell Receptor-Derived C Regions. Biochem. Biophys. Res. Commun. 1987, 149, 960–968. [Google Scholar] [CrossRef]
- Brocker, T.; Karjalainen, K. Signals through T Cell Receptor-Zeta Chain Alone Are Insufficient to Prime Resting T Lymphocytes. J. Exp. Med. 1995, 181, 1653–1659. [Google Scholar] [CrossRef]
- Gong, M.C.; Latouche, J.B.; Krause, A.; Heston, W.D.; Bander, N.H.; Sadelain, M. Cancer Patient T Cells Genetically Targeted to Prostate-Specific Membrane Antigen Specifically Lyse Prostate Cancer Cells and Release Cytokines in Response to Prostate-Specific Membrane Antigen. Neoplasia 1999, 1, 123–127. [Google Scholar] [CrossRef]
- Till, B.G.; Jensen, M.C.; Wang, J.; Chen, E.Y.; Wood, B.L.; Greisman, H.A.; Qian, X.; James, S.E.; Raubitschek, A.; Forman, S.J.; et al. Adoptive Immunotherapy for Indolent Non-Hodgkin Lymphoma and Mantle Cell Lymphoma Using Genetically Modified Autologous CD20-Specific T Cells. Blood 2008, 112, 2261–2271. [Google Scholar] [CrossRef]
- Krause, A.; Guo, H.F.; Latouche, J.B.; Tan, C.; Cheung, N.K.; Sadelain, M. Antigen-Dependent CD28 Signaling Selectively Enhances Survival and Proliferation in Genetically Modified Activated Human Primary T Lymphocytes. J. Exp. Med. 1998, 188, 619–626. [Google Scholar] [CrossRef]
- Wang, L.-C.S.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting Fibroblast Activation Protein in Tumor Stroma with Chimeric Antigen Receptor T Cells Can Inhibit Tumor Growth and Augment Host Immunity without Severe Toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Curran, K.J. Novel Cellular Therapies for Leukemia: CAR-Modified T Cells Targeted to the CD19 Antigen. Hematol. Am. Soc. Hematol. Educ. Program 2012, 2012, 143–151. [Google Scholar] [CrossRef]
- Mullard, A. FDA Approves First CAR T Therapy. Nat. Rev. Drug Discov. 2017, 16, 669. [Google Scholar] [CrossRef] [PubMed]
- FDA Okays Second CAR-T for Kite. Nat. Biotechnol. 2020, 38, 1012. [CrossRef] [PubMed]
- Mullard, A. FDA Approves Fourth CAR-T Cell Therapy. Nat. Rev. Drug Discov. 2021, 20, 166. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. FDA Approves Second BCMA-Targeted CAR-T Cell Therapy. Nat. Rev. Drug Discov. 2022, 21, 249. [Google Scholar] [CrossRef] [PubMed]
- First CAR-T Therapy to Target BCMA Gets FDA Nod. Nat. Biotechnol. 2021, 39, 531. [CrossRef]
- Gill, S.; Maus, M.V.; Porter, D.L. Chimeric Antigen Receptor T Cell Therapy: 25years in the Making. Blood Rev. 2016, 30, 157–167. [Google Scholar] [CrossRef]
- Fousek, K.; Ahmed, N. The Evolution of T-Cell Therapies for Solid Malignancies. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 3384–3392. [Google Scholar] [CrossRef]
- Newick, K.; O’Brien, S.; Moon, E.; Albelda, S.M. CAR T Cell Therapy for Solid Tumors. Annu. Rev. Med. 2017, 68, 139–152. [Google Scholar] [CrossRef]
- Jin, L.; Tao, H.; Karachi, A.; Long, Y.; Hou, A.Y.; Na, M.; Dyson, K.A.; Grippin, A.J.; Deleyrolle, L.P.; Zhang, W.; et al. CXCR1- or CXCR2-Modified CAR T Cells Co-Opt IL-8 for Maximal Antitumor Efficacy in Solid Tumors. Nat. Commun. 2019, 10, 4016. [Google Scholar] [CrossRef]
