
Radiotherapy, PARP Inhibition, and Immune-Checkpoint Blockade: A Triad to Overcome the Double-Edged Effects of Each Single Player


Abstract
:Simple Summary
Abstract
1. Introduction
2. Radiotherapy
3. Immuno-Stimulating Effects of Radiotherapy
4. Immuno-Depressing Effects of Radiotherapy
5. PARP Inhibitors
6. Synergy between PARPi and Radiotherapy
7. Synergic Immunological Effects of RT and PARPi
8. Immune-Checkpoint Inhibitors
9. Synergy between Radiotherapy and ICI
10. Synergy between PARPi and ICI
11. Conclusions: Combining RT, PARPis, and ICIs to Overcome Respective Limitations
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Global Burden of Disease 2019 Cancer Collaboration; Kocarnik, J.M.; Compton, K.; Dean, F.E.; Fu, W.; Gaw, B.L.; Harvey, J.D.; Henrikson, H.J.; Lu, D.; Pennini, A.; et al. Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life Years for 29 Cancer Groups From 2010 to 2019: A Systematic Analysis for the Global Burden of Disease Study 2019. JAMA Oncol. 2022, 8, 420–444. [Google Scholar] [CrossRef]
- Baumann, M.; Krause, M.; Overgaard, J.; Debus, J.; Bentzen, S.M.; Daartz, J.; Richter, C.; Zips, D.; Bortfeld, T. Radiation oncology in the era of precision medicine. Nat. Rev. Cancer 2016, 16, 234–249. [Google Scholar] [CrossRef]
- Sutherland, B.M.; Bennett, P.V.; Sutherland, J.C.; Laval, J. Clustered DNA damages induced by x rays in human cells. Radiat. Res. 2002, 157, 611–616. [Google Scholar] [CrossRef]
- Goodhead, D.T. Initial events in the cellular effects of ionizing radiations: Clustered damage in DNA. Int. J. Radiat. Biol. 1994, 65, 7–17. [Google Scholar] [CrossRef]
- Lu, Z.; Zheng, X.; Ding, C.; Zou, Z.; Liang, Y.; Zhou, Y.; Li, X. Deciphering the Biological Effects of Radiotherapy in Cancer Cells. Biomolecules 2022, 12, 1167. [Google Scholar] [CrossRef]
- Alterio, D.; Gugliandolo, S.G.; Augugliaro, M.; Marvaso, G.; Gandini, S.; Bellerba, F.; Russell-Edu, S.W.; De Simone, I.; Cinquini, M.; Starzyńska, A.; et al. IMRT versus 2D/3D conformal RT in oropharyngeal cancer: A review of the literature and meta-analysis. Oral Dis. 2021, 27, 1644–1653. [Google Scholar] [CrossRef] [PubMed]
- Marta, G.N.; Silva, V.; de Andrade Carvalho, H.; de Arruda, F.F.; Hanna, S.A.; Gadia, R.; da Silva, J.L.; Correa, S.F.; Vita Abreu, C.E.; Riera, R. Intensity-modulated radiation therapy for head and neck cancer: Systematic review and meta-analysis. Radiother. Oncol. 2014, 110, 9–15. [Google Scholar] [CrossRef]
- Byun, H.K.; Han, M.C.; Yang, K.; Kim, J.S.; Yoo, G.S.; Koom, W.S.; Kim, Y.B. Physical and Biological Characteristics of Particle Therapy for Oncologists. Cancer Res. Treat. 2021, 53, 611–620. [Google Scholar] [CrossRef]
- Yuan, T.Z.; Zhan, Z.J.; Qian, C.N. New frontiers in proton therapy: Applications in cancers. Cancer Commun. 2019, 39, 61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Hu, W.; Hu, J.; Gao, J.; Yang, J.; Kong, L.; Lu, J.J. Carbon ion radiation therapy for sinonasal malignancies: Promising results from 2282 cases from the real world. Cancer Sci. 2020, 111, 4465–4479. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Motevaseli, E.; Shirazi, A.; Geraily, G.; Rezaeyan, A.; Norouzi, F.; Rezapoor, S.; Abdollahi, H. Mechanisms of inflammatory responses to radiation and normal tissues toxicity: Clinical implications. Int. J. Radiat. Biol. 2018, 94, 335–356. [Google Scholar] [CrossRef]
- Rodríguez-Ruiz, M.E.; Vanpouille-Box, C.; Melero, I.; Formenti, S.C.; Demaria, S. Immunological Mechanisms Responsible for Radiation-Induced Abscopal Effect. Trends Immunol. 2018, 39, 644–655. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.-D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef]
- Yamazaki, T.; Galluzzi, L. Mitochondrial control of innate immune signaling by irradiated cancer cells. OncoImmunology 2020, 9, 1797292. [Google Scholar] [CrossRef]
- Burnette, B.C.; Liang, H.; Lee, Y.; Chlewicki, L.; Khodarev, N.N.; Weichselbaum, R.R.; Fu, Y.-X.; Auh, S.L. The Efficacy of Radiotherapy Relies upon Induction of Type I Interferon–Dependent Innate and Adaptive Immunity. Cancer Res. 2011, 71, 2488–2496. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Kageyama, S.I.; Hirata, H.; Motegi, A.; Nakamura, M.; Hirano, Y.; Okumura, M.; Yamashita, R.; Tsuchihara, K.; Hojo, H.; et al. Comparative analysis of the immune responses in cancer cells irradiated with X-ray, proton and carbon-ion beams. Biochem. Biophys. Res. Commun. 2021, 585, 55–60. [Google Scholar] [CrossRef]
- Cytlak, U.M.; Dyer, D.P.; Honeychurch, J.; Williams, K.J.; Travis, M.A.; Illidge, T.M. Immunomodulation by radiotherapy in tumour control and normal tissue toxicity. Nat. Rev. Immunol. 2022, 22, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Connolly, K.A.; Belt, B.A.; Figueroa, N.M.; Murthy, A.; Patel, A.; Kim, M.; Lord, E.M.; Linehan, D.C.; Gerber, S.A. Increasing the efficacy of radiotherapy by modulating the CCR2/CCR5 chemokine axes. Oncotarget 2016, 7, 86522–86535. [Google Scholar] [CrossRef] [PubMed]
- Barker, H.E.; Paget, J.T.; Khan, A.A.; Harrington, K.J. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Rev. Cancer 2015, 15, 409–425. [Google Scholar] [CrossRef]
- Li, H.; Chen, X.; Zeng, W.; Zhou, W.; Zhou, Q.; Wang, Z.; Jiang, W.; Xie, B.; Sun, L.Q. Radiation-Enhanced Expression of CCL22 in Nasopharyngeal Carcinoma is Associated With CCR4(+) CD8 T Cell Recruitment. Int. J. Radiat. Oncol. Biol. Phys. 2020, 108, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, S.; Wang, B.; Kawashima, N.; Braunstein, S.; Badura, M.; Cameron, T.O.; Babb, J.S.; Schneider, R.J.; Formenti, S.C.; Dustin, M.L.; et al. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J. Immunol. 2008, 181, 3099–3107. [Google Scholar] [CrossRef]
