
Antigene MYCN Silencing by BGA002 Inhibits SCLC Progression Blocking mTOR Pathway and Overcomes Multidrug Resistance

 , ,
, ,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Cell Line Treatment
2.3. Quantitative Real-Time PCR
2.4. Cell Viability Assay and Western Blot Analysis
2.5. Apoptosis Analysis
2.6. Transmission Electron Microscopy
2.7. Lysosomes Distribution Analysis
2.8. Xenograft Ectotopic SCLC Mouse Models
2.9. Statistical Analysis
3. Results
3.1. BGA002, a Specific MYCN Antigene Oligonucleotide, Strongly Inhibits MYCN Tumorigenic Activity in SCLC Cells
3.2. BGA002 Treatment Increases Lysosome Number and Modifies Their Distribution in SCLC Cells
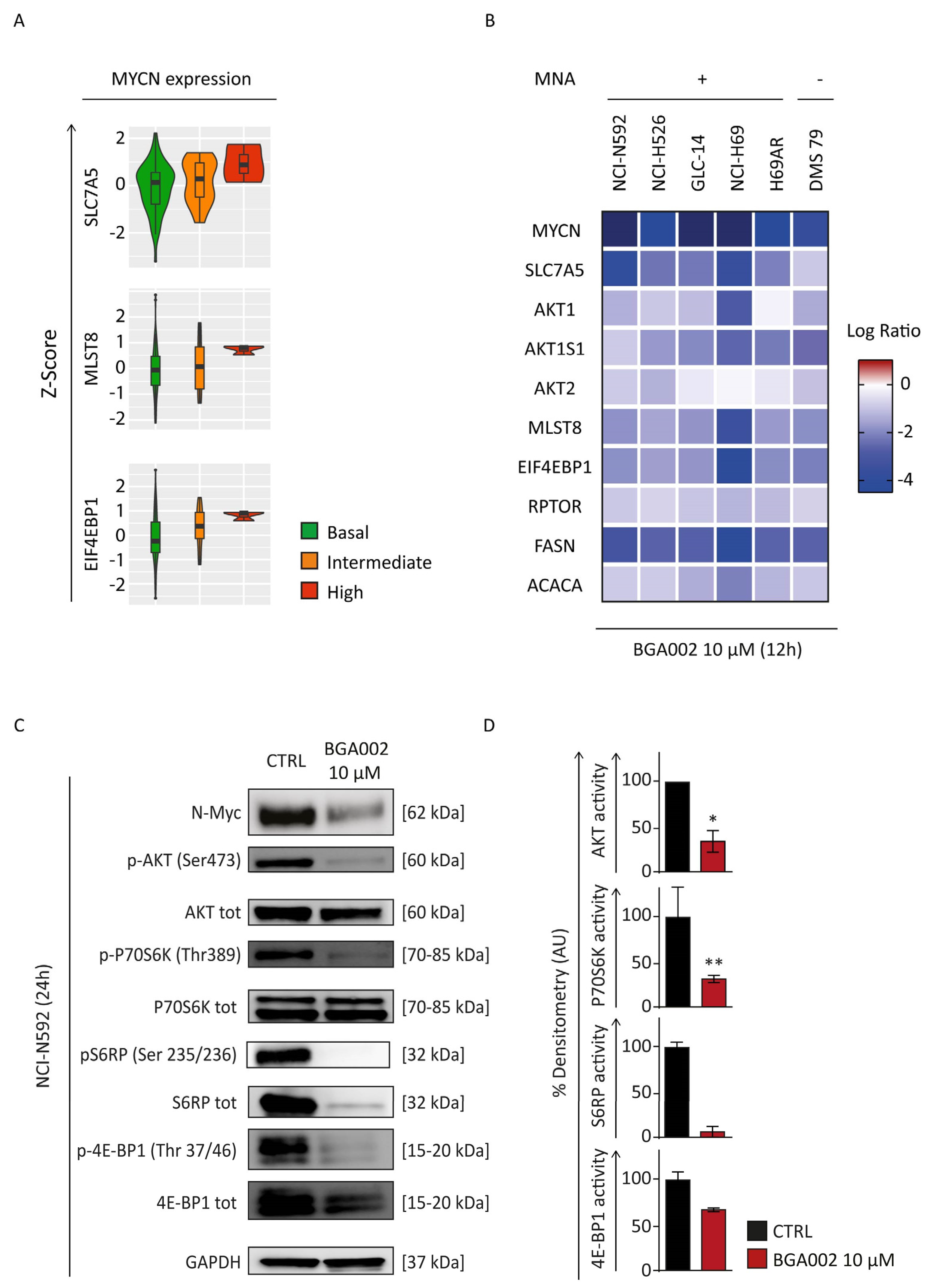
3.3. MYCN Inhibition Blocks mTOR Complex in SCLC Cells
3.4. BGA002 Regulates Multiple Cancer Related Pathways in SCLC Cells
3.5. BGA002 Improves Survival in MNA-SCLC Xenograft Mouse Models, Independently of Multidrug Resistance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morabito, A.; Carillio, G.; Daniele, G.; Piccirillo, M.C.; Montanino, A.; Costanzo, R.; Sandomenico, C.; Giordano, P.; Normanno, N.; Perrone, F.; et al. Treatment of Small Cell Lung Cancer. Crit. Rev. Oncol. Hematol. 2014, 91, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Thai, A.A.; Solomon, B.J.; Sequist, L.V.; Gainor, J.F.; Heist, R.S. Lung Cancer. Lancet 2021, 398, 535–554. [Google Scholar] [CrossRef] [PubMed]
- Peifer, M.; Fernández-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative Genome Analyses Identify Key Somatic Driver Mutations of Small-Cell Lung Cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- van Meerbeeck, J.P.; Fennell, D.A.; De Ruysscher, D.K. Small-Cell Lung Cancer. Lancet 2011, 378, 1741–1755. [Google Scholar] [CrossRef]
- Califano, R.; Abidin, A.Z.; Peck, R.; Faivre-Finn, C.; Lorigan, P. Management of Small Cell Lung Cancer: Recent Developments for Optimal Care. Drugs 2012, 72, 471–490. [Google Scholar] [CrossRef]
- Gazdar, A.F.; Bunn, P.A.; Minna, J.D. Small-Cell Lung Cancer: What We Know, What We Need to Know and the Path Forward. Nat. Rev. Cancer 2017, 17, 725–737. [Google Scholar] [CrossRef]
- Poirier, J.T.; George, J.; Owonikoko, T.K.; Berns, A.; Brambilla, E.; Byers, L.A.; Carbone, D.; Chen, H.J.; Christensen, C.L.; Dive, C.; et al. New Approaches to SCLC Therapy: From the Laboratory to the Clinic. J. Thorac. Oncol. 2020, 15, 520–540. [Google Scholar] [CrossRef]
