
Heat-Shock Proteins in Leukemia and Lymphoma: Multitargets for Innovative Therapeutic Approaches

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. HSP Families’ Overview
2.1. Heat-Shock Factor
2.2. HSP110 Family
2.3. HSP90 Family
2.4. HSP70 Family
2.5. Small HSPs Family
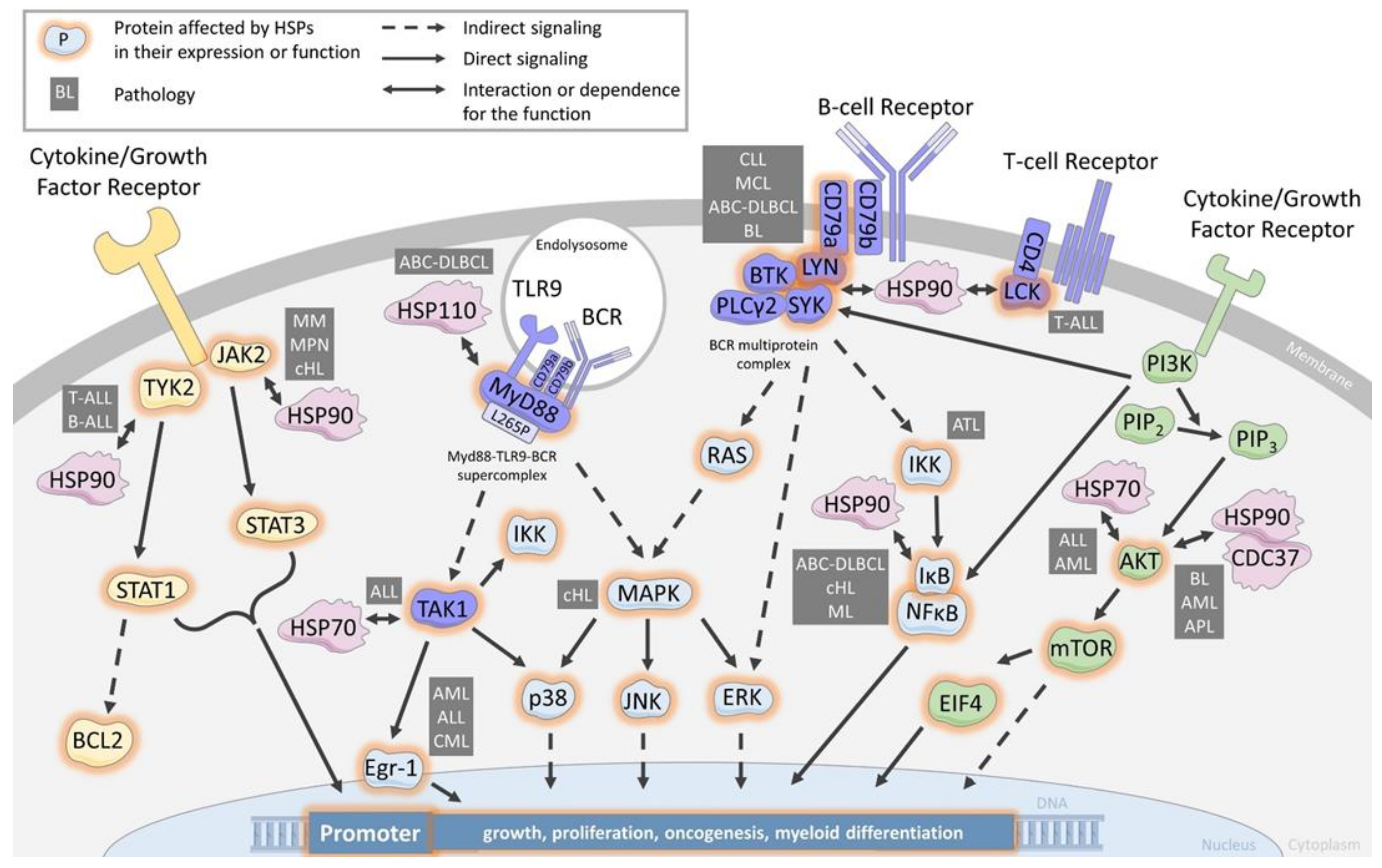
3. HSPs as Chaperons of Cell Signaling Proteins
3.1. Chaperoning the B-Cell Receptor and the T-Cell-Receptor
3.2. Chaperoning the JAK/STAT Pathway
3.3. Chaperoning the PI3K/AKT Pathway
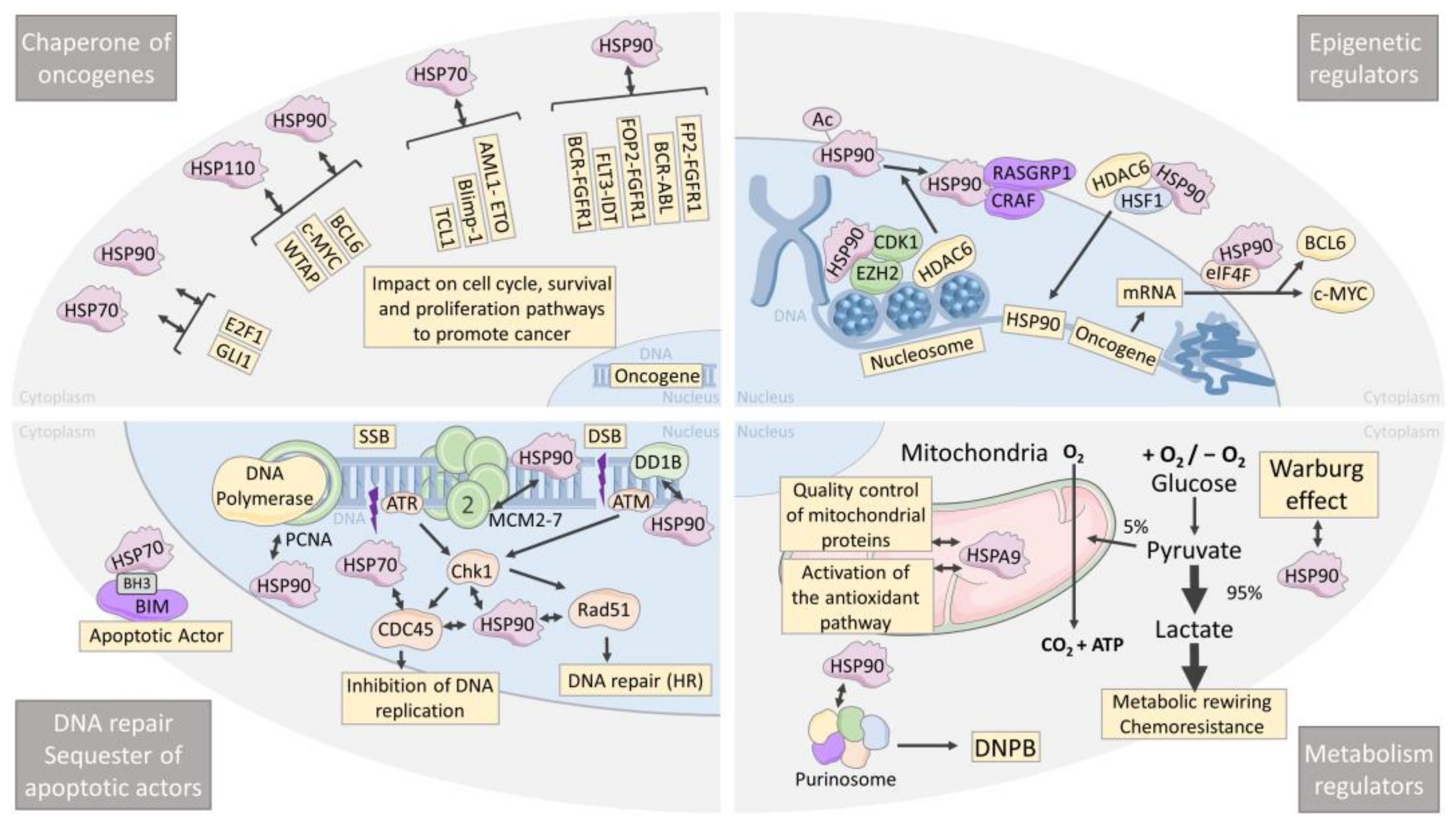
4. HSPs Are Chaperones of Oncogenes
5. HSPs Are Epigenetic Regulators
6. HSPs Are Leukemia and Lymphoma Metabolism Regulators
7. HSP Are Chaperones of DNA Repair and Sequesters of Apoptotic Actors
8. Recent Progress in Clinical Targeting of HSP90
9. Discussion
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Moraes Hungria, V.T.; Chiattone, C.; Pavlovsky, M.; Abenoza, L.M.; Agreda, G.P.; Armenta, J.; Arrais, C.; Flores, O.A.; Barroso, F.; Basquiera, A.L.; et al. Epidemiology of hematologic malignancies in real-world settings: Findings from the hemato-oncology Latin america observational registry study. J. Glob. Oncol. 2019, 2019, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Sinha, S.; Aldape, K.; Hannenhalli, S.; Sahinalp, C.; Ruppin, E. Big data in basic and translational cancer research. Nat. Rev. Cancer 2022, 22, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Thomas, X.; Campos, L.; Mounier, C.; Cornillon, J.; Flandrin, P.; Le, Q.H.; Piselli, S.; Guyotat, D. Expression of heat-shock proteins is associated with major adverse prognostic factors in acute myeloid leukemia. Leuk. Res. 2005, 29, 1049–1058. [Google Scholar] [CrossRef]
- Pocaly, M.; Lagarde, V.; Etienne, G.; Ribeil, J.A.; Claverol, S.; Bonneu, M.; Moreau-Gaudry, F.; Guyonnet-Duperat, V.; Hermine, O.; Melo, J.V.; et al. Overexpression of the heat-shock protein 70 is associated to imatinib resistance in chronic myeloid leukemia. Leukemia 2007, 21, 93–101. [Google Scholar] [CrossRef]
- Frezzato, F.; Raggi, F.; Martini, V.; Severin, F.; Trimarco, V.; Visentin, A.; Scomazzon, E.; Accordi, B.; Bresolin, S.; Piazza, F.; et al. HSP70/HSF1 axis, regulated via a PI3K/AKT pathway, is a druggable target in chronic lymphocytic leukemia. Int. J. Cancer 2019, 145, 3089–3100. [Google Scholar] [CrossRef]
- Sun, Q.; Ye, Y.; Gui, A.; Sun, X.; Xie, S.; Zhan, Y.; Chen, R.; Yan, Y.; Gu, J.; Qiu, S.; et al. MORTALIN-Ca2+ axis drives innate rituximab resistance in diffuse large B-cell lymphoma. Cancer Lett. 2022, 537, 215678. [Google Scholar] [CrossRef]
- Frezzato, F.; Visentin, A.; Severin, F.; Pizzo, S.; Ruggeri, E.; Mouawad, N.; Martinello, L.; Pagnin, E.; Trimarco, V.; Tonini, A.; et al. Targeting of HSP70/HSF1 axis abrogates in vitro ibrutinib-resistance in chronic lymphocytic leukemia. Cancers 2021, 13, 5453. [Google Scholar] [CrossRef]
- Yang, L.; Cao, L.; Yang, M.; Tang, D.; Kang, R.; Min, X.; Zhu, S.; Yu, Y. Hsp27: A novel therapeutic target for pediatric M4/M5 acute myeloid leukemia. Oncol. Rep. 2013, 29, 1459–1466. [Google Scholar] [CrossRef] [Green Version]
- Shevtsov, M.; Huile, G.; Multhoff, G. Membrane heat shock protein 70: A theranostic target for cancer therapy. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20160526. [Google Scholar] [CrossRef]
- Steiner, K.; Graf, M.; Hecht, K.; Reif, S.; Rossbacher, L.; Pfister, K.; Kolb, H.J.; Schmetzer, H.M.; Multhoff, G. High HSP70-membrane expression on leukemic cells from patients with acute myeloid leukemia is associated with a worse prognosis. Leukemia 2006, 20, 2076–2079. [Google Scholar] [CrossRef]
- Yeh, C.H.; Tseng, R.; Hannah, A.; Estrov, Z.; Estey, E.; Kantarjian, H.; Albitar, M. Clinical correlation of circulating heat shock protein 70 in acute leukemia. Leuk. Res. 2010, 34, 605–609. [Google Scholar] [CrossRef]
- Berthenet, K.; Boudesco, C.; Collura, A.; Svrcek, M.; Richaud, S.; Hammann, A.; Causse, S.; Yousfi, N.; Wanherdrick, K.; Duplomb, L.; et al. Extracellular HSP110 skews macrophage polarization in colorectal cancer. Oncoimmunology 2016, 5, e1170264. [Google Scholar] [CrossRef]
- Höpken, U.E.; Rehm, A. Targeting the Tumor Microenvironment of Leukemia and Lymphoma. Trends Cancer 2019, 5, 351–364. [Google Scholar] [CrossRef]
- Yufu, Y.; Nishimura, J.; Nawata, H. High constitutive expression of heat shock protein 90α in human acute leukemia cells. Leuk. Res. 1992, 16, 597–605. [Google Scholar] [CrossRef]
- Trentin, L.; Frasson, M.; Donella-Deana, A.; Frezzato, F.; Pagano, M.A.; Tibaldi, E.; Gattazzo, C.; Zambello, R.; Semenzato, G.; Brunati, A.M. Geldanamycin-induced Lyn dissociation from aberrant Hsp90-stabilized cytosolic complex is an early event in apoptotic mechanisms in B-chronic lymphocytic leukemia. Blood 2008, 112, 4665–4674. [Google Scholar] [CrossRef]
- Hacıhanefioglu, A.; Gonullu, E.; Mehtap, O.; Keski, H.; Yavuz, M.; Ercin, C. Effect of heat shock protein-90 (HSP90) and vascular endothelial growth factor (VEGF) on survival in acute lymphoblastic leukemia: An immunohistochemical study. Med. Oncol. 2011, 28, 846–851. [Google Scholar] [CrossRef]
- Milani, M.; Laranjeira, A.B.A.; De Vasconcellos, J.F.; Brandalise, S.R.; Nowill, A.E.; Yunes, J.A. Plasma Hsp90 Level as a Marker of Early Acute Lymphoblastic Leukemia Engraftment and Progression in Mice. PLoS ONE 2015, 10, e0129298. [Google Scholar]
- Žáčková, M.; Moučková, D.; Lopotová, T.; Ondráčková, Z.; Klamová, H.; Moravcová, J. Hsp90—A potential prognostic marker in CML. Blood Cells, Mol. Dis. 2013, 50, 184–189. [Google Scholar] [CrossRef]
- Guo, A.; Lu, P.; Lee, J.; Zhen, C.; Chiosis, G.; Wang, Y.L. HSP90 stabilizes B-cell receptor kinases in a multi-client interactome: PU-H71 induces CLL apoptosis in a cytoprotective microenvironment. Oncogene 2017, 36, 3441–3449. [Google Scholar] [CrossRef] [PubMed]
- Zappasodi, R.; Bongarzone, I.; Ghedini, G.C.; Castagnoli, L.; Cabras, A.D.; Messina, A.; Tortoreto, M.; Tripodo, C.; Magni, M.; Carlo-Stella, C.; et al. Serological identification of HSP105 as a novel non-Hodgkin lymphoma therapeutic target. Blood 2011, 118, 4421–4430. [Google Scholar] [CrossRef] [PubMed]