- Cheng, C.C.; Chang, Y.F.; Ho, A.S.; Sie, Z.L.; Chang, J.S.; Peng, C.L.; Chang, C.C. Irradiation Mediates IFNα and CXCL9 Expression in Non-Small Cell Lung Cancer to Stimulate CD8(+) T Cells Activity and Migration toward Tumors. Biomedicines 2021, 9, 1349. [Google Scholar] [CrossRef]
- Hallahan, D.; Kuchibhotla, J.; Wyble, C. Cell adhesion molecules mediate radiation-induced leukocyte adhesion to the vascular endothelium. Cancer Res. 1996, 56, 5150–5155. [Google Scholar]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2019, 20, 95–112. [Google Scholar] [CrossRef]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Gold, L.I.; Eggleton, P.; Sweetwyne, M.T.; Van Duyn, L.B.; Greives, M.R.; Naylor, S.M.; Michalak, M.; Murphy-Ullrich, J.E. Calreticulin: Non-endoplasmic reticulum functions in physiology and disease. FASEB J. 2010, 24, 665–683. [Google Scholar] [CrossRef]
- Gardai, S.J.; McPhillips, K.A.; Frasch, S.C.; Janssen, W.J.; Starefeldt, A.; Murphy-Ullrich, J.E.; Bratton, D.L.; Oldenborg, P.A.; Michalak, M.; Henson, P.M. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 2005, 123, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Gameiro, S.R.; Malamas, A.S.; Bernstein, M.B.; Tsang, K.Y.; Vassantachart, A.; Sahoo, N.; Tailor, R.; Pidikiti, R.; Guha, C.P.; Hahn, S.M.; et al. Tumor Cells Surviving Exposure to Proton or Photon Radiation Share a Common Immunogenic Modulation Signature, Rendering Them More Sensitive to T Cell–Mediated Killing. Int. J. Radiat. Oncol. Biol. 2016, 95, 120–130. [Google Scholar] [CrossRef]
- Böttcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis e Sousa, C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krombach, J.; Hennel, R.; Brix, N.; Orth, M.; Schoetz, U.; Ernst, A.; Schuster, J.; Zuchtriegel, G.; Reichel, C.A.; Bierschenk, S.; et al. Priming anti-tumor immunity by radiotherapy: Dying tumor cell-derived DAMPs trigger endothelial cell activation and recruitment of myeloid cells. Oncoimmunology 2019, 8, e1523097. [Google Scholar] [CrossRef] [PubMed]
- Reits, E.A.; Hodge, J.W.; Herberts, C.A.; Groothuis, T.A.; Chakraborty, M.K.; Wansley, E.; Camphausen, K.; Luiten, R.M.; de Ru, A.H.; Neijssen, J.; et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J. Exp. Med. 2006, 203, 1259–1271. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Auh, S.L.; Wang, Y.; Burnette, B.; Wang, Y.; Meng, Y.; Beckett, M.; Sharma, R.; Chin, R.; Tu, T.; et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: Changing strategies for cancer treatment. Blood 2009, 114, 589–595. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Procureur, A.; Simonaggio, A.; Bibault, J.E.; Oudard, S.; Vano, Y.A. Enhance the Immune Checkpoint Inhibitors Efficacy with Radiotherapy Induced Immunogenic Cell Death: A Comprehensive Review and Latest Developments. Cancers 2021, 13, 678. [Google Scholar] [CrossRef]
- Mauch, P.; Constine, L.; Greenberger, J.; Knospe, W.; Sullivan, J.; Liesveld, J.L.; Deeg, H.J. Hematopoietic stem cell compartment: Acute and late effects of radiation therapy and chemotherapy. Int. J. Radiat. Oncol. Biol. Phys. 1995, 31, 1319–1339. [Google Scholar] [CrossRef]
- Frasca, D.; Guidi, F.; Arbitrio, M.; Pioli, C.; Poccia, F.; Cicconi, R.; Doria, G. Hematopoietic reconstitution after lethal irradiation and bone marrow transplantation: Effects of different hematopoietic cytokines on the recovery of thymus, spleen and blood cells. Bone Marrow Transplant. 2000, 25, 427–433. [Google Scholar] [CrossRef]
- Frasca, D.; Pioli, C.; Guidi, F.; Pucci, S.; Arbitrio, M.; Leter, G.; Doria, G. IL-11 synergizes with IL-3 in promoting the recovery of the immune system after irradiation. Int. Immunol. 1996, 8, 1651–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heylmann, D.; Rödel, F.; Kindler, T.; Kaina, B. Radiation sensitivity of human and murine peripheral blood lymphocytes, stem and progenitor cells. Biochim. Biophys. Acta 2014, 1846, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Berte, N.; Eich, M.; Heylmann, D.; Koks, C.; Van Gool, S.W.; Kaina, B. Impaired DNA repair in mouse monocytes compared to macrophages and precursors. DNA Repair 2021, 98, 103037. [Google Scholar] [CrossRef]
- Leblond, M.M.; Pérès, E.A.; Helaine, C.; Gérault, A.N.; Moulin, D.; Anfray, C.; Divoux, D.; Petit, E.; Bernaudin, M.; Valable, S. M2 macrophages are more resistant than M1 macrophages following radiation therapy in the context of glioblastoma. Oncotarget 2017, 8, 72597–72612. [Google Scholar] [CrossRef]
- Groves, A.M.; Johnston, C.J.; Misra, R.S.; Williams, J.P.; Finkelstein, J.N. Effects of IL-4 on pulmonary fibrosis and the accumulation and phenotype of macrophage subpopulations following thoracic irradiation. Int. J. Radiat. Biol. 2016, 92, 754–765. [Google Scholar] [CrossRef]
- Arina, A.; Beckett, M.; Fernandez, C.; Zheng, W.; Pitroda, S.; Chmura, S.J.; Luke, J.J.; Forde, M.; Hou, Y.; Burnette, B.; et al. Tumor-reprogrammed resident T cells resist radiation to control tumors. Nat. Commun. 2019, 10, 3959. [Google Scholar] [CrossRef]
- Qinfeng, S.; Depu, W.; Xiaofeng, Y.; Shah, W.; Hongwei, C.; Yili, W. In situ observation of the effects of local irradiation on cytotoxic and regulatory T lymphocytes in cervical cancer tissue. Radiat. Res. 2013, 179, 584–589. [Google Scholar] [CrossRef]
- Qu, Y.; Jin, S.; Zhang, A.; Zhang, B.; Shi, X.; Wang, J.; Zhao, Y. Gamma-ray resistance of regulatory CD4+CD25+Foxp3+ T cells in mice. Radiat. Res. 2010, 173, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Kachikwu, E.L.; Iwamoto, K.S.; Liao, Y.P.; DeMarco, J.J.; Agazaryan, N.; Economou, J.S.; McBride, W.H.; Schaue, D. Radiation enhances regulatory T cell representation. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 1128–1135. [Google Scholar] [CrossRef]