- Bunn, P.A.; Minna, J.D.; Augustyn, A.; Gazdar, A.F.; Ouadah, Y.; Krasnow, M.A.; Berns, A.; Brambilla, E.; Rekhtman, N.; Massion, P.P.; et al. Small Cell Lung Cancer: Can Recent Advances in Biology and Molecular Biology Be Translated into Improved Outcomes? J. Thorac. Oncol. 2016, 11, 453–474. [Google Scholar] [CrossRef]
- Wang, Y.; Zou, S.; Zhao, Z.; Liu, P.; Ke, C.; Xu, S. New Insights into Small-Cell Lung Cancer Development and Therapy. Cell Biol. Int. 2020, 44, 1564–1576. [Google Scholar] [CrossRef]
- Qin, A.; Kalemkerian, G.P. Treatment Options for Relapsed Small-Cell Lung Cancer: What Progress Have We Made? J. Oncol. Pract. 2018, 14, 369–370. [Google Scholar] [CrossRef]
- Tariq, S.; Kim, S.Y.; Monteiro de Oliveira Novaes, J.; Cheng, H. Update 2021: Management of Small Cell Lung Cancer. Lung 2021, 199, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, A.S.; Holliday, E.B.; Allen, P.K.; Wei, X.; Cox, J.D.; Komaki, R. Prophylactic Cranial Irradiation after Definitive Chemoradiotherapy for Limited-Stage Small Cell Lung Cancer: Do All Patients Benefit? Radiother. Oncol. 2017, 122, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.Y.; Sio, T.T.; Ernani, V.; Savvides, P.; Schild, S.E. Role of Prophylactic Cranial Irradiation in Extensive-Stage Small Cell Lung Cancer. J. Natl. Compr. Canc. Netw. 2021, 19, 1465–1469. [Google Scholar] [CrossRef]
- Asai, N.; Ohkuni, Y.; Kaneko, N.; Yamaguchi, E.; Kubo, A. Relapsed Small Cell Lung Cancer: Treatment Options and Latest Developments. Ther. Adv. Med. Oncol. 2014, 6, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Salgia, R. Managing Patients With Relapsed Small-Cell Lung Cancer. J. Oncol. Pract. 2018, 14, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Zugazagoitia, J.; Paz-Ares, L. Extensive-Stage Small-Cell Lung Cancer: First-Line and Second-Line Treatment Options. J. Clin. Oncol. 2022, 40, 671–680. [Google Scholar] [CrossRef]
- Horita, N.; Yamamoto, M.; Sato, T.; Tsukahara, T.; Nagakura, H.; Tashiro, K.; Shibata, Y.; Watanabe, H.; Nagai, K.; Inoue, M.; et al. Topotecan for Relapsed Small-Cell Lung Cancer: Systematic Review and Meta-Analysis of 1347 Patients. Sci. Rep. 2015, 5, 15437. [Google Scholar] [CrossRef]
- Singh, S.; Jaigirdar, A.A.; Mulkey, F.; Cheng, J.; Hamed, S.S.; Li, Y.; Liu, J.; Zhao, H.; Goheer, A.; Helms, W.S.; et al. FDA Approval Summary: Lurbinectedin for the Treatment of Metastatic Small Cell Lung Cancer. Clin. Cancer Res. 2021, 27, 2378–2382. [Google Scholar] [CrossRef]
- Patel, S.; Petty, W.J.; Sands, J.M. An Overview of Lurbinectedin as a New Second-Line Treatment Option for Small Cell Lung Cancer. Ther. Adv. Med. Oncol. 2021, 13, 17588359211020528. [Google Scholar] [CrossRef]