- Zappasodi, R.; Ruggiero, G.; Guarnotta, C.; Tortoreto, M.; Tringali, C.; Cavanè, A.; Cabras, A.D.; Castagnoli, L.; Venerando, B.; Zaffaroni, N.; et al. HSPH1 inhibition downregulates Bcl-6 and c-Myc and hampers the growth of human aggressive B-cell non-Hodgkin lymphoma. Blood 2015, 125, 1768–1771. [Google Scholar] [CrossRef] [PubMed]
- Boudesco, C.; Verhoeyen, E.; Martin, L.; Chassagne-Clement, C.; Salmi, L.; Mhaidly, R.; Pangault, C.; Fest, T.; Ramla, S.; Jardin, F.; et al. HSP110 sustains chronic NF-kB signaling in activated B-cell diffuse large B-cell lymphoma through MyD88 stabilization. Blood 2018, 132, 510–520. [Google Scholar] [CrossRef]
- Boudesco, C.; Rattier, T.; Garrido, C.; Jego, G. Do not stress, just differentiate: Role of stress proteins in hematopoiesis. Cell Death Dis. 2015, 6. [Google Scholar] [CrossRef]
- Syafruddin, S.E.; Ling, S.; Low, T.Y.; Aiman Mohtar, M. More Than Meets the Eye: Revisiting the Roles of Heat Shock Factor 4 in Health and Diseases. Biomolecules 2021, 11, 523. [Google Scholar] [CrossRef]
- Gomez-Pastor, R.; Burchfiel, E.T.; Thiele, D.J. Regulation of heat shock transcription factors and their roles in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 4. [Google Scholar] [CrossRef]
- Morán Luengo, T.; Mayer, M.P.; Rüdiger, S.G.D. The Hsp70–Hsp90 Chaperone Cascade in Protein Folding. Trends Cell Biol. 2019, 29, 164–177. [Google Scholar] [CrossRef]
- Lang, B.J.; Guerrero, M.E.; Prince, T.L.; Okusha, Y.; Bonorino, C.; Calderwood, S.K. The functions and regulation of heat shock proteins; key orchestrators of proteostasis and the heat shock response. Arch. Toxicol. 2021, 95, 1943–1970. [Google Scholar] [CrossRef]
- Kruta, M.; Sunshine, M.J.; Chua, B.A.; Fu, Y.; Chawla, A.; Dillingham, C.H.; Hidalgo San Jose, L.; De Jong, B.; Zhou, F.J.; Signer, R.A.J. Hsf1 promotes hematopoietic stem cell fitness and proteostasis in response to ex vivo culture stress and aging. Cell Stem Cell 2021, 28, 1950–1965.e6. [Google Scholar] [CrossRef]
- Dong, Q.; Xiu, Y.; Wang, Y.; Hodgson, C.; Borcherding, N.; Jordan, C.; Buchanan, J.; Taylor, E.; Wagner, B.; Leidinger, M.; et al. HSF1 is a driver of leukemia stem cell self-renewal in acute myeloid leukemia. Nat. Commun. 2022, 13, 6107. [Google Scholar] [CrossRef]
- Nishida, Y.; Zhao, R.; Heese, L.E.; Akiyama, H.; Patel, S.; Jaeger, A.M.; Jacamo, R.O.; Kojima, K.; Ma, M.C.J.; Ruvolo, V.R.; et al. Inhibition of translation initiation factor eIF4a inactivates heat shock factor 1 (HSF1) and exerts anti-leukemia activity in AML. Leukemia 2021, 35, 2469–2481. [Google Scholar] [CrossRef]
- Cyran, A.M.; Zhitkovich, A. Heat Shock Proteins and HSF1 in Cancer. Front. Oncol. 2022, 12, 860320. [Google Scholar] [CrossRef]
- Yang, Z.; Wan, W.; Zhang, P.; Wang, S.; Zhao, Z.; Xue, J.; Yao, M.; Zhao, Y.; Zheng, W.; Niu, B.; et al. Crosstalk between HSF1 and STAT3 mediated by IL-8 autocrine signaling maintains the cancer stem cell phenotype in liver cancer. J. Gastroenterol. Hepatol. 2022, 38, 138–152. [Google Scholar] [CrossRef]
- Qin, T.; Chen, K.; Li, J.; Qian, W.; Xiao, Y.; Wu, E.; Ma, J.; Chen, Z.; Wang, Z.; Ma, Q.; et al. Heat shock factor 1 inhibition sensitizes pancreatic cancer to gemcitabine via the suppression of cancer stem cell-like properties. Biomed. Pharmacother. 2022, 148, 112713. [Google Scholar] [CrossRef]
- Kabiri Renani, M.; Yousefi, R.; Koohkan, F.; Heidari, M.; Asad, S.; Hosseinzadeh, S.; Nazaran, M.H. Knockdown of HSF1 sensitizes resistant prostate cancer cell line to chemotherapy. Mod. Med. Lab. J. 2022, 5, 47–55. [Google Scholar] [CrossRef]
- Östling, P.; Björk, J.K.; Roos-Mattjus, P.; Mezger, V.; Sistonen, L. Heat Shock Factor 2 (HSF2) contributes to inducible expression of hsp genes through interplay with HSF1. J. Biol. Chem. 2007, 282, 7077–7086. [Google Scholar] [CrossRef]
- Smith, R.S.; Takagishi, S.R.; Amici, D.R.; Metz, K.; Gayatri, S.; Alasady, M.J.; Wu, Y.; Brockway, S.; Taiberg, S.L.; Khalatyan, N.; et al. HSF2 cooperates with HSF1 to drive a transcriptional program critical for the malignant state. Sci. Adv. 2022, 8, 6526. [Google Scholar] [CrossRef]
- Tokunaga, Y.; Otsuyama, K.-I.; Kakuta, S.; Hayashida, N. Heat Shock Transcription Factor 2 Is Significantly Involved in Neurodegenerative Diseases, Inflammatory Bowel Disease, Cancer, Male Infertility, and Fetal Alcohol Spectrum Disorder: The Novel Mechanisms of Several Severe Diseases. Int. J. Mol. Sci. 2022, 23, 13763. [Google Scholar] [CrossRef]
- Wang, X.Y.; Subjeck, J.R. High molecular weight stress proteins: Identification, cloning and utilisation in cancer immunotherapy. Int. J. Hyperth. 2013, 29, 364–375. [Google Scholar] [CrossRef]
- Liu, T.; Daniels, C.K.; Cao, S. Comprehensive review on the HSC70 functions, interactions with related molecules and involvement in clinical diseases and therapeutic potential. Pharmacol. Ther. 2012, 136, 354–374. [Google Scholar] [CrossRef] [PubMed]
- Mattoo, R.U.H.; Sharma, S.K.; Priya, S.; Finka, A.; Goloubinoff, P. Hsp110 is a bona fide chaperone using ATP to unfold stable misfolded polypeptides and reciprocally collaborate with Hsp70 to solubilize protein aggregates. J. Biol. Chem. 2013, 288, 21399–21411. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, T.; Rios, Z.; Mei, Q.; Lin, X.; Cao, S. Heat Shock Proteins and Cancer. Trends Pharmacol. Sci. 2017, 38, 226–256. [Google Scholar] [CrossRef]
- Wang, L.; Xu, X.; Jiang, Z.; You, Q. Modulation of protein fate decision by small molecules: Targeting molecular chaperone machinery. Acta Pharm. Sin. B 2020, 10, 1904–1925. [Google Scholar] [CrossRef]
- Treweek, T.M.; Meehan, S.; Ecroyd, H.; Carver, J.A. Small heat-shock proteins: Important players in regulating cellular proteostasis. Cell. Mol. Life Sci. 2015, 72, 429–451. [Google Scholar] [CrossRef]
- Chelouche-Lev, D.; Kluger, H.M.; Berger, A.J.; Rimm, D.L.; Price, J.E. αB-crystallin as a marker of lymph node involvement in breast carcinoma. Cancer 2004, 100, 2543–2548. [Google Scholar] [CrossRef]
- Ivanov, O.; Chen, F.; Wiley, E.L.; Keswani, A.; Diaz, L.K.; Memmel, H.C.; Rademaker, A.; Gradishar, W.J.; Morrow, M.; Khan, S.A.; et al. αB-crystallin is a novel predictor of resistance to neoadjuvant chemotherapy in breast cancer. Breast Cancer Res. Treat. 2008, 111, 411–417. [Google Scholar] [CrossRef]
