


Explainable AI for Estimating Pathogenicity of Genetic Variants Using Large-Scale Knowledge Graphs

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
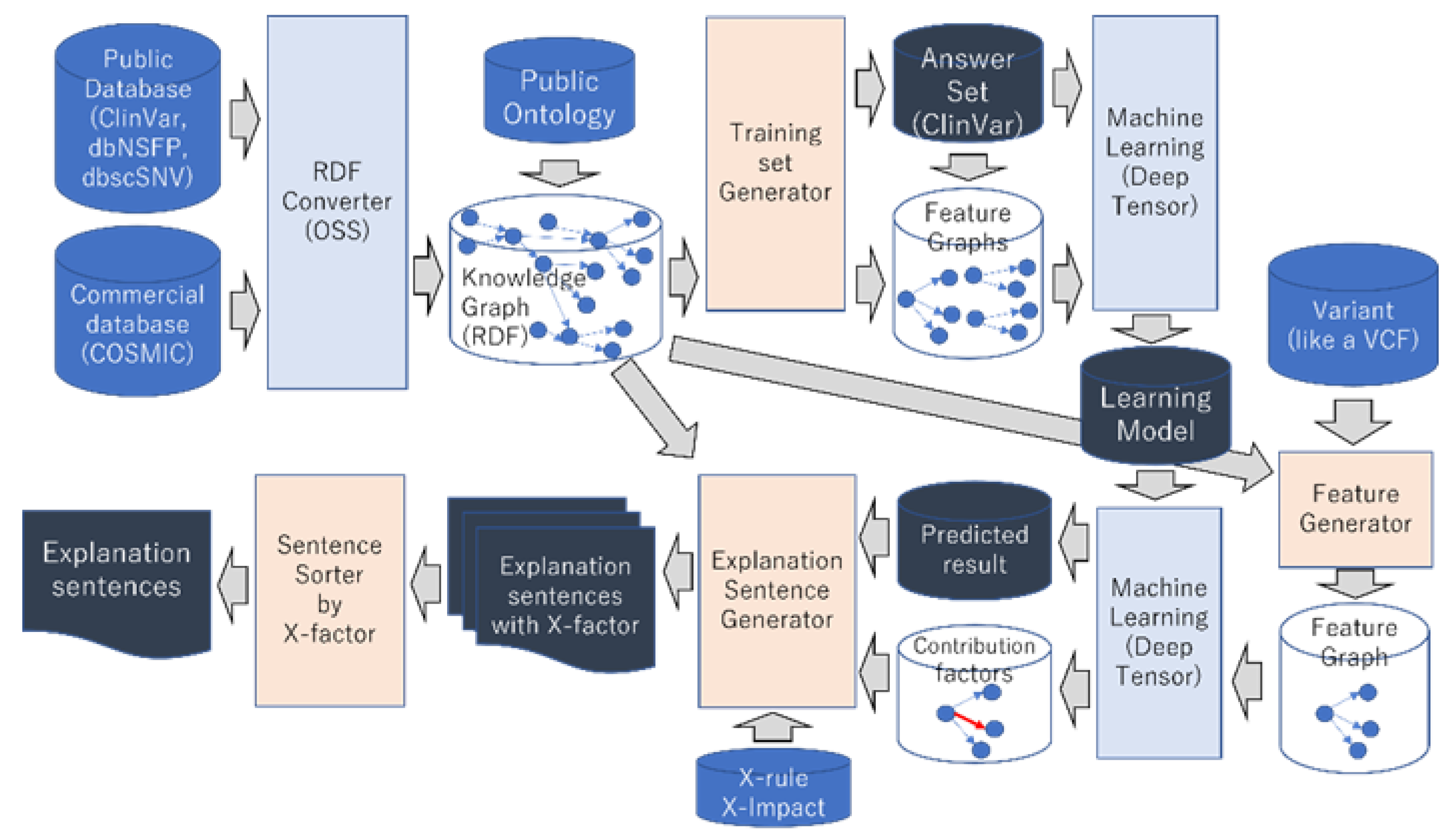
2.1. Knowledge Graph–Integrating Genome Databases
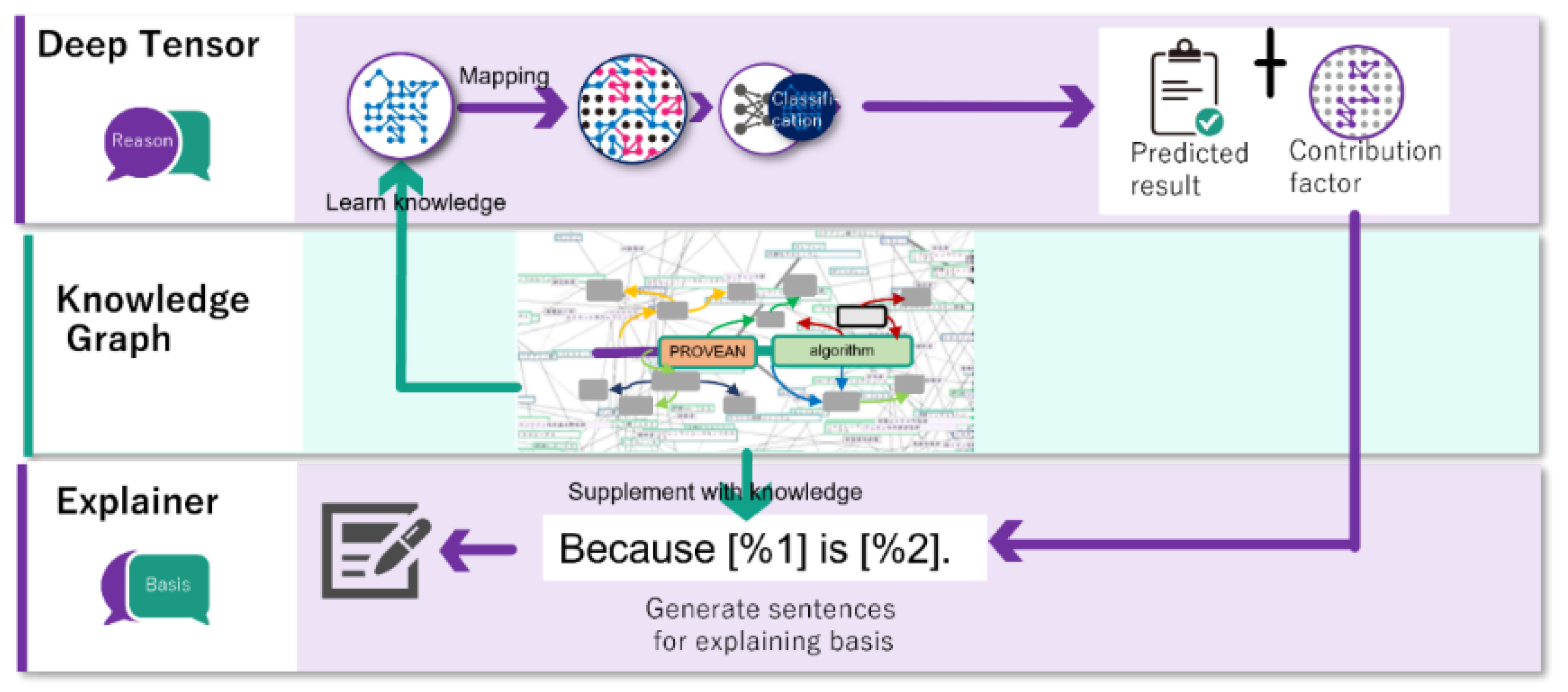
2.2. Explainable AI
- Lack of explanatory information: Presenting only the highly contributing nodes is insufficient as an explanation, and there are often cases where physicians cannot understand the meaning of the nodes or why they are the reason for the estimation result unless related nodes are presented simultaneously. For example, if the contribution of a node in pathogenicity is high, presenting only this node does not improve comprehensibility for physicians. In this case, it is necessary to explain the name of the variant to which the pathogenic node is connected and the fact that this information is from the ClinVar database, thereby simultaneously presenting the related information.
- Lack of consideration of importance: It is important to present the information that physicians consider important. For example, if a variant is listed in ClinVar or COSMIC, the fact that it is listed provides important information to the physician.