- Arriola, E.; González-Cao, M.; Domine, M.; De Castro, J.; Cobo, M.; Bernabé, R.; Navarro, A.; Sullivan, I.; Trigo, J.M.; Mosquera, J.; et al. Addition of Immune Checkpoint Inhibitors to Chemotherapy vs. Chemotherapy Alone as First-Line Treatment in Extensive-Stage Small-Cell Lung Carcinoma: A Systematic Review and Meta-Analysis. Oncol. Ther. 2022, 10, 167–184. [Google Scholar] [CrossRef]
- Hou, W.; Zhou, X.; Yi, C.; Zhu, H. Immune Check Point Inhibitors and Immune-Related Adverse Events in Small Cell Lung Cancer. Front. Oncol. 2021, 11, 604227. [Google Scholar] [CrossRef] [PubMed]
- Niu, Z.; Guo, S.; Cao, J.; Zhang, Y.; Guo, X.; Grossi, F.; Ichiki, Y.; Li, Y.; Wang, Z. Immune Checkpoint Inhibitors for Treatment of Small-Cell Lung Cancer: A Systematic Review and Meta-Analysis. Ann. Transl. Med. 2021, 9, 705. [Google Scholar] [CrossRef]
- Sher, T.; Dy, G.K.; Adjei, A.A. Small Cell Lung Cancer. Mayo Clin. Proc. 2008, 83, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Altan, M.; Chiang, A.C. Management of Small Cell Lung Cancer: Progress and Updates. Cancer J. 2015, 21, 425. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Pérez, M.V.; Henley, A.B.; Arsenian-Henriksson, M. The MYCN Protein in Health and Disease. Genes 2017, 8, 113. [Google Scholar] [CrossRef]
- Massó-Vallés, D.; Beaulieu, M.-E.; Soucek, L. MYC, MYCL, and MYCN as Therapeutic Targets in Lung Cancer. Expert Opin. Ther. Targets 2020, 24, 101–114. [Google Scholar] [CrossRef]
- Beltran, H. The N-Myc Oncogene: Maximizing Its Targets, Regulation, and Therapeutic Potential. Mol. Cancer. Res. 2014, 12, 815–822. [Google Scholar] [CrossRef]
- Grunblatt, E.; Wu, N.; Zhang, H.; Liu, X.; Norton, J.P.; Ohol, Y.; Leger, P.; Hiatt, J.B.; Eastwood, E.C.; Thomas, R.; et al. MYCN Drives Chemoresistance in Small Cell Lung Cancer While USP7 Inhibition Can Restore Chemosensitivity. Genes Dev. 2020, 34, 1210–1226. [Google Scholar] [CrossRef]
- Lissa, D.; Takahashi, N.; Desai, P.; Manukyan, I.; Schultz, C.W.; Rajapakse, V.; Velez, M.J.; Mulford, D.; Roper, N.; Nichols, S.; et al. Heterogeneity of Neuroendocrine Transcriptional States in Metastatic Small Cell Lung Cancers and Patient-Derived Models. Nat. Commun. 2022, 13, 2023. [Google Scholar] [CrossRef]
- Patel, A.S.; Yoo, S.; Kong, R.; Sato, T.; Sinha, A.; Karam, S.; Bao, L.; Fridrikh, M.; Emoto, K.; Nudelman, G.; et al. Prototypical Oncogene Family Myc Defines Unappreciated Distinct Lineage States of Small Cell Lung Cancer. Sci. Adv. 2021, 7, eabc2578. [Google Scholar] [CrossRef]