- Takashi, M.; Katsuno, S.; Sakata, T.; Ohshima, S.; Kato, K. Different concentrations of two small stress proteins, alphaB crystallin and HSP27 in human urological tumor tissues. Urol. Res. 1998, 26, 395–399. [Google Scholar] [CrossRef]
- Liu, X.L.; Guo, K.P.; Ma, F.; Xie, G.Y.; He, Y.; Hu, C.H. Expression profile of heat shock proteins in tissues and cells of lung adenocarcinoma. J. Cent. South Univ. Medical Sci. 2007, 32, 660–664. [Google Scholar]
- Tang, Q.; Liu, Y.F.; Zhu, X.J.; Li, Y.H.; Zhu, J.; Zhang, J.P.; Feng, Z.Q.; Guan, X.H. Expression and prognostic significance of the alpha B-crystallin gene in human hepatocellular carcinoma. Hum. Pathol. 2009, 40, 300–305. [Google Scholar] [CrossRef]
- Profitós-Pelejà, N.; Santos, J.C.; Marín-Niebla, A.; Roué, G.; Ribeiro, M.L. Regulation of B-Cell Receptor Signaling and Its Therapeutic Relevance in Aggressive B-Cell Lymphomas. Cancers 2022, 14, 860. [Google Scholar] [CrossRef]
- Löwenberg, M.; Verhaar, A.P.; Bilderbeek, J.; van Marle, J.; Buttgereit, F.; Peppelenbosch, M.P.; van Deventer, S.J.; Hommes, D.W. Glucocorticoids cause rapid dissociation of a T-cell-receptor-associated protein complex containing LCK and FYN. EMBO Rep. 2006, 7, 1023–1029. [Google Scholar] [CrossRef]
- Nika, K.; Soldani, C.; Salek, M.; Paster, W.; Gray, A.; Etzensperger, R.; Fugger, L.; Polzella, P.; Cerundolo, V.; Dushek, O.; et al. Constitutively active Lck kinase in T cells drives antigen receptor signal transduction. Immunity 2010, 32, 766–777. [Google Scholar] [CrossRef]
- Serafin, V.; Lissandron, V.; Buldini, B.; Bresolin, S.; Paganin, M.; Grillo, F.; Andriano, N.; Palmi, C.; Cazzaniga, G.; Marmiroli, S.; et al. Phosphoproteomic analysis reveals hyperactivation of mTOR/STAT3 and LCK/Calcineurin axes in pediatric early T-cell precursor ALL. Leukemia 2017, 31, 1007–1011. [Google Scholar] [CrossRef]
- Buffière, A.; Accogli, T.; Saint-Paul, L.; Lucchi, G.; Uzan, B.; Ballerini, P.; Bastie, J.N.; Delva, L.; Pflumio, F.; Quéré, R. Saracatinib impairs maintenance of human T-ALL by targeting the LCK tyrosine kinase in cells displaying high level of lipid rafts. Leukemia 2018, 32, 2062–2065. [Google Scholar] [CrossRef]
- Serafin, V.; Capuzzo, G.; Milani, G.; Minuzzo, S.A.; Pinazza, M.; Bortolozzi, R.; Bresolin, S.; Porcù, E.; Frasson, C.; Indraccolo, S.; et al. Glucocorticoid resistance is reverted by LCK inhibition in pediatric T-cell acute lymphoblastic leukemia. Blood 2017, 130, 2750–2761. [Google Scholar] [CrossRef]
- Taipale, M.; Krykbaeva, I.; Koeva, M.; Kayatekin, C.; Westover, K.D.; Karras, G.I.; Lindquist, S. Quantitative Analysis of Hsp90-Client Interactions Reveals Principles of Substrate Recognition. Cell 2012, 150, 987–1001. [Google Scholar] [CrossRef]
- Mshaik, R.; Simonet, J.; Georgievski, A.; Jamal, L.; Bechoua, S.; Ballerini, P.; Bellaye, P.S.; Mlamla, Z.; Pais de Barros, J.P.; Geissler, A.; et al. HSP90 inhibitor NVP-BEP800 affects stability of SRC kinases and growth of T-cell and B-cell acute lymphoblastic leukemias. Blood Cancer J. 2021, 11, 61. [Google Scholar] [CrossRef]
- Harr, M.W.; Caimi, P.F.; McColl, K.S.; Zhong, F.; Patel, S.N.; Barr, P.M.; Distelhorst, C.W. Inhibition of Lck enhances glucocorticoid sensitivity and apoptosis in lymphoid cell lines and in chronic lymphocytic leukemia. Cell Death Differ. 2010, 17, 1381–1391. [Google Scholar] [CrossRef]
- Mahmud, H.; Mendez, M.; Mukhopadhyay, B.; Holter-Chakrabarty, J.; Ghosh, A.K. HSP90 overexpression potentiates the B-cell receptor and fibroblast growth factor receptor survival signals in chronic lymphocytic leukemia cells. Oncotarget 2020, 11, 2037. [Google Scholar] [CrossRef]
- Jacobson, C.; Kopp, N.; Layer, J.V.; Redd, R.A.; Tschuri, S.; Haebe, S.; Van Bodegom, D.; Bird, L.; Christie, A.L.; Christodoulou, A.; et al. HSP90 inhibition overcomes ibrutinib resistance in mantle cell lymphoma. Blood 2016, 128, 2517–2526. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, R.L.; Yang, S.N.; Taldone, T.; Chang, B.; Gerecitano, J.; Elenitoba-Johnson, K.; Shaknovich, R.; Tam, W.; Leonard, J.P.; Chiosis, G.; et al. Pharmacoproteomics identifies combinatorial therapy targets for diffuse large B cell lymphoma. J. Clin. Invest. 2015, 125, 4559–4571. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.L.; Harrington, B.; Truxall, J.; Wasmuth, R.; Prouty, A.; Sloan, S.; Lehman, A.M.; Sampath, D.; Orlemans, E.; Baiocchi, R.A.; et al. Preclinical evaluation of the Hsp90 inhibitor SNX-5422 in ibrutinib resistant CLL. J. Hematol. Oncol. 2021, 14, 36. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.H.; Young, R.M.; Schmitz, R.; Yang, Y.; Pittaluga, S.; Wright, G.; Lih, C.J.; Williams, P.M.; Shaffer, A.L.; Gerecitano, J.; et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat. Med. 2015, 21, 922–926. [Google Scholar] [CrossRef]
- Phelan, J.D.; Young, R.M.; Webster, D.E.; Roulland, S.; Wright, G.W.; Kasbekar, M.; Shaffer, A.L.; Ceribelli, M.; Wang, J.Q.; Schmitz, R.; et al. A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature 2018, 560, 387–391. [Google Scholar] [CrossRef]
- Gozzi, G.J.; Gonzalez, D.; Boudesco, C.; Dias, A.M.M.; Gotthard, G.; Uyanik, B.; Dondaine, L.; Marcion, G.; Hermetet, F.; Denis, C.; et al. Selecting the first chemical molecule inhibitor of HSP110 for colorectal cancer therapy. Cell Death Differ. 2019, 27, 117–129. [Google Scholar] [CrossRef]
- Walter, R.; Pan, K.T.; Doebele, C.; Comoglio, F.; Tomska, K.; Bohnenberger, H.; Young, R.M.; Jacobs, L.; Keller, U.; Bönig, H.; et al. HSP90 promotes Burkitt lymphoma cell survival by maintaining tonic B-cell receptor signaling. Blood 2017, 129, 598–608. [Google Scholar] [CrossRef]
- Segges, P.; Corrêa, S.; Du Rocher, B.; Vera-Lozada, G.; Krsticevic, F.; Arce, D.; Sternberg, C.; Abdelhay, E.; Hassan, R. Targeting Hodgkin and Reed–Sternberg Cells with an Inhibitor of Heat-Shock Protein 90: Molecular Pathways of Response and Potential Mechanisms of Resistance. Int. J. Mol. Sci. 2018, 19, 836. [Google Scholar] [CrossRef]
- Taniguchi, H.; Hasegawa, H.; Sasaki, D.; Ando, K.; Sawayama, Y.; Imanishi, D.; Taguchi, J.; Imaizumi, Y.; Hata, T.; Tsukasaki, K.; et al. Heat shock protein 90 inhibitor NVP-AUY922 exerts potent activity against adult T-cell leukemia-lymphoma cells. Cancer Sci. 2014, 105, 1601–1608. [Google Scholar] [CrossRef]
