
Niclosamide Revitalizes Sorafenib through Insulin-like Growth Factor 1 Receptor (IGF-1R)/Stemness and Metabolic Changes in Hepatocellular Carcinoma


 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. RNA Isolation and Real-Time Reverse-Transcription Polymerase Chain Reaction
2.3. Western Blot Analysis
2.4. HCC Organoid Generation
2.5. Cell Viability Assay
2.6. Mitochondria Membrane Potential Assay
2.7. Tumor Xenograft Mouse Model
2.8. TUNEL Assay
2.9. Drug Combination Index
2.10. Statistical Analyses
3. Results
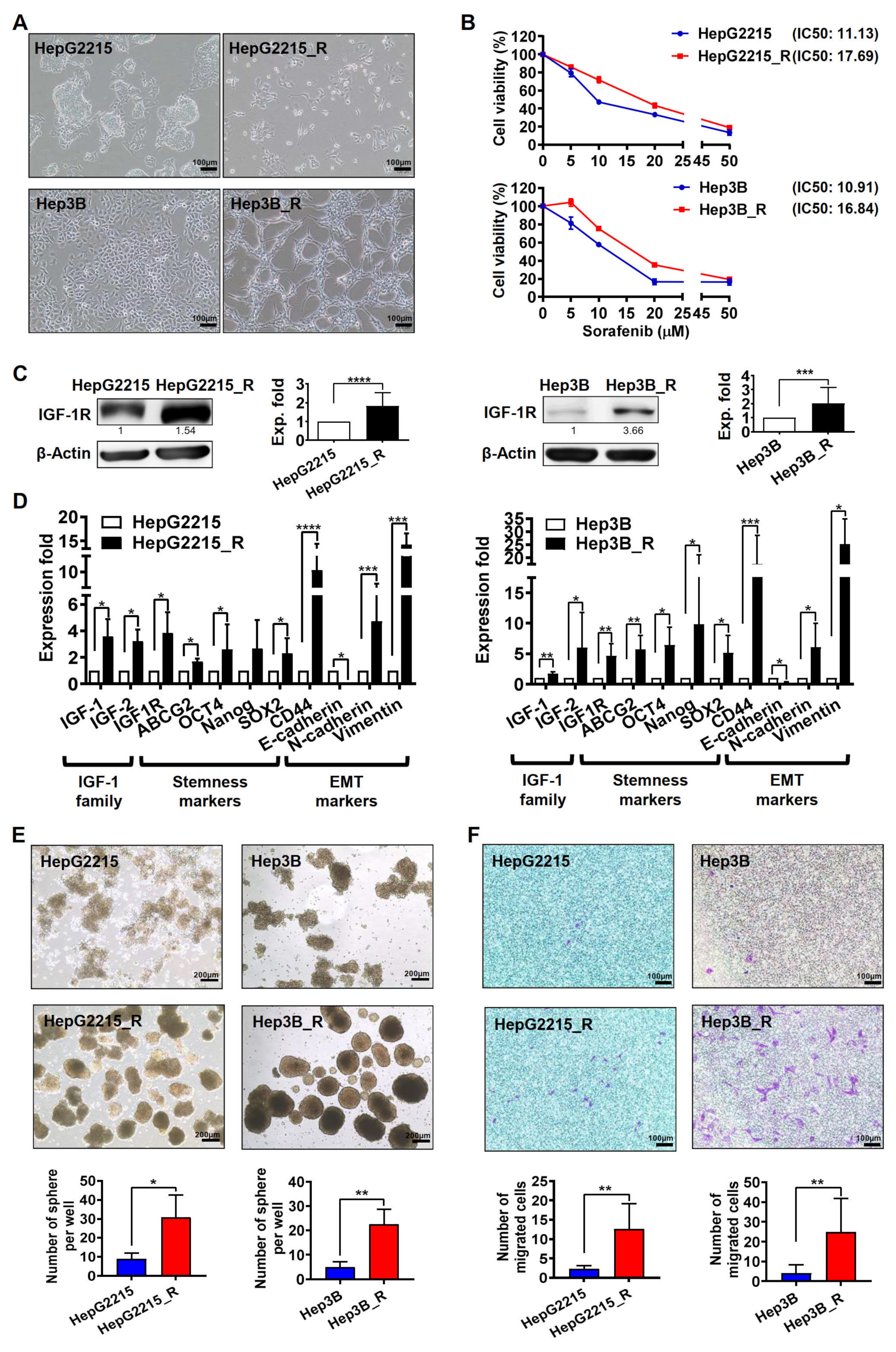
3.1. Sorafenib-Resistant HCC Cells Exhibited Higher IGF-1R Expression Levels and Stronger Stemness/EMT-Related Properties
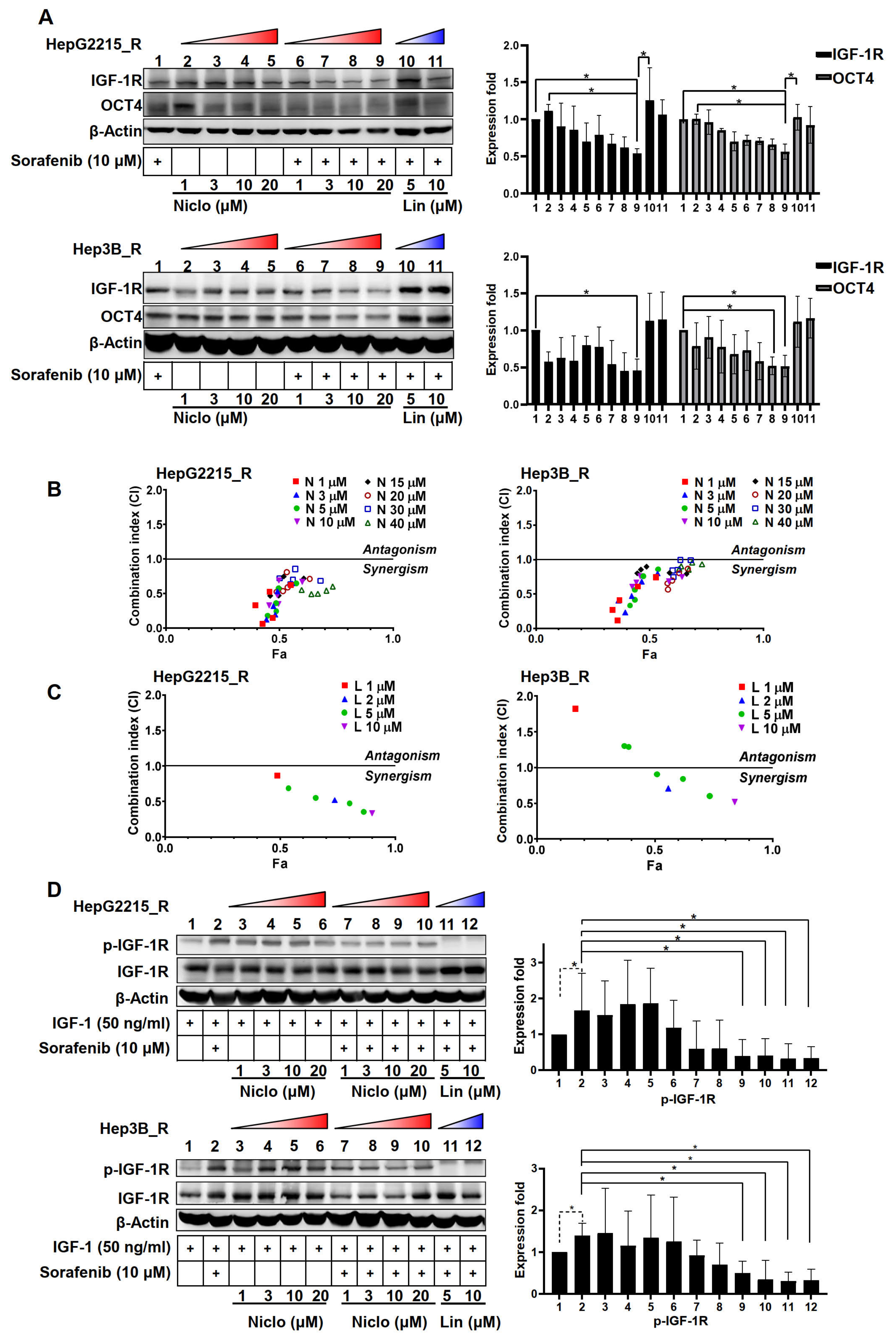
3.2. Niclosamide Significantly Enhanced the Sorafenib Sensitivity (as Indicated by Cell Viability) and Suppressed the Stemness-Related Properties of Sorafenib-Resistant HCC Cells
3.3. The Combination of Niclosamide and Sorafenib Reduced the Cell Viability of Sorafenib-Resistant HCC-Derived Organoids
3.4. Niclosamide Effectively Mitigated IGF-1R and OCT4 Expressions as Well as Sorafenib-Induced IGF-1R Phosphorylation in the Sorafenib-Resistant HCC Cells under IGF-1 Treatment
3.5. The Combination Niclosamide and Sorafenib Suppressed the Sorafenib-Induced Glycolysis-Related Genes in Sorafenib-Resistant HCC Cells and Their Organoids
3.6. Niclosamide Enhanced the Ability of Sorafenib to Reduce Mitochondrial Membrane Potential in Sorafenib-Resistant HCC Cells
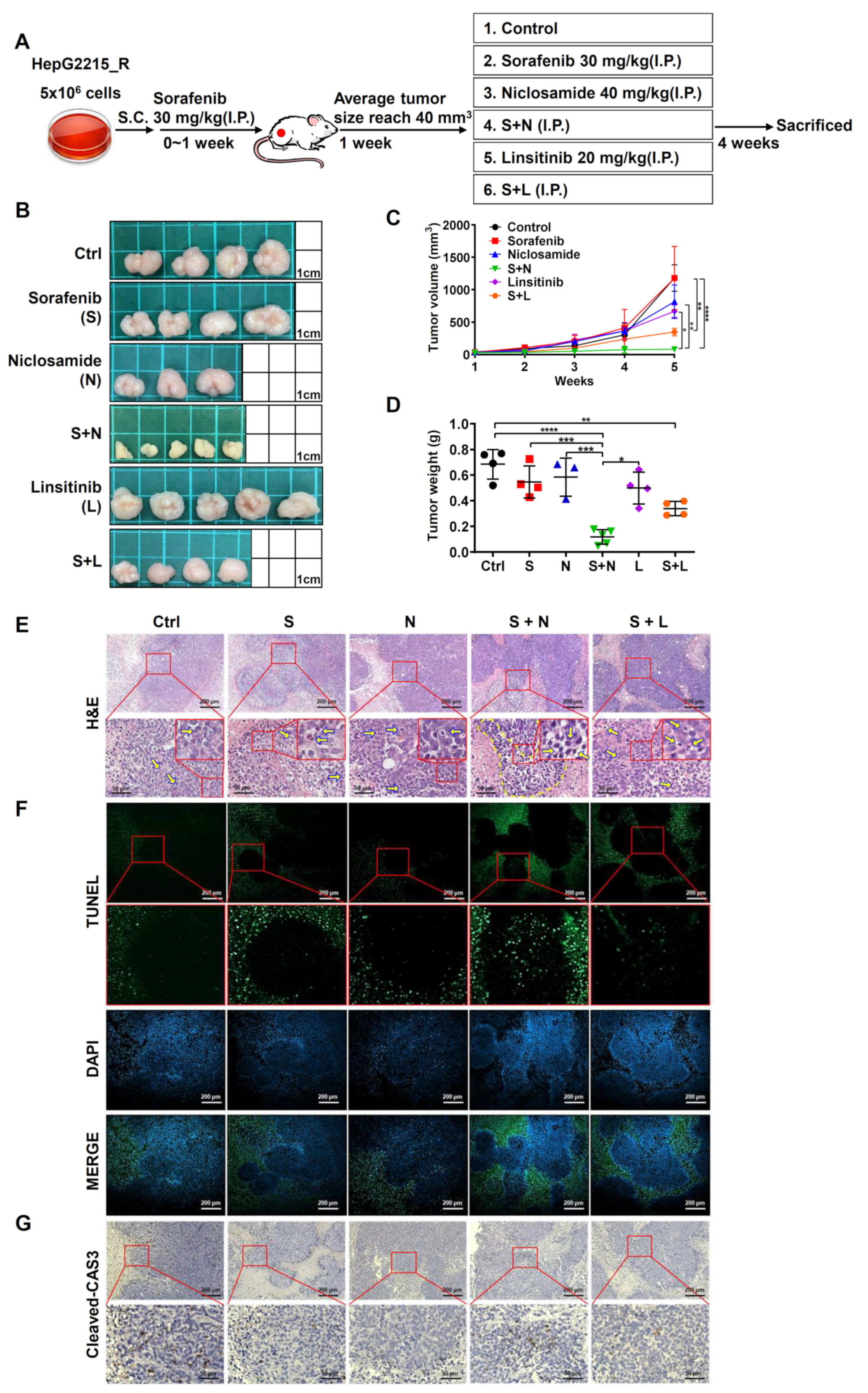
3.7. The Combination of Niclosamide and Sorafenib Suppressed Tumor Growth by Increasing the Cell Apoptosis in Sorafenib-Resistant HCC Cells In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef] [PubMed]
- Altekruse, S.F.; McGlynn, K.A.; Reichman, M.E. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J. Clin. Oncol. 2009, 27, 1485–1491. [Google Scholar] [CrossRef] [PubMed]
- Reig, M.; Forner, A.; Rimola, J.; Ferrer-Fàbrega, J.; Burrel, M.; Garcia-Criado, Á.; Kelley, R.K.; Galle, P.R.; Mazzaferro, V.; Salem, R.; et al. BCLC strategy for prognosis prediction and treatment recommendation: The 2022 update. J. Hepatol. 2022, 76, 681–693. [Google Scholar] [CrossRef]
