
Mechanism of cis-Nerolidol-Induced Bladder Carcinoma Cell Death


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture
2.3. Cell Counting and Trypan Blue Staining
2.4. Annexin-V/PI Staining
2.5. Hydrogen Peroxide Levels in Nerolidol-Treated Cells (H2O2-Glo)
2.6. Measurement of Cytosolic Ca2+
2.7. Determination of ATP
2.8. Measurement of cAMP
2.9. SDS-PAGE and Western Blot Analysis
2.10. Alkaline Comet Assay
2.11. Statistical Analysis
3. Results
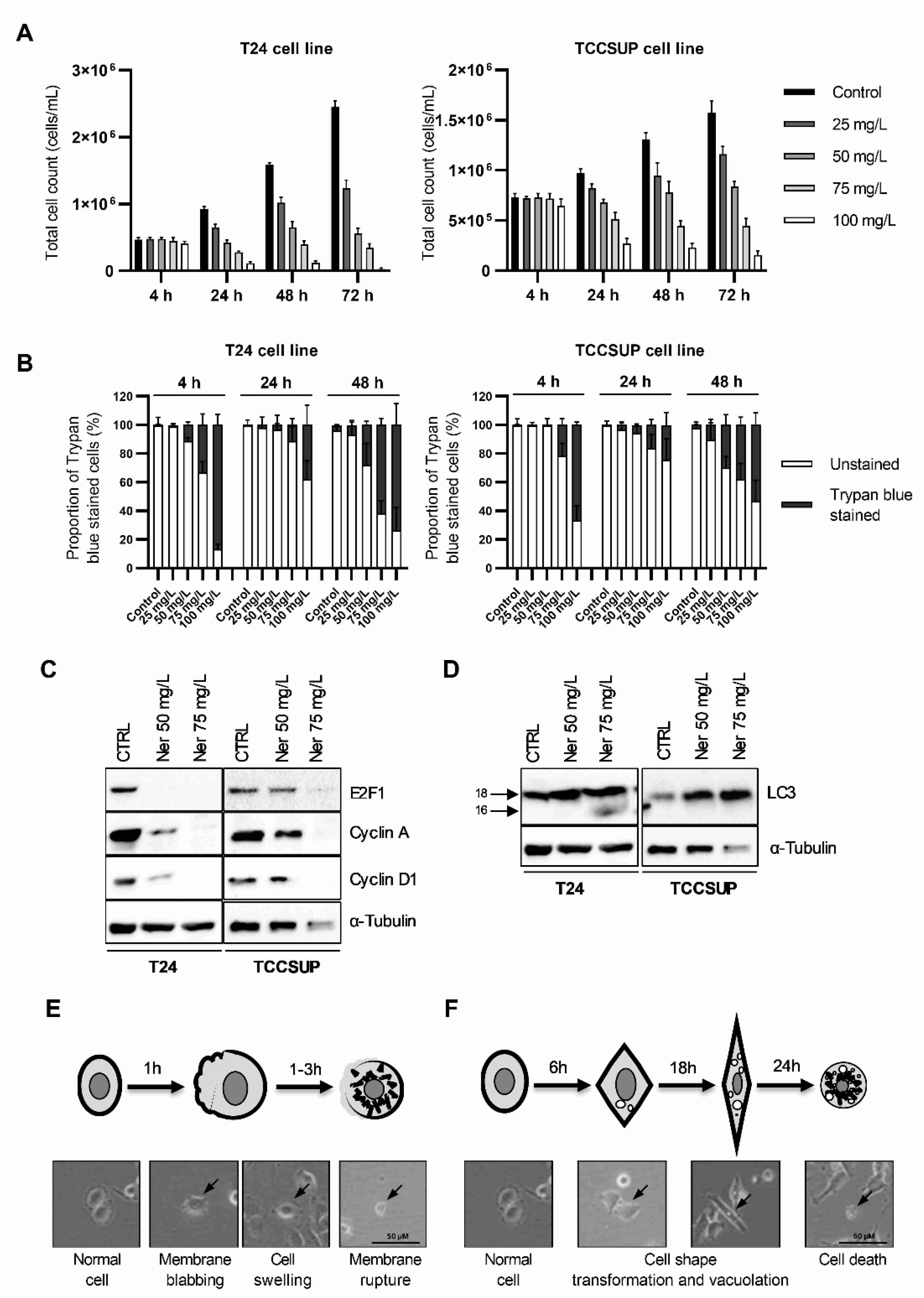
3.1. Nerolidol Induces Two Different Mechanisms of Cell Death with Distinct Cell Morphologies
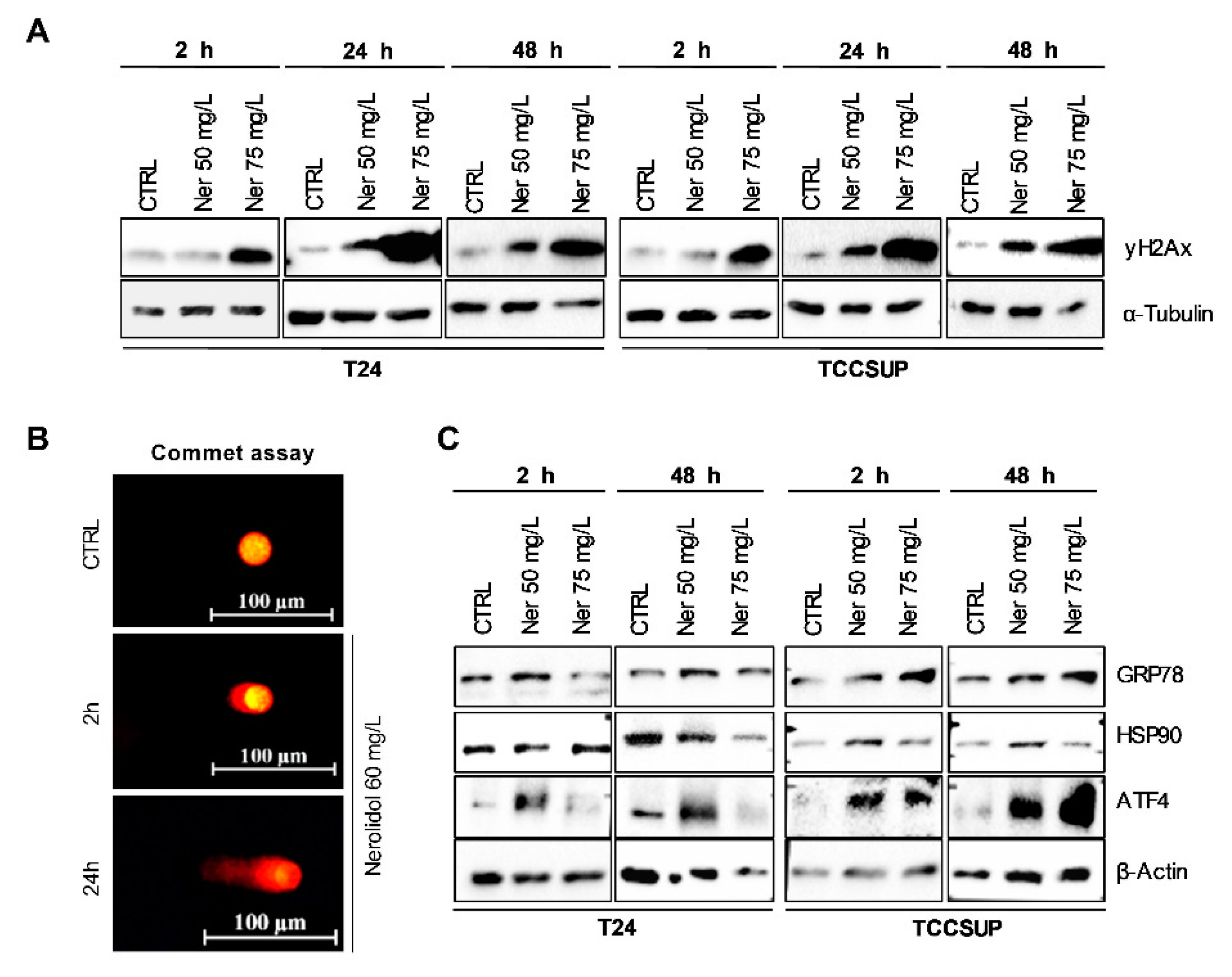
3.2. Prolonged Nerolidol Exposure Caused Increasing Levels of DNA Damage and ER Stress
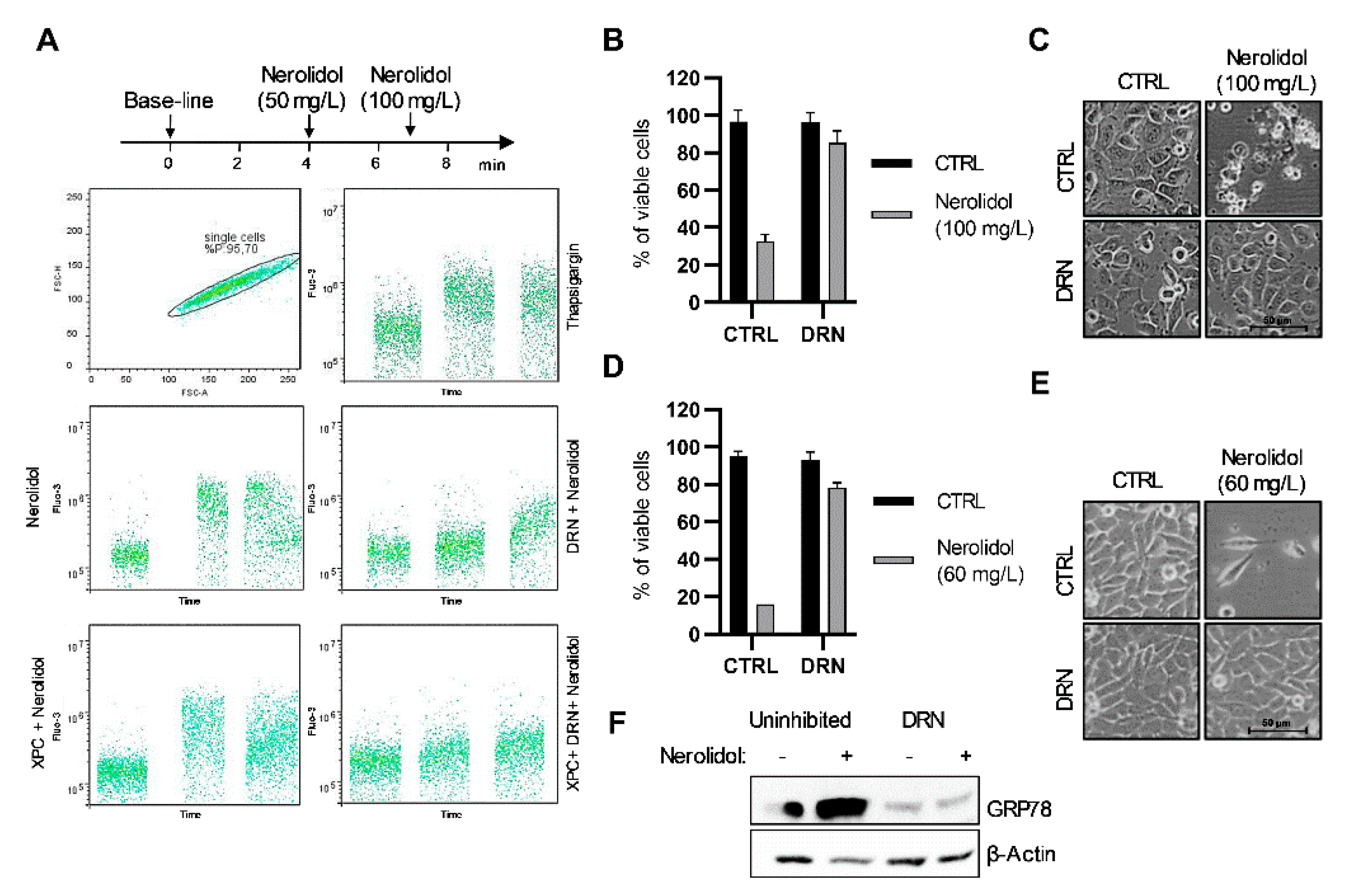
3.3. Nerolidol Cytotoxicity Originates from Ca2+ Depletion in the ER through Ryanodine Receptors (RYR)
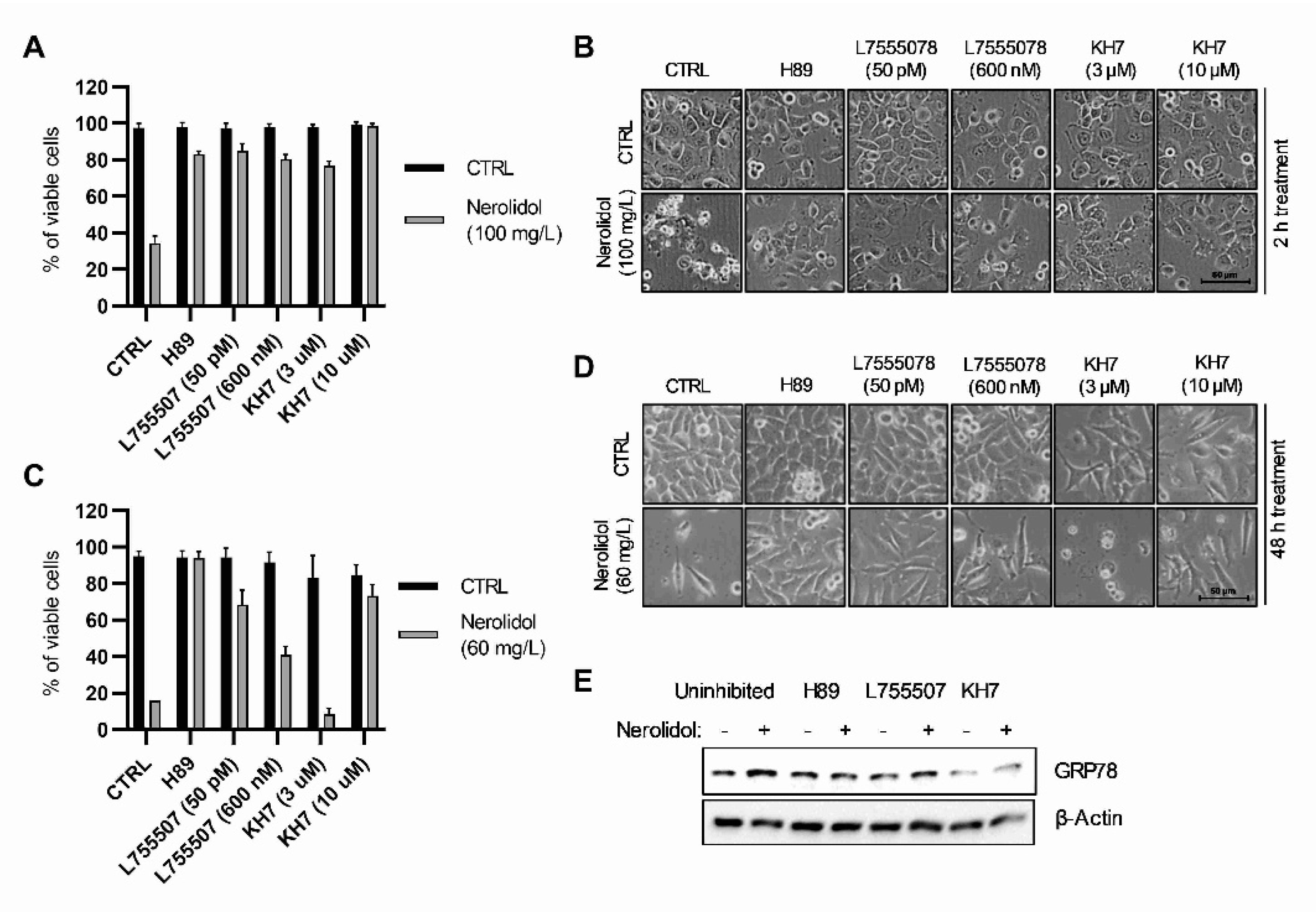
3.4. The Upstream Signals of Both Cell Death Pathways Include cAMP-Dependent Signal Transduction through β-Adrenergic Receptors, PKA, and Soluble Adenylyl Cyclases
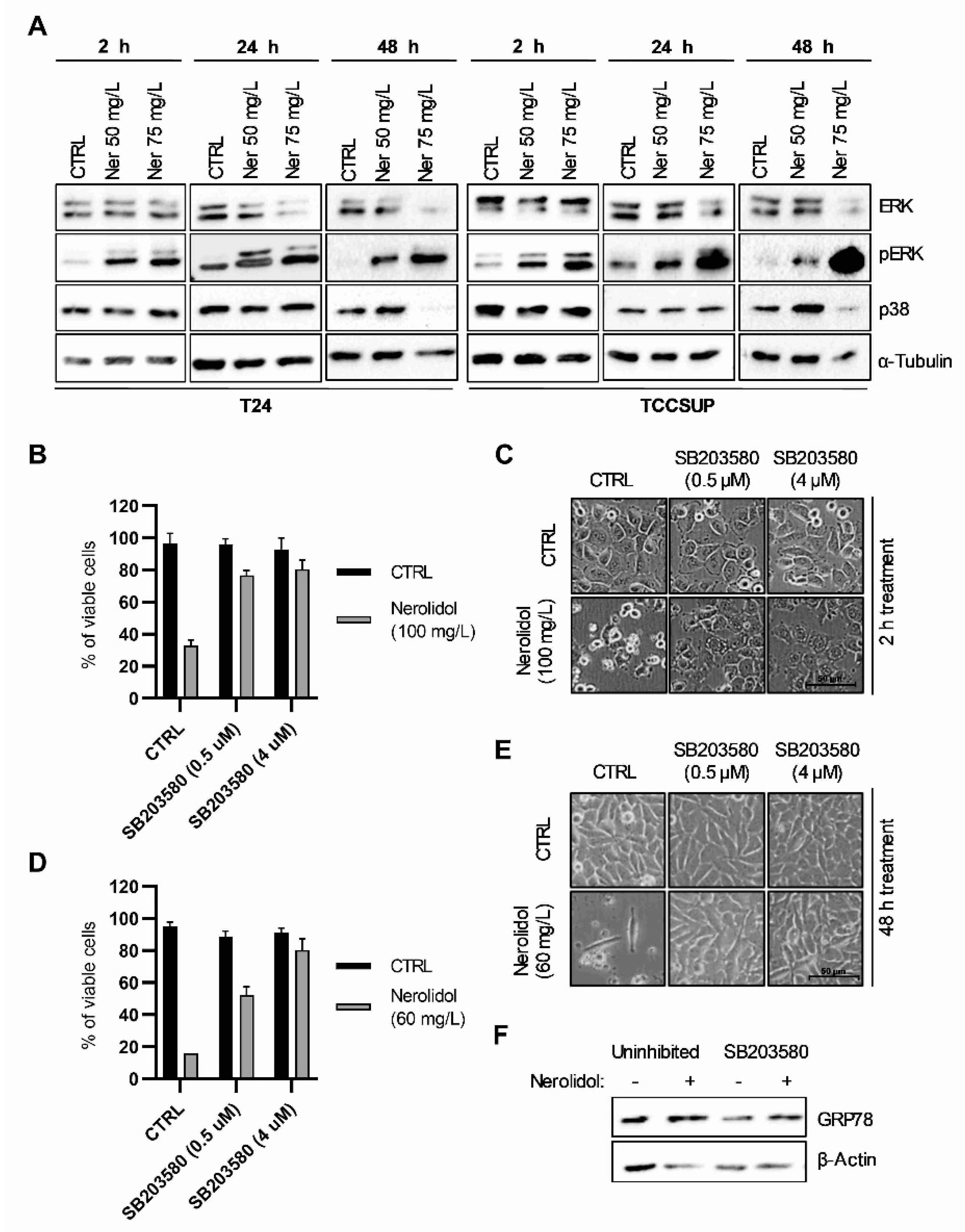
3.5. Nerolidol Cytotoxicity Is Dependent on MAPK Activation Downstream of ER Stress
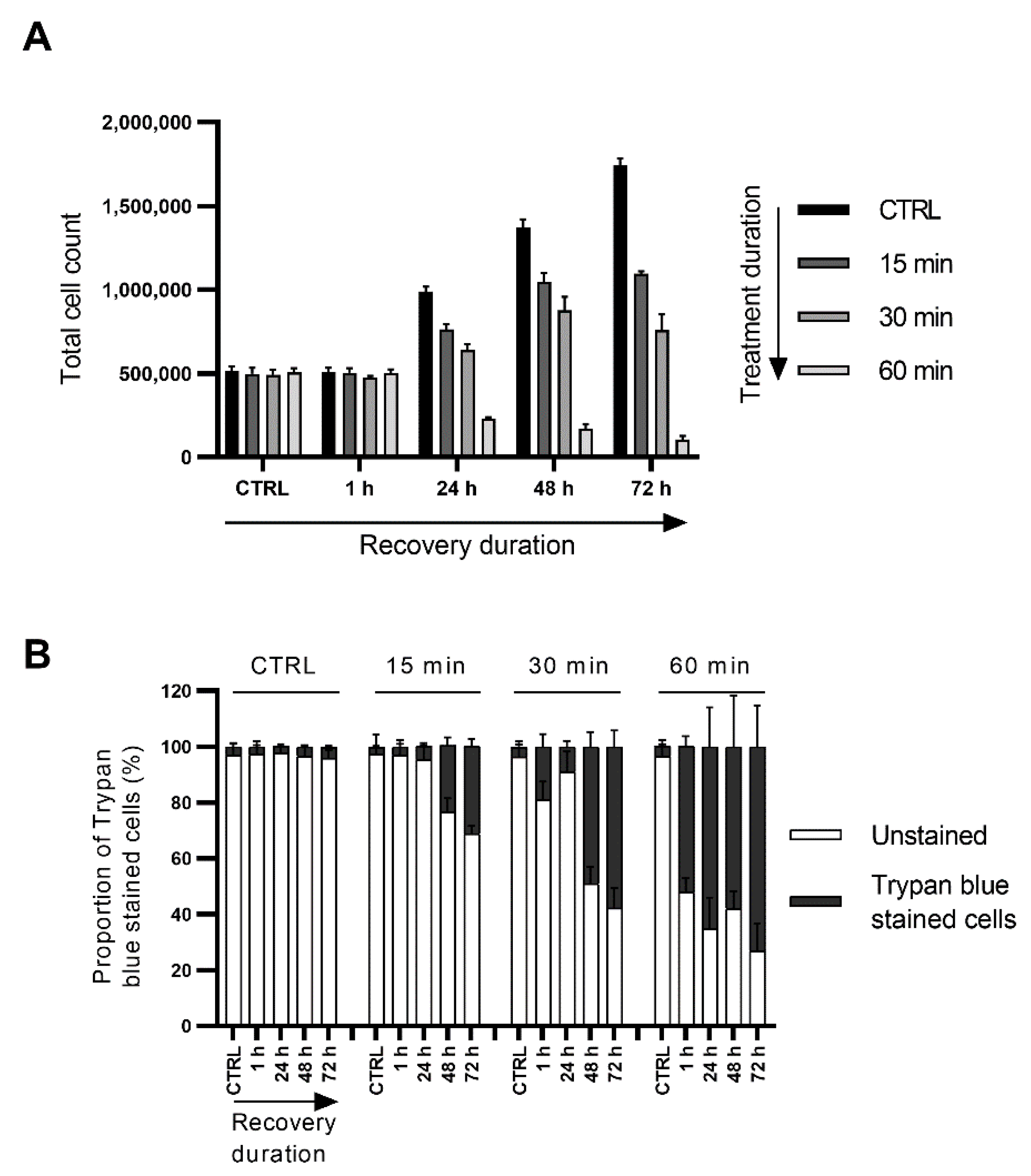
3.6. Short Exposure to Nerolidol Induces Similar Cytotoxic Effect on Cells as Continuous Treatments
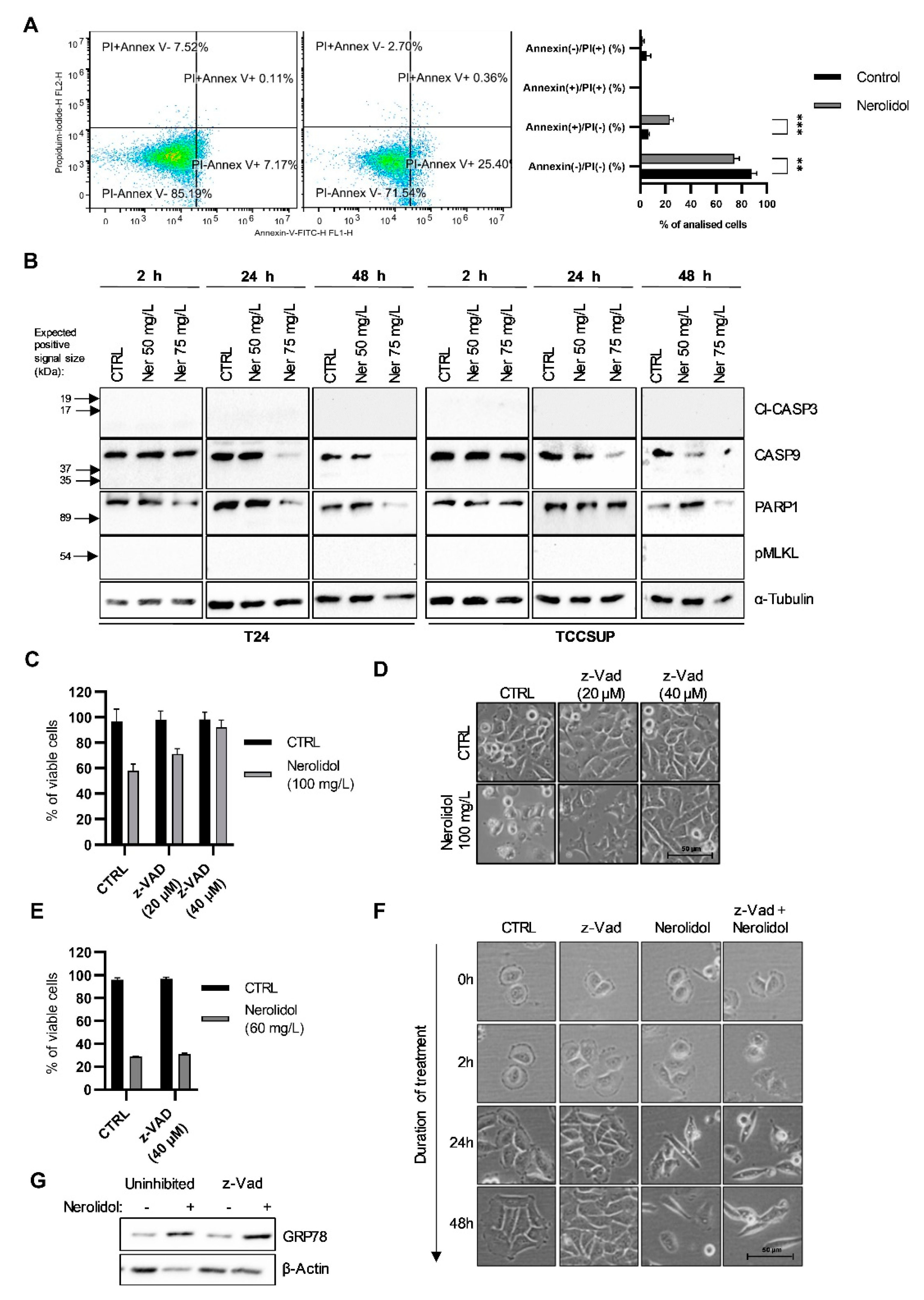
3.7. The Two Pathways of Cell Death Differ by Their Involvement of Caspases
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schubert, V.; Dietrich, A.; Ulrich, T.; Mosandl, A. The Stereoisomers of Nerolidol: Separation, Analysis and Olfactoric Properties. Zeitschrift Naturforsch. C 1992, 47, 304–307. [Google Scholar] [CrossRef]
- Chan, W.-K.; Tan, L.; Chan, K.-G.; Lee, L.-H.; Goh, B.-H. Nerolidol: A Sesquiterpene Alcohol with Multi-Faceted Pharmacological and Biological Activities. Molecules 2016, 21, 529. [Google Scholar] [CrossRef]
- Lapczynski, A.; Bhatia, S.P.; Letizia, C.S.; Api, A.M. Fragrance Material Review on Nerolidol (Isomer Unspecified). Food Chem. Toxicol. 2008, 46, S247–S250. [Google Scholar] [CrossRef]