- Hertlein, E.; Wagner, A.J.; Jones, J.; Lin, T.S.; Maddocks, K.J.; Towns, W.H.; Goettl, V.M.; Zhang, X.; Jarjoura, D.; Raymond, C.A.; et al. 17-DMAG targets the nuclear factor-κB family of proteins to induce apoptosis in chronic lymphocytic leukemia: Clinical implications of HSP90 inhibition. Blood 2010, 116, 45–53. [Google Scholar] [CrossRef]
- Chen, I.T.; Hsu, P.H.; Hsu, W.C.; Chen, N.J.; Tseng, P.H. Polyubiquitination of Transforming Growth Factor β-activated Kinase 1 (TAK1) at Lysine 562 Residue Regulates TLR4-mediated JNK and p38 MAPK Activation. Sci. Rep. 2015, 5, 12300. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.; Zhang, A.; Huang, J.; Suo, M.; Zhong, Y.; Liang, Y. Suppression of HSP70 inhibits the development of acute lymphoblastic leukemia via TAK1/Egr-1. Biomed. Pharmacother. 2019, 119, 109399. [Google Scholar] [CrossRef]
- Desterke, C.; Bennaceur-Griscelli, A.; Turhan, A.G. EGR1 dysregulation defines an inflammatory and leukemic program in cell trajectory of human-aged hematopoietic stem cells (HSC). Stem Cell Res. Ther. 2021, 12, 419. [Google Scholar] [CrossRef]
- Kulkarni, R. Early Growth Response Factor 1 in Aging Hematopoietic Stem Cells and Leukemia. Front. Cell Dev. Biol. 2022, 10, 925761. [Google Scholar] [CrossRef]
- Sanda, T.; Tyner, J.W.; Gutierrez, A.; Ngo, V.N.; Glover, J.; Chang, B.H.; Yost, A.; Ma, W.; Fleischman, A.G.; Zhou, W.; et al. TYK2-STAT1-BCL2 pathway dependence in T-cell acute lymphoblastic leukemia. Cancer Discov. 2013, 3, 564–577. [Google Scholar] [CrossRef]
- Wöss, K.; Simonović, N.; Strobl, B.; Macho-Maschler, S.; Müller, M. Tyk2: An upstream kinase of stats in cancer. Cancers 2019, 11, 1728. [Google Scholar] [CrossRef]
- Akahane, K.; Sanda, T.; Mansour, M.R.; Radimerski, T.; Deangelo, D.J.; Weinstock, D.M.; Look, A.T. HSP90 inhibition leads to degradation of the TYK2 kinase and apoptotic cell death in T-cell acute lymphoblastic leukemia. Leukemia 2016, 30, 219–228. [Google Scholar] [CrossRef]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Brkic, S.; Stivala, S.; Santopolo, A.; Szybinski, J.; Jungius, S.; Passweg, J.R.; Tsakiris, D.; Dirnhofer, S.; Hutter, G.; Leonards, K.; et al. Dual targeting of JAK2 and ERK interferes with the myeloproliferative neoplasm clone and enhances therapeutic efficacy. Leukemia 2021, 35, 2875–2884. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Verstovsek, S.; Manshouri, T.; Rao, R.; Balusu, R.; Venkannagari, S.; Nalabothula, N.R.; Ha, K.; Smith, J.E.; Hembruff, S.L.; et al. Heat shock protein 90 inhibitor is synergistic with JAK2 inhibitor and overcomes resistance to JAK2-TKI in human myeloproliferative neoplasm cells. Clin. Cancer Res. 2011, 17, 7347–7358. [Google Scholar] [CrossRef] [PubMed]
- Marubayashi, S.; Koppikar, P.; Taldone, T.; Abdel-Wahab, O.; West, N.; Bhagwat, N.; Caldas-Lopes, E.; Ross, K.N.; Gönen, M.; Gozman, A.; et al. HSP90 is a therapeutic target in JAK2-dependent myeloproliferative neoplasms in mice and humans. J. Clin. Invest. 2010, 120, 3578–3593. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, G.; Litvin, R.; Ahn, J.; Mckenney, A.S.; Mauro, M.J.; Tallman, M.S.; Berman, E.; Heaney, M.L.; Levine, R.L.; Rampal, R.K. AUY922, a Heat Shock Protein 90 (Hsp90) Inhibitor, Demonstrates Activity in Patients with Myeloproliferative Neoplasms (MPNs). Blood 2015, 126, 4075. [Google Scholar] [CrossRef]
- Weigert, O.; Lane, A.A.; Bird, L.; Kopp, N.; Chapuy, B.; van Bodegom, D.; Toms, A.V.; Marubayashi, S.; Christie, A.L.; McKeown, M.; et al. Genetic resistance to JAK2 enzymatic inhibitors is overcome by HSP90 inhibition. J. Exp. Med. 2012, 209, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Sevin, M.; Kubovcakova, L.; Pernet, N.; Causse, S.; Vitte, F.; Villeval, J.L.; Lacout, C.; Cordonnier, M.; Rodrigues-Lima, F.; Chanteloup, G.; et al. HSP27 is a partner of JAK2-STAT5 and a potential therapeutic target in myelofibrosis. Nat. Commun. 2018, 9, 1431. [Google Scholar] [CrossRef]
- Schoof, N.; Von Bonin, F.; Trümper, L.; Kube, D. HSP90 is essential for Jak-STAT signaling in classical Hodgkin lymphoma cells. Cell Commun. Signal. 2009, 7, 17. [Google Scholar] [CrossRef]
- Khong, T.; Spencer, A. Targeting HSP 90 induces apoptosis and inhibits critical survival and proliferation pathways in multiple myeloma. Mol. Cancer Ther. 2011, 10, 1909–1917. [Google Scholar] [CrossRef]
- Richardson, P.G.; Mitsiades, C.S.; Laubach, J.P.; Lonial, S.; Chanan-Khan, A.A.; Anderson, K.C. Inhibition of heat shock protein 90 (HSP90) as a therapeutic strategy for the treatment of myeloma and other cancers. Br. J. Haematol. 2011, 152, 367–379. [Google Scholar] [CrossRef]
- Xu, W.; Berning, P.; Lenz, G. Targeting B-cell receptor and PI3K signaling in diffuse large B-cell lymphoma. Blood 2021, 138, 1110–1119. [Google Scholar] [CrossRef]
- Shouse, G.; Danilova, O.V.; Danilov, A.V. Current status of phosphoinotiside-3 kinase inhibitors in blood cancers. Curr. Opin. Oncol. 2022, 34, 540–545. [Google Scholar] [CrossRef]
- Sato, S.; Fujita, N.; Tsuruo, T. Modulation of Akt kinase activity by binding to Hsp90. Proc. Natl. Acad. Sci. USA 2000, 97, 10832–10837. [Google Scholar] [CrossRef]
- Basso, A.D.; Solit, D.B.; Chiosis, G.; Giri, B.; Tsichlis, P.; Rosen, N. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J. Biol. Chem. 2002, 277, 39858–39866. [Google Scholar] [CrossRef] [Green Version]
- Giulino-Roth, L.; Van Besien, H.J.; Dalton, T.; Totonchy, J.E.; Rodina, A.; Taldone, T.; Bolaender, A.; Erdjument-Bromage, H.; Sadek, J.; Chadburn, A.; et al. Inhibition of Hsp90 suppresses PI3K/AKT/mTOR signaling and has antitumor activity in Burkitt lymphoma. Mol. Cancer Ther. 2017, 16, 1779–1790. [Google Scholar] [CrossRef]
- Lazenby, M.; Hills, R.; Burnett, A.K.; Zabkiewicz, J. The HSP90 inhibitor ganetespib: A potential effective agent for Acute Myeloid Leukemia in combination with cytarabine. Leuk. Res. 2015, 39, 617–624. [Google Scholar] [CrossRef]
- Chen, L.; Monti, S.; Juszczynski, P.; Daley, J.; Chen, W.; Witzig, T.E.; Habermann, T.M.; Kutok, J.L.; Shipp, M.A. SYK-dependent tonic B-cell receptor signaling is a rational treatment target in diffuse large B-cell lymphoma. Blood 2008, 111, 2230–2237. [Google Scholar] [CrossRef]
- Havranek, O.; Xu, J.; Köhrer, S.; Wang, Z.; Becker, L.; Comer, J.M.; Henderson, J.; Ma, W.; Ma, J.M.C.; Westin, J.R.; et al. Tonic B-cell receptor signaling in diffuse large B-cell lymphoma. Blood 2017, 130, 995–1006. [Google Scholar] [CrossRef]