- Asokan, S.; Bandapalli, O.R. CXCL8 Signaling in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2021, 1302, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T Cells in Solid Tumors: Challenges and Opportunities. Stem Cell Res. Ther. 2021, 12, 81. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current Concepts in the Diagnosis and Management of Cytokine Release Syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric Antigen Receptor T-Cell Therapy—Assessment and Management of Toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Kochenderfer, J.N. Toxicities of Chimeric Antigen Receptor T Cells: Recognition and Management. Blood 2016, 127, 3321–3330. [Google Scholar] [CrossRef] [PubMed]
- Hay, K.A.; Hanafi, L.-A.; Li, D.; Gust, J.; Liles, W.C.; Wurfel, M.M.; López, J.A.; Chen, J.; Chung, D.; Harju-Baker, S.; et al. Kinetics and Biomarkers of Severe Cytokine Release Syndrome after CD19 Chimeric Antigen Receptor-Modified T-Cell Therapy. Blood 2017, 130, 2295–2306. [Google Scholar] [CrossRef]
- Teachey, D.T.; Lacey, S.F.; Shaw, P.A.; Melenhorst, J.J.; Maude, S.L.; Frey, N.; Pequignot, E.; Gonzalez, V.E.; Chen, F.; Finklestein, J.; et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-Cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov. 2016, 6, 664–679. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.-L.; Rohrbach, R.; Schmitt, M.; Stein-Thoeringer, C.K. The Potential Role of the Intestinal Micromilieu and Individual Microbes in the Immunobiology of Chimeric Antigen Receptor T-Cell Therapy. Front. Immunol. 2021, 12, 670286. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Hunter, B.D.; Jacobson, C.A. CAR T-Cell Associated Neurotoxicity: Mechanisms, Clinicopathologic Correlates, and Future Directions. JNCI J. Natl. Cancer Inst. 2019, 111, 646–654. [Google Scholar] [CrossRef]
- Nastoupil, L.J.; Jain, M.D.; Feng, L.; Spiegel, J.Y.; Ghobadi, A.; Lin, Y.; Dahiya, S.; Lunning, M.; Lekakis, L.; Reagan, P.; et al. Standard-of-Care Axicabtagene Ciloleucel for Relapsed or Refractory Large B-Cell Lymphoma: Results From the US Lymphoma CAR T Consortium. J. Clin. Oncol. 2020, 38, 3119–3128. [Google Scholar] [CrossRef] [PubMed]
- Gust, J.; Hay, K.A.; Hanafi, L.-A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Liles, W.C.; Wurfel, M.; Lopez, J.A.; et al. Endothelial Activation and Blood–Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017, 7, 1404–1419. [Google Scholar] [CrossRef] [PubMed]
- Gust, J.; Finney, O.C.; Li, D.; Brakke, H.M.; Hicks, R.M.; Futrell, R.B.; Gamble, D.N.; Rawlings-Rhea, S.D.; Khalatbari, H.K.; Ishak, G.E.; et al. Glial Injury in Neurotoxicity after Pediatric CD19-directed Chimeric Antigen Receptor T Cell Therapy. Ann. Neurol. 2019; 86, 42–54. [Google Scholar] [CrossRef]
- Santomasso, B.D.; Park, J.H.; Salloum, D.; Riviere, I.; Flynn, J.; Mead, E.; Halton, E.; Wang, X.; Senechal, B.; Purdon, T.; et al. Clinical and Biological Correlates of Neurotoxicity Associated with CAR T-Cell Therapy in Patients with B-Cell Acute Lymphoblastic Leukemia. Cancer Discov. 2018, 8, 958–971. [Google Scholar] [CrossRef]