- Marciscano, A.E.; Ghasemzadeh, A.; Nirschl, T.R.; Theodros, D.; Kochel, C.M.; Francica, B.J.; Muroyama, Y.; Anders, R.A.; Sharabi, A.B.; Velarde, E.; et al. Elective Nodal Irradiation Attenuates the Combinatorial Efficacy of Stereotactic Radiation Therapy and Immunotherapy. Clin. Cancer Res. 2018, 24, 5058–5071. [Google Scholar] [CrossRef] [PubMed]
- Yovino, S.; Kleinberg, L.; Grossman, S.A.; Narayanan, M.; Ford, E. The Etiology of Treatment-related Lymphopenia in Patients with Malignant Gliomas: Modeling Radiation Dose to Circulating Lymphocytes Explains Clinical Observations and Suggests Methods of Modifying the Impact of Radiation on Immune Cells. Cancer Investig. 2013, 31, 140–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, D.; An, D.; Wan, C.; Yang, K. Radiotherapy: Brightness and darkness in the era of immunotherapy. Transl. Oncol. 2022, 19, 101366. [Google Scholar] [CrossRef]
- Derer, A.; Spiljar, M.; Bäumler, M.; Hecht, M.; Fietkau, R.; Frey, B.; Gaipl, U.S. Chemoradiation Increases PD-L1 Expression in Certain Melanoma and Glioblastoma Cells. Front. Immunol. 2016, 7, 610. [Google Scholar] [CrossRef]
- Gao, Y.; Li, Y.; Lin, Z.; Zeng, Y.; Huang, Z.; Han, L.; Zhong, Y.; Gong, Y.; Wu, Q.; Xie, C. Ataxia telangiectasia mutated kinase inhibition promotes irradiation-induced PD-L1 expression in tumour-associated macrophages through IFN-I/JAK signalling pathway. Immunology 2022, 168, 346–361. [Google Scholar] [CrossRef] [PubMed]
- Mondini, M.; Loyher, P.-L.; Hamon, P.; Gerbé de Thoré, M.; Laviron, M.; Berthelot, K.; Clémenson, C.; Salomon, B.L.; Combadière, C.; Deutsch, E.; et al. CCR2-Dependent Recruitment of Tregs and Monocytes Following Radiotherapy Is Associated with TNFα-Mediated Resistance. Cancer Immunol. Res. 2019, 7, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Deng, L.; Hou, Y.; Meng, X.; Huang, X.; Rao, E.; Zheng, W.; Mauceri, H.; Mack, M.; Xu, M.; et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat. Commun. 2017, 8, 1736. [Google Scholar] [CrossRef] [PubMed]
- Jobling, M.F.; Mott, J.D.; Finnegan, M.T.; Jurukovski, V.; Erickson, A.C.; Walian, P.J.; Taylor, S.E.; Ledbetter, S.; Lawrence, C.M.; Rifkin, D.B.; et al. Isoform-specific activation of latent transforming growth factor beta (LTGF-beta) by reactive oxygen species. Radiat. Res. 2006, 166, 839–848. [Google Scholar] [CrossRef]
- Chiang, C.S.; Fu, S.Y.; Wang, S.C.; Yu, C.F.; Chen, F.H.; Lin, C.M.; Hong, J.H. Irradiation promotes an m2 macrophage phenotype in tumor hypoxia. Front. Oncol. 2012, 2, 89. [Google Scholar] [CrossRef]
- Farhood, B.; Khodamoradi, E.; Hoseini-Ghahfarokhi, M.; Motevaseli, E.; Mirtavoos-Mahyari, H.; Eleojo Musa, A.; Najafi, M. TGF-β in radiotherapy: Mechanisms of tumor resistance and normal tissues injury. Pharmacol. Res. 2020, 155, 104745. [Google Scholar] [CrossRef]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef]
- Liao, Y.; Liu, S.; Fu, S.; Wu, J. HMGB1 in Radiotherapy: A Two Headed Signal Regulating Tumor Radiosensitivity and Immunity. OncoTargets Ther. 2020, 13, 6859–6871. [Google Scholar] [CrossRef]
- Shrivastava, S.; Mansure, J.J.; Almajed, W.; Cury, F.; Ferbeyre, G.; Popovic, M.; Seuntjens, J.; Kassouf, W. The Role of HMGB1 in Radioresistance of Bladder Cancer. Mol. Cancer Ther. 2016, 15, 471–479. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Cheng, J.; Sun, L.; Wang, Y.; Wang, C.; Liu, X.; Zhang, Z.; Zhao, M.; Luo, Y.; Tian, L.; et al. HMGB1 released by irradiated tumor cells promotes living tumor cell proliferation via paracrine effect. Cell Death Dis. 2018, 9, 648. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Gabrilovich, D.I. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011, 32, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Beavis, P.A.; Stagg, J.; Darcy, P.K.; Smyth, M.J. CD73: A potent suppressor of antitumor immune responses. Trends Immunol. 2012, 33, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A.; Gorelik, E.; Prasad, S.J.; Ronchese, F.; Lukashev, D.; Wong, M.K.; Huang, X.; Caldwell, S.; Liu, K.; Smith, P.; et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13132–13137. [Google Scholar] [CrossRef] [PubMed]
- Stagg, J.; Beavis, P.A.; Divisekera, U.; Liu, M.C.; Möller, A.; Darcy, P.K.; Smyth, M.J. CD73-deficient mice are resistant to carcinogenesis. Cancer Res. 2012, 72, 2190–2196. [Google Scholar] [CrossRef] [PubMed]
- Michaud, M.; Sukkurwala, A.Q.; Martins, I.; Shen, S.; Zitvogel, L.; Kroemer, G. Subversion of the chemotherapy-induced anticancer immune response by the ecto-ATPase CD39. Oncoimmunology 2012, 1, 393–395. [Google Scholar] [CrossRef]
- Krishnakumar, R.; Kraus, W.L. The PARP side of the nucleus: Molecular actions, physiological outcomes, and clinical targets. Mol. Cell 2010, 39, 8–24. [Google Scholar] [CrossRef]
- El-Khamisy, S.F.; Masutani, M.; Suzuki, H.; Caldecott, K.W. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003, 31, 5526–5533. [Google Scholar] [CrossRef]
- Mladenov, E.; Iliakis, G. Induction and repair of DNA double strand breaks: The increasing spectrum of non-homologous end joining pathways. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2011, 711, 61–72. [Google Scholar] [CrossRef]