- Vinholes, J.; Gonçalves, P.; Martel, F.; Coimbra, M.A.; Rocha, S.M. Assessment of the Antioxidant and Antiproliferative Effects of Sesquiterpenic Compounds in in Vitro Caco-2 Cell Models. Food Chem. 2014, 156, 204–211. [Google Scholar] [CrossRef]
- Javed, H.; Azimullah, S.; Abul Khair, S.B.; Ojha, S.; Haque, M.E. Neuroprotective Effect of Nerolidol against Neuroinflammation and Oxidative Stress Induced by Rotenone. BMC Neurosci. 2016, 17, 58. [Google Scholar] [CrossRef]
- Arunachalam, S.; Nagoor Meeran, M.F.; Azimullah, S.; Sharma, C.; Goyal, S.N.; Ojha, S. Nerolidol Attenuates Oxidative Stress, Inflammation, and Apoptosis by Modulating Nrf2/MAPK Signaling Pathways in Doxorubicin-Induced Acute Cardiotoxicity in Rats. Antioxidants 2021, 10, 984. [Google Scholar] [CrossRef]
- Ni, Y.-L.; Shen, H.-T.; Su, C.-H.; Chen, W.-Y.; Huang-Liu, R.; Chen, C.-J.; Chen, S.-P.; Kuan, Y.-H. Nerolidol Suppresses the Inflammatory Response during Lipopolysaccharide-Induced Acute Lung Injury via the Modulation of Antioxidant Enzymes and the AMPK/Nrf-2/HO-1 Pathway. Oxid. Med. Cell Longev. 2019, 2019, 9605980. [Google Scholar] [CrossRef]
- Fonsêca, D.V.; Salgado, P.R.R.; de Carvalho, F.L.; Salvadori, M.G.S.S.; Penha, A.R.S.; Leite, F.C.; Borges, C.J.S.; Piuvezam, M.R.; de Morais Pordeus, L.C.; Sousa, D.P.; et al. Nerolidol Exhibits Antinociceptive and Anti-Inflammatory Activity: Involvement of the GABAergic System and Proinflammatory Cytokines. Fundam. Clin. Pharmacol. 2016, 30, 14–22. [Google Scholar] [CrossRef]
- Brehm-Stecher, B.F.; Johnson, E.A. Sensitization of Staphylococcus Aureus and Escherichia Coli to Antibiotics by the Sesquiterpenoids Nerolidol, Farnesol, Bisabolol, and Apritone. Antimicrob. Agents Chemother. 2003, 47, 3357–3360. [Google Scholar] [CrossRef]
- Ryabchenko, B.; Tulupova, E.; Schmidt, E.; Wlcek, K.; Buchbauer, G.; Jirovetz, L. Investigation of Anticancer and Antiviral Properties of Selected Aroma Samples. Nat. Prod. Commun. 2008, 3, 1085–1088. [Google Scholar] [CrossRef]
- Fernandes, F.H.; Guterres, Z.d.R.; Violante, I.M.P.; Lopes, T.F.S.; Garcez, W.S.; Garcez, F.R. Evaluation of Mutagenic and Antimicrobial Properties of Brown Propolis Essential Oil from the Brazilian Cerrado Biome. Toxicol. Rep. 2015, 2, 1482–1488. [Google Scholar] [CrossRef]