- Schubert, M.-L.; Schmitt, M.; Wang, L.; Ramos, C.A.; Jordan, K.; Müller-Tidow, C.; Dreger, P. Side-Effect Management of Chimeric Antigen Receptor (CAR) T-Cell Therapy. Ann. Oncol. 2021, 32, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Wudhikarn, K.; Palomba, M.L.; Pennisi, M.; Garcia-Recio, M.; Flynn, J.R.; Devlin, S.M.; Afuye, A.; Silverberg, M.L.; Maloy, M.A.; Shah, G.L.; et al. Infection during the First Year in Patients Treated with CD19 CAR T Cells for Diffuse Large B Cell Lymphoma. Blood Cancer J. 2020, 10, 79. [Google Scholar] [CrossRef] [PubMed]
- Tamburini, F.B.; Andermann, T.M.; Tkachenko, E.; Senchyna, F.; Banaei, N.; Bhatt, A.S. Precision Identification of Diverse Bloodstream Pathogens in the Gut Microbiome. Nat. Med. 2018, 24, 1809–1814. [Google Scholar] [CrossRef]
- Eshel, A.; Sharon, I.; Nagler, A.; Bomze, D.; Danylesko, I.; Fein, J.A.; Geva, M.; Henig, I.; Shimoni, A.; Zuckerman, T.; et al. Origins of Bloodstream Infections Following Fecal Microbiota Transplantation: A Strain-Level Analysis. Blood Adv. 2022, 6, 568–573. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between Microbiota and Immunity in Health and Disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Ivanov, I.I.; Tuganbaev, T.; Skelly, A.N.; Honda, K. T Cell Responses to the Microbiota. Annu. Rev. Immunol. 2022, 40, 559–587. [Google Scholar] [CrossRef]
- Viaud, S.; Saccheri, F.; Mignot, G.; Yamazaki, T.; Daillère, R.; Hannani, D.; Enot, D.P.; Pfirschke, C.; Engblom, C.; Pittet, M.J.; et al. The Intestinal Microbiota Modulates the Anticancer Immune Effects of Cyclophosphamide. Science 2013, 342, 971–976. [Google Scholar] [CrossRef]
- Yang, K.; Hou, Y.; Zhang, Y.; Liang, H.; Sharma, A.; Zheng, W.; Wang, L.; Torres, R.; Tatebe, K.; Chmura, S.J.; et al. Suppression of Local Type I Interferon by Gut Microbiota–Derived Butyrate Impairs Antitumor Effects of Ionizing Radiation. J. Exp. Med. 2021, 218, e20201915. [Google Scholar] [CrossRef]
- Andrews, M.C.; Duong, C.P.M.; Gopalakrishnan, V.; Iebba, V.; Chen, W.-S.; Derosa, L.; Khan, M.A.W.; Cogdill, A.P.; White, M.G.; Wong, M.C.; et al. Gut Microbiota Signatures Are Associated with Toxicity to Combined CTLA-4 and PD-1 Blockade. Nat. Med. 2021, 27, 1432–1441. [Google Scholar] [CrossRef] [PubMed]
- Paulos, C.M.; Wrzesinski, C.; Kaiser, A.; Hinrichs, C.S.; Chieppa, M.; Cassard, L.; Palmer, D.C.; Boni, A.; Muranski, P.; Yu, Z.; et al. Microbial Translocation Augments the Function of Adoptively Transferred Self/Tumor-Specific CD8+ T Cells via TLR4 Signaling. J. Clin. Investig. 2007, 117, 2197–2204. [Google Scholar] [CrossRef] [PubMed]
- Peled, J.U.; Gomes, A.L.C.; Devlin, S.M.; Littmann, E.R.; Taur, Y.; Sung, A.D.; Weber, D.; Hashimoto, D.; Slingerland, A.E.; Slingerland, J.B.; et al. Microbiota as Predictor of Mortality in Allogeneic Hematopoietic-Cell Transplantation. N. Engl. J. Med. 2020, 382, 822–834. [Google Scholar] [CrossRef] [PubMed]