- Langelier, M.F.; Planck, J.L.; Roy, S.; Pascal, J.M. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science 2012, 336, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006, 34, 6170–6182. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, E.A.; Alzahrani, A.M.; Scherthan, H. Parp1-Dependent DNA Double-Strand Break Repair in Irradiated Late Pachytene Spermatocytes. DNA Cell Biol. 2021, 40, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Pilié, P.G.; Gay, C.M.; Byers, L.A.; O’Connor, M.J.; Yap, T.A. PARP Inhibitors: Extending Benefit Beyond BRCA-Mutant Cancers. Clin. Cancer Res. 2019, 25, 3759–3771. [Google Scholar] [CrossRef] [Green Version]
- Zandarashvili, L.; Langelier, M.F.; Velagapudi, U.K.; Hancock, M.A.; Steffen, J.D.; Billur, R.; Hannan, Z.M.; Wicks, A.J.; Krastev, D.B.; Pettitt, S.J.; et al. Structural basis for allosteric PARP-1 retention on DNA breaks. Science 2020, 368, eaax6367. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Christie, E.L.; Fereday, S.; Doig, K.; Pattnaik, S.; Dawson, S.J.; Bowtell, D.D.L. Reversion of BRCA1/2 Germline Mutations Detected in Circulating Tumor DNA From Patients With High-Grade Serous Ovarian Cancer. J. Clin. Oncol. 2017, 35, 1274–1280. [Google Scholar] [CrossRef]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef]
- Park, P.H.; Yamamoto, T.M.; Li, H.; Alcivar, A.L.; Xia, B.; Wang, Y.; Bernhardy, A.J.; Turner, K.M.; Kossenkov, A.V.; Watson, Z.L.; et al. Amplification of the Mutation-Carrying BRCA2 Allele Promotes RAD51 Loading and PARP Inhibitor Resistance in the Absence of Reversion Mutations. Mol. Cancer Ther. 2020, 19, 602–613. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef]
- Kupczyk, P.; Simiczyjew, A.; Marczuk, J.; Dratkiewicz, E.; Beberok, A.; Rok, J.; Pieniazek, M.; Biecek, P.; Nevozhay, D.; Slowikowski, B.; et al. PARP1 as a Marker of an Aggressive Clinical Phenotype in Cutaneous Melanoma—A Clinical and an In Vitro Study. Cells 2021, 10, 286. [Google Scholar] [CrossRef] [PubMed]
- Raleigh, D.; Ahmed, K.M.; Zhang, H.; Ziaee, S.; Park, C.C. PARP-1 modulates β1-integrin/NF-κB-mediated radioresistance in human breast cancer. J. Cancer Ther. Res. 2016, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Cerrato, A.; Morra, F.; Celetti, A. Use of poly ADP-ribose polymerase [PARP] inhibitors in cancer cells bearing DDR defects: The rationale for their inclusion in the clinic. J. Exp. Clin. Cancer Res. 2016, 35, 179. [Google Scholar] [CrossRef]
- Zhao, W.; Hu, H.; Mo, Q.; Guan, Y.; Li, Y.; Du, Y.; Li, L. Function and mechanism of combined PARP-1 and BRCA genes in regulating the radiosensitivity of breast cancer cells. Int. J. Clin. Exp. Pathol. 2019, 12, 3915–3920. [Google Scholar] [PubMed]
- Dungey, F.A.; Löser, D.A.; Chalmers, A.J. Replication-dependent radiosensitization of human glioma cells by inhibition of poly(ADP-Ribose) polymerase: Mechanisms and therapeutic potential. Int. J. Radiat. Oncol. Biol. Phys. 2008, 72, 1188–1197. [Google Scholar] [CrossRef]
- Elser, M.; Borsig, L.; Hassa, P.O.; Erener, S.; Messner, S.; Valovka, T.; Keller, S.; Gassmann, M.; Hottiger, M.O. Poly(ADP-Ribose) Polymerase 1 Promotes Tumor Cell Survival by Coactivating Hypoxia-Inducible Factor-1–Dependent Gene Expression. Mol. Cancer Res. 2008, 6, 282–290. [Google Scholar] [CrossRef]
- Gonzalez-Flores, A.; Aguilar-Quesada, R.; Siles, E.; Pozo, S.; Rodríguez-Lara, M.I.; López-Jiménez, L.; López-Rodríguez, M.; Peralta-Leal, A.; Villar, D.; Martín-Oliva, D.; et al. Interaction between PARP-1 and HIF-2α in the hypoxic response. Oncogene 2014, 33, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Lesueur, P.; Chevalier, F.; Austry, J.B.; Waissi, W.; Burckel, H.; Noël, G.; Habrand, J.L.; Saintigny, Y.; Joly, F. Poly-(ADP-ribose)-polymerase inhibitors as radiosensitizers: A systematic review of pre-clinical and clinical human studies. Oncotarget 2017, 8, 69105–69124. [Google Scholar] [CrossRef] [PubMed]
- Barcellini, A.; Loap, P.; Murata, K.; Villa, R.; Kirova, Y.; Okonogi, N.; Orlandi, E. PARP Inhibitors in Combination with Radiotherapy: To Do or Not to Do? Cancers 2021, 13, 5380. [Google Scholar] [CrossRef] [PubMed]
- Rosado, M.M.; Bennici, E.; Novelli, F.; Pioli, C. Beyond DNA repair, the immunological role of PARP-1 and its siblings. Immunology 2013, 139, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Rosado, M.M.; Pioli, C. ADP-ribosylation in evasion, promotion and exacerbation of immune responses. Immunology 2021, 164, 15–30. [Google Scholar] [CrossRef]
- Chabanon, R.M.; Muirhead, G.; Krastev, D.B.; Adam, J.; Morel, D.; Garrido, M.; Lamb, A.; Hénon, C.; Dorvault, N.; Rouanne, M.; et al. PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. J. Clin. Investig. 2019, 129, 1211–1228. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.Y.H.; Gerber, S.A.; Murphy, S.P.; Lord, E.M. Type I interferons induced by radiation therapy mediate recruitment and effector function of CD8+ T cells. Cancer Immunol. Immunother. 2014, 63, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Pantelidou, C.; Sonzogni, O.; De Oliveria Taveira, M.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP Inhibitor Efficacy Depends on CD8(+) T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef]
- Huang, J.; Wang, L.; Cong, Z.; Amoozgar, Z.; Kiner, E.; Xing, D.; Orsulic, S.; Matulonis, U.; Goldberg, M.S. The PARP1 inhibitor BMN 673 exhibits immunoregulatory effects in a Brca1−/− murine model of ovarian cancer. Biochem. Biophys. Res. Commun. 2015, 463, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Thorén, F.B.; Romero, A.I.; Hellstrand, K. Oxygen Radicals Induce Poly(ADP-Ribose) Polymerase-Dependent Cell Death in Cytotoxic Lymphocytes1. J. Immunol. 2006, 176, 7301–7307. [Google Scholar] [CrossRef] [PubMed]