- Poon, R.T.; Fan, S.T.; Lo, C.M.; Liu, C.L.; Wong, J. Long-term survival and pattern of recurrence after resection of small hepatocellular carcinoma in patients with preserved liver function: Implications for a strategy of salvage transplantation. Ann. Surg. 2002, 235, 373–382. [Google Scholar] [CrossRef]
- Wang, J.; Pillai, A. Systemic therapy for hepatocellular carcinoma. Clin. Liver Dis. 2021, 17, 337–340. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Bruix, J.; Takayama, T.; Mazzaferro, V.; Chau, G.Y.; Yang, J.; Kudo, M.; Cai, J.; Poon, R.T.; Han, K.H.; Tak, W.Y.; et al. Adjuvant sorafenib for hepatocellular carcinoma after resection or ablation (STORM): A phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2015, 16, 1344–1354. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.W.; et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- Zhu, Y.J.; Zheng, B.; Wang, H.Y.; Chen, L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol. Sin. 2017, 38, 614–622. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Chan, S.L.; Kudo, M.; Lau, G.; Kelley, R.K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.-K.; Dao, T.V.; Toni, E.N.D.; et al. Phase 3 randomized, open-label, multicenter study of tremelimumab (T) and durvalumab (D) as first-line therapy in patients (pts) with unresectable hepatocellular carcinoma (uHCC): HIMALAYA. J. Clin. Oncol. 2022, 40, 379. [Google Scholar] [CrossRef]
- Hosseinzadeh, F.; Verdi, J.; Ai, J.; Hajighasemlou, S.; Seyhoun, I.; Parvizpour, F.; Hosseinzadeh, F.; Iranikhah, A.; Shirian, S. Combinational immune-cell therapy of natural killer cells and sorafenib for advanced hepatocellular carcinoma: A review. Cancer Cell Int. 2018, 18, 133. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef]
- Chen, W.; Mook, R.A., Jr.; Premont, R.T.; Wang, J. Niclosamide: Beyond an antihelminthic drug. Cell Signal. 2018, 41, 89–96. [Google Scholar] [CrossRef]