- Mutombo, A.B.; Tozin, R.; Kanyiki, H.; Van Geertruyden, J.-P.; Jacquemyn, Y. Impact of Antiviral AV2 in the Topical Treatment of HPV-Associated Lesions of the Cervix: Results of a Phase III Randomized Placebo-Controlled Trial. Contemp. Clin. Trials Commun. 2019, 15, 100377. [Google Scholar] [CrossRef]
- Martins, M.; Henriques, M.; Azeredo, J.; Rocha, S.M.; Coimbra, M.A.; Oliveira, R. Morphogenesis Control in Candida Albicans and Candida Dubliniensis through Signaling Molecules Produced by Planktonic and Biofilm Cells. Eukaryot. Cell 2007, 6, 2429–2436. [Google Scholar] [CrossRef]
- Camargos, H.S.; Moreira, R.A.; Mendanha, S.A.; Fernandes, K.S.; Dorta, M.L.; Alonso, A. Terpenes Increase the Lipid Dynamics in the Leishmania Plasma Membrane at Concentrations Similar to Their IC50 Values. PLoS ONE 2014, 9, e104429. [Google Scholar] [CrossRef] [PubMed]
- Alonso, L.; Fernandes, K.S.; Mendanha, S.A.; Gonçalves, P.J.; Gomes, R.S.; Dorta, M.L.; Alonso, A. In Vitro Antileishmanial and Cytotoxic Activities of Nerolidol Are Associated with Changes in Plasma Membrane Dynamics. Biochim. Biophys. Acta Biomembr. 2019, 1861, 1049–1056. [Google Scholar] [CrossRef]
- Silva, M.; Oliveira, G.; de Carvalho, R.; de Sousa, D.; Freitas, R.; Pinto, P.; Moraes, J. Antischistosomal Activity of the Terpene Nerolidol. Molecules 2014, 19, 3793–3803. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.Y.; Marin Rodriguez, A.A.; Menchaca Vega, D.S.; Sussmann, R.A.C.; Kimura, E.A.; Katzin, A.M. Antimalarial Activity of the Terpene Nerolidol. Int. J. Antimicrob. Agents 2016, 48, 641–646. [Google Scholar] [CrossRef]
- Ambrož, M.; Matoušková, P.; Skarka, A.; Zajdlová, M.; Žáková, K.; Skálová, L. The Effects of Selected Sesquiterpenes from Myrica Rubra Essential Oil on the Efficacy of Doxorubicin in Sensitive and Resistant Cancer Cell Lines. Molecules 2017, 22, 1021. [Google Scholar] [CrossRef]
- Biazi, B.I.; Zanetti, T.A.; Baranoski, A.; Corveloni, A.C.; Mantovani, M.S. Cis-Nerolidol Induces Endoplasmic Reticulum Stress and Cell Death in Human Hepatocellular Carcinoma Cells through Extensive CYP2C19 and CYP1A2 Oxidation. Basic Clin. Pharmacol. Toxicol. 2017, 121, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Hanušova, V.; Skalova, L.; Ambrož, M.; Kralova, V.; Longhansova, L.; Matouškova, P. The Effect of Myrica Rubra Essential Oil and Its Components α-Humulene and Trans-Nerolidol on Adhesion and Apoptosis of Colorectal Cancer Cells. Cancer Cell Microenviron. 2015, 2, e1058. [Google Scholar] [CrossRef]
