



Unique Metabolic Contexts Sensitize Cancer Cells and Discriminate between Glycolytic Tumor Types


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Reagents
2.2. Viability, Cell Number, and ATP/Cell Measurements
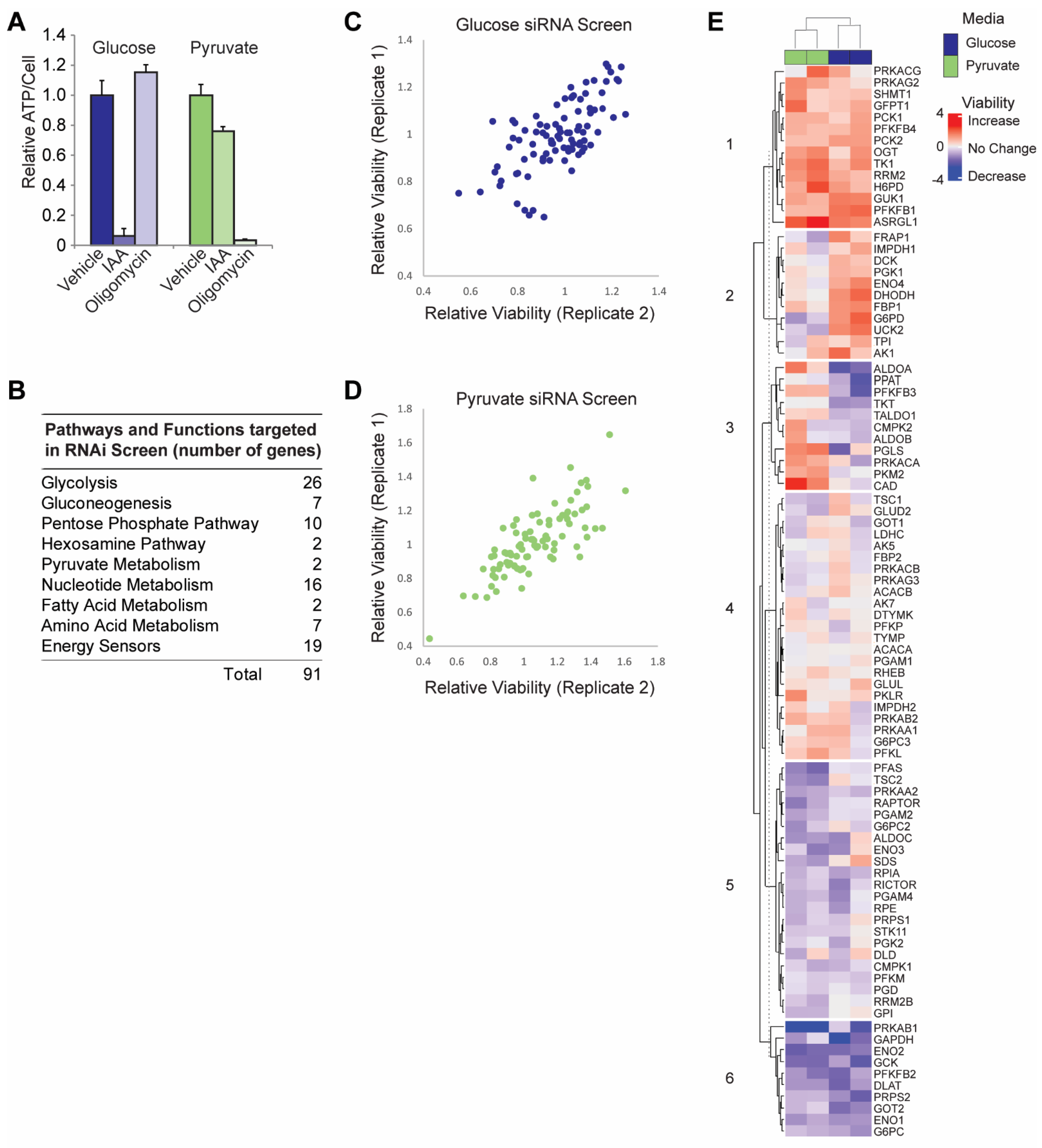
2.3. RNAi Screen
2.4. Hierarchical Clustering
2.5. cBioPortal Data Query and Processing
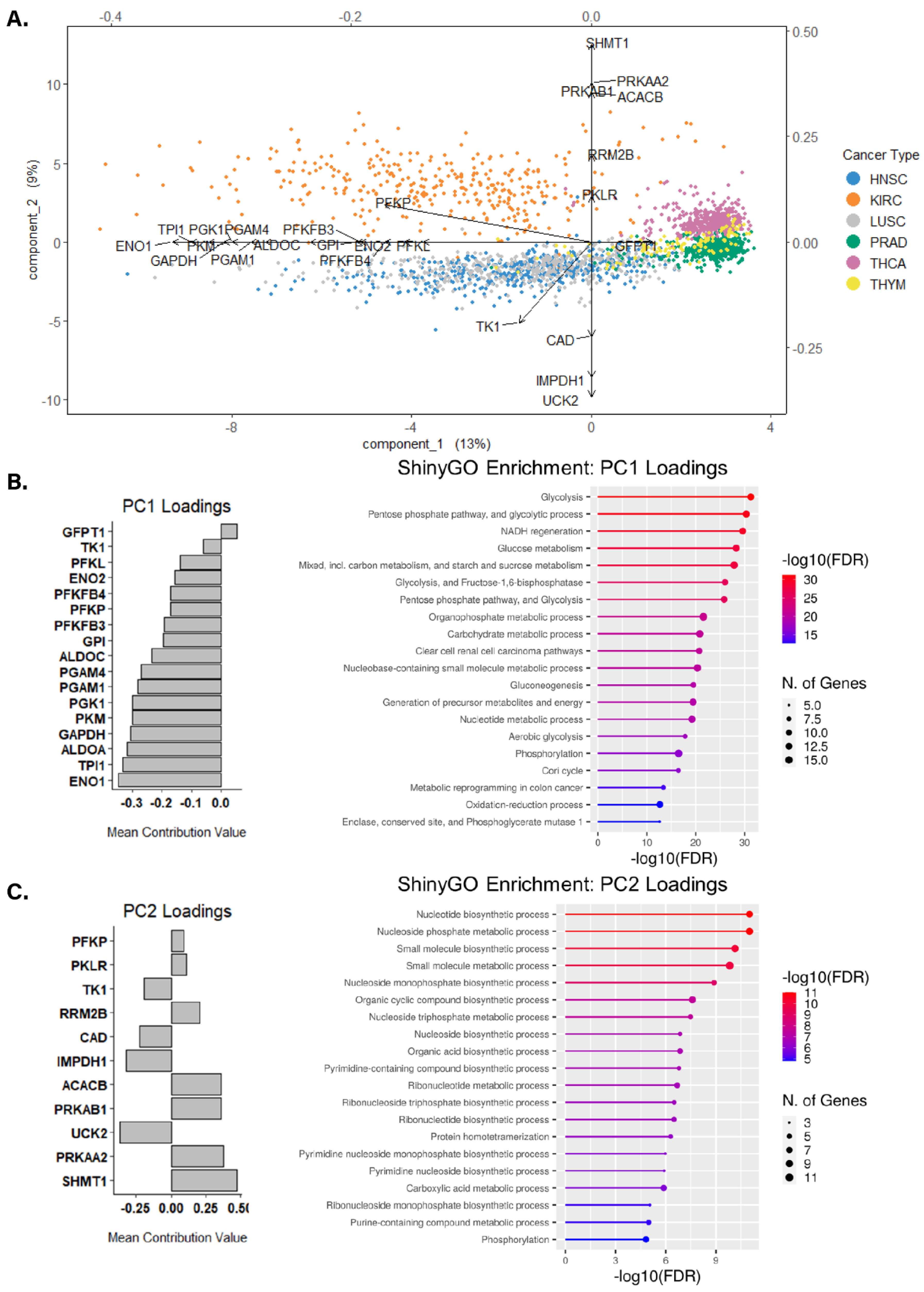
2.6. Sparse Principal Component Analysis
2.7. Copy Number Alteration Analysis
2.8. Pathway Enrichment Analysis
3. Results
3.1. siRNA Screen in Glycolytic versus OXPHOS Conditions
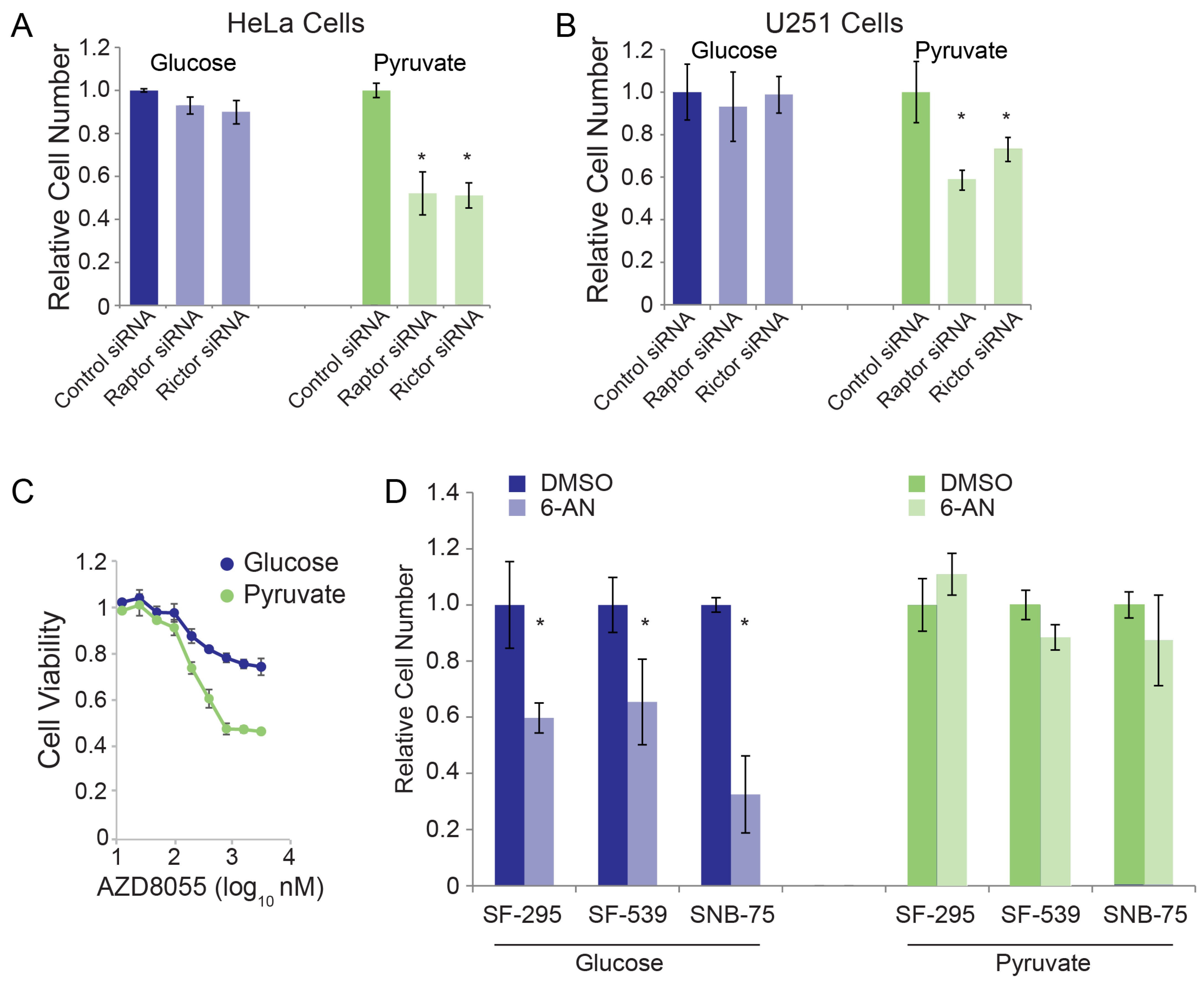
3.2. Validation of RNAi Screen Hits
3.3. Identifying Altered Transcript & Genetic Profiles in Cancers with High or Low Glycolytic Profiles
3.4. Assessing Biological Themes in Genes Clustering Variably Glycolytic Cancers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic Reprogramming and Cancer Progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Kumar, R.; Mishra, A.; Gautam, P.; Feroz, Z.; Vijayaraghavalu, S.; Likos, E.M.; Shukla, G.C.; Kumar, M. Metabolic Pathways, Enzymes, and