- Gay, C.M.; Stewart, C.A.; Park, E.M.; Diao, L.; Groves, S.M.; Heeke, S.; Nabet, B.Y.; Fujimoto, J.; Solis, L.M.; Lu, W.; et al. Patterns of Transcription Factor Programs and Immune Pathway Activation Define Four Major Subtypes of SCLC with Distinct Therapeutic Vulnerabilities. Cancer Cell 2021, 39, 346–360. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, S.S.; Clarke, S.; Veschi, V.; Thiele, C.J. Targeting MYCN in Pediatric and Adult Cancers. Front. Oncol. 2021, 10, 623679. [Google Scholar] [CrossRef]
- Tonelli, R.; Purgato, S.; Camerin, C.; Fronza, R.; Bologna, F.; Alboresi, S.; Franzoni, M.; Corradini, R.; Sforza, S.; Faccini, A.; et al. Anti-Gene Peptide Nucleic Acid Specifically Inhibits MYCN Expression in Human Neuroblastoma Cells Leading to Cell Growth Inhibition and Apoptosis. Mol. Cancer Ther. 2005, 4, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, R.; McIntyre, A.; Camerin, C.; Walters, Z.S.; Di Leo, K.; Selfe, J.; Purgato, S.; Missiaglia, E.; Tortori, A.; Renshaw, J.; et al. Antitumor Activity of Sustained N-Myc Reduction in Rhabdomyosarcomas and Transcriptional Block by Antigene Therapy. Clin. Cancer Res. 2012, 18, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Montemurro, L.; Raieli, S.; Angelucci, S.; Bartolucci, D.; Amadesi, C.; Lampis, S.; Scardovi, A.L.; Venturelli, L.; Nieddu, G.; Cerisoli, L.; et al. A Novel MYCN-Specific Antigene Oligonucleotide Deregulates Mitochondria and Inhibits Tumor Growth in MYCN-Amplified Neuroblastoma. Cancer Res. 2019, 79, 6166–6177. [Google Scholar] [CrossRef] [PubMed]
- Lampis, S.; Raieli, S.; Montemurro, L.; Bartolucci, D.; Amadesi, C.; Bortolotti, S.; Angelucci, S.; Scardovi, A.L.; Nieddu, G.; Cerisoli, L.; et al. The MYCN Inhibitor BGA002 Restores the Retinoic Acid Response Leading to Differentiation or Apoptosis by the MTOR Block in MYCN-Amplified Neuroblastoma. J. Exp. Clin. Cancer Res. 2022, 41, 160. [Google Scholar] [CrossRef]
- Evangelisti, C.; Paganelli, F.; Giuntini, G.; Mattioli, E.; Cappellini, A.; Ramazzotti, G.; Faenza, I.; Maltarello, M.; Martelli, A.; Scotlandi, K.; et al. Lamin A and Prelamin A Counteract Migration of Osteosarcoma Cells. Cells 2020, 9, 774. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef]
- Circu, M.; Cardelli, J.; Barr, M.; O’Byrne, K.; Mills, G.; El-Osta, H. Modulating Lysosomal Function through Lysosome Membrane Permeabilization or Autophagy Suppression Restores Sensitivity to Cisplatin in Refractory Non-Small-Cell Lung Cancer Cells. PLoS ONE 2017, 12, e0184922. [Google Scholar] [CrossRef]
