
Identification of T Cell Receptors Targeting a Neoantigen Derived from Recurrently Mutated FGFR3
 , , ,
, , , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Antibodies
2.2. Prediction of Potential Shared Neoantigens
2.3. Induction of Neoantigen-Reactive CD8+ T Cells Using Peripheral Blood Mononuclear Cells (PBMCs) from Healthy Donors
2.4. ELISPOT Assay and Enzyme-Linked Immunosorbent Assay (ELISA)
2.5. TCR Sequencing Analysis
2.6. TCR-Engineered T Cells
2.7. Cytotoxicity Assay
2.8. Statistical Analysis
3. Results
3.1. Screening of Shared Neoantigens from TCGA Exome Sequencing Data
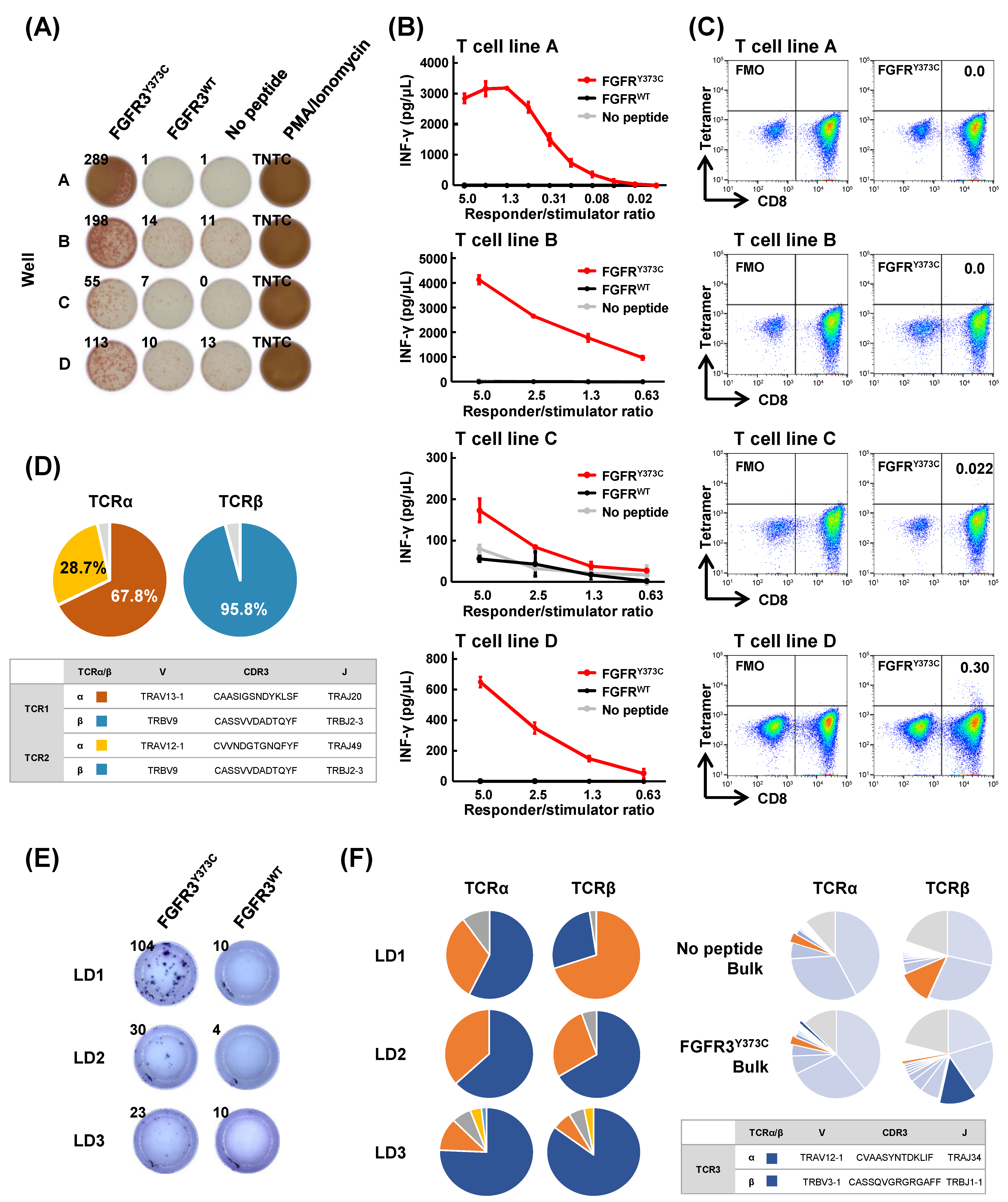
3.2. Induction of Shared Neoantigen-Reactive CD8+ T Cells Using HLA-Matched Healthy Donors’ Blood
3.3. Isolation of FGFR3Y373C-specific CD8+ T Cell Clones and Identification of Their TCR Sequences
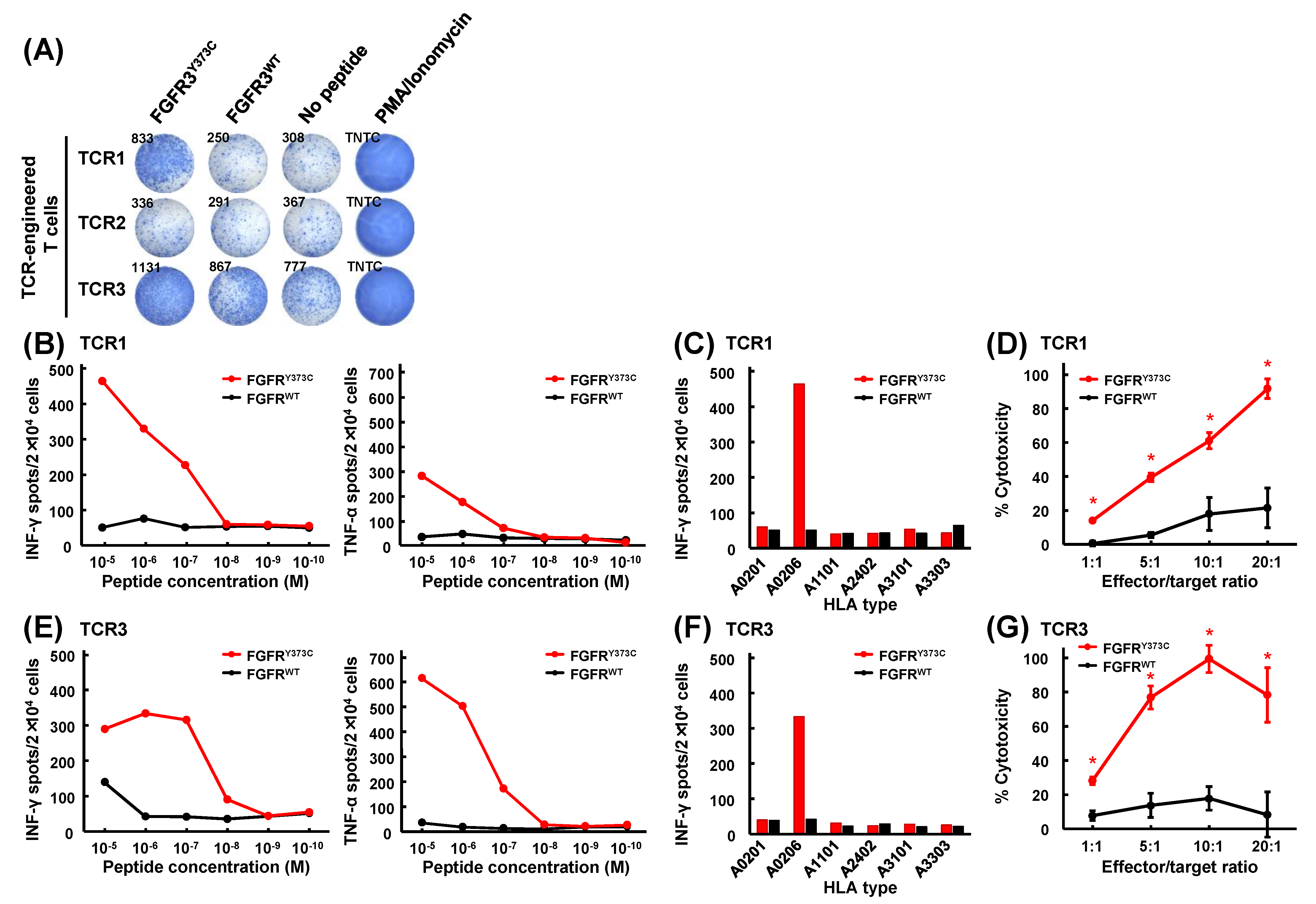
3.4. Mutant Peptide-Specific Recognition and Cytotoxicity of FGFR3Y373C-specific TCR-engineered T Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune checkpoint blockade in cancer therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Shalabi, A.; Hubbard-Lucey, V.M. Comprehensive analysis of the clinical immuno-oncology landscape. Ann. Oncol. 2018, 29, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Matsuda, T.; Ikeda, Y.; Park, J.H.; Leisegang, M.; Yoshimura, S.; Hikichi, T.; Harada, M.; Zewde, M.; Sato, S.; et al. Effective screening of T cells recognizing neoantigens and construction of T-cell receptor-engineered T cells. Oncotarget 2018, 9, 11009–11019. [Google Scholar] [CrossRef]
- Matsuda, T.; Leisegang, M.; Park, J.H.; Ren, L.; Kato, T.; Ikeda, Y.; Harada, M.; Kiyotani, K.; Lengyel, E.; Fleming, G.F.; et al. Induction of Neoantigen-Specific Cytotoxic T Cells and Construction of T-cell Receptor-Engineered T Cells for Ovarian Cancer. Clin. Cancer Res. 2018, 24, 5357–5367. [Google Scholar] [CrossRef]
- Ren, L.; Leisegang, M.; Deng, B.; Matsuda, T.; Kiyotani, K.; Kato, T.; Harada, M.; Park, J.H.; Saloura, V.; Seiwert, T.; et al. Identification of neoantigen-specific T cells and their targets: Implications for immunotherapy of head and neck squamous cell carcinoma. Oncoimmunology 2019, 8, e1568813. [Google Scholar] [CrossRef]