Metabolites: Opportunities in Cancer Therapy. Cancers 2022, 14, 5268. [Google Scholar] [CrossRef] [PubMed]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.K.; Jang, H.G.; Jha, A.K.; et al. Functional Genomics Reveal That the Serine Synthesis Pathway Is Essential in Breast Cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Chacon-Barahona, J.A.; Salladay-Perez, I.A.; Lanning, N.J. Lung Adenocarcinoma Transcriptomic Analysis Predicts Adenylate Kinase Signatures Contributing to Tumor Progression and Negative Patient Prognosis. Metabolites 2021, 11, 859. [Google Scholar] [CrossRef]
- Xie, Y.; Ma, S.; Tong, M. Metabolic Plasticity of Cancer Stem Cells in Response to Microenvironmental Cues. Cancers 2022, 14, 5345. [Google Scholar] [CrossRef]
- Rossi, M.; Altea-Manzano, P.; Demicco, M.; Doglioni, G.; Bornes, L.; Fukano, M.; Vandekeere, A.; Cuadros, A.M.; Fernández-García, J.; Riera-Domingo, C.; et al. PHGDH Heterogeneity Potentiates Cancer Cell Dissemination and Metastasis. Nature 2022, 605, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Lien, E.C.; Westermark, A.M.; Zhang, Y.; Yuan, C.; Li, Z.; Lau, A.N.; Sapp, K.M.; Wolpin, B.M.; vander