- Groth-Pedersen, L.; Jäättelä, M. Combating Apoptosis and Multidrug Resistant Cancers by Targeting Lysosomes. Cancer Lett. 2013, 332, 265–274. [Google Scholar] [CrossRef]
- Chen, Q.-Y.; Shi, J.-G.; Yao, Q.-H.; Jiao, D.-M.; Wang, Y.-Y.; Hu, H.-Z.; Wu, Y.-Q.; Song, J.; Yan, J.; Wu, L.-J. Lysosomal Membrane Permeabilization Is Involved in Curcumin-Induced Apoptosis of A549 Lung Carcinoma Cells. Mol. Cell Biochem. 2012, 359, 389–398. [Google Scholar] [CrossRef]
- Enzenmüller, S.; Gonzalez, P.; Debatin, K.-M.; Fulda, S. Chloroquine Overcomes Resistance of Lung Carcinoma Cells to the Dual PI3K/MTOR Inhibitor PI103 by Lysosome-Mediated Apoptosis. Anti Cancer Drugs 2013, 24, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Zong, D.; Zielinska-Chomej, K.; Juntti, T.; Mörk, B.; Lewensohn, R.; Hååg, P.; Viktorsson, K. Harnessing the Lysosome-Dependent Antitumor Activity of Phenothiazines in Human Small Cell Lung Cancer. Cell Death Dis. 2014, 5, e1111. [Google Scholar] [CrossRef] [PubMed]
- Araujo, M.E.G.; Liebscher, G.; Hess, M.W.; Huber, L.A. Lysosomal Size Matters. Traffic 2020, 21, 60–75. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Y.; Sabatini, D.M. MTOR at the Nexus of Nutrition, Growth, Ageing and Disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Zhang, W.; Wu, L.; Liu, L.; Men, Y.; Wang, J.; Liang, J.; Hui, Z.; Zhou, Z.; Bi, N.; et al. MicroRNA-Related Polymorphisms in PI3K/Akt/MTOR Pathway Genes Are Predictive of Limited-Disease Small Cell Lung Cancer Treatment Outcomes. Biomed. Res. Int. 2017, 2017, 1–10. [Google Scholar] [CrossRef]
- Umemura, S.; Mimaki, S.; Makinoshima, H.; Tada, S.; Ishii, G.; Ohmatsu, H.; Niho, S.; Yoh, K.; Matsumoto, S.; Takahashi, A.; et al. Therapeutic Priority of the PI3K/AKT/MTOR Pathway in Small Cell Lung Cancers as Revealed by a Comprehensive Genomic Analysis. J. Thorac. Oncol. 2014, 9, 1324–1331. [Google Scholar] [CrossRef]
- Krencz, I.; Sztankovics, D.; Danko, T.; Sebestyen, A.; Khoor, A. Progression and Metastasis of Small Cell Lung Carcinoma: The Role of the PI3K/Akt/MTOR Pathway and Metabolic Alterations. Cancer Metastasis Rev. 2021, 40, 1141–1157. [Google Scholar] [CrossRef]
- Marinov, M.; Fischer, B.; Arcaro, A. Targeting MTOR Signaling in Lung Cancer. Crit. Rev. Oncol. Hematol. 2007, 63, 172–182. [Google Scholar] [CrossRef]