- Shono, Y.; Docampo, M.D.; Peled, J.U.; Perobelli, S.M.; Velardi, E.; Tsai, J.J.; Slingerland, A.E.; Smith, O.M.; Young, L.F.; Gupta, J.; et al. Increased GVHD-Related Mortality with Broad-Spectrum Antibiotic Use after Allogeneic Hematopoietic Stem Cell Transplantation in Human Patients and Mice. Sci. Transl. Med. 2016, 8, 339ra71. [Google Scholar] [CrossRef] [PubMed]
- Stein-Thoeringer, C.K.; Nichols, K.B.; Lazrak, A.; Docampo, M.D.; Slingerland, A.E.; Slingerland, J.B.; Clurman, A.G.; Armijo, G.; Gomes, A.L.C.; Shono, Y.; et al. Lactose Drives Enterococcus Expansion to Promote Graft-versus-Host Disease. Science 2019, 366, 1143–1149. [Google Scholar] [CrossRef]
- Peled, J.U.; Devlin, S.M.; Staffas, A.; Lumish, M.; Khanin, R.; Littmann, E.R.; Ling, L.; Kosuri, S.; Maloy, M.; Slingerland, J.B.; et al. Intestinal Microbiota and Relapse After Hematopoietic-Cell Transplantation. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 1650–1659. [Google Scholar] [CrossRef]
- Cullin, N.; Azevedo Antunes, C.; Straussman, R.; Stein-Thoeringer, C.K.; Elinav, E. Microbiome and Cancer. Cancer Cell 2021, 39, 1317–1341. [Google Scholar] [CrossRef]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.-L.; et al. Commensal Bifidobacterium Promotes Antitumor Immunity and Facilitates Anti-PD-L1 Efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.M.; et al. Anticancer Immunotherapy by CTLA-4 Blockade Relies on the Gut Microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef]
- Tanoue, T.; Morita, S.; Plichta, D.R.; Skelly, A.N.; Suda, W.; Sugiura, Y.; Narushima, S.; Vlamakis, H.; Motoo, I.; Sugita, K.; et al. A Defined Commensal Consortium Elicits CD8 T Cells and Anti-Cancer Immunity. Nature 2019, 565, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.; Dai, A.; Ghilardi, G.; Amelsberg, K.V.; Devlin, S.M.; Pajarillo, R.; Slingerland, J.B.; Beghi, S.; Herrera, P.S.; Giardina, P.; et al. Gut Microbiome Correlates of Response and Toxicity Following Anti-CD19 CAR T Cell Therapy. Nat. Med. 2022, 28, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Kamada, N.; Seo, S.-U.; Chen, G.Y.; Núñez, G. Role of the Gut Microbiota in Immunity and Inflammatory Disease. Nat. Rev. Immunol. 2013, 13, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, J.; Ni, F.; Yang, Z.; Gui, X.; Bao, Z.; Zhao, H.; Wei, G.; Wang, Y.; Zhang, M.; et al. CAR-T Cell Therapy-Related Cytokine Release Syndrome and Therapeutic Response Is Modulated by the Gut Microbiome in Hematologic Malignancies. Nat. Commun. 2022, 13, 5313. [Google Scholar] [CrossRef]
- Blumenberg, V.; Schubert, M.-L.; Zamir, E.; Schmidt, S.; Rohrbach, R.; Waldhoff, P.; Bozic, D.; Pock, H.; Elinav, E.; Schmidt, C.; et al. Antibiotic Therapy and Low Gut Microbiome Diversity Is Associated with Decreased Response and High Toxicity in BCP-ALL and DLBCL Patients after Treatment with CD19. CAR T-Cells. Blood 2020, 136, 33–34. [Google Scholar] [CrossRef]