- Kiyotani, K.; Chan, H.T.; Nakamura, Y. Immunopharmacogenomics towards personalized cancer immunotherapy targeting neoantigens. Cancer Sci. 2018, 109, 542–549. [Google Scholar] [CrossRef]
- Kiyotani, K.; Toyoshima, Y.; Nakamura, Y. Immunogenomics in personalized cancer treatments. J. Hum. Genet. 2021, 66, 901–907. [Google Scholar] [CrossRef]
- Schumacher, T.; Bunse, L.; Pusch, S.; Sahm, F.; Wiestler, B.; Quandt, J.; Menn, O.; Osswald, M.; Oezen, I.; Ott, M.; et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 2014, 512, 324–327. [Google Scholar] [CrossRef]
- Tran, E.; Ahmadzadeh, M.; Lu, Y.C.; Gros, A.; Turcotte, S.; Robbins, P.F.; Gartner, J.J.; Zheng, Z.; Li, Y.F.; Ray, S.; et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015, 350, 1387–1390. [Google Scholar] [CrossRef]
- Deniger, D.C.; Pasetto, A.; Robbins, P.F.; Gartner, J.J.; Prickett, T.D.; Paria, B.C.; Malekzadeh, P.; Jia, L.; Yossef, R.; Langhan, M.M.; et al. T-cell responses to TP53 “hotspot” mutations and unique neoantigens expressed by human ovarian cancers. Clin Cancer Res. 2018, 24, 5562–5573. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Lu, Y.C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-cell transfer therapy targeting mutant KRAS in cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef]
- Leidner, R.; Sanjuan Silva, N.; Huang, H.; Sprott, D.; Zheng, C.; Shih, Y.P.; Leung, A.; Payne, R.; Sutcliffe, K.; Cramer, J.; et al. Neoantigen T-cell receptor gene therapy in pancreatic cancer. N. Engl. J. Med. 2022, 386, 2112–2119. [Google Scholar] [CrossRef]
- Lo, W.; Parkhurst, M.; Robbins, P.F.; Tran, E.; Lu, Y.C.; Jia, L.; Gartner, J.J.; Pasetto, A.; Deniger, D.; Malekzadeh, P.; et al. Immunologic recognition of a shared p53 mutated neoantigen in a patient with metastatic colorectal cancer. Cancer Immunol. Res. 2019, 7, 534–543. [Google Scholar] [CrossRef]
- Malekzadeh, P.; Pasetto, A.; Robbins, P.F.; Parkhurst, M.R.; Paria, B.C.; Jia, L.; Gartner, J.J.; Hill, V.; Yu, Z.; Restifo, N.P.; et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J. Clin. Investig. 2019, 129, 1109–1114. [Google Scholar] [CrossRef]