- Unfamiliar format: Normally, physicians report the results of their literature searches and verify medical information in writing. Likewise, the literature and guidelines to which physicians refer are written, and physicians rarely have the opportunity to refer to graphical data. Therefore, graphical data are a form of expression unfamiliar to physicians, and explanations using graphical data may be difficult for physicians to understand. Explanations using text, a data format familiar to physicians, are important. Thus, our XAI generates text from graph data and presents explanations.
- Explain the node with the highest contribution to deep learning.
- When explaining a node, the node necessary for understanding should be explained simultaneously.
- The information that physicians consider important should also be explained.
- Explain according to the guideline of ACMG.
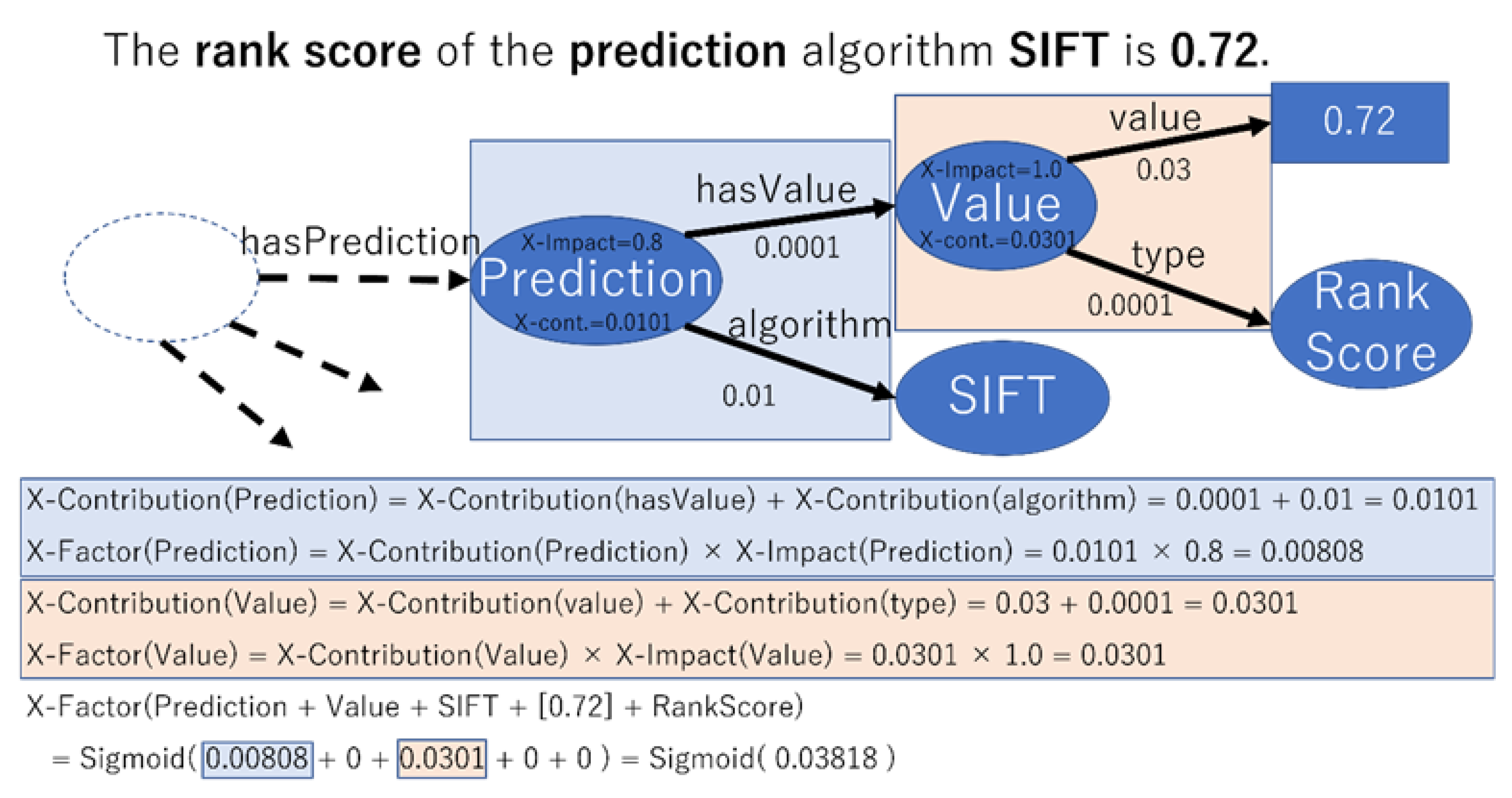
- Importance of each node to the physician (X-Impact).
- A rule (X-Rule) consisting of a combination of the following information:
- A rule for the set of nodes required to describe any node.
- Rules for generating sentences from node sets.
2.3. Evaluation Method
2.3.1. Target of Evaluation
2.3.2. Correct Answer Set
2.3.3. Five-Fold Cross-Validation
2.3.4. Balancing the Number of Pathogenic and Benign Variants
2.3.5. Trained Model
2.3.6. Correct Answer Set for Explanation
2.3.7. Features for Estimation
- ClinVar: clinical significance, review status, year of last update, number of submissions, publications, and sources of publications;
- COSMIC: registration status, number of samples, and publications.
2.3.8. Features for Explanation
- ClinVar: clinical significance, review status, year of last update, number of submissions, publications, and sources of publications;
- dbNSFP: scores for each algorithm;
- DbscSNV: Scores of each algorithm;
- AdaBoost;
- Random forest.
2.3.9. Methods of Comparison
- Deep tensor: A black-box AI and deep learning method that uses graph data. We used this in our proposed method;
- Decision trees: A white-box AI said to have excellent explanatory properties;
- Random forests: A black-box AI with excellent estimation performance.
2.3.10. Features for Decision Trees and Random Forests
- If number of samples = 1, aggregate to the number 0;
- If 1 < number of samples ≤ 10, aggregate to the number 1;
- If 10 < number of samples ≤ 100, aggregate to the number 2;
- If 100 < number of samples ≤ 1000, aggregate to the number 3;
- If 1000 < number of samples ≤ 10,000, aggregate to the number 4;
- If 10,000 < number of samples, aggregate to the number 5; All cases greater than 10,000 were aggregated to 5.
- VariantImpact and clinicalsignificance;
- VariantImpact, reviewStatus, and clinicalsignificance.
2.3.11. Other Experimental Conditions
3. Results
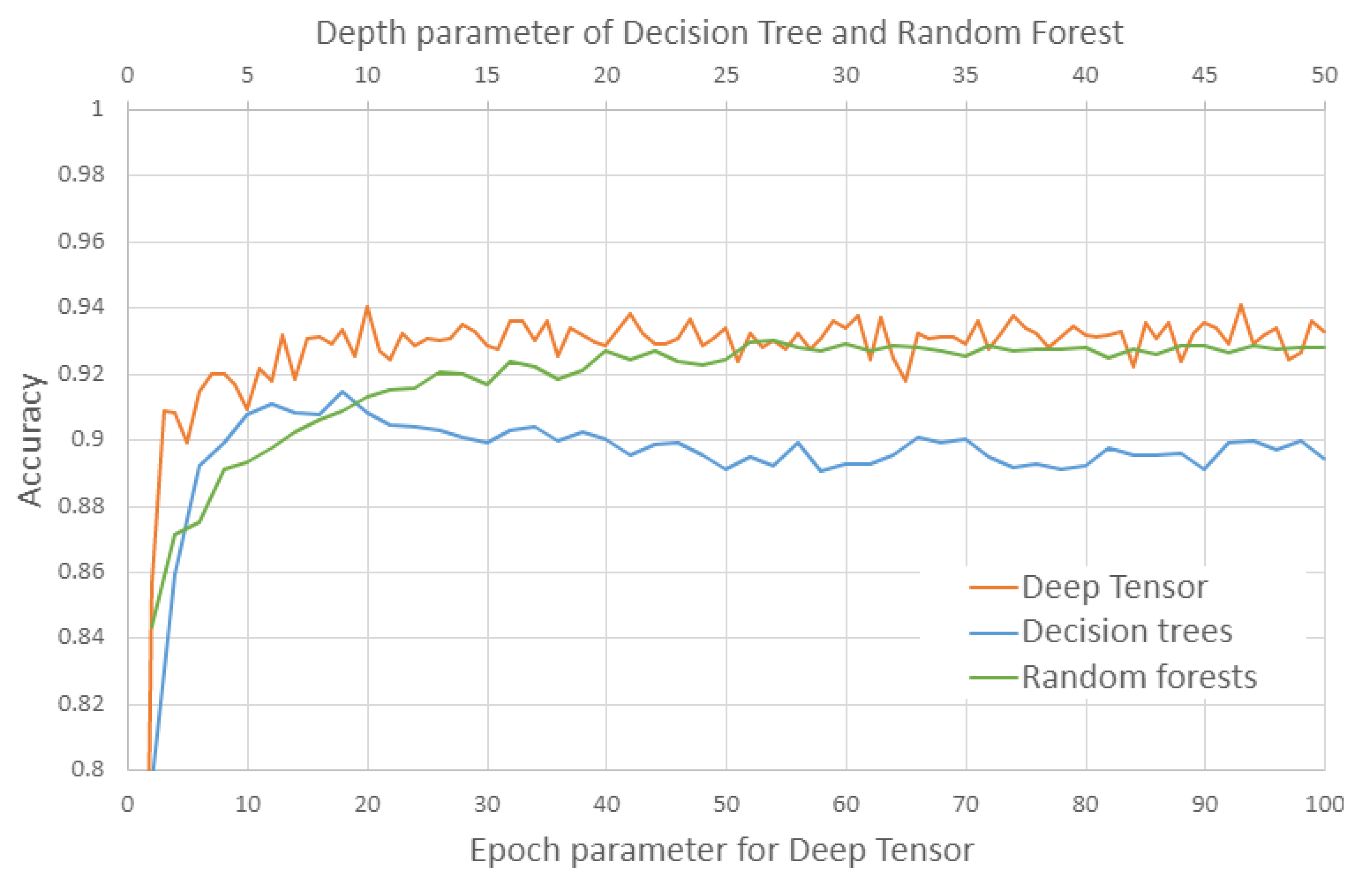
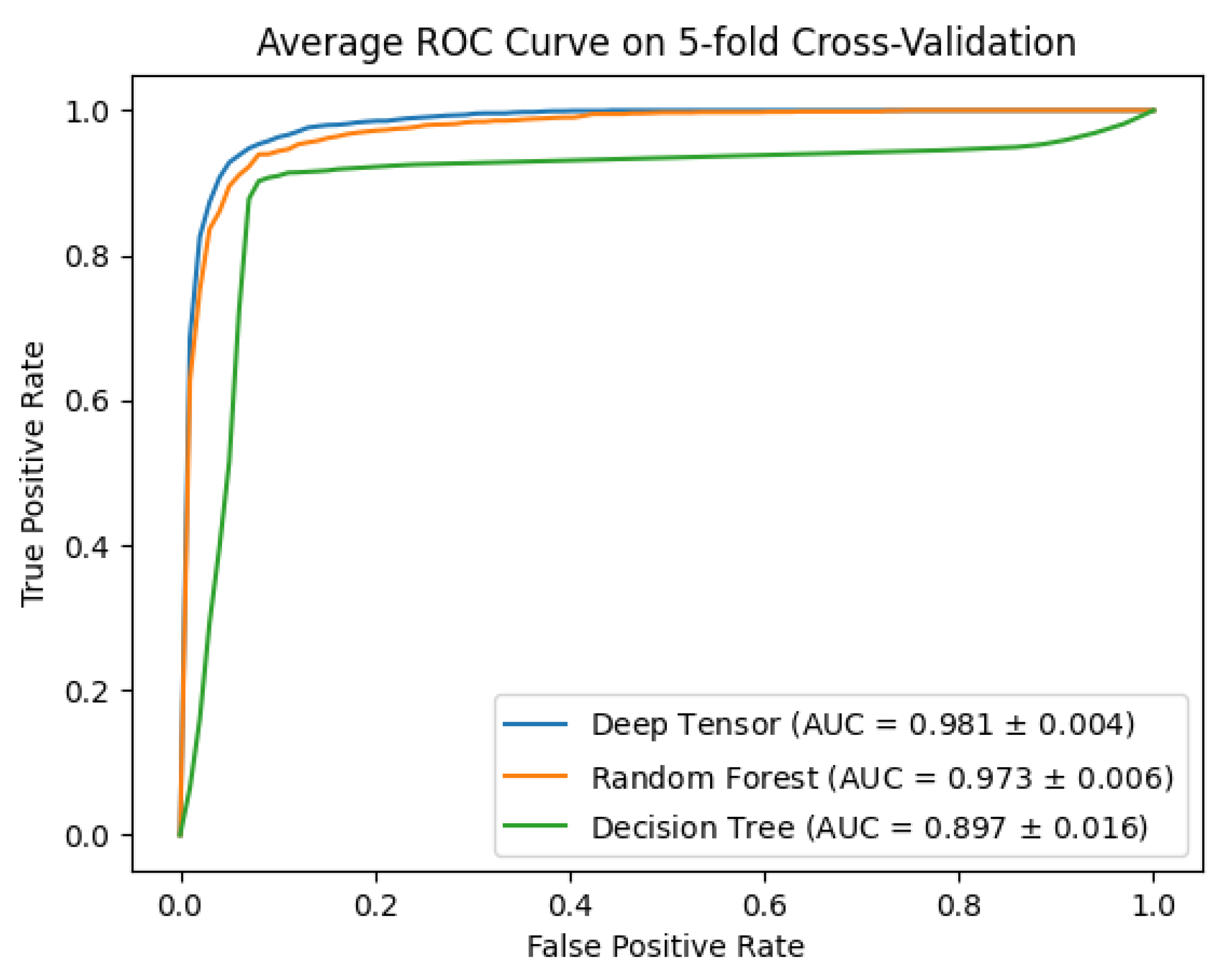
3.1. Evaluation of Estimation
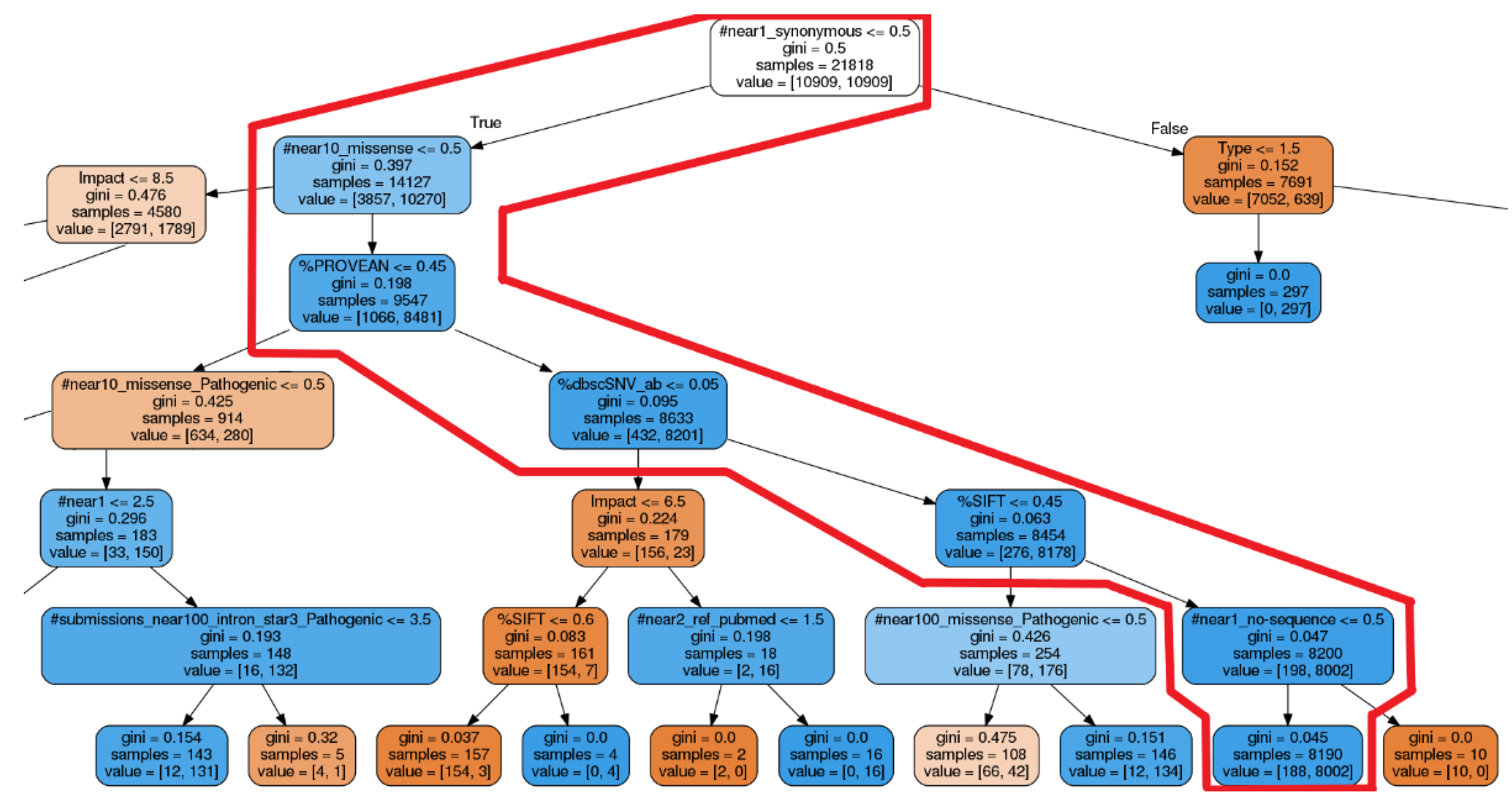
3.2. Evaluation of Explicability
4. Discussion
4.1. Results of the Performance Evaluation
4.2. Comparison of Explanations
4.3. Estimation Performance of Other Methods
- The AUC listed in the ClinPred paper is 0.98, and the AUC of the proposed method is also 0.98. From this point of view, the performance is equivalent.
- The mutations in the test results of ClinPred were matched with the mutations in the test results of the five-fold cross-validation of the proposed method, and the performance of ClinPred was calculated only from the mutations that could be matched. The mutations that could not be matched were 30 of 863, 8 of 863, 14 of 863, 8 of 863, and 6 of 864, respectively. Since only a few mutations could not be matched, we do not believe that they had a significant impact on the performance measures. The performance of ClinPred in these results was accuracy 0.96, precision 0.99, recall 0.93, F1 score 0.96, and AUC 0.99. This performance is higher than the performance of ClinPred described in their paper [15]. We believe that differences in training data are the cause of the differences in performance. ClinPred has excluded some mutations from the test to avoid false performance increases due to type 1 circularity [15,33]. On the other hand, the proposed method excludes such mutations from the training data for the same reason. From this, we believe that the increase in performance of ClinPred is due to the fact that it differs from the test set used in their paper [15].