- Karimi, K.; Karimi, Y.; Chan, J.; Boudreau, J.E.; Basset, J.; Chew, M.V.; Reid, S.; Bramson, J.L.; Wan, Y.; Ashkar, A.A. Type I IFN signaling on dendritic cells is required for NK cell-mediated anti-tumor immunity. Innate Immun. 2015, 21, 626–634. [Google Scholar] [CrossRef]
- Müller, E.; Speth, M.; Christopoulos, P.F.; Lunde, A.; Avdagic, A.; Øynebråten, I.; Corthay, A. Both Type I and Type II Interferons Can Activate Antitumor M1 Macrophages When Combined With TLR Stimulation. Front. Immunol. 2018, 9, 2520. [Google Scholar] [CrossRef]
- Yarchoan, M.; Johnson, B.A.; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef]
- Germano, G.; Lamba, S.; Rospo, G.; Barault, L.; Magrì, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 2017, 552, 116–120. [Google Scholar] [CrossRef]
- Marcus, L.; Lemery, S.J.; Keegan, P.; Pazdur, R. FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clin. Cancer Res. 2019, 25, 3753–3758. [Google Scholar] [CrossRef]
- Zhang, N.; Gao, Y.; Zeng, Z.; Luo, Y.; Jiang, X.; Zhang, J.; Li, J.; Zhang, J.; Gong, Y.; Xie, C. PARP inhibitor niraparib as a radiosensitizer promotes antitumor immunity of radiotherapy in EGFR-mutated non-small cell lung cancer. Clin. Transl. Oncol. 2021, 23, 1827–1837. [Google Scholar] [CrossRef] [PubMed]
- Seyedin, S.N.; Hasibuzzaman, M.M.; Pham, V.; Petronek, M.S.; Callaghan, C.; Kalen, A.L.; Mapuskar, K.A.; Mott, S.L.; Spitz, D.R.; Allen, B.G.; et al. Combination Therapy with Radiation and PARP Inhibition Enhances Responsiveness to Anti-PD-1 Therapy in Colorectal Tumor Models. Int. J. Radiat. Oncol. Biol. Phys. 2020, 108, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Nasta, F.; Laudisi, F.; Sambucci, M.; Rosado, M.M.; Pioli, C. Increased Foxp3+ regulatory T cells in poly(ADP-Ribose) polymerase-1 deficiency. J. Immunol. 2010, 184, 3470–3477. [Google Scholar] [CrossRef] [PubMed]
- Angioni, R.; Sánchez-Rodríguez, R.; Viola, A.; Molon, B. TGF-β in Cancer: Metabolic Driver of the Tolerogenic Crosstalk in the Tumor Microenvironment. Cancers 2021, 13, 401. [Google Scholar] [CrossRef]
- Li, C.-W.; Lim, S.-O.; Xia, W.; Lee, H.-H.; Chan, L.-C.; Kuo, C.-W.; Khoo, K.-H.; Chang, S.-S.; Cha, J.-H.; Kim, T.; et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 2016, 7, 12632. [Google Scholar] [CrossRef]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef]
- Wolchok, J. Putting the Immunologic Brakes on Cancer. Cell 2018, 175, 1452–1454. [Google Scholar] [CrossRef] [PubMed]
- Brunet, J.F.; Denizot, F.; Luciani, M.F.; Roux-Dosseto, M.; Suzan, M.; Mattei, M.G.; Golstein, P. A new member of the immunoglobulin superfamily--CTLA-4. Nature 1987, 328, 267–270. [Google Scholar] [CrossRef]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef]
- Pioli, C.; Gatta, L.; Frasca, D.; Doria, G. Cytotoxic T lymphocyte antigen 4 (CTLA-4) inhibits CD28-induced IkappaBalpha degradation and RelA activation. Eur. J. Immunol. 1999, 29, 856–863. [Google Scholar] [CrossRef]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1994, 1, 405–413. [Google Scholar] [CrossRef]
- Gatta, L.; Calviello, G.; Di Nicuolo, F.; Pace, L.; Ubaldi, V.; Doria, G.; Pioli, C. Cytotoxic T lymphocyte-associated antigen-4 inhibits integrin-mediated stimulation. Immunology 2002, 107, 209–216. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Peggs, K.S.; Quezada, S.A.; Chambers, C.A.; Korman, A.J.; Allison, J.P. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J. Exp. Med. 2009, 206, 1717–1725. [Google Scholar] [CrossRef]
- Grosso, J.F.; Jure-Kunkel, M.N. CTLA-4 blockade in tumor models: An overview of preclinical and translational research. Cancer Immun. 2013, 13, 5. [Google Scholar]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [PubMed]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Hirano, F.; Kaneko, K.; Tamura, H.; Dong, H.; Wang, S.; Ichikawa, M.; Rietz, C.; Flies, D.B.; Lau, J.S.; Zhu, G.; et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005, 65, 1089–1096. [Google Scholar] [CrossRef]
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019, 20, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Kim, K.; Park, H.J.; Kim, G.-R.; Hong, K.H.; Oh, J.H.; Son, J.; Park, D.J.; Kim, D.; Choi, J.-M.; et al. Deletion of PD-1 destabilizes the lineage identity and metabolic fitness of tumor-infiltrating regulatory T cells. Nat. Immunol. 2023, 24, 148–161. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef]
- Huang, Q.; Zheng, Y.; Gao, Z.; Yuan, L.; Sun, Y.; Chen, H. Comparative Efficacy and Safety of PD-1/PD-L1 Inhibitors for Patients with Solid Tumors: A Systematic Review and Bayesian Network Meta-analysis. J. Cancer 2021, 12, 1133–1143. [Google Scholar] [CrossRef]
- Vuky, J.; Balar, A.V.; Castellano, D.; O’Donnell, P.H.; Grivas, P.; Bellmunt, J.; Powles, T.; Bajorin, D.; Hahn, N.M.; Savage, M.J.; et al. Long-Term Outcomes in KEYNOTE-052: Phase II Study Investigating First-Line Pembrolizumab in Cisplatin-Ineligible Patients With Locally Advanced or Metastatic Urothelial Cancer. J. Clin. Oncol. 2020, 38, 2658–2666. [Google Scholar] [CrossRef]
- Shiravand, Y.; Khodadadi, F.; Kashani, S.M.A.; Hosseini-Fard, S.R.; Hosseini, S.; Sadeghirad, H.; Ladwa, R.; O’Byrne, K.; Kulasinghe, A. Immune Checkpoint Inhibitors in Cancer Therapy. Curr. Oncol. 2022, 29, 3044–3060. [Google Scholar] [CrossRef]