- Boris, R.; Elena, T.; Erich, S.; Walter, J.; Gerhard, B.; Leopold, J. Cytotoxic Properties of Selected Sesquiterpene Alcohols on Human Cervix Carcinoma Cell Lines. J. Essent. Oil Bear. Plants 2011, 14, 316–319. [Google Scholar] [CrossRef]
- Ferreira, F.M.; Palmeira, C.M.; Oliveira, M.M.; Santos, D.; Simões, A.M.; Rocha, S.M.; Coimbra, M.A.; Peixoto, F. Nerolidol Effects on Mitochondrial and Cellular Energetics. Toxicol. Vitr. 2012, 26, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Ambrož, M.; Šmatová, M.; Šadibolová, M.; Pospíšilová, E.; Hadravská, P.; Kašparová, M.; Skarková, V.H.; Králová, V.; Skálová, L. Sesquiterpenes α-Humulene and β-Caryophyllene Oxide Enhance the Efficacy of 5-Fluorouracil and Oxaliplatin in Colon Cancer Cells. Acta Pharm. 2019, 69, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Mendanha, S.A.; Moura, S.S.; Anjos, J.L.V.; Valadares, M.C.; Alonso, A. Toxicity of Terpenes on Fibroblast Cells Compared to Their Hemolytic Potential and Increase in Erythrocyte Membrane Fluidity. Toxicol. Vitr. 2013, 27, 323–329. [Google Scholar] [CrossRef]
- Wattenberg, L.W. Inhibition of Azoxymethane-Induced Neoplasia of the Large Bowel by 3-Hydroxy-3,7,11-Trimethyl-l,6,10-Dodecatriene (Nerolidol). Carcinogenesis 1991, 12, 151–152. [Google Scholar] [CrossRef] [PubMed]
- Nogueira Neto, J.D.; de Almeida, A.A.C.; da Silva Oliveira, J.; dos Santos, P.S.; de Sousa, D.P.; de Freitas, R.M. Antioxidant Effects of Nerolidol in Mice Hippocampus After Open Field Test. Neurochem. Res. 2013, 38, 1861–1870. [Google Scholar] [CrossRef]
- Saginala, K.; Barsouk, A.; Aluru, J.S.; Rawla, P.; Padala, S.A.; Barsouk, A. Epidemiology of Bladder Cancer. Med. Sci. 2020, 8, 15. [Google Scholar] [CrossRef]
- Tran, L.; Xiao, J.-F.; Agarwal, N.; Duex, J.E.; Theodorescu, D. Advances in Bladder Cancer Biology and Therapy. Nat. Rev. Cancer 2021, 21, 104–121. [Google Scholar] [CrossRef]