4.4. Realization of Explanations by Random Forests
4.5. Another Knowledge Graph
4.6. Application to Genome Medicine
4.7. Application of AI in Future Genomic Medicine
4.8. Application to Other Fields
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mosele, F.; Remon, J.; Mateo, J.; Westphalen, C.B.; Barlesi, F.; Lolkema, M.P.; Normanno, N.; Scarpa, A.; Robson, M.; Meric-Bernstam, F.; et al. Recommendations for the Use of Next-Generation Sequencing (NGS) for Patients with Metastatic Cancers: A Report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2020, 31, 1491–1505. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Fujita, M. Whole Genome Sequencing Analysis for Cancer Genomics and Precision Medicine. Cancer Sci. 2018, 109, 513–522. [Google Scholar] [CrossRef] [PubMed]
- El-Sappagh, S.; Ali, F.; Abuhmed, T.; Singh, J.; Alonso, J.M. Automatic Detection of Alzheimer’s Disease Progression: An Efficient Information Fusion Approach with Heterogeneous Ensemble Classifiers. Neurocomputing 2022, 512, 203–224. [Google Scholar] [CrossRef]
- Vulli, A.; Srinivasu, P.N.; Sashank, M.S.K.; Shafi, J.; Choi, J.; Ijaz, M.F. Fine-Tuned DenseNet-169 for Breast Cancer Metastasis Prediction Using FastAI and 1-Cycle Policy. Sensors 2022, 22, 2988. [Google Scholar] [CrossRef]
- Quinodoz, M.; Peter, V.G.; Cisarova, K.; Royer-Bertrand, B.; Stenson, P.D.; Cooper, D.N.; Unger, S.; Superti-Furga, A.; Rivolta, C. Analysis of Missense Variants in the Human Genome Reveals Widespread Gene-Specific Clustering and Improves Prediction of Pathogenicity. Am. J. Hum. Genet. 2022, 109, 457–470. [Google Scholar] [CrossRef]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef]
- Livesey, B.J.; Marsh, J.A. Interpreting Protein Variant Effects with Computational Predictors and Deep Mutational Scanning. Dis. Model. Mech. 2022, 15, dmm049510. [Google Scholar] [CrossRef]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and Integration of Deleteriousness Prediction Methods for Nonsynonymous SNVs in Whole Exome Sequencing Studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef]
- Xu, J.; Yang, P.; Xue, S.; Sharma, B.; Sanchez-Martin, M.; Wang, F.; Beaty, K.A.; Dehan, E.; Parikh, B. Translating Cancer Genomics into Precision Medicine with Artificial Intelligence: Applications, Challenges and Future Perspectives. Hum. Genet. 2019, 138, 109–124. [Google Scholar] [CrossRef]
- Sakai, A.; Komatsu, M.; Komatsu, R.; Matsuoka, R.; Yasutomi, S.; Dozen, A.; Shozu, K.; Arakaki, T.; Machino, H.; Asada, K.; et al. Medical Professional Enhancement Using Explainable Artificial Intelligence in Fetal Cardiac Ultrasound Screening. Biomedicines 2022, 10, 551. [Google Scholar] [CrossRef]
- Gunning, D.; Aha, D. DARPA’s Explainable Artificial Intelligence (XAI) Program. AI Mag. 2019, 40, 44–58. [Google Scholar] [CrossRef]
- Barredo Arrieta, A.; Díaz-Rodríguez, N.; Del Ser, J.; Bennetot, A.; Tabik, S.; Barbado, A.; Garcia, S.; Gil-Lopez, S.; Molina, D.; Benjamins, R.; et al. Explainable Artificial Intelligence (XAI): Concepts, Taxonomies, Opportunities and Challenges toward Responsible AI. Inf. Fusion 2020, 58, 82–115. [Google Scholar] [CrossRef]
- Islam, M.R.; Ahmed, M.U.; Barua, S.; Begum, S. A Systematic Review of Explainable Artificial Intelligence in Terms of Different Application Domains and Tasks. Appl. Sci. 2022, 12, 1353. [Google Scholar] [CrossRef]
- Vilone, G.; Longo, L. Explainable Artificial Intelligence: A Systematic Review. arXiv 2020, arXiv:2006.00093. [Google Scholar] [CrossRef]
- Alirezaie, N.; Kernohan, K.D.; Hartley, T.; Majewski, J.; Hocking, T.D. ClinPred: Prediction Tool to Identify Disease-Relevant Nonsynonymous Single-Nucleotide Variants. Am. J. Hum. Genet. 2018, 103, 474–483. [Google Scholar] [CrossRef]
- Dias, R.; Torkamani, A. Artificial Intelligence in Clinical and Genomic Diagnostics. Genome Med. 2019, 11, 70. [Google Scholar] [CrossRef]
- Moradi, M.; Samwald, M. Deep Learning, Natural Language Processing, and Explainable Artificial Intelligence in the Biomedical Domain. arXiv 2022, arXiv:2202.12678. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Guidelines for the Practice of Hematopoietic Tumors, 2018 Revised Edition. Available online: http://www.jshem.or.jp/gui-hemali/table.html (accessed on 18 November 2022).