- Zhang, Z.; Hernandez, K.; Savage, J.; Li, S.; Miller, D.; Agrawal, S.; Ortuno, F.; Staudt, L.M.; Heath, A.; Grossman, R.L. Uniform genomic data analysis in the NCI Genomic Data Commons. Nat. Commun. 2021, 12, 1226. [Google Scholar] [CrossRef]
- Lundegaard, C.; Lamberth, K.; Harndahl, M.; Buus, S.; Lund, O.; Nielsen, M. NetMHC-3.0: Accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8-11. Nucleic Acids Res. 2008, 36, W509–W512. [Google Scholar] [CrossRef]
- Nielsen, M.; Lundegaard, C.; Blicher, T.; Lamberth, K.; Harndahl, M.; Justesen, S.; Roder, G.; Peters, B.; Sette, A.; Lund, O.; et al. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS ONE 2007, 2, e796. [Google Scholar] [CrossRef]
- Choudhury, N.J.; Kiyotani, K.; Yap, K.L.; Campanile, A.; Antic, T.; Yew, P.Y.; Steinberg, G.; Park, J.H.; Nakamura, Y.; O’Donnell, P.H. Low T-cell Receptor Diversity, High Somatic Mutation Burden, and High Neoantigen Load as Predictors of Clinical Outcome in Muscle-invasive Bladder Cancer. Eur. Urol. Focus 2016, 2, 445–452. [Google Scholar] [CrossRef]
- Hirata, J.; Hosomichi, K.; Sakaue, S.; Kanai, M.; Nakaoka, H.; Ishigaki, K.; Suzuki, K.; Akiyama, M.; Kishikawa, T.; Ogawa, K.; et al. Genetic and phenotypic landscape of the major histocompatibilty complex region in the Japanese population. Nat. Genet. 2019, 51, 470–480. [Google Scholar] [CrossRef]
- Kiyotani, K.; Toyoshima, Y.; Nemoto, K.; Nakamura, Y. Bioinformatic prediction of potential T cell epitopes for SARS-Cov-2. J. Hum. Genet. 2020, 65, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, S.; Tsunoda, T.; Osawa, R.; Harada, M.; Watanabe, T.; Hikichi, T.; Katsuda, M.; Miyazawa, M.; Tani, M.; Iwahashi, M.; et al. Identification of an HLA-A2-restricted epitope peptide derived from hypoxia-inducible protein 2 (HIG2). PLoS ONE 2014, 9, e85267. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Yamaguchi, R.; Liu, X.; Daigo, Y.; Yew, P.Y.; Tanikawa, C.; Matsuda, K.; Imoto, S.; Miyano, S.; Nakamura, Y. Quantitative T cell repertoire analysis by deep cDNA sequencing of T cell receptor alpha and beta chains using next-generation sequencing (NGS). Oncoimmunology 2014, 3, e968467. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.J.; Zhao, Y.; Zheng, Z.; Rosenberg, S.A.; Morgan, R.A. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. 2006, 66, 8878–8886. [Google Scholar] [CrossRef]