- Alì, G.; Boldrini, L.; Capodanno, A.; Pelliccioni, S.; Servadio, A.; Crisman, G.; Picchi, A.; Davini, F.; Mussi, A.; Fontanini, G. Expression of P-AKT and p-MTOR in a Large Series of Bronchopulmonary Neuroendocrine Tumors. Exp. Ther. Med. 2011, 2, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.A.; Kim, J.; Foster, D.G.; Mishra, R.; Gardner, E.E.; Poirier, J.T.; Rivard, C.; Yu, H.; Finigan, J.H.; Dowlati, A.; et al. Role of MTOR as an Essential Kinase in SCLC. J. Thorac. Oncol. 2020, 15, 1522–1534. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Marquez, C.P.; Kostyrko, K.; Koehne, A.L.; Marini, K.; Simpson, D.R.; Lee, A.G.; Leung, S.G.; Sayles, L.C.; Shrager, J.; et al. Antitumor Activity of an Engineered Decoy Receptor Targeting CLCF1-CNTFR Signaling in Lung Adenocarcinoma. Nat. Med. 2019, 25, 1783–1795. [Google Scholar] [CrossRef] [PubMed]
- Marquez, C.; Kim, J.; Giaccia, A.; Cochran, J.; Sweet-Cordero, A. Abstract 3918: Targeting the CLCF1-CNTFR Signaling Axis Using Directed Evolution for Lung Cancer Therapy. Cancer Res. 2018, 78, 3918. [Google Scholar] [CrossRef]
- Vicent, S.; Sayles, L.C.; Vaka, D.; Khatri, P.; Gevaert, O.; Chen, R.; Zheng, Y.; Gillespie, A.K.; Clarke, N.; Xu, Y.; et al. Cross-Species Functional Analysis of Cancer-Associated Fibroblasts Identifies a Critical Role for CLCF1 and IL-6 in Non–Small Cell Lung Cancer In Vivo. Cancer Res. 2012, 72, 5744–5756. [Google Scholar] [CrossRef]
- Kato, T.; Sato, N.; Takano, A.; Miyamoto, M.; Nishimura, H.; Tsuchiya, E.; Kondo, S.; Nakamura, Y.; Daigo, Y. Activation of Placenta-Specific Transcription Factor Distal-Less Homeobox 5 Predicts Clinical Outcome in Primary Lung Cancer Patients. Clin. Cancer Res. 2008, 14, 2363–2370. [Google Scholar] [CrossRef]
- Das, T.; Chen, Z.; Hendriks, R.W.; Kool, M. A20/Tumor Necrosis Factor α-Induced Protein 3 in Immune Cells Controls Development of Autoinflammation and Autoimmunity: Lessons from Mouse Models. Front. Immunol. 2018, 9, 104. [Google Scholar] [CrossRef]
- Chu, Y.; Vahl, J.C.; Kumar, D.; Heger, K.; Bertossi, A.; Wójtowicz, E.; Soberon, V.; Schenten, D.; Mack, B.; Reutelshöfer, M.; et al. B Cells Lacking the Tumor Suppressor TNFAIP3/A20 Display Impaired Differentiation and Hyperactivation and Cause Inflammation and Autoimmunity in Aged Mice. Blood 2011, 117, 2227–2236. [Google Scholar] [CrossRef]
- Yang, H.-C.; Wu, Y.-H.; Yen, W.-C.; Liu, H.-Y.; Hwang, T.-L.; Stern, A.; Chiu, D.T.-Y. The Redox Role of G6PD in Cell Growth, Cell Death, and Cancer. Cells 2019, 8, 1055. [Google Scholar] [CrossRef]