- Wang, C.; Zhou, X.; Xu, H.; Shi, X.; Zhao, J.; Yang, M.; Zhang, L.; Jin, X.; Hu, Y.; Li, X.; et al. Niclosamide inhibits cell growth and enhances drug sensitivity of hepatocellular carcinoma cells via STAT3 signaling pathway. J. Cancer 2018, 9, 4150–4155. [Google Scholar] [CrossRef]
- Liu, C.; Lou, W.; Armstrong, C.; Zhu, Y.; Evans, C.P.; Gao, A.C. Niclosamide suppresses cell migration and invasion in enzalutamide resistant prostate cancer cells via Stat3-AR axis inhibition. Prostate 2015, 75, 1341–1353. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.C.; Chen, Y.K.; Hsu, Y.J.; Lin, B.R. Niclosamide inhibits the cell proliferation and enhances the responsiveness of esophageal cancer cells to chemotherapeutic agents. Oncol. Rep. 2020, 43, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Ngo, M.T.; Jeng, H.Y.; Kuo, Y.C.; Diony Nanda, J.; Brahmadhi, A.; Ling, T.Y.; Chang, T.S.; Huang, Y.H. The role of IGF/IGF-1R signaling in hepatocellular carcinomas: Stemness-related properties and drug resistance. Int. J. Mol. Sci 2021, 22, 1931. [Google Scholar] [CrossRef] [PubMed]
- Ngo, M.T.; Peng, S.W.; Kuo, Y.C.; Lin, C.Y.; Wu, M.H.; Chuang, C.H.; Kao, C.X.; Jeng, H.Y.; Lin, G.W.; Ling, T.Y.; et al. A yes-associated protein (YAP) and insulin-like growth factor 1 receptor (IGF-1R) signaling loop is involved in sorafenib resistance in hepatocellular carcinoma. Cancers 2021, 13, 3812. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, B.D.; Diering, G.H.; Bidinosti, M.A.; Dalal, K.; Alain, T.; Balgi, A.D.; Forestieri, R.; Nodwell, M.; Rajadurai, C.V.; Gunaratnam, C.; et al. Structure-activity analysis of niclosamide reveals potential role for cytoplasmic pH in control of mammalian target of rapamycin complex 1 (mTORC1) signaling. J. Biol. Chem. 2012, 287, 17530–17545. [Google Scholar] [CrossRef]
- Balgi, A.D.; Fonseca, B.D.; Donohue, E.; Tsang, T.C.; Lajoie, P.; Proud, C.G.; Nabi, I.R.; Roberge, M. Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling. PLoS ONE 2009, 4, e7124. [Google Scholar] [CrossRef]
- Ren, X.; Duan, L.; He, Q.; Zhang, Z.; Zhou, Y.; Wu, D.; Pan, J.; Pei, D.; Ding, K. Identification of niclosamide as a new small-molecule inhibitor of the STAT3 signaling pathway. ACS Med. Chem. Lett. 2010, 1, 454–459. [Google Scholar] [CrossRef]