- Leisegang, M.; Engels, B.; Meyerhuber, P.; Kieback, E.; Sommermeyer, D.; Xue, S.A.; Reuss, S.; Stauss, H.; Uckert, W. Enhanced functionality of T cell receptor-redirected T cells is defined by the transgene cassette. J. Mol. Med. 2008, 86, 573–583. [Google Scholar] [CrossRef]
- Padovan, E.; Casorati, G.; Dellabona, P.; Meyer, S.; Brockhaus, M.; Lanzavecchia, A. Expression of two T cell receptor alpha chains: Dual receptor T cells. Science 1993, 262, 422–424. [Google Scholar] [CrossRef]
- Coulie, P.G.; Van den Eynde, B.J.; van der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Lower, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrors, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanovic, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; van der Burg, S.H.; et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu-Lieskovan, S.; Chmielowski, B.; Govindan, R.; Naing, A.; Bhardwaj, N.; Margolin, K.; Awad, M.M.; Hellmann, M.D.; Lin, J.J.; et al. A phase Ib trial of personalized neoantigen therapy plus anti-PD-1 in patients with advanced melanoma, non-small cell lung cancer, or bladder cancer. Cell 2020, 183, 347–362.e324. [Google Scholar] [CrossRef]
- Greulich, H.; Pollock, P.M. Targeting mutant fibroblast growth factor receptors in cancer. Trends Mol. Med. 2011, 17, 283–292. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Sidney, J.; Peters, B.; Frahm, N.; Brander, C.; Sette, A. HLA class I supertypes: A revised and updated classification. BMC Immunol. 2008, 9, 1. [Google Scholar] [CrossRef]
- Hikichi, T.; Sakamoto, M.; Harada, M.; Saito, M.; Yamane, Y.; Tokumura, K.; Nakamura, Y. Identification of cytotoxic T cells and their T cell receptor sequences targeting COVID-19 using MHC class I-binding peptides. J. Hum. Genet. 2022, 67, 411–419. [Google Scholar] [CrossRef]
- Fleischhauer, K.; Tanzarella, S.; Russo, V.; Sensi, M.L.; van der Bruggen, P.; Bordignon, C.; Traversari, C. Functional heterogeneity of HLA-A*02 subtypes revealed by presentation of a MAGE-3-encoded peptide to cytotoxic T cell clones. J. Immunol. 1997, 159, 2513–2521. [Google Scholar] [CrossRef]