- Zhu, K.; Deng, C.; Du, P.; Liu, T.; Piao, J.; Piao, Y.; Yang, M.; Chen, L. G6PC Indicated Poor Prognosis in Cervical Cancer and Promoted Cervical Carcinogenesis in Vitro and in Vivo. Reprod. Biol. Endocrinol. 2022, 20, 50. [Google Scholar] [CrossRef]
- Uchimura, T.; Hollander, J.M.; Nakamura, D.S.; Liu, Z.; Rosen, C.J.; Georgakoudi, I.; Zeng, L. An Essential Role for IGF2 in Cartilage Development and Glucose Metabolism during Postnatal Long Bone Growth. Development 2017, 144, 3533–3546. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Garg, M.; Wang, Y.; Wang, W.; Godbout, R. DEAD Box 1 (DDX1) Protein Binds to and Protects Cytoplasmic Stress Response MRNAs in Cells Exposed to Oxidative Stress. J. Biol. Chem. 2022, 298, 102180. [Google Scholar] [CrossRef] [PubMed]
- Kaehler, C.; Isensee, J.; Nonhoff, U.; Terrey, M.; Hucho, T.; Lehrach, H.; Krobitsch, S. Ataxin-2-Like Is a Regulator of Stress Granules and Processing Bodies. PLoS ONE 2012, 7, e50134. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Alvarez, A.J.; Pascual, M.L.; Boccaccio, G.L.; Thomas, M.G. Smaug Variants in Neural and Non-Neuronal Cells. Commun. Integr. Biol. 2016, 9, e1139252. [Google Scholar] [CrossRef] [PubMed]
- Ujhazy, P.; Lindwasser, O.W. Small Cell Lung Cancer: Updates and New Concepts. Transl. Lung Cancer Res. 2018, 7, 1–3. [Google Scholar] [CrossRef]
- De Cássia, S.; Alves, R.; Meurer, R.T.; Roehe, A.V. MYC Amplification Is Associated with Poor Survival in Small Cell Lung Cancer: A Chromogenic in Situ Hybridization Study. J. Cancer Res. Clin. Oncol. 2014, 140, 2021–2025. [Google Scholar] [CrossRef]
- Nau, M.M.; Carney, D.N.; Battey, J.; Johnson, B.; Little, C.; Gazdar, A.; Minna, J.D. Amplification, Expression and Rearrangement of c-Myc and N-Myc Oncogenes in Human Lung Cancer. Curr. Top. Microbiol. Immunol. 1984, 113, 172–177. [Google Scholar] [CrossRef]
- Funa, K.; Steinholtz, L.; Nou, E.; Bergh, J. Increased Expression of N-Myc in Human Small Cell Lung Cancer Biopsies Predicts Lack of Response to Chemotherapy and Poor Prognosis. Am. J. Clin. Pathol. 1987, 88, 216–220. [Google Scholar] [CrossRef]
- Simpson, K.L.; Stoney, R.; Frese, K.K.; Simms, N.; Rowe, W.; Pearce, S.P.; Humphrey, S.; Booth, L.; Morgan, D.; Dynowski, M.; et al. A Biobank of Small Cell Lung Cancer CDX Models Elucidates Inter- and Intratumoral Phenotypic Heterogeneity. Nat. Cancer 2020, 1, 437–451. [Google Scholar] [CrossRef]