- Jin, Y.; Lu, Z.; Ding, K.; Li, J.; Du, X.; Chen, C.; Sun, X.; Wu, Y.; Zhou, J.; Pan, J. Antineoplastic mechanisms of niclosamide in acute myelogenous leukemia stem cells: Inactivation of the NF-kappaB pathway and generation of reactive oxygen species. Cancer Res. 2010, 70, 2516–2527. [Google Scholar] [CrossRef]
- Sack, U.; Walther, W.; Scudiero, D.; Selby, M.; Kobelt, D.; Lemm, M.; Fichtner, I.; Schlag, P.M.; Shoemaker, R.H.; Stein, U. Novel effect of antihelminthic niclosamide on S100A4-mediated metastatic progression in colon cancer. J. Natl. Cancer Inst. 2011, 103, 1018–1036. [Google Scholar] [CrossRef]
- Li, Y.; Li, P.K.; Roberts, M.J.; Arend, R.C.; Samant, R.S.; Buchsbaum, D.J. Multi-targeted therapy of cancer by niclosamide: A new application for an old drug. Cancer Lett. 2014, 349, 8–14. [Google Scholar] [CrossRef]
- Yo, Y.T.; Lin, Y.W.; Wang, Y.C.; Balch, C.; Huang, R.L.; Chan, M.W.; Sytwu, H.K.; Chen, C.K.; Chang, C.C.; Nephew, K.P.; et al. Growth inhibition of ovarian tumor-initiating cells by niclosamide. Mol. Cancer Ther. 2012, 11, 1703–1712. [Google Scholar] [CrossRef] [Green Version]
- Suliman, M.A.; Zhang, Z.; Na, H.; Ribeiro, A.L.; Zhang, Y.; Niang, B.; Hamid, A.S.; Zhang, H.; Xu, L.; Zuo, Y. Niclosamide inhibits colon cancer progression through downregulation of the Notch pathway and upregulation of the tumor suppressor miR-200 family. Int. J. Mol. Med. 2016, 38, 776–784. [Google Scholar] [CrossRef]
- Chen, B.; Wei, W.; Ma, L.; Yang, B.; Gill, R.M.; Chua, M.S.; Butte, A.J.; So, S. Computational discovery of niclosamide ethanolamine, a repurposed drug candidate that reduces growth of hepatocellular carcinoma cells in vitro and in mice by inhibiting cell division cycle 37 signaling. Gastroenterology 2017, 152, 2022–2036. [Google Scholar] [CrossRef]
- Zhao, J.; He, Q.; Gong, Z.; Chen, S.; Cui, L. Niclosamide suppresses renal cell carcinoma by inhibiting Wnt/β-catenin and inducing mitochondrial dysfunctions. Springerplus 2016, 5, 1436. [Google Scholar] [CrossRef]