- Mullard, A. Second CTLA4-targeted checkpoint inhibitor secures FDA approval. Nat. Rev. Drug Discov. 2022, 21, 868. [Google Scholar] [CrossRef]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.M.; Hwu, W.-J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Forde, P.M.; Chaft, J.E.; Smith, K.N.; Anagnostou, V.; Cottrell, T.R.; Hellmann, M.D.; Zahurak, M.; Yang, S.C.; Jones, D.R.; Broderick, S.; et al. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N. Engl. J. Med. 2018, 378, 1976–1986. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Nebot-Bral, L.; Brandao, D.; Verlingue, L.; Rouleau, E.; Caron, O.; Despras, E.; El-Dakdouki, Y.; Champiat, S.; Aoufouchi, S.; Leary, A.; et al. Hypermutated tumours in the era of immunotherapy: The paradigm of personalised medicine. Eur. J. Cancer 2017, 84, 290–303. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Geukes Foppen, M.H.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Wang, S.; Xie, K.; Liu, T. Cancer Immunotherapies: From Efficacy to Resistance Mechanisms—Not Only Checkpoint Matters. Front. Immunol. 2021, 12, 690112. [Google Scholar] [CrossRef] [PubMed]
- Fay, E.K.; Graff, J.N. Immunotherapy in Prostate Cancer. Cancers 2020, 12, 1752. [Google Scholar] [CrossRef] [PubMed]
- Emens, L.A. Breast Cancer Immunotherapy: Facts and Hopes. Clin. Cancer Res. 2018, 24, 511–520. [Google Scholar] [CrossRef]
- Saleh, R.; Toor, S.M.; Khalaf, S.; Elkord, E. Breast Cancer Cells and PD-1/PD-L1 Blockade Upregulate the Expression of PD-1, CTLA-4, TIM-3 and LAG-3 Immune Checkpoints in CD4(+) T Cells. Vaccines 2019, 7, 149. [Google Scholar] [CrossRef]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef]
- Mohsenzadegan, M.; Bavandpour, P.; Nowroozi, M.R.; Amini, E.; Kourosh-Arami, M.; Momeni, S.A.; Bokaie, S.; Sharifi, L. The Potential of T Cell Immunoglobulin and Mucin-Domain Containing-3 (Tim-3) in Designing Novel Immunotherapy for Bladder Cancer. Endocr. Metab. Immune Disord. Drug Targets 2021, 21, 2131–2146. [Google Scholar] [CrossRef] [PubMed]
- Abuodeh, Y.; Venkat, P.; Kim, S. Systematic review of case reports on the abscopal effect. Curr. Probl. Cancer 2016, 40, 25–37. [Google Scholar] [CrossRef]
- Janopaul-Naylor, J.R.; Shen, Y.; Qian, D.C.; Buchwald, Z.S. The Abscopal Effect: A Review of Pre-Clinical and Clinical Advances. Int. J. Mol. Sci. 2021, 22, 11061. [Google Scholar] [CrossRef]
- Dewan, M.Z.; Galloway, A.E.; Kawashima, N.; Dewyngaert, J.K.; Babb, J.S.; Formenti, S.C.; Demaria, S. Fractionated but Not Single-Dose Radiotherapy Induces an Immune-Mediated Abscopal Effect when Combined with Anti–CTLA-4 Antibody. Clin. Cancer Res. 2009, 15, 5379–5388. [Google Scholar] [CrossRef]
- Demaria, S.; Kawashima, N.; Yang, A.M.; Devitt, M.L.; Babb, J.S.; Allison, J.P.; Formenti, S.C. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin. Cancer Res. 2005, 11, 728–734. [Google Scholar] [CrossRef]
- Arnold, K.M.; Flynn, N.J.; Raben, A.; Romak, L.; Yu, Y.; Dicker, A.P.; Mourtada, F.; Sims-Mourtada, J. The Impact of Radiation on the Tumor Microenvironment: Effect of Dose and Fractionation Schedules. Cancer Growth Metastasis 2018, 11, 1179064418761639. [Google Scholar] [CrossRef]
- Rudqvist, N.-P.; Pilones, K.A.; Lhuillier, C.; Wennerberg, E.; Sidhom, J.-W.; Emerson, R.O.; Robins, H.S.; Schneck, J.; Formenti, S.C.; Demaria, S. Radiotherapy and CTLA-4 Blockade Shape the TCR Repertoire of Tumor-Infiltrating T Cells. Cancer Immunol. Res. 2018, 6, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Gong, X.; Li, X.; Jiang, T.; Xie, H.; Zhu, Z.; Zhou, F.; Zhou, C. Combined Radiotherapy and Anti–PD-L1 Antibody Synergistically Enhances Antitumor Effect in Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 1085–1097. [Google Scholar] [CrossRef] [PubMed]
- Pilones, K.A.; Hensler, M.; Daviaud, C.; Kraynak, J.; Fucikova, J.; Galluzzi, L.; Demaria, S.; Formenti, S.C. Converging focal radiation and immunotherapy in a preclinical model of triple negative breast cancer: Contribution of VISTA blockade. Oncoimmunology 2020, 9, 1830524. [Google Scholar] [CrossRef]
- Zeng, J.; See, A.P.; Phallen, J.; Jackson, C.M.; Belcaid, Z.; Ruzevick, J.; Durham, N.; Meyer, C.; Harris, T.J.; Albesiano, E.; et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int. J. Radiat. Oncol. Biol. Phys 2013, 86, 343–349. [Google Scholar] [CrossRef]
- Wei, J.; Montalvo-Ortiz, W.; Yu, L.; Krasco, A.; Ebstein, S.; Cortez, C.; Lowy, I.; Murphy, A.J.; Sleeman, M.A.; Skokos, D. Sequence of αPD-1 relative to local tumor irradiation determines the induction of abscopal antitumor immune responses. Sci. Immunol. 2021, 6, eabg0117. [Google Scholar] [CrossRef] [PubMed]
- Dovedi, S.J.; Adlard, A.L.; Lipowska-Bhalla, G.; McKenna, C.; Jones, S.; Cheadle, E.J.; Stratford, I.J.; Poon, E.; Morrow, M.; Stewart, R.; et al. Acquired Resistance to Fractionated Radiotherapy Can Be Overcome by Concurrent PD-L1 Blockade. Cancer Res. 2014, 74, 5458–5468. [Google Scholar] [CrossRef]
- Young, K.H.; Baird, J.R.; Savage, T.; Cottam, B.; Friedman, D.; Bambina, S.; Messenheimer, D.J.; Fox, B.; Newell, P.; Bahjat, K.S.; et al. Optimizing Timing of Immunotherapy Improves Control of Tumors by Hypofractionated Radiation Therapy. PLoS ONE 2016, 11, e0157164. [Google Scholar] [CrossRef] [PubMed]
- Golden, E.B.; Demaria, S.; Schiff, P.B.; Chachoua, A.; Formenti, S.C. An abscopal response to radiation and ipilimumab in a patient with metastatic non-small cell lung cancer. Cancer Immunol. Res. 2013, 1, 365–372. [Google Scholar] [CrossRef]
- Stamell, E.F.; Wolchok, J.D.; Gnjatic, S.; Lee, N.Y.; Brownell, I. The abscopal effect associated with a systemic anti-melanoma immune response. Int. J. Radiat. Oncol. Biol. Phys. 2013, 85, 293–295. [Google Scholar] [CrossRef]
- Postow, M.A.; Callahan, M.K.; Barker, C.A.; Yamada, Y.; Yuan, J.; Kitano, S.; Mu, Z.; Rasalan, T.; Adamow, M.; Ritter, E.; et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N. Engl. J. Med. 2012, 366, 925–931. [Google Scholar] [CrossRef]