- Raieli, S.; Di Renzo, D.; Lampis, S.; Amadesi, C.; Montemurro, L.; Pession, A.; Hrelia, P.; Fischer, M.; Tonelli, R. MYCN Drives a Tumor Immunosuppressive Environment Which Impacts Survival in Neuroblastoma. Front. Oncol. 2021, 11, 625207. [Google Scholar] [CrossRef]
- Magaway, C.; Kim, E.; Jacinto, E. Targeting MTOR and Metabolism in Cancer: Lessons and Innovations. Cells 2019, 8, 1584. [Google Scholar] [CrossRef] [PubMed]
- Soefje, S.A.; Karnad, A.; Brenner, A.J. Common Toxicities of Mammalian Target of Rapamycin Inhibitors. Target. Oncol. 2011, 6, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, E.; Himmelmann, A.; Malipiero, U.; Tobler, A.; Stahel, R.; Fontana, A. Human Small Cell Lung Cancer Expresses the Octamer DNA-Binding and Nervous System-Specific Transcription Factor N-Oct 3 (Brain-2)1. Cancer Res. 1992, 52, 6121–6124. [Google Scholar] [PubMed]
- Olsen, R.R.; Otero, J.H.; García-López, J.; Wallace, K.; Finkelstein, D.; Rehg, J.E.; Yin, Z.; Wang, Y.-D.; Freeman, K.W. MYCN Induces Neuroblastoma in Primary Neural Crest Cells. Oncogene 2017, 36, 5075–5082. [Google Scholar] [CrossRef]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bortolotti, S.; Angelucci, S.; Montemurro, L.; Bartolucci, D.; Raieli, S.; Lampis, S.; Amadesi, C.; Scardovi, A.; Nieddu, G.; Cerisoli, L.; et al. Antigene MYCN Silencing by BGA002 Inhibits SCLC Progression Blocking mTOR Pathway and Overcomes Multidrug Resistance. Cancers 2023, 15, 990. https://doi.org/10.3390/cancers15030990
Bortolotti S, Angelucci S, Montemurro L, Bartolucci D, Raieli S, Lampis S, Amadesi C, Scardovi A, Nieddu G, Cerisoli L, et al. Antigene MYCN Silencing by BGA002 Inhibits SCLC Progression Blocking mTOR Pathway and Overcomes Multidrug Resistance. Cancers. 2023; 15(3):990. https://doi.org/10.3390/cancers15030990
Chicago/Turabian StyleBortolotti, Sonia, Silvia Angelucci, Luca Montemurro, Damiano Bartolucci, Salvatore Raieli, Silvia Lampis, Camilla Amadesi, Annalisa Scardovi, Giammario Nieddu, Lucia Cerisoli, and et al. 2023. "Antigene MYCN Silencing by BGA002 Inhibits SCLC Progression Blocking mTOR Pathway and Overcomes Multidrug Resistance" Cancers 15, no. 3: 990. https://doi.org/10.3390/cancers15030990
APA StyleBortolotti, S., Angelucci, S., Montemurro, L., Bartolucci, D., Raieli, S., Lampis, S., Amadesi, C., Scardovi, A., Nieddu, G., Cerisoli, L., Paganelli, F., Chiarini, F., Teti, G., Falconi, M., Pession, A., Hrelia, P., & Tonelli, R. (2023). Antigene MYCN Silencing by BGA002 Inhibits SCLC Progression Blocking mTOR Pathway and Overcomes Multidrug Resistance. Cancers, 15(3), 990. https://doi.org/10.3390/cancers15030990