- Chowdhury, M.K.; Turner, N.; Bentley, N.L.; Das, A.; Wu, L.E.; Richani, D.; Bustamante, S.; Gilchrist, R.B.; Morris, M.J.; Shepherd, P.R.; et al. Niclosamide reduces glucagon sensitivity via hepatic PKA inhibition in obese mice: Implications for glucose metabolism improvements in type 2 diabetes. Sci. Rep. 2017, 7, 40159. [Google Scholar] [CrossRef]
- Niu, L.; Liu, L.; Yang, S.; Ren, J.; Lai, P.B.S.; Chen, G.G. New insights into sorafenib resistance in hepatocellular carcinoma: Responsible mechanisms and promising strategies. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 564–570. [Google Scholar] [CrossRef]
- Zhai, B.; Hu, F.; Jiang, X.; Xu, J.; Zhao, D.; Liu, B.; Pan, S.; Dong, X.; Tan, G.; Wei, Z.; et al. Inhibition of Akt reverses the acquired resistance to sorafenib by switching protective autophagy to autophagic cell death in hepatocellular carcinoma. Mol. Cancer Ther. 2014, 13, 1589–1598. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Q.; Liu, J.; Cao, H. Inhibition of the PI3K/Akt signaling pathway reverses sorafenib-derived chemo-resistance in hepatocellular carcinoma. Oncol. Lett. 2018, 15, 9377–9384. [Google Scholar] [CrossRef]
- Wang, C.; Jin, H.; Gao, D.; Lieftink, C.; Evers, B.; Jin, G.; Xue, Z.; Wang, L.; Beijersbergen, R.L.; Qin, W.; et al. Phospho-ERK is a biomarker of response to a synthetic lethal drug combination of sorafenib and MEK inhibition in liver cancer. J. Hepatol. 2018, 69, 1057–1065. [Google Scholar] [CrossRef]
- Xie, L.; Zeng, Y.; Dai, Z.; He, W.; Ke, H.; Lin, Q.; Chen, Y.; Bu, J.; Lin, D.; Zheng, M. Chemical and genetic inhibition of STAT3 sensitizes hepatocellular carcinoma cells to sorafenib induced cell death. Int. J. Biol. Sci. 2018, 14, 577–585. [Google Scholar] [CrossRef]
- Su, J.C.; Tseng, P.H.; Wu, S.H.; Hsu, C.Y.; Tai, W.T.; Li, Y.S.; Chen, I.T.; Liu, C.Y.; Chen, K.F.; Shiau, C.W. SC-2001 overcomes STAT3-mediated sorafenib resistance through RFX-1/SHP-1 activation in hepatocellular carcinoma. Neoplasia 2014, 16, 595–605. [Google Scholar] [CrossRef] [Green Version]