- Stein-Thoeringer, C.K.; Saini, N.Y.; Zamir, E.; Blumenberg, V.; Schubert, M.-L.; Mor, U.; Fante, M.A.; Schmidt, S.; Hayase, E.; Hayase, T.; et al. A Non-Antibiotic-Disrupted Gut Microbiome Predicts Clinical Responses to CD19-Targeted CAR-T Cell Cancer Immunotherapy across International Cohorts. Nat. Med. 2023, in press. [Google Scholar]
- Luu, M.; Riester, Z.; Baldrich, A.; Reichardt, N.; Yuille, S.; Busetti, A.; Klein, M.; Wempe, A.; Leister, H.; Raifer, H.; et al. Microbial Short-Chain Fatty Acids Modulate CD8+ T Cell Responses and Improve Adoptive Immunotherapy for Cancer. Nat. Commun. 2021, 12, 4077. [Google Scholar] [CrossRef] [PubMed]
- Rangan, P.; Mondino, A. Microbial Short-Chain Fatty Acids: A Strategy to Tune Adoptive T Cell Therapy. J. Immunother. Cancer 2022, 10, e004147. [Google Scholar] [CrossRef]
- Baruch, E.N.; Youngster, I.; Ben-Betzalel, G.; Ortenberg, R.; Lahat, A.; Katz, L.; Adler, K.; Dick-Necula, D.; Raskin, S.; Bloch, N.; et al. Fecal Microbiota Transplant Promotes Response in Immunotherapy-Refractory Melanoma Patients. Science 2021, 371, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Davar, D.; Dzutsev, A.K.; McCulloch, J.A.; Rodrigues, R.R.; Chauvin, J.-M.; Morrison, R.M.; Deblasio, R.N.; Menna, C.; Ding, Q.; Pagliano, O.; et al. Fecal Microbiota Transplant Overcomes Resistance to Anti-PD-1 Therapy in Melanoma Patients. Science 2021, 371, 595–602. [Google Scholar] [CrossRef]
- Docampo, M.D.; da Silva, M.B.; Lazrak, A.; Nichols, K.B.; Lieberman, S.R.; Slingerland, A.E.; Armijo, G.K.; Shono, Y.; Nguyen, C.; Monette, S.; et al. Alloreactive T Cells Deficient of the Short-Chain Fatty Acid Receptor GPR109A Induce Less Graft-versus-Host Disease. Blood 2022, 139, 2392–2405. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, N.; Zhou, L.; Wang, J.; Zhou, Y.; Zhang, T.; Fang, Y.; Deng, J.; Gao, Y.; Liang, X.; et al. IL-2 Regulates Tumor-Reactive CD8+ T Cell Exhaustion by Activating the Aryl Hydrocarbon Receptor. Nat. Immunol. 2021, 22, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Gargaro, M.; Manni, G.; Scalisi, G.; Puccetti, P.; Fallarino, F. Tryptophan Metabolites at the Crossroad of Immune-Cell Interaction via the Aryl Hydrocarbon Receptor: Implications for Tumor Immunotherapy. Int. J. Mol. Sci. 2021, 22, 4644. [Google Scholar] [CrossRef] [PubMed]
- Zeevi, D.; Korem, T.; Zmora, N.; Israeli, D.; Rothschild, D.; Weinberger, A.; Ben-Yacov, O.; Lador, D.; Avnit-Sagi, T.; Lotan-Pompan, M.; et al. Personalized Nutrition by Prediction of Glycemic Responses. Cell 2015, 163, 1079–1094. [Google Scholar] [CrossRef] [PubMed]
- Thaiss, C.A.; Zeevi, D.; Levy, M.; Zilberman-Schapira, G.; Suez, J.; Tengeler, A.C.; Abramson, L.; Katz, M.N.; Korem, T.; Zmora, N.; et al. Transkingdom Control of Microbiota Diurnal Oscillations Promotes Metabolic Homeostasis. Cell 2014, 159, 514–529. [Google Scholar] [CrossRef]