Heiden, M.G. Low Glycaemic Diets Alter Lipid Metabolism to Influence Tumour Growth. Nature 2021, 599, 302–307. [Google Scholar] [CrossRef]
- Lanning, N.J.; Looyenga, B.D.; Kauffman, A.L.; Niemi, N.M.; Sudderth, J.; DeBerardinis, R.J.; MacKeigan, J.P. A Mitochondrial RNAi Screen Defines Cellular Bioenergetic Determinants and Identifies an Adenylate Kinase as a Key Regulator of ATP Levels. Cell Rep. 2014, 7, 907–917. [Google Scholar] [CrossRef]
- Feng, J.; Li, J.; Wu, L.; Yu, Q.; Ji, J.; Wu, J.; Dai, W.; Guo, C. Emerging Roles and the Regulation of Aerobic Glycolysis in Hepatocellular Carcinoma. J. Exp. Clin. Cancer Res. 2020, 39, 126. [Google Scholar] [CrossRef]
- Bonatelli, M.; Silva, E.C.A.; Cárcano, F.M.; Zaia, M.G.; Lopes, L.F.; Scapulatempo-Neto, C.; Pinheiro, C. The Warburg Effect Is Associated with Tumor Aggressiveness in Testicular Germ Cell Tumors. Front. Endocrinol. 2019, 10, 417. [Google Scholar] [CrossRef]