- Méndez-Blanco, C.; Fondevila, F.; García-Palomo, A.; González-Gallego, J.; Mauriz, J.L. Sorafenib resistance in hepatocarcinoma: Role of hypoxia-inducible factors. Exp. Mol. Med. 2018, 50, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mir, N.; Jayachandran, A.; Dhungel, B.; Shrestha, R.; Steel, J.C. Epithelial-to-mesenchymal transition: A mediator of sorafenib resistance in advanced hepatocellular carcinoma. Curr. Cancer Drug Targets 2017, 17, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Wu, Y.; Yang, L.; Du, J.; Gong, K.; Chen, W.; Dai, J.; Li, X.; Xi, S. Repression of YAP by NCTD disrupts NSCLC progression. Oncotarget 2017, 8, 2307–2319. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Pan, Y.; Liang, Y.; Xu, J.; Cai, X. The microenvironmental and metabolic aspects of sorafenib resistance in hepatocellular carcinoma. EBioMedicine 2020, 51, 102610. [Google Scholar] [CrossRef]
- Ashokachakkaravarthy, K.; Pottakkat, B. Sorafenib resistance and autophagy in hepatocellular carcinoma: A concealed threat. J. Cancer Res. Pract. 2019, 6, 107–116. [Google Scholar]
- Al-Gayyar, M.M.H.; Bagalagel, A.; Noor, A.O.; Almasri, D.M.; Diri, R. The therapeutic effects of nicotinamide in hepatocellular carcinoma through blocking IGF-1 and effecting the balance between Nrf2 and PKB. Biomed. Pharmacother. 2019, 112, 108653. [Google Scholar] [CrossRef]
- Ren, Y.; Gu, Y.K.; Li, Z.; Xu, G.Z.; Zhang, Y.M.; Dong, M.X.; Wang, Y.; Zhou, X.B. CXCR3 confers sorafenib resistance of HCC cells through regulating metabolic alteration and AMPK pathway. Am. J. Transl. Res. 2020, 12, 825–836. [Google Scholar]
- Tang, W.; Chen, Z.; Zhang, W.; Cheng, Y.; Zhang, B.; Wu, F.; Wang, Q.; Wang, S.; Rong, D.; Reiter, F.P.; et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: Theoretical basis and therapeutic aspects. Signal. Transduct. Target. Ther. 2020, 5, 87. [Google Scholar] [CrossRef]
- Imamura, Y.; Mukohara, T.; Shimono, Y.; Funakoshi, Y.; Chayahara, N.; Toyoda, M.; Kiyota, N.; Takao, S.; Kono, S.; Nakatsura, T.; et al. Comparison of 2D- and 3D-culture models as drug-testing platforms in breast cancer. Oncol. Rep. 2015, 33, 1837–1843. [Google Scholar] [CrossRef]