- Kaiser, M.; Kühnl, A.; Reins, J.; Fischer, S.; Ortiz-Tanchez, J.; Schlee, C.; Mochmann, L.H.; Heesch, S.; Benlasfer, O.; Hofmann, W.K.; et al. Antileukemic activity of the HSP70 inhibitor pifithrin-l in acute leukemia. Blood Cancer J. 2011, 1, e28. [Google Scholar] [CrossRef]
- Peng, Y.; Huang, Z.; Zhou, F.; Wang, T.; Mou, K.; Feng, W. Effect of HSP90AB1 and CC domain interaction on Bcr-Abl protein cytoplasm localization and function in chronic myeloid leukemia cells. Cell Commun. Signal. 2021, 19, 71. [Google Scholar] [CrossRef]
- Ju, H.Q.; Wang, S.X.; Xiang, Y.F.; Liu, Z.; Liu, J.Y.; Chen, Z.P.; Zeng, F.L.; Xia, M.; Liu, Z.H.; Xing, G.W.; et al. BJ-B11, a novel Hsp90 inhibitor, induces apoptosis in human chronic myeloid leukemia K562 cells through the mitochondria-dependent pathway. Eur. J. Pharmacol. 2011, 666, 26–34. [Google Scholar] [CrossRef]
- Bhatia, S.; Diedrich, D.; Frieg, B.; Ahlert, H.; Stein, S.; Bopp, B.; Lang, F.; Zang, T.; Kröger, T.; Ernst, T.; et al. Targeting HSP90 dimerization via the C terminus is effective in imatinib-resistant CML and lacks the heat shock response. Blood 2018, 132, 307–320. [Google Scholar] [CrossRef]
- Jinwal, U.K.; Trotter, J.H.; Abisambra, J.F.; Koren, J.; Lawson, L.Y.; Vestal, G.D.; O’Leary, J.C.; Johnson, A.G.; Jin, Y.; Jones, J.R.; et al. The Hsp90 kinase co-chaperone Cdc37 regulates tau stability and phosphorylation dynamics. J. Biol. Chem. 2011, 286, 16976–16983. [Google Scholar] [CrossRef] [PubMed]
- Reikvam, H.; Hatfield, K.J.; Ersvær, E.; Hovland, R.; Skavland, J.; Gjertsen, B.T.; Petersen, K.; Bruserud, Ø. Expression profile of heat shock proteins in acute myeloid leukaemia patients reveals a distinct signature strongly associated with FLT3 mutation status—Consequences and potentials for pharmacological intervention. Br. J. Haematol. 2012, 156, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Peiris, M.N.; Meyer, A.N.; Nelson, K.N.; Bisom-Rapp, E.W.; Donoghue, D.J. Oncogenic fusion protein BCR-FGFR1 requires the breakpoint cluster region-mediated oligomerization and chaperonin Hsp90 for activation. Haematologica 2020, 105, 1262–1273. [Google Scholar] [CrossRef] [PubMed]
- Wendel, T.; Zhen, Y.; Suo, Z.; Bruheim, S.; Wiedlocha, A. The novel HSP90 inhibitor NVP-AUY922 shows synergistic anti-leukemic activity with cytarabine in vivo. Exp. Cell Res. 2016, 340, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zhuang, H.; Yu, Q.; Zhang, X.; Jiang, X.; Lu, X.; Xu, Y.; Yang, L.; Wu, B.; Ma, A.; et al. Homoharringtonine Combined with the Heat Shock Protein 90 Inhibitor IPI504 in the Treatment of FLT3-ITD Acute Myeloid Leukemia. Transl. Oncol. 2019, 12, 801–809. [Google Scholar] [CrossRef]
- Katayama, K.; Noguchi, K.; Sugimoto, Y. Heat shock protein 90 inhibitors overcome the resistance to Fms-like tyrosine kinase 3 inhibitors in acute myeloid leukemia. Oncotarget 2018, 9, 34240–34258. [Google Scholar] [CrossRef]
- Roh, S.H.; Kasembeli, M.; Galaz-Montoya, J.G.; Trnka, M.; Lau, W.C.Y.; Burlingame, A.; Chiu, W.; Tweardy, D.J. Chaperonin TRiC/CCT modulates the folding and activity of leukemogenic fusion oncoprotein AML1-ETO. J. Biol. Chem. 2016, 291, 4732–4741. [Google Scholar] [CrossRef]
- Cerchietti, L.C.; Lopes, E.C.; Yang, S.N.; Hatzi, K.; Bunting, K.L.; Tsikitas, L.A.; Mallik, A.; Robles, A.I.; Walling, J.; Varticovski, L.; et al. A purine scaffold Hsp90 inhibitor destabilizes BCL-6 and has specific antitumor activity in BCL-6-dependent B cell lymphomas. Nat. Med. 2009, 15, 1369–1376. [Google Scholar] [CrossRef]
- Kuai, Y.; Gong, X.; Ding, L.; Li, F.; Lei, L.; Gong, Y.; Liu, Q.; Tan, H.; Zhang, X.; Liu, D.; et al. Wilms’ tumor 1-associating protein plays an aggressive role in diffuse large B-cell lymphoma and forms a complex with BCL6 via Hsp90. Cell Commun. Signal. 2018, 16, 50. [Google Scholar] [CrossRef]
- Lee, J.; Zhang, L.L.; Wu, W.; Guo, H.; Li, Y.; Sukhanova, M.; Venkataraman, G.; Zhang, H.; Alikhan, M.; Lu, P.; et al. Activation of MYC, a bona fide client of HSP90, contributes to intrinsic ibrutinib resistance in mantle cell lymphoma. Blood Adv. 2018, 2, 2039–2051. [Google Scholar] [CrossRef]
- Poole, C.J.; Zheng, W.; Lee, H.; Young, D.; Lodh, A.; Chadli, A.; van Riggelen, J. Targeting the MYC oncogene in burkitt lymphoma through HSP90 inhibition. Cancers 2018, 10, 448. [Google Scholar] [CrossRef]
- Liu, H.; Lu, Z.; Shi, X.; Liu, L.; Zhang, P.; Golemis, E.A.; Tu, Z. HSP90 inhibition downregulates DNA replication and repair genes via E2F1 repression. J. Biol. Chem. 2021, 297, 100996. [Google Scholar] [CrossRef]
- Freisleben, F.; Modemann, F.; Muschhammer, J.; Stamm, H.; Brauneck, F.; Krispien, A.; Bokemeyer, C.; Kirschner, K.N.; Wellbrock, J.; Fiedler, W. Mebendazole mediates proteasomal degradation of GLI transcription factors in acute myeloid leukemia. Int. J. Mol. Sci. 2021, 22, 10670. [Google Scholar] [CrossRef]
- Wang, W.F.; Yan, L.; Liu, Z.; Liu, L.X.; Lin, J.; Liu, Z.Y.; Chen, X.P.; Zhang, W.; Xu, Z.Z.; Shi, T.; et al. HSP70-Hrd1 axis precludes the oncorepressor potential of N-terminal misfolded Blimp-1s in lymphoma cells. Nat. Commun. 2017, 8, 363. [Google Scholar] [CrossRef]
- Frisan, E.; Vandekerckhove, J.; De Thonel, A.; Pierre-Eugène, C.; Sternberg, A.; Arlet, J.B.; Floquet, C.; Gyan, E.; Kosmider, O.; Dreyfus, F.; et al. Defective nuclear localization of Hsp70 is associated with dyserythropoiesis and GATA-1 cleavage in myelodysplastic syndromes. Blood 2012, 119, 1532–1542. [Google Scholar] [CrossRef]
- Pekarsky, Y.; Palamarchuk, A.; Maximov, V.; Efanov, A.; Nazaryan, N.; Santanam, U.; Rassenti, L.; Kipps, T.; Croce, C.M. Tcl1 functions as a transcriptional regulator and is directly involved in the pathogenesis of CLL. Proc. Natl. Acad. Sci. USA 2008, 105, 19643–19648. [Google Scholar] [CrossRef]
- Paduano, F.; Gaudio, E.; Mensah, A.A.; Pinton, S.; Bertoni, F.; Trapasso, F. T-Cell Leukemia/Lymphoma 1 (TCL1): An oncogene regulating multiple signaling pathways. Front. Oncol. 2018, 8, 317. [Google Scholar] [CrossRef]
- Gaudio, E.; Paduano, F.; Ngankeu, A.; Lovat, F.; Fabbri, M.; Sun, H.L.; Gasparini, P.; Efanov, A.; Peng, Y.; Zanesi, N.; et al. Heat shock protein 70 regulates Tcl1 expression in leukemia and lymphomas. Blood 2013, 121, 351–359. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef]