- de Wit, M.; Retz, M.M.; Rödel, C.; Gschwend, J.E. The Diagnosis and Treatment of Patients with Bladder Carcinoma. Dtsch. Arztebl. Int. 2021, 118, 169–176. [Google Scholar] [CrossRef]
- Huang, B.; Huang, G.; Li, W.; Chen, L.; Mao, X.; Chen, J. Intra-Arterial Chemotherapy Combined with Intravesical Chemotherapy Compared with Intravesical BCG Immunotherapy Retrospectively in High-Risk Non-Muscle-Invasive Bladder Cancer after Transurethral Resection of the Bladder Tumor. J. Cancer Res. Clin. Oncol. 2021, 147, 1781–1788. [Google Scholar] [CrossRef]
- Porten, S.; Leapman, M.; Greene, K. Intravesical Chemotherapy in Non-Muscle-Invasive Bladder Cancer. Indian J. Urol. 2015, 31, 297. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Chen, M.; Wen, X.; Gao, Y.; Huang, D.; Cao, H.; Peng, Y.; Guo, N.; Ni, J.; Zhang, S. Endoplasmic Reticulum Stress and Tumor Microenvironment in Bladder Cancer: The Missing Link. Front. Cell Dev. Biol. 2021, 9, 683940. [Google Scholar] [CrossRef] [PubMed]
- Gild, P.; Nguyen, D.-D.; Fletcher, S.A.; Cole, A.P.; Lipsitz, S.R.; Kibel, A.S.; Fisch, M.; Preston, M.A.; Trinh, Q.-D. Contemporary Survival Rates for Muscle-Invasive Bladder Cancer Treated With Definitive or Non-Definitive Therapy. Clin. Genitourin. Cancer 2019, 17, e488–e493. [Google Scholar] [CrossRef]
- Vines, A.; McBean, G.J.; Blanco-Fernández, A. A Flow-Cytometric Method for Continuous Measurement of Intracellular Ca2+ Concentration. Cytom. Part A 2010, 77A, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.-H.; Leem, D.-G.; Shin, J.-S.; Kim, J.-I.; Kim, K.-T.; Choi, S.Y.; Lee, M.-H.; Choi, J.-H.; Lee, K.-T. Compound K Induced Apoptosis via Endoplasmic Reticulum Ca2+ Release through Ryanodine Receptor in Human Lung Cancer Cells. J. Ginseng Res. 2018, 42, 165–174. [Google Scholar] [CrossRef]
- Wang, F.; Yu, J.; Lin, P.; Sigalas, C.; Zhang, S.; Gong, Y.; Sitsapesan, R.; Song, L. The Ryanodine Receptor Mutational Characteristics and Its Indication for Cancer Prognosis. Sci. Rep. 2022, 12, 16113. [Google Scholar] [CrossRef]
- Wen, J.; Chen, Z.; Zhao, M.; Zu, S.; Zhao, S.; Wang, S.; Zhang, X. Cell Deformation at the Air-Liquid Interface Evokes Intracellular Ca2+ Increase and ATP Release in Cultured Rat Urothelial Cells. Front. Physiol. 2021, 12, 631022. [Google Scholar] [CrossRef]