- Alasadi, A.; Chen, M.; Swapna, G.V.T.; Tao, H.; Guo, J.; Collantes, J.; Fadhil, N.; Montelione, G.T.; Jin, S. Effect of mitochondrial uncouplers niclosamide ethanolamine (NEN) and oxyclozanide on hepatic metastasis of colon cancer. Cell Death Dis. 2018, 9, 215. [Google Scholar] [CrossRef]
- Tao, H.; Zhang, Y.; Zeng, X.; Shulman, G.I.; Jin, S. Niclosamide ethanolamine-induced mild mitochondrial uncoupling improves diabetic symptoms in mice. Nat. Med. 2014, 20, 1263–1269. [Google Scholar] [CrossRef]
- Wei, W.; Liu, H.; Yuan, J.; Yao, Y. Targeting Wnt/β-catenin by anthelmintic drug niclosamide overcomes paclitaxel resistance in esophageal cancer. Fundam. Clin. Pharmacol. 2021, 35, 165–173. [Google Scholar] [CrossRef]
- Chang, T.S.; Wu, Y.C.; Chi, C.C.; Su, W.C.; Chang, P.J.; Lee, K.F.; Tung, T.H.; Wang, J.; Liu, J.J.; Tung, S.Y.; et al. Activation of IL6/IGFIR confers poor prognosis of HBV-related hepatocellular carcinoma through induction of OCT4/NANOG expression. Clin. Cancer Res. 2015, 21, 201–210. [Google Scholar] [CrossRef]
- Lai, S.C.; Su, Y.T.; Chi, C.C.; Kuo, Y.C.; Lee, K.F.; Wu, Y.C.; Lan, P.C.; Yang, M.H.; Chang, T.S.; Huang, Y.H. DNMT3b/OCT4 expression confers sorafenib resistance and poor prognosis of hepatocellular carcinoma through IL-6/STAT3 regulation. J. Exp. Clin. Cancer Res. 2019, 38, 474. [Google Scholar] [CrossRef]
- Chen, P.C.; Kuo, Y.C.; Chuong, C.M.; Huang, Y.H. Niche modulation of IGF-1R signaling: Its role in stem cell pluripotency, cancer reprogramming, and therapeutic applications. Front. Cell Dev. Biol. 2020, 8, 625943. [Google Scholar] [CrossRef]
- Shan, Y.; Lu, C.; Wang, J.; Li, M.; Ye, S.; Wu, S.; Huang, J.; Bu, S.; Wang, F. IGF-1 contributes to liver cancer development in diabetes patients by promoting autophagy. Ann. Hepatol. 2022, 27, 100697. [Google Scholar] [CrossRef]
- Wang, F.; Bank, T.; Malnassy, G.; Arteaga, M.; Shang, N.; Dalheim, A.; Ding, X.; Cotler, S.J.; Denning, M.F.; Nishimura, M.I.; et al. Inhibition of insulin-like growth factor 1 receptor enhances the efficacy of sorafenib in inhibiting hepatocellular carcinoma cell growth and survival. Hepatol. Commun. 2018, 2, 732–746. [Google Scholar] [CrossRef]