- Tan, A.T.; Loggi, E.; Boni, C.; Chia, A.; Gehring, A.J.; Sastry, K.S.; Goh, V.; Fisicaro, P.; Andreone, P.; Brander, C.; et al. Host ethnicity and virus genotype shape the hepatitis B virus-specific T-cell repertoire. J. Virol. 2008, 82, 10986–10997. [Google Scholar] [CrossRef]
- van Buuren, M.M.; Dijkgraaf, F.E.; Linnemann, C.; Toebes, M.; Chang, C.X.; Mok, J.Y.; Nguyen, M.; van Esch, W.J.; Kvistborg, P.; Grotenbreg, G.M.; et al. HLA micropolymorphisms strongly affect peptide-MHC multimer-based monitoring of antigen-specific CD8+ T cell responses. J. Immunol. 2014, 192, 641–648. [Google Scholar] [CrossRef]
- Leisegang, M.; Engels, B.; Schreiber, K.; Yew, P.Y.; Kiyotani, K.; Idel, C.; Arina, A.; Duraiswamy, J.; Weichselbaum, R.R.; Uckert, W.; et al. Eradication of large solid tumors by gene therapy with a T-cell receptor targeting a single cancer-specific point mutation. Clin. Cancer Res. 2016, 22, 2734–2743. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Leisegang, M.; Xia, M.; Kiyotani, K.; Li, N.; Zeng, C.; Deng, C.; Jiang, J.; Harada, M.; Agrawal, N.; et al. Generation of neoantigen-specific T cells for adoptive cell transfer for treating head and neck squamous cell carcinoma. Oncoimmunology 2021, 10, 1929726. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Cancer Types | Sample Size | SNVs | Recurrent SNVs | Shared Neoantigen Peptides |
|---|---|---|---|---|
| ACC (Adrenocortical carcinoma) | 92 | 5575 | 1 | 7 |
| BLCA (Bladder urothelial carcinoma) | 412 | 74,185 | 13 | 79 |
| BRCA (Breast invasive carcinoma) | 986 | 61,609 | 5 | 27 |
| CESC (Cervical squamous cell carcinoma) | 289 | 46,622 | 7 | 40 |
| CHOL (Cholangiocarcinoma) | 51 | 2174 | 9 | 49 |
| COAD (Colon adenocarcinoma) | 399 | 127,752 | 22 | 138 |
| DLBC (Diffuse large B-cell lymphoma) | 37 | 3354 | 3 | 9 |
| ESCA (Esophageal carcinoma) | 184 | 20,159 | 19 | 160 |
| GBM (Glioblastoma multiforme) | 393 | 47,827 | 6 | 35 |
| HNSC (Head and neck squamous cell carcinoma) | 508 | 56,481 | 5 | 19 |
| KICH (Kidney chromophobe) | 66 | 1546 | 2 | 6 |
| KIRC (Kidney renal clear cell carcinoma) | 336 | 12,977 | 0 | 0 |
| KIRP (Kidney renal papillary cell carcinoma) | 281 | 11,707 | 0 | 0 |
| LAML (Acute myeloid leukemia) | 143 | 5417 | 9 | 70 |
| LGG (Brain lower-grade glioma) | 508 | 19,586 | 10 | 54 |