- Chicas-Sett, R.; Morales-Orue, I.; Rodriguez-Abreu, D.; Lara-Jimenez, P. Combining radiotherapy and ipilimumab induces clinically relevant radiation-induced abscopal effects in metastatic melanoma patients: A systematic review. Clin. Transl. Radiat. Oncol. 2018, 9, 5–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koller, K.M.; Mackley, H.B.; Liu, J.; Wagner, H.; Talamo, G.; Schell, T.D.; Pameijer, C.; Neves, R.I.; Anderson, B.; Kokolus, K.M.; et al. Improved survival and complete response rates in patients with advanced melanoma treated with concurrent ipilimumab and radiotherapy versus ipilimumab alone. Cancer Biol. Ther. 2017, 18, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; Rudqvist, N.P.; Golden, E.; Cooper, B.; Wennerberg, E.; Lhuillier, C.; Vanpouille-Box, C.; Friedman, K.; Ferrari de Andrade, L.; Wucherpfennig, K.W.; et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat. Med. 2018, 24, 1845–1851. [Google Scholar] [CrossRef] [PubMed]
- Bestvina, C.M.; Pointer, K.B.; Karrison, T.; Al-Hallaq, H.; Hoffman, P.C.; Jelinek, M.J.; Juloori, A.; Melotek, J.M.; Murgu, S.; Partouche, J.; et al. A Phase 1 Trial of Concurrent or Sequential Ipilimumab, Nivolumab, and Stereotactic Body Radiotherapy in Patients With Stage IV NSCLC Study. J. Thorac. Oncol. 2022, 17, 130–140. [Google Scholar] [CrossRef]
- Antonia, S.J.; Balmanoukian, A.; Brahmer, J.; Ou, S.I.; Hellmann, M.D.; Kim, S.W.; Ahn, M.J.; Kim, D.W.; Gutierrez, M.; Liu, S.V.; et al. Clinical Activity, Tolerability, and Long-Term Follow-Up of Durvalumab in Patients With Advanced NSCLC. J. Thorac. Oncol. 2019, 14, 1794–1806. [Google Scholar] [CrossRef]
- Yamaguchi, O.; Kaira, K.; Hashimoto, K.; Mouri, A.; Miura, Y.; Shiono, A.; Nishihara, F.; Murayama, Y.; Noda, S.E.; Kato, S.; et al. Radiotherapy is an independent prognostic marker of favorable prognosis in non-small cell lung cancer patients after treatment with the immune checkpoint inhibitor, nivolumab. Thorac. Cancer 2019, 10, 992–1000. [Google Scholar] [CrossRef]
- Shaverdian, N.; Lisberg, A.E.; Bornazyan, K.; Veruttipong, D.; Goldman, J.W.; Formenti, S.C.; Garon, E.B.; Lee, P. Previous radiotherapy and the clinical activity and toxicity of pembrolizumab in the treatment of non-small-cell lung cancer: A secondary analysis of the KEYNOTE-001 phase 1 trial. Lancet Oncol. 2017, 18, 895–903. [Google Scholar] [CrossRef]
- De Ruysscher, D.; Ramalingam, S.; Urbanic, J.; Gerber, D.E.; Tan, D.S.W.; Cai, J.; Li, A.; Peters, S. CheckMate 73L: A Phase 3 Study Comparing Nivolumab Plus Concurrent Chemoradiotherapy Followed by Nivolumab With or Without Ipilimumab Versus Concurrent Chemoradiotherapy Followed by Durvalumab for Previously Untreated, Locally Advanced Stage III Non-Small-Cell Lung Cancer. Clin. Lung Cancer 2022, 23, e264–e268. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.; Nowak, A.K.; Bowyer, S.; Endersby, R.; Ebert, M.A.; Cook, A. Clinical evidence for synergy between immunotherapy and radiotherapy (SITAR). J. Med. Imaging Radiat. Oncol. 2022, 66, 881–895. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, T.; Flies, D.B.; Marjon, N.A.; Mantia-Smaldone, G.; Ronner, L.; Gimotty, P.A.; Adams, S.F. CTLA-4 Blockade Synergizes Therapeutically with PARP Inhibition in BRCA1-Deficient Ovarian Cancer. Cancer Immunol. Res. 2015, 3, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Kim, H.J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980.e5. [Google Scholar] [CrossRef]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef]
- Sen, T.; Rodriguez, B.L.; Chen, L.; Corte, C.M.D.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L.; et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef]
- Adams, S.F.; Rixe, O.; Lee, J.-H.; McCance, D.J.; Westgate, S.; Eberhardt, S.C.; Rutledge, T.; Muller, C. Phase I study combining olaparib and tremelimumab for the treatment of women with BRCA-deficient recurrent ovarian cancer. J. Clin. Oncol. 2017, 35, e17052. [Google Scholar] [CrossRef]
- Lee, J.-M.; Cimino-Mathews, A.; Peer, C.J.; Zimmer, A.; Lipkowitz, S.; Annunziata, C.M.; Cao, L.; Harrell, M.I.; Swisher, E.M.; Houston, N.; et al. Safety and Clinical Activity of the Programmed Death-Ligand 1 Inhibitor Durvalumab in Combination With Poly (ADP-Ribose) Polymerase Inhibitor Olaparib or Vascular Endothelial Growth Factor Receptor 1-3 Inhibitor Cediranib in Women’s Cancers: A Dose-Escalation, Phase I Study. J. Clin. Oncol. 2017, 35, 2193–2202. [Google Scholar] [CrossRef]
- Friedlander, M.; Meniawy, T.; Markman, B.; Mileshkin, L.; Harnett, P.; Millward, M.; Lundy, J.; Freimund, A.; Norris, C.; Mu, S.; et al. Pamiparib in combination with tislelizumab in patients with advanced solid tumours: Results from the dose-escalation stage of a multicentre, open-label, phase 1a/b trial. Lancet Oncol. 2019, 20, 1306–1315. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination With Pembrolizumab in Patients With Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019, 5, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Lampert, E.J.; Zimmer, A.; Padget, M.; Cimino-Mathews, A.; Nair, J.R.; Liu, Y.; Swisher, E.M.; Hodge, J.W.; Nixon, A.B.; Nichols, E.; et al. Combination of PARP Inhibitor Olaparib, and PD-L1 Inhibitor Durvalumab, in Recurrent Ovarian Cancer: A Proof-of-Concept Phase II Study. Clin. Cancer Res. 2020, 26, 4268–4279. [Google Scholar] [CrossRef] [PubMed]
- Färkkilä, A.; Gulhan, D.C.; Casado, J.; Jacobson, C.A.; Nguyen, H.; Kochupurakkal, B.; Maliga, Z.; Yapp, C.; Chen, Y.A.; Schapiro, D.; et al. Immunogenomic profiling determines responses to combined PARP and PD-1 inhibition in ovarian cancer. Nat. Commun. 2020, 11, 1459. [Google Scholar] [CrossRef]