- ASH Clinical Practice Guidelines. Available online: https://www.hematology.org:443/education/clinicians/guidelines-and-quality-care/clinical-practice-guidelines (accessed on 18 November 2022).
- NCCN Guidelines. Available online: https://www.nccn.org/guidelines/category_1 (accessed on 18 November 2022).
- Resource Description Framework (RDF): Concepts and Abstract Syntax. Available online: https://www.w3.org/TR/rdf-concepts/ (accessed on 18 November 2022).
- ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/ (accessed on 18 November 2022).
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. DbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef]
- Cosmic COSMIC—Catalogue of Somatic Mutations in Cancer. Available online: https://cancer.sanger.ac.uk/cosmic (accessed on 18 November 2022).
- Med2rdf-Ontology. Available online: https://github.com/med2rdf/med2rdf-ontology (accessed on 18 November 2022).
- Semanticscience Integrated Ontology. Available online: https://bioportal.bioontology.org/ontologies/SIO (accessed on 18 November 2022).
- Human Chromosome Ontology. Available online: https://github.com/med2rdf/hco (accessed on 18 November 2022).
- Bolleman, J.T.; Mungall, C.J.; Strozzi, F.; Baran, J.; Dumontier, M.; Bonnal, R.J.P.; Buels, R.; Hoehndorf, R.; Fujisawa, T.; Katayama, T.; et al. FALDO: A Semantic Standard for Describing the Location of Nucleotide and Protein Feature Annotation. J. Biomed. Semant. 2016, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Auer, S.; Bizer, C.; Kobilarov, G.; Lehmann, J.; Cyganiak, R.; Ives, Z. DBpedia: A Nucleus for a Web of Open Data. In The Semantic Web; Aberer, K., Choi, K.-S., Noy, N., Allemang, D., Lee, K.-I., Nixon, L., Golbeck, J., Mika, P., Maynard, D., Mizoguchi, R., et al., Eds.; Lecture Notes in Computer Science; Springer: Berlin/Heidelberg, Germany, 2007; Volume 4825, pp. 722–735. ISBN 978-3-540-76297-3. [Google Scholar]
- Maruhashi, K.; Todoriki, M.; Ohwa, T.; Goto, K.; Hasegawa, Y.; Inakoshi, H.; Anai, H. Learning Multi-Way Relations via Tensor Decomposition With Neural Networks. In Proceedings of the Thirty-Second AAAI Conference on Artificial Intelligence, New Orleans, LA, USA, 2–7 February 2018; Volume 32. [Google Scholar] [CrossRef]
- Martelotto, L.G.; Ng, C.K.; De Filippo, M.R.; Zhang, Y.; Piscuoglio, S.; Lim, R.S.; Shen, R.; Norton, L.; Reis-Filho, J.S.; Weigelt, B. Benchmarking Mutation Effect Prediction Algorithms Using Functionally Validated Cancer-Related Missense Mutations. Genome Biol. 2014, 15, 484. [Google Scholar] [CrossRef] [PubMed]
- Grimm, D.G.; Azencott, C.; Aicheler, F.; Gieraths, U.; MacArthur, D.G.; Samocha, K.E.; Cooper, D.N.; Stenson, P.D.; Daly, M.J.; Smoller, J.W.; et al. The Evaluation of Tools Used to Predict the Impact of Missense Variants Is Hindered by Two Types of Circularity. Hum. Mutat. 2015, 36, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. Predicting Deleterious Amino Acid Substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Chun, S.; Fay, J.C. Identification of Deleterious Mutations within Three Human Genomes. Genome Res. 2009, 19, 1553–1561. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Chan, A.P. PROVEAN Web Server: A Tool to Predict the Functional Effect of Amino Acid Substitutions and Indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef]
- Siepel, A.; Pollard, K.S.; Haussler, D. New Methods for Detecting Lineage-Specific Selection. In Research in Computational Molecular Biology; Apostolico, A., Guerra, C., Istrail, S., Pevzner, P.A., Waterman, M., Eds.; Lecture Notes in Computer Science; Springer: Berlin/Heidelberg, Germany, 2006; Volume 3909, pp. 190–205. ISBN 978-3-540-33295-4. [Google Scholar]
- Davydov, E.V.; Goode, D.L.; Sirota, M.; Cooper, G.M.; Sidow, A.; Batzoglou, S. Identifying a High Fraction of the Human Genome to Be under Selective Constraint Using GERP++. PLoS Comput. Biol. 2010, 6, e1001025. [Google Scholar] [CrossRef]
- Garber, M.; Guttman, M.; Clamp, M.; Zody, M.C.; Friedman, N.; Xie, X. Identifying Novel Constrained Elements by Exploiting Biased Substitution Patterns. Bioinformatics 2009, 25, i54–i62. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the Deleteriousness of Variants throughout the Human Genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Quang, D.; Chen, Y.; Xie, X. DANN: A Deep Learning Approach for Annotating the Pathogenicity of Genetic Variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef]
- Shihab, H.A.; Gough, J.; Mort, M.; Cooper, D.N.; Day, I.N.; Gaunt, T.R. Ranking Non-Synonymous Single Nucleotide Polymorphisms Based on Disease Concepts. Hum. Genom. 2014, 8, 11. [Google Scholar] [CrossRef]
- Jagadeesh, K.A.; Wenger, A.M.; Berger, M.J.; Guturu, H.; Stenson, P.D.; Cooper, D.N.; Bernstein, J.A.; Bejerano, G. M-CAP Eliminates a Majority of Variants of Uncertain Significance in Clinical Exomes at High Sensitivity. Nat. Genet. 2016, 48, 1581–1586. [Google Scholar] [CrossRef] [PubMed]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.-J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. Inferring the Molecular and Phenotypic Impact of Amino Acid Variants with MutPred2. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation Prediction for the Deep-Sequencing Age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; et al. Evolutionarily Conserved Elements in Vertebrate, Insect, Worm, and Yeast Genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef]
- Scikit-Learn. Available online: https://scikit-learn.org/stable/ (accessed on 18 November 2022).