| LIHC (Liver hepatocellular carcinoma) | 364 | 27,874 | 3 | 18 |
| LUAD (Lung adenocarcinoma) | 567 | 120,229 | 6 | 48 |
| LUSC (Lung squamous cell carcinoma) | 492 | 103,323 | 5 | 38 |
| MESO (Mesothelioma) | 82 | 1905 | 0 | 0 |
| OV (Ovarian serous cystadenocarcinoma) | 436 | 39,436 | 6 | 29 |
| PAAD (Pancreatic adenocarcinoma) | 178 | 17,272 | 13 | 80 |
| PCPG (Pheochromocytoma and paraganglioma) | 179 | 1335 | 2 | 14 |
| PRAD (Prostate adenocarcinoma) | 495 | 16,337 | 1 | 6 |
| READ (Rectum adenocarcinoma) | 137 | 35,319 | 29 | 207 |
| SARC (Sarcoma) | 237 | 12,742 | 1 | 4 |
| SKCM (Skin cutaneous melanoma) | 467 | 190,460 | 40 | 296 |
| STAD (Stomach adenocarcinoma) | 437 | 107,714 | 10 | 75 |
| TGCT (Testicular germ cell tumors) | 144 | 1645 | 4 | 33 |
| THCA (Thyroid carcinoma) | 492 | 4087 | 4 | 17 |
| THYM (Thymoma) | 123 | 1859 | 3 | 19 |
| UCEC (Uterine corpus endometrial carcinoma) | 530 | 391,093 | 34 | 254 |
| UCS (Uterine carcinosarcoma) | 57 | 5970 | 28 | 203 |
| UVM (Uveal melanoma) | 80 | 1024 | 6 | 60 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tate, T.; Matsumoto, S.; Nemoto, K.; Leisegang, M.; Nagayama, S.; Obama, K.; Nakamura, Y.; Kiyotani, K. Identification of T Cell Receptors Targeting a Neoantigen Derived from Recurrently Mutated FGFR3. Cancers 2023, 15, 1031. https://doi.org/10.3390/cancers15041031
Tate T, Matsumoto S, Nemoto K, Leisegang M, Nagayama S, Obama K, Nakamura Y, Kiyotani K. Identification of T Cell Receptors Targeting a Neoantigen Derived from Recurrently Mutated FGFR3. Cancers. 2023; 15(4):1031. https://doi.org/10.3390/cancers15041031
Chicago/Turabian StyleTate, Tomohiro, Saki Matsumoto, Kensaku Nemoto, Matthias Leisegang, Satoshi Nagayama, Kazutaka Obama, Yusuke Nakamura, and Kazuma Kiyotani. 2023. "Identification of T Cell Receptors Targeting a Neoantigen Derived from Recurrently Mutated FGFR3" Cancers 15, no. 4: 1031. https://doi.org/10.3390/cancers15041031
APA StyleTate, T., Matsumoto, S., Nemoto, K., Leisegang, M., Nagayama, S., Obama, K., Nakamura, Y., & Kiyotani, K. (2023). Identification of T Cell Receptors Targeting a Neoantigen Derived from Recurrently Mutated FGFR3. Cancers, 15(4), 1031. https://doi.org/10.3390/cancers15041031