- Zhang, N.; Gao, Y.; Huang, Z.; Dai, P.; Luo, Y.; Wu, Q.; Jiang, X.; Sun, W.; Zhang, J.; Han, L.; et al. PARP inhibitor plus radiotherapy reshapes an inflamed tumor microenvironment that sensitizes small cell lung cancer to the anti-PD-1 immunotherapy. Cancer Lett. 2022, 545, 215852. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, S.K.; Cho, B.C.; Bria, E.; Kato, T.; Bhosle, J.; Gainor, J.F.; Reguart, N.; Wang, L.; Morgensztern, D.; Shentu, Y.; et al. Rationale and Design of the Phase III KEYLYNK-012 Study of Pembrolizumab and Concurrent Chemoradiotherapy Followed by Pembrolizumab With or Without Olaparib for Stage III Non-Small-Cell Lung Cancer. Clin. Lung Cancer 2022, 23, e342–e346. [Google Scholar] [CrossRef]

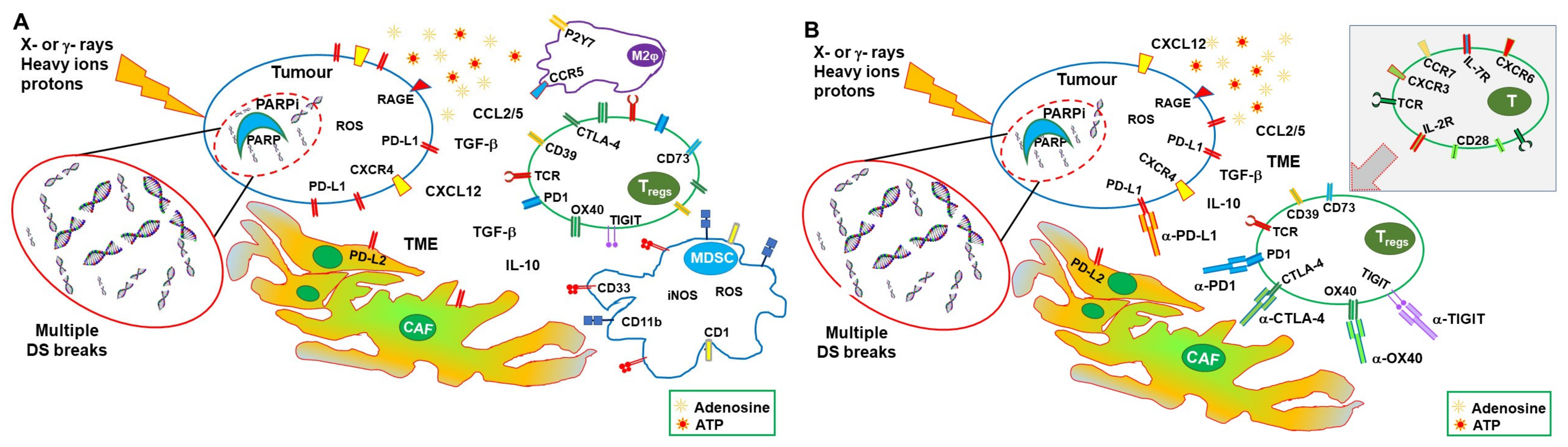

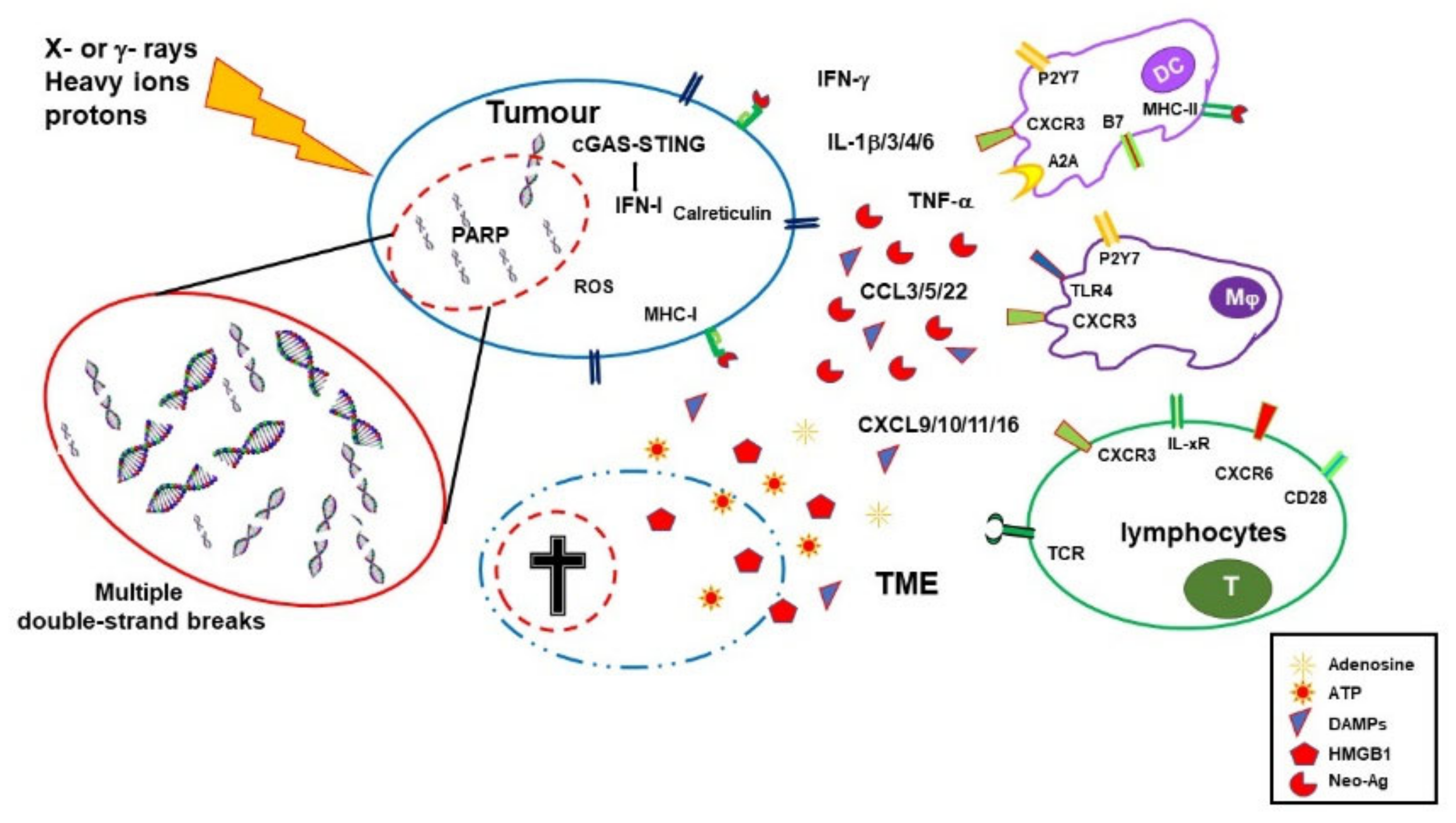{kind=link}
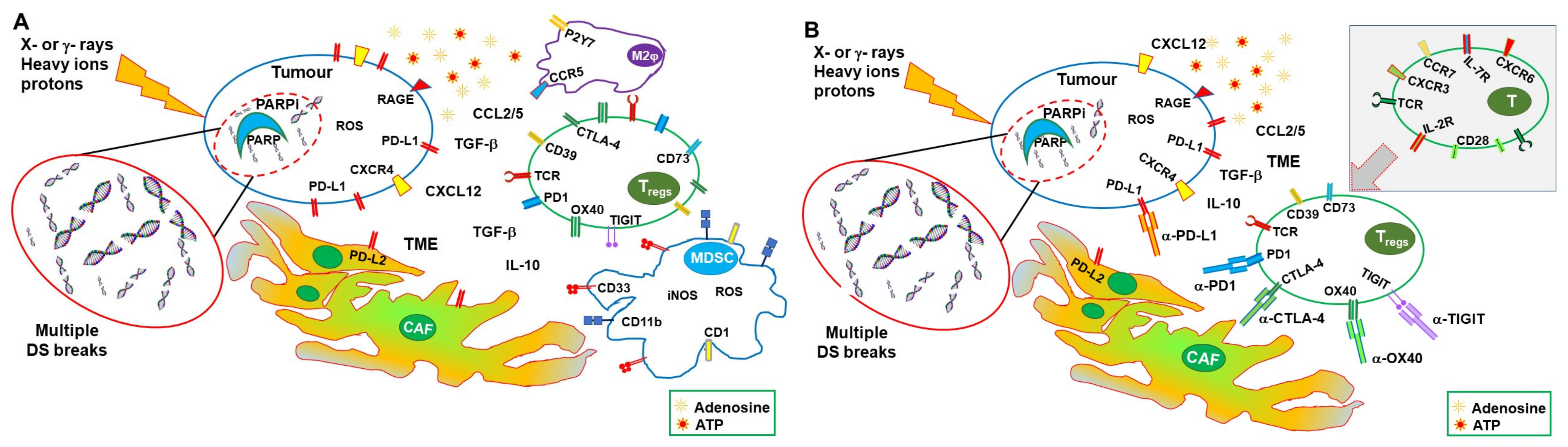{kind=link}
| Title | Conditions | Therapies | Phase | Estimated Enrollment (Patients) | Status | Estimated Completion Dates | NCT Number | Last Update Posted |
|---|---|---|---|---|---|---|---|---|
| Testing the safety of the anticancer drugs durvalumab and olaparib during radiation therapy for locally advanced unresectable pancreatic cancer | Locally advanced pancreatic carcinoma Stage II or III pancreatic cancer Unresectable pancreatic carcinoma | Durvalumab Olaparib RT | I | 18 | Recruiting | Primary and final: 31 March 2024 | 05411094 | 1 December 2022 |
| A safety study adding niraparib and dostarlimab to radiation therapy for rectal cancers | Rectal neoplasms Rectal neoplasm malignant | Niraparib Dostarlimab Short course RT | I–II | 38 | Recruiting | Primary: 31 December 2024 Final: 31 December 2026 | 04926324 | 26 July 2022 |
| Niraparib + dostarlimab + RT in pancreatic cancer | Pancreatic cancer Metastatic pancreatic cancer | Niraparib Dostarlimab RT | II | 25 | Active, not recruiting | Primary: 19 January 2022 Final: October 2026 | 04409002 | 8 September 2022 |
| Radiation, immunotherapy, and PARP inhibitor in triple-negative breast cancer | Breast cancer TNBC | Niraparib Dostarlimab RT | II | 32 | Recruiting | Primary: 1 April 2023 Final: 1 December 2029 | 04837209 | 23 December 2022 |
| Radiotherapy and durvalumab/durvalumab combo (tremelimumab/olaparid) for small-cell lung cancer | SCLC extensive stage SCLC | Durvalumab Tremelimumab Olaparib Thoracic RT | I | 25 | Active, not recruiting | Primary and final: 1 June 2023 | 03923270 | 6 January 2023 |
| A study of radiation therapy with pembrolizumab and olaparib or other radiosensitizers in women who have triple-negative or hormone-receptor positive/HER2 negative breast cancer | TNBC Metastatic breast cancer | Pembrolizumab Olaparib RT | II | 34 | Recruiting | Primary and final: January 2025 | 04683679 | 21 October 2022 |
| Pembro with radiation with or without olaparib | Prostate cancer | Pembrolizumab Olaparib Androgen deprivation therapy RT | II | 64 | Not yet recruiting | Primary: 2 January 2025 Final: 2 January 2028 | 05568550 | 5 October 2022 |
| Olaparib and durvalumab with carboplatin, etoposide, and/or radiation therapy for the treatment of extensive-stage small-cell lung cancer, PRIO trial | Extensive-stage SCLC Stage IV lung cancer Stage IVA lung cancer Stage IVB lung cancer | Carboplatin Durvalumab Etoposide Olaparib RT | I–II | 63 | Recruiting | Primary and final: 31 January 2024 | 04728230 | 9 November 2022 |
| Study of SBRT/olaparib followed by pembrolizumab/olaparib in gastric cancers | Gastric cancer Gastroesophageal cancer | Pembrolizumab Olaparib SBRT | II | 26 | Recruiting | Primary: December 2025 Final: December 2028 | 05379972 | 5 January 2023 |
| Placebo-controlled study of concurrent chemoradiation therapy with pembrolizumab followed by pembrolizumab and olaparib in newly diagnosed treatment-naïve limited-stage small-cell lung cancer (LS-SCLC) (MK 7339-013/KEYLYNK-013) | SCLC | Pembrolizumab (2 doses) Olaparib Etoposide Platinum Standard thoracic RT Prophylactic cranial irradiation | III | 672 | Recruiting | Primary and final: 28 October 2027 | 04624204 | 23 December 2022 |
| Pembrolizumab plus olaparib in LA-HNSCC | Head and neck squamous cell carcinoma | Pembrolizumab Olaparib Cisplatin IMRT | II | 45 | Recruiting | Primary: 31 October 2024 Final: 31 October 2025 | 05366166 | 28 October 2022 |
| Study of pembrolizumab with concurrent chemoradiation therapy, followed by pembrolizumab with or without olaparib in stage III non-small-cell lung cancer (NSCLC) (MK-7339-012/KEYLYNK-012) | Lung neoplasms NSCLC | Pembrolizumab Olaparib Etoposide Carboplatin Cisplatin Paclitaxel Pemetrexed Thoracic RT Durvalumab | III | 870 | Recruiting | Primary and final: 6 July 2026 | 04380636 | 30 November 2022 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosado, M.M.; Pioli, C. Radiotherapy, PARP Inhibition, and Immune-Checkpoint Blockade: A Triad to Overcome the Double-Edged Effects of Each Single Player. Cancers 2023, 15, 1093. https://doi.org/10.3390/cancers15041093
Rosado MM, Pioli C. Radiotherapy, PARP Inhibition, and Immune-Checkpoint Blockade: A Triad to Overcome the Double-Edged Effects of Each Single Player. Cancers. 2023; 15(4):1093. https://doi.org/10.3390/cancers15041093
Chicago/Turabian StyleRosado, Maria Manuela, and Claudio Pioli. 2023. "Radiotherapy, PARP Inhibition, and Immune-Checkpoint Blockade: A Triad to Overcome the Double-Edged Effects of Each Single Player" Cancers 15, no. 4: 1093. https://doi.org/10.3390/cancers15041093
APA StyleRosado, M. M., & Pioli, C. (2023). Radiotherapy, PARP Inhibition, and Immune-Checkpoint Blockade: A Triad to Overcome the Double-Edged Effects of Each Single Player. Cancers, 15(4), 1093. https://doi.org/10.3390/cancers15041093