- Niggli, E.; Ullrich, N.D.; Gutierrez, D.; Kyrychenko, S.; Poláková, E.; Shirokova, N. Posttranslational Modifications of Cardiac Ryanodine Receptors: Ca2+ Signaling and EC-Coupling. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 866–875. [Google Scholar] [CrossRef]
- George, I.; Sabbah, H.N.; Xu, K.; Wang, N.; Wang, J. β-Adrenergic Receptor Blockade Reduces Endoplasmic Reticulum Stress and Normalizes Calcium Handling in a Coronary Embolization Model of Heart Failure in Canines. Cardiovasc. Res. 2011, 91, 447–455. [Google Scholar] [CrossRef]
- Dalal, S.; Foster, C.R.; Das, B.C.; Singh, M.; Singh, K. β-Adrenergic Receptor Stimulation Induces Endoplasmic Reticulum Stress in Adult Cardiac Myocytes: Role in Apoptosis. Mol. Cell. Biochem. 2012, 364, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Sassone-Corsi, P. The Cyclic AMP Pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011148. [Google Scholar] [CrossRef] [PubMed]
- Steegborn, C. Structure, Mechanism, and Regulation of Soluble Adenylyl Cyclases—Similarities and Differences to Transmembrane Adenylyl Cyclases. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 2535–2547. [Google Scholar] [CrossRef]
- Jiang, Q.; Li, F.; Shi, K.; Wu, P.; An, J.; Yang, Y.; Xu, C. Involvement of P38 in Signal Switching from Autophagy to Apoptosis via the PERK/EIF2α/ATF4 Axis in Selenite-Treated NB4 Cells. Cell Death Dis. 2014, 5, e1270. [Google Scholar] [CrossRef] [PubMed]
- Denton, D.; Kumar, S. Autophagy-Dependent Cell Death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Lee, D.M.; Seo, M.J.; Lee, H.J.; Choi, K.S. Intracellular Ca2+ Imbalance Critically Contributes to Paraptosis. Front. Cell Dev. Biol. 2021, 8, 607844. [Google Scholar] [CrossRef]
- Zhou, W.; Yuan, J. Necroptosis in Health and Diseases. Semin. Cell Dev. Biol. 2014, 35, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Shubin, A.V.; Demidyuk, I.V.; Komissarov, A.A.; Rafieva, L.M.; Kostrov, S.V. Cytoplasmic Vacuolization in Cell Death and Survival. Oncotarget 2016, 7, 55863–55889. [Google Scholar] [CrossRef]