- Kempf, J.M.; Weser, S.; Bartoschek, M.D.; Metzeler, K.H.; Vick, B.; Herold, T.; Völse, K.; Mattes, R.; Scholz, M.; Wange, L.E.; et al. Loss-of-function mutations in the histone methyltransferase EZH2 promote chemotherapy resistance in AML. Sci. Rep. 2021, 11, 5838. [Google Scholar] [CrossRef]
- Göllner, S.; Oellerich, T.; Agrawal-Singh, S.; Schenk, T.; Klein, H.U.; Rohde, C.; Pabst, C.; Sauer, T.; Lerdrup, M.; Tavor, S.; et al. Loss of the histone methyltransferase EZH2 induces resistance to multiple drugs in acute myeloid leukemia. Nat. Med. 2017, 23, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Bali, P.; Pranpat, M.; Bradner, J.; Balasis, M.; Fiskus, W.; Guo, F.; Rocha, K.; Kumaraswamy, S.; Boyapalle, S.; Atadja, P.; et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: A novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 2005, 280, 26729–26734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Xiao, J.; Wang, Y.; Song, X.; Huang, L.; Ren, Z.; Kitazato, K.; Wang, Y. Posttranslational modification and beyond: Interplay between histone deacetylase 6 and heat-shock protein 90. Mol. Med. 2021, 27, 110. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Peterson, K.L.; Correia, C.; Koh, B.; Schneider, P.A.; Nowakowski, G.S.; Kaufmann, S.H. Histone deacetylase inhibitors interrupt HSP90•RASGRP1 and HSP90•CRAF interactions to upregulate BIM and circumvent drug resistance in lymphoma cells. Leukemia 2017, 31, 1593–1602. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.; Fiskus, W.; Yang, Y.; Lee, P.; Joshi, R.; Fernandez, P.; Mandawat, A.; Atadja, P.; Bradner, J.E.; Bhalla, K. HDAC6 inhibition enhances 17-AAG mediated abrogation of hsp90 chaperone function in human leukemia cells. Blood 2008, 112, 1886–1893. [Google Scholar] [CrossRef]
- Ojha, R.; Huang, H.L.; HuangFu, W.C.; Wu, Y.W.; Nepali, K.; Lai, M.J.; Su, C.J.; Sung, T.Y.; Chen, Y.L.; Pan, S.L.; et al. 1-Aroylindoline-hydroxamic acids as anticancer agents, inhibitors of HSP90 and HDAC. Eur. J. Med. Chem. 2018, 150, 667–677. [Google Scholar] [CrossRef]
- Culjkovic-Kraljacic, B.; Fernando, T.M.; Marullo, R.; Calvo-Vidal, N.; Verma, A.; Yang, S.N.; Tabbò, F.; Gaudiano, M.; Zahreddine, H.; Goldstein, R.L.; et al. Combinatorial targeting of nuclear export and translation of RNA inhibits aggressive B-cell lymphomas. Blood 2016, 127, 858–868. [Google Scholar] [CrossRef]
- Isaacs, J.S. Hsp90 as a “Chaperone” of the Epigenome: Insights and Opportunities for Cancer Therapy. In Advances in Cancer Research; Academic Press: Cambridge, MA, USA, 2016; Volume 129, pp. 107–140. ISBN 9780128022900. [Google Scholar]
- Kirsch, B.J.; Chang, S.J.; Betenbaugh, M.J.; Le, A. Non-Hodgkin Lymphoma Metabolism. Adv. Exp. Med. Biol. 2021, 1311, 103–116. [Google Scholar]
- Calvo-Vidal, M.N.; Zamponi, N.; Krumsiek, J.; Stockslager, M.A.; Revuelta, M.V.; Phillip, J.M.; Marullo, R.; Tikhonova, E.; Kotlov, N.; Patel, J.; et al. Oncogenic HSP90 facilitates metabolic alterations in aggressive B-cell lymphomas. Cancer Res. 2021, 81, 5202. [Google Scholar] [CrossRef]
- Pedley, A.M.; Boylan, J.P.; Chan, C.Y.; Kennedy, E.L.; Kyoung, M.; Benkovic, S.J. Purine biosynthetic enzymes assemble into liquid-like condensates dependent on the activity of chaperone protein HSP90. J. Biol. Chem. 2022, 298, 101845. [Google Scholar] [CrossRef]
- Tai-Nagara, I.; Matsuoka, S.; Ariga, H.; Suda, T. Mortalin and DJ-1 coordinately regulate hematopoietic stem cell function through the control of oxidative stress. Blood 2014, 123, 41–50. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Detappe, A.; Cai, K.; Keys, H.R.; Brune, Z.; Ying, W.; Thiru, P.; Reidy, M.; Kugener, G.; Rossen, J.; et al. Mitochondrial metabolism promotes adaptation to proteotoxic stress. Nat. Chem. Biol. 2019, 15, 681–689. [Google Scholar] [CrossRef]
- Ferguson, I.D.; Lin, Y.H.T.; Lam, C.; Shao, H.; Tharp, K.M.; Hale, M.; Kasap, C.; Mariano, M.C.; Kishishita, A.; Patiño Escobar, B.; et al. Allosteric HSP70 inhibitors perturb mitochondrial proteostasis and overcome proteasome inhibitor resistance in multiple myeloma. Cell Chem. Biol. 2022, 29, 1288–1302.e7. [Google Scholar] [CrossRef]
- Huang, L.; Wang, Y.; Bai, J.; Yang, Y.; Wang, F.; Feng, Y.; Zhang, R.; Li, F.; Zhang, P.; Lv, N.; et al. Blockade of HSP70 by VER-155008 synergistically enhances bortezomib-induced cytotoxicity in multiple myeloma. Cell Stress Chaperones 2020, 25, 357–367. [Google Scholar] [CrossRef]
- Li, J.; Zhang, X.; Shen, H.; Guo, J.; Wang, X.; Liu, J. Bortezomib promotes apoptosis of multiple myeloma cells by regulating HSP27. Mol. Med. Rep. 2019, 20, 2410–2418. [Google Scholar] [CrossRef]
- Dubrez, L.; Causse, S.; Borges Bonan, N.; Dumétier, B.; Garrido, C. Heat-shock proteins: Chaperoning DNA repair. Oncogene 2020, 39, 516–529. [Google Scholar] [CrossRef]
- Sottile, M.L.; Nadin, S.B. Heat shock proteins and DNA repair mechanisms: An updated overview. Cell Stress Chaperones 2018, 23, 303–315. [Google Scholar] [CrossRef]
- Chao, H.X.; Poovey, C.E.; Privette, A.A.; Grant, G.D.; Chao, H.Y.; Cook, J.G.; Purvis, J.E. Orchestration of DNA Damage Checkpoint Dynamics across the Human Cell Cycle. Cell Syst. 2017, 5, 445–459.e5. [Google Scholar] [CrossRef]
- Che, Y.; Best, O.G.; Zhong, L.; Kaufman, K.L.; MacTier, S.; Raftery, M.; Graves, L.M.; Mulligan, S.P.; Christopherson, R.I. Hsp90 inhibitor SNX-7081 dysregulates proteins involved with DNA repair and replication and the cell cycle in human chronic lymphocytic leukemia (CLL) cells. J. Proteome Res. 2013, 12, 1710–1722. [Google Scholar] [CrossRef]
- Best, O.G.; Che, Y.; Singh, N.; Forsyth, C.; Christopherson, R.I.; Mulligan, S.P. The Hsp90 inhibitor SNX-7081 synergizes with and restores sensitivity to fludarabine in chronic lymphocytic leukemia cells with lesions in the TP53 pathway: A potential treatment strategy for fludarabine refractory disease. Leuk. Lymphoma 2012, 53, 1367–1375. [Google Scholar] [CrossRef]