- Bultman, S.J. Molecular Pathways: Gene-Environment Interactions Regulating Dietary Fiber Induction of Proliferation and Apoptosis via Butyrate for Cancer Prevention. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Mantrana, I.; Selma-Royo, M.; Alcantara, C.; Collado, M.C. Shifts on Gut Microbiota Associated to Mediterranean Diet Adherence and Specific Dietary Intakes on General Adult Population. Front. Microbiol. 2018, 9, 890. [Google Scholar] [CrossRef]
- Donohoe, D.R.; Holley, D.; Collins, L.B.; Montgomery, S.A.; Whitmore, A.C.; Hillhouse, A.; Curry, K.P.; Renner, S.W.; Greenwalt, A.; Ryan, E.P.; et al. A Gnotobiotic Mouse Model Demonstrates That Dietary Fiber Protects against Colorectal Tumorigenesis in a Microbiota- and Butyrate-Dependent Manner. Cancer Discov. 2014, 4, 1387–1397. [Google Scholar] [CrossRef]
- Berry, S.E.; Valdes, A.M.; Drew, D.A.; Asnicar, F.; Mazidi, M.; Wolf, J.; Capdevila, J.; Hadjigeorgiou, G.; Davies, R.; Al Khatib, H.; et al. Human Postprandial Responses to Food and Potential for Precision Nutrition. Nat. Med. 2020, 26, 964–973. [Google Scholar] [CrossRef]
- Federici, S.; Kredo-Russo, S.; Valdés-Mas, R.; Kviatcovsky, D.; Weinstock, E.; Matiuhin, Y.; Silberberg, Y.; Atarashi, K.; Furuichi, M.; Oka, A.; et al. Targeted Suppression of Human IBD-Associated Gut Microbiota Commensals by Phage Consortia for Treatment of Intestinal Inflammation. Cell 2022, 185, 2879–2898.e24. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut Microbiome Modulates Response to Anti-PD-1 Immunotherapy in Melanoma Patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.-L.; Luke, J.J.; Gajewski, T.F. The Commensal Microbiome Is Associated with Anti-PD-1 Efficacy in Metastatic Melanoma Patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut Microbiome Influences Efficacy of PD-1-Based Immunotherapy against Epithelial Tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.A.; Thomas, A.M.; Bolte, L.A.; Björk, J.R.; de Ruijter, L.K.; Armanini, F.; Asnicar, F.; Blanco-Miguez, A.; Board, R.; Calbet-Llopart, N.; et al. Cross-Cohort Gut Microbiome Associations with Immune Checkpoint Inhibitor Response in Advanced Melanoma. Nat. Med. 2022, 28, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Villéger, R.; Lopès, A.; Carrier, G.; Veziant, J.; Billard, E.; Barnich, N.; Gagnière, J.; Vazeille, E.; Bonnet, M. Intestinal Microbiota: A Novel Target to Improve Anti-Tumor Treatment? Int. J. Mol. Sci. 2019, 20, 4584. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asokan, S.; Cullin, N.; Stein-Thoeringer, C.K.; Elinav, E. CAR-T Cell Therapy and the Gut Microbiota. Cancers 2023, 15, 794. https://doi.org/10.3390/cancers15030794
Asokan S, Cullin N, Stein-Thoeringer CK, Elinav E. CAR-T Cell Therapy and the Gut Microbiota. Cancers. 2023; 15(3):794. https://doi.org/10.3390/cancers15030794
Chicago/Turabian StyleAsokan, Sahana, Nyssa Cullin, Christoph K. Stein-Thoeringer, and Eran Elinav. 2023. "CAR-T Cell Therapy and the Gut Microbiota" Cancers 15, no. 3: 794. https://doi.org/10.3390/cancers15030794
APA StyleAsokan, S., Cullin, N., Stein-Thoeringer, C. K., & Elinav, E. (2023). CAR-T Cell Therapy and the Gut Microbiota. Cancers, 15(3), 794. https://doi.org/10.3390/cancers15030794