- Mimnaugh, E.G.; Xu, W.; Vos, M.; Yuan, X.; Neckers, L. Endoplasmic Reticulum Vacuolization and Valosin-Containing Protein Relocalization Result from Simultaneous Hsp90 Inhibition by Geldanamycin and Proteasome Inhibition by Velcade. Mol. Cancer Res. 2006, 4, 667–681. [Google Scholar] [CrossRef]
- Han, Y.; Yuan, M.; Guo, Y.-S.; Shen, X.-Y.; Gao, Z.-K.; Bi, X. Mechanism of Endoplasmic Reticulum Stress in Cerebral Ischemia. Front. Cell. Neurosci. 2021, 15, 704334. [Google Scholar] [CrossRef]
- Nirmala, J.G.; Lopus, M. Cell Death Mechanisms in Eukaryotes. Cell Biol. Toxicol. 2020, 36, 145–164. [Google Scholar] [CrossRef]
- Zheng, Z.; Li, G. Mechanisms and Therapeutic Regulation of Pyroptosis in Inflammatory Diseases and Cancer. Int. J. Mol. Sci. 2020, 21, 1456. [Google Scholar] [CrossRef]
- Zheng, Z.; Wang, T.; Chen, J.; Qiu, H.; Zhang, C.; Liu, W.; Qin, S.; Tian, J.; Guo, J. Inflammasome-Induced Osmotic Pressure and the Mechanical Mechanisms Underlying Astrocytic Swelling and Membrane Blebbing in Pyroptosis. Front. Immunol. 2021, 12, 688674. [Google Scholar] [CrossRef]
- Li, D.; Ren, W.; Jiang, Z.; Zhu, L. Regulation of the NLRP3 Inflammasome and Macrophage Pyroptosis by the P38 MAPK Signaling Pathway in a Mouse Model of Acute Lung Injury. Mol. Med. Rep. 2018, 18, 4399–4409. [Google Scholar] [CrossRef] [PubMed]
- Bröker, L.E.; Kruyt, F.A.E.; Giaccone, G. Cell Death Independent of Caspases: A Review. Clin. Cancer Res. 2005, 11, 3155–3162. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wen, X.; Zhang, N.; Wang, L.; Hao, D.; Jiang, X.; He, G. Small-Molecule Compounds Target Paraptosis to Improve Cancer Therapy. Biomed. Pharmacother. 2019, 118, 109203. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Gu, Y.; Bian, Y.; Cai, D.; Li, Y.; Zhao, Y.; Zhang, Z.; Xue, M.; Zhang, L. Honokiol Induces Paraptosis-like Cell Death of Acute Promyelocytic Leukemia via MTOR & MAPK Signaling Pathways Activation. Apoptosis 2021, 26, 195–208. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.-S. Calcium, ATP, and ROS: A Mitochondrial Love-Hate Triangle. Am. J. Physiol. Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Blankenberg, F.G. In Vivo Imaging of Apoptosis. Cancer Biol. Ther. 2008, 7, 1525–1532. [Google Scholar] [CrossRef]