- Vlassenko, A.G.; McConathy, J.; Couture, L.E.; Su, Y.; Massoumzadeh, P.; Leeds, H.S.; Chicoine, M.R.; Tran, D.D.; Huang, J.; Dahiya, S.; et al. Aerobic Glycolysis as a Marker of Tumor Aggressiveness: Preliminary Data in High Grade Human Brain Tumors. Dis. Markers 2015, 2015, 874904. [Google Scholar] [CrossRef]
- Alfarouk, K.O.; Ahmed, S.B.M.; Elliott, R.L.; Benoit, A.; Alqahtani, S.S.; Ibrahim, M.E.; Bashir, A.H.H.; Alhoufie, S.T.S.; Elhassan, G.O.; Wales, C.C.; et al. The Pentose Phosphate Pathway Dynamics in Cancer and Its Dependency on Intracellular pH. Metabolites 2020, 10, 285. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. We Need to Talk about the Warburg Effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef]
- Shestov, A.A.; Liu, X.; Ser, Z.; Cluntun, A.A.; Hung, Y.P.; Huang, L.; Kim, D.; Le, A.; Yellen, G.; Albeck, J.G.; et al. Quantitative Determinants of Aerobic Glycolysis Identify Flux through the Enzyme GAPDH as a Limiting Step. eLife 2014, 3, e03342. [Google Scholar] [CrossRef]
- Slavov, N.; Budnik, B.A.; Schwab, D.; Airoldi, E.M.; van Oudenaarden, A. Constant Growth Rate Can Be Supported by Decreasing Energy Flux and Increasing Aerobic Glycolysis