- Suemura, S.; Kodama, T.; Myojin, Y.; Yamada, R.; Shigekawa, M.; Hikita, H.; Sakamori, R.; Tatsumi, T.; Takehara, T. CRISPR loss-of-function screen identifies the hippo signaling pathway as the mediator of regorafenib efficacy in hepatocellular carcinoma. Cancers 2019, 11, 1362. [Google Scholar] [CrossRef]
- Lippolis, C.; Refolo, M.G.; D’Alessandro, R.; Carella, N.; Messa, C.; Cavallini, A.; Carr, B.I. Resistance to multikinase inhibitor actions mediated by insulin like growth factor-1. J. Exp. Clin. Cancer Res. 2015, 34, 90. [Google Scholar] [CrossRef] [Green Version]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, S.-W.; Ngo, M.-H.T.; Kuo, Y.-C.; Teng, M.-H.; Guo, C.-L.; Lai, H.-C.; Chang, T.-S.; Huang, Y.-H. Niclosamide Revitalizes Sorafenib through Insulin-like Growth Factor 1 Receptor (IGF-1R)/Stemness and Metabolic Changes in Hepatocellular Carcinoma. Cancers 2023, 15, 931. https://doi.org/10.3390/cancers15030931
Peng S-W, Ngo M-HT, Kuo Y-C, Teng M-H, Guo C-L, Lai H-C, Chang T-S, Huang Y-H. Niclosamide Revitalizes Sorafenib through Insulin-like Growth Factor 1 Receptor (IGF-1R)/Stemness and Metabolic Changes in Hepatocellular Carcinoma. Cancers. 2023; 15(3):931. https://doi.org/10.3390/cancers15030931
Chicago/Turabian StylePeng, Syue-Wei, Mai-Huong T. Ngo, Yung-Che Kuo, Ming-Hao Teng, Chin-Lin Guo, Hung-Cheng Lai, Te-Sheng Chang, and Yen-Hua Huang. 2023. "Niclosamide Revitalizes Sorafenib through Insulin-like Growth Factor 1 Receptor (IGF-1R)/Stemness and Metabolic Changes in Hepatocellular Carcinoma" Cancers 15, no. 3: 931. https://doi.org/10.3390/cancers15030931
APA StylePeng, S.-W., Ngo, M.-H. T., Kuo, Y.-C., Teng, M.-H., Guo, C.-L., Lai, H.-C., Chang, T.-S., & Huang, Y.-H. (2023). Niclosamide Revitalizes Sorafenib through Insulin-like Growth Factor 1 Receptor (IGF-1R)/Stemness and Metabolic Changes in Hepatocellular Carcinoma. Cancers, 15(3), 931. https://doi.org/10.3390/cancers15030931