- Chao, H.X.; Poovey, C.E.; Privette, A.A.; Grant, G.D.; Chao, H.Y.; Cook, J.G.; Purvis, J.E. Orchestration of DNA Damage Checkpoint Dynamics across the Human Cell Cycle. Cell Syst. 2017, 5, 445–459.e5. [Google Scholar] [CrossRef]
- Dong, J.-R.; Chang, W.-W.; Chen, S.-M. Nerolidol Inhibits Proliferation of Leiomyoma Cells via Reactive Oxygen Species-Induced DNA Damage and Downregulation of the ATM/Akt Pathway. Phytochemistry 2021, 191, 112901. [Google Scholar] [CrossRef]
- Tyagi, P.; Thomas, C.A.; Yoshimura, N.; Chancellor, M.B. Investigations into the Presence of Functional SS1, SS2 and SS3-Adrenoceptors in Urothelium and Detrusor of Human Bladder. Int. Braz J. Urol. 2009, 35, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Birder, L.A.; Nealen, M.L.; Kiss, S.; de Groat, W.C.; Caterina, M.J.; Wang, E.; Apodaca, G.; Kanai, A.J. β-Adrenoceptor Agonists Stimulate Endothelial Nitric Oxide Synthase in Rat Urinary Bladder Urothelial Cells. J. Neurosci. 2002, 22, 8063–8070. [Google Scholar] [CrossRef] [PubMed]
- Rains, S.L.; Amaya, C.N.; Bryan, B.A. Beta-Adrenergic Receptors Are Expressed across Diverse Cancers. Oncoscience 2017, 4, 95–105. [Google Scholar] [CrossRef]
- Cole, S.W.; Sood, A.K. Molecular Pathways: Beta-Adrenergic Signaling in Cancer. Clin. Cancer Res. 2012, 18, 1201–1206. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glumac, M.; Čikeš Čulić, V.; Marinović-Terzić, I.; Radan, M. Mechanism of cis-Nerolidol-Induced Bladder Carcinoma Cell Death. Cancers 2023, 15, 981. https://doi.org/10.3390/cancers15030981
Glumac M, Čikeš Čulić V, Marinović-Terzić I, Radan M. Mechanism of cis-Nerolidol-Induced Bladder Carcinoma Cell Death. Cancers. 2023; 15(3):981. https://doi.org/10.3390/cancers15030981
Chicago/Turabian StyleGlumac, Mateo, Vedrana Čikeš Čulić, Ivana Marinović-Terzić, and Mila Radan. 2023. "Mechanism of cis-Nerolidol-Induced Bladder Carcinoma Cell Death" Cancers 15, no. 3: 981. https://doi.org/10.3390/cancers15030981
APA StyleGlumac, M., Čikeš Čulić, V., Marinović-Terzić, I., & Radan, M. (2023). Mechanism of cis-Nerolidol-Induced Bladder Carcinoma Cell Death. Cancers, 15(3), 981. https://doi.org/10.3390/cancers15030981