- Ribeiro, M.T.; Singh, S.; Guestrin, C. “Why Should I Trust You?”: Explaining the Predictions of Any Classifier. In Proceedings of the Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 1135–1144. [Google Scholar]
- Lundberg, S.M.; Lee, S.-I. A Unified Approach to Interpreting Model Predictions. In Proceedings of the 31st International Conference on Neural Information Processing Systems, Long Beach, CA, USA, 4–9 December 2017; Curran Associates Inc.: Red Hook, NY, USA, 2017; pp. 4768–4777. [Google Scholar]
- Santos, A.; Colaço, A.R.; Nielsen, A.B.; Niu, L.; Geyer, P.E.; Coscia, F.; Albrechtsen, N.J.W.; Mundt, F.; Jensen, L.J.; Mann, M. Clinical Knowledge Graph Integrates Proteomics Data into Clinical Decision-Making. Bioinformatics 2020. [Google Scholar] [CrossRef]
- FDA Center for Devices and Radiological Health. FoundationOne CDx—P170019/S014; FDA: Silver Spring, MD, USA, 2022. [Google Scholar]
- FDA Center for Devices and Radiological Health. OncomineTM Dx Target Test—P160045/S035; FDA: Silver Spring, MD, USA, 2022. [Google Scholar]
- Sakai, K.; Takeda, M.; Shimizu, S.; Takahama, T.; Yoshida, T.; Watanabe, S.; Iwasa, T.; Yonesaka, K.; Suzuki, S.; Hayashi, H.; et al. A Comparative Study of Curated Contents by Knowledge-Based Curation System in Cancer Clinical Sequencing. Sci. Rep. 2019, 9, 11340. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| X-Rule Name | Sample Sentence |
|---|---|
| Neighboring variants in ClinVar | Fourteen neighboring variants are “missense variant”, three of which are registered as “Pathogenic” in ClinVar. |
| Outstanding algorithms in dbNSFP | This variant has dbNSFP algorithms: phyloP100way_vertebrate is high (0.97), SiPhy_29way_logOdds is high (0.88), SIFT is high (0.72), PROVEAN is high (0.71), and LRT is moderate (0.63). |
| Average of algorithms in dbNSFP | This variant has an average score of 0.84 for 14 prediction algorithms, and 0.79 for 4 conservation algorithms from dbNSFP. |
| Same amino acid variants | Neighboring variants in the same base are registered as "VUS" in ClinVar. |
| Neighboring variant papers | Eight neighboring variants are reported in twenty-six papers. |
| Variants in COSMIC | A neighboring variant, p.R1861*, is registered in COSMIC. |
| Variants in dbNSFP | This variant has a score of 0.88 according to the SIFT algorithm of dbNSFP. |
| Variants reference | This variant is explained in https://pubmed.ncbi.nlm.nih.gov/28492532/ (accessed on 2 December 2022). |
| Variants in ClinVar | This variant is considered pathogenic with 3 stars in ClinVar, and the report has 3 submissions. |
| Variants type | This variant is a missense variant. |
| Method | Accuracy | Precision | Recall | F1 Score | AUC |
|---|---|---|---|---|---|
| Deep tensor | 0.94 | 0.94 | 0.94 | 0.94 | 0.98 |
| Random forests | 0.93 | 0.93 | 0.92 | 0.92 | 0.97 |
| Decision trees | 0.91 | 0.92 | 0.90 | 0.91 | 0.90 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abe, S.; Tago, S.; Yokoyama, K.; Ogawa, M.; Takei, T.; Imoto, S.; Fuji, M. Explainable AI for Estimating Pathogenicity of Genetic Variants Using Large-Scale Knowledge Graphs. Cancers 2023, 15, 1118. https://doi.org/10.3390/cancers15041118
Abe S, Tago S, Yokoyama K, Ogawa M, Takei T, Imoto S, Fuji M. Explainable AI for Estimating Pathogenicity of Genetic Variants Using Large-Scale Knowledge Graphs. Cancers. 2023; 15(4):1118. https://doi.org/10.3390/cancers15041118
Chicago/Turabian StyleAbe, Shuya, Shinichiro Tago, Kazuaki Yokoyama, Miho Ogawa, Tomomi Takei, Seiya Imoto, and Masaru Fuji. 2023. "Explainable AI for Estimating Pathogenicity of Genetic Variants Using Large-Scale Knowledge Graphs" Cancers 15, no. 4: 1118. https://doi.org/10.3390/cancers15041118
APA StyleAbe, S., Tago, S., Yokoyama, K., Ogawa, M., Takei, T., Imoto, S., & Fuji, M. (2023). Explainable AI for Estimating Pathogenicity of Genetic Variants Using Large-Scale Knowledge Graphs. Cancers, 15(4), 1118. https://doi.org/10.3390/cancers15041118