- Kaufman, K.L.; Jenkins, Y.; Alomari, M.; Mirzaei, M.; Giles Best, O.; Pascovici, D.; Mactier, S.; Mulligan, S.P.; Haynes, P.A.; Christopherson, R.I. The Hsp90 inhibitor SNX-7081 is synergistic with fludarabine nucleoside via DNA damage and repair mechanisms in human, p53-negative chronic lymphocytic leukemia. Oncotarget 2015, 6, 40981–40997. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.H.; Mitchell, S.; Wu, P.J.; Orwick, S.; Liu, C.; Ravikrishnan, J.; Woyach, J.; Mims, A.; Plunkett, W.; Puduvalli, V.K.; et al. HSP90 inhibition depletes DNA repair proteins to sensitize acute myelogenous leukemia to nucleoside analog chemotherapeutics. Leuk. Lymphoma 2019, 60, 2308–2311. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Song, T.; Wang, Z.; Lin, D.; Cao, K.; Liu, P.; Feng, Y.; Zhang, X.; Wang, P.; Yin, F.; et al. The chaperone Hsp70 is a BH3 receptor activated by the pro-apoptotic Bim to stabilize anti-apoptotic clients. J. Biol. Chem. 2020, 295, 12900–12909. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Guo, Y.; Xue, Z.; Guo, Z.; Wang, Z.; Lin, D.; Zhang, H.; Pan, H.; Zhang, X.; Yin, F.; et al. Small-molecule inhibitor targeting the Hsp70-Bim protein–protein interaction in CML cells overcomes BCR-ABL-independent TKI resistance. Leukemia 2021, 35, 2862–2874. [Google Scholar] [CrossRef]
- Eyre, T.A.; Walter, H.S.; Iyengar, S.; Follows, G.; Cross, M.; Fox, C.P.; Hodson, A.; Coats, J.; Narat, S.; Morley, N.; et al. Efficacy of venetoclax monotherapy in patients with relapsed, refractory mantle cell lymphoma after Bruton tyrosine kinase inhibitor therapy. Haematologica 2019, 104, e68. [Google Scholar] [CrossRef]
- Che, Y.; Wang, W.; Liu, Y.; Lee, H.-H.; Li, Y.; Chen, Z.; McIntosh, J.; Jiang, C.; Yao, Y.; Wang, M. HSP27 Promotes Mantle Cell Lymphoma Growth and Mediates Resistance to Venetoclax. Blood 2022, 140, 6405–6406. [Google Scholar] [CrossRef]
- Beeharry, N.; Landrette, S.; Grotzke, J.; Gayle, S.; Hernandez, M.; Halene, S.; Young, P.R.; Miller, L.L.; Xu, T.; Rothberg, J.; et al. LAM-003, a Novel Oral Heat Shock Protein 90 Inhibitor for Treatment of Acute Myeloid Leukemia, Including Wild-Type and FMS-like Tyrosine Kinase 3 (FLT3)-Mutant Disease. Blood 2019, 134, 2664. [Google Scholar] [CrossRef]
- Lu, Z.; Wang, Z.; Tu, Z.; Liu, H. HSP90 Inhibitor Ganetespib Enhances the Sensitivity of Mantle Cell Lymphoma to Bruton’s Tyrosine Kinase Inhibitor Ibrutinib. Front. Pharmacol. 2022, 13, 864194. [Google Scholar] [CrossRef]
- Albakova, Z.; Mangasarova, Y.; Albakov, A.; Nikulina, E.; Kravchenko, S.; Sapozhnikov, A. Aberrant HSP90 Expression in Lymphocytes and HSP90 Response to Anti-PD-1 Therapy in Lymphoma Patients. Front. Immunol. 2022, 13, 893137. [Google Scholar] [CrossRef]
- Schmidt, L.; Issa, I.I.; Haraldsdóttir, H.; Hald, J.L.; Schmitz, A.; Due, H.; Dybkær, K. Hsp90 inhibition sensitizes DLBCL cells to cisplatin. Cancer Chemother. Pharmacol. 2022, 89, 431. [Google Scholar] [CrossRef]
- Lancet, J.E.; Gojo, I.; Burton, M.; Quinn, M.; Tighe, S.M.; Kersey, K.; Zhong, Z.; Albitar, M.X.; Bhalla, K.; Hannah, A.L.; et al. Phase I study of the heat shock protein 90 inhibitor alvespimycin (KOS-1022, 17-DMAG) administered intravenously twice weekly to patients with acute myeloid leukemia. Leukemia 2010, 24, 699–705. [Google Scholar] [CrossRef]
- George, P.; Bali, P.; Cohen, P.; Tao, J.; Guo, F.; Sigua, C.; Vishvanath, A.; Fiskus, W.; Scuto, A.; Annavarapu, S.; et al. Cotreatment with 17-allylamino-demethoxygeldanamycin and FLT-3 kinase inhibitor PKC412 is highly effective against human acute myelogenous leukemia cells with mutant FLT-3. Cancer Res. 2004, 64, 3645–3652. [Google Scholar] [CrossRef]
- George, P.; Bali, P.; Annavarapu, S.; Scuto, A.; Fiskus, W.; Guo, F.; Sigua, C.; Sondarva, G.; Moscinski, L.; Atadja, P.; et al. Combination of the histone deacetylase inhibitor LBH589 and the hsp90 inhibitor 17-AAG is highly active against human CML-BC cells and AML cells with activating mutation of FLT-3. Blood 2005, 105, 1768–1776. [Google Scholar] [CrossRef] [Green Version]
- Khajapeer, K.V.; Baskaran, R. Hsp90 Inhibitors for the Treatment of Chronic Myeloid Leukemia. Leuk. Res. Treatment 2015, 2015. [Google Scholar] [CrossRef]
- Qin, Y.; Liang, Y.; Jiang, G.; Peng, Y.; Feng, W. ACY-1215 suppresses the proliferation and induces apoptosis of chronic myeloid leukemia cells via the ROS/PTEN/Akt pathway. Cell Stress Chaperones 2022, 27, 383–396. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, J.; Zhang, T.; Shi, M.; Chen, X.; Chen, Y.; Yu, J. Destabilization of ROR1 enhances activity of Ibrutinib against chronic lymphocytic leukemia in vivo. Pharmacol. Res. 2020, 151, 104512. [Google Scholar] [CrossRef]
- Kourtis, N.; Lazaris, C.; Hockemeyer, K.; Balandrán, J.C.; Jimenez, A.R.; Mullenders, J.; Gong, Y.; Trimarchi, T.; Bhatt, K.; Hu, H.; et al. Oncogenic hijacking of the stress response machinery in T cell acute lymphoblastic leukemia. Nat. Med. 2018, 24, 1157. [Google Scholar] [CrossRef]
- Ikebe, E.; Shimosaki, S.; Hasegawa, H.; Iha, H.; Tsukamoto, Y.; Wang, Y.; Sasaki, D.; Imaizumi, Y.; Miyazaki, Y.; Yanagihara, K.; et al. TAS-116 (pimitespib), a heat shock protein 90 inhibitor, shows efficacy in preclinical models of adult T-cell leukemia. Cancer Sci. 2022, 113, 684–696. [Google Scholar] [CrossRef]
- Gvozdenov, Z.; Kolhe, J.; Freeman, B.C. The nuclear and DNA-associated molecular chaperone network. Cold Spring Harb. Perspect. Biol. 2019, 11, a034009. [Google Scholar] [CrossRef]
- Ren, X.; Li, T.; Zhang, W.; Yang, X. Targeting Heat-Shock Protein 90 in Cancer: An Update on Combination Therapy. Cells 2022, 11, 2556. [Google Scholar] [CrossRef]
- Wang, Y.; Koay, Y.C.; McAlpine, S.R. How selective are Hsp90 inhibitors for cancer cells over normal cells? ChemMedChem 2017, 12, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Birbo, B.; Madu, E.E.; Madu, C.O.; Jain, A.; Lu, Y. Role of hsp90 in cancer. Int. J. Mol. Sci. 2021, 22, 10317. [Google Scholar] [CrossRef] [PubMed]
- Mendillo, M.L.; Santagata, S.; Koeva, M.; Bell, G.W.; Hu, R.; Tamimi, R.M.; Fraenkel, E.; Ince, T.A.; Whitesell, L.; Lindquist, S. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 2012, 150, 549–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jego, G.; Lanneau, D.; De Thonel, A.; Berthenet, K.; Hazoumé, A.; Droin, N.; Hamman, A.; Girodon, F.; Bellaye, P.S.; Wettstein, G.; et al. Dual regulation of SPI1/PU.1 transcription factor by heat shock factor 1 (HSF1) during macrophage differentiation of monocytes. Leukemia 2014, 28, 1676–1686. [Google Scholar] [CrossRef]
- Jin, X.; Moskophidis, D.; Mivechi, N.F. Heat Shock Transcription Factor 1 Is a Key Determinant of HCC Development by Regulating Hepatic Steatosis and Metabolic Syndrome. Cell Metab. 2011, 14, 91–103. [Google Scholar] [CrossRef]
- Dong, B.; Jaeger, A.M.; Hughes, P.F.; Loiselle, D.R.; Spencer Hauck, J.; Fu, Y.; Haystead, T.A.; Huang, J.; Thiele, D.J. Targeting therapy-resistant prostate cancer via a direct inhibitor of the human heat shock transcription factor 1. Sci. Transl. Med. 2020, 12, eabb5647. [Google Scholar] [CrossRef]
- Jego, G.; Chiron, D.; Berthenet, K.; Pellat-Deceunynck, C. Modulation of normal and malignant plasma cells function by toll-like receptors. Front. Biosci—Elit. 2012, 4, 2289–2301. [Google Scholar] [CrossRef]
- Chalmin, F.; Ladoire, S.; Mignot, G.; Vincent, J.; Bruchard, M.; Remy-Martin, J.P.; Boireau, W.; Rouleau, A.; Simon, B.; Lanneau, D.; et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J. Clin. Invest. 2010, 120, 457–471. [Google Scholar] [CrossRef]
- Gobbo, J.; Marcion, G.; Cordonnier, M.; Dias, A.M.M.M.; Pernet, N.; Hammann, A.; Richaud, S.; Mjahed, H.; Isambert, N.; Clausse, V.; et al. Restoring Anticancer Immune Response by Targeting Tumor-Derived Exosomes With a HSP70 Peptide Aptamer. J. Natl. Cancer Inst. 2016, 108, djv330. [Google Scholar] [CrossRef]
- Marcion, G.; Hermetet, F.; Neiers, F.; Uyanik, B.; Dondaine, L.; Dias, A.M.M.; Da Costa, L.; Moreau, M.; Bellaye, P.S.; Collin, B.; et al. Nanofitins targeting heat shock protein 110: An innovative immunotherapeutic modality in cancer. Int. J. Cancer 2021, 148, 3019–3031. [Google Scholar] [CrossRef]
- Regimbeau, M.; Abrey, J.; Vautrot, V.; Causse, S.; Gobbo, J.; Garrido, C. Heat shock proteins and exosomes in cancer theranostics. Semin. Cancer Biol. 2022, 86, 46–57. [Google Scholar] [CrossRef]
- Provencio, M.; Rodríguez, M.; Cantos, B.; Sabín, P.; Quero, C.; García-Arroyo, F.R.; Rueda, A.; Maximiano, C.; Rodríguez-Abreu, D.; Sánchez, A.; et al. mRNA in exosomas as a liquid biopsy in non-Hodgkin Lymphoma: A multicentric study by the Spanish lymphoma oncology group. Oncotarget 2017, 8, 50949–50957. [Google Scholar] [CrossRef]
- Cordonnier, M.; Chanteloup, G.; Isambert, N.; Seigneuric, R.; Fumoleau, P.; Garrido, C.; Gobbo, J. Exosomes in cancer theranostic: Diamonds in the rough. Cell Adhes. Migr. 2017, 11, 151–163. [Google Scholar] [CrossRef] [Green Version]
- Chanteloup, G.; Cordonnier, M.; Isambert, N.; Bertaut, A.; Hervieu, A.; Hennequin, A.; Luu, M.; Zanetta, S.; Coudert, B.; Bengrine, L.; et al. Monitoring HSP70 exosomes in cancer patients’ follow up: A clinical prospective pilot study. J. Extracell. Vesicles 2020, 9, 1766192. [Google Scholar] [CrossRef]
- Cordonnier, M.; Nardin, C.; Chanteloup, G.; Derangere, V.; Algros, M.P.; Arnould, L.; Garrido, C.; Aubin, F.; Gobbo, J. Tracking the evolution of circulating exosomal-PD-L1 to monitor melanoma patients. J. Extracell. Vesicles 2020, 9, 1710899. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| HSP Inhibitors | Target | Mechanism of Action | Phase of Development | Hematological Malignancies |
|---|---|---|---|---|
| Foldamers 33 and 52 | HSP110 | Binding to the ATP binding domain | Pre-clinical research | ABC DLBCL [67] |
| PUH71 (Zelavespib) | HSP90 | Binding to the ATP binding domain | Clinical: Phase I | NHL NCT01269593/MPN NCT01393509 |
| Pre-clinical research (with Ibrutinib) | ALL/ABC DLBCL [63] | |||
| Pre-clinical research (with PI3K/mTOR inhibitors) | BL [93] | |||
| SNX-5422 (PF-04929113) | HSP90 | Convert to SNX-2112 and binding to the ATP binding domain | Clinical: Phase I (with Ibrutinib) | CLL [64,157] |
| SNX-7081 | HSP90 | Binding to the ATP binding domain | Pre-clinical research (with Fludarabine) | CLL [140] |
| NVP-AUY922 (Luminespib) | HSP90 | Binding to the ATP binding domain | Pre-clinical research (with Cytarabine) | AML [94,104] |
| Aminoxyrone | HSP90 | Binding to C-terminal dimerization domain | Pre-clinical research | CML [100] |
| 17-AAG (Tanespimycin) | HSP90 | Binding to the ATP binding domain | Clinical: Phase II | Hodgkin/MCL/ALCL/MM (NCT00117988) |
| Pre-clinical research (with HDAC inhibitors) | AML/CML [125] | |||
| LAM-003 (MPC-3100) | HSP90 | Convert to LAM-003A | Clinical: Phase I | AML [148] |
| Ganetespib | HSP90 | Binding to the ATP binding domain | Clinical: Phase I | MM (NCT01485835) |
| Pre-clinical research (with Ibrutinib) | MCL [149] | |||
| Pre-clinical research (with cytarabine) | CML [94] | |||
| 17-DMAG (Alvespimycin) | HSP90 | Binding to the ATP binding domain | Clinical: Phase I | Lymphoma & leukemia (NCT00089271) |
| Pre-clinical research | AML CLL [71] | |||
| TAS-116 (Pimitespib) | HSP90 | Binding to the N-terminal domain of HSP90 | Pre-clinical research | ALL [159] |
| PFT-µ (Pifithrin-μ) | HSP70 | Binding to the C-terminal domain of HSP70 | Pre-clinical research | ALL/AML [97] |
| VER-155008 | HSP70 | Binding to the ATP binding domain | Pre-clinical research | MM [135] |
| JG-98 | HSP70 | Allosteric HSP70 inhibitors | Pre-clinical research | MM [134] |
| DTHIB | HSF1 | Binding to the DNA binding domain | Pre-clinical research | AML [31] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabaud-Gibouin, V.; Durand, M.; Quéré, R.; Girodon, F.; Garrido, C.; Jego, G. Heat-Shock Proteins in Leukemia and Lymphoma: Multitargets for Innovative Therapeutic Approaches. Cancers 2023, 15, 984. https://doi.org/10.3390/cancers15030984
Cabaud-Gibouin V, Durand M, Quéré R, Girodon F, Garrido C, Jego G. Heat-Shock Proteins in Leukemia and Lymphoma: Multitargets for Innovative Therapeutic Approaches. Cancers. 2023; 15(3):984. https://doi.org/10.3390/cancers15030984
Chicago/Turabian StyleCabaud-Gibouin, Vincent, Manon Durand, Ronan Quéré, François Girodon, Carmen Garrido, and Gaëtan Jego. 2023. "Heat-Shock Proteins in Leukemia and Lymphoma: Multitargets for Innovative Therapeutic Approaches" Cancers 15, no. 3: 984. https://doi.org/10.3390/cancers15030984
APA StyleCabaud-Gibouin, V., Durand, M., Quéré, R., Girodon, F., Garrido, C., & Jego, G. (2023). Heat-Shock Proteins in Leukemia and Lymphoma: Multitargets for Innovative Therapeutic Approaches. Cancers, 15(3), 984. https://doi.org/10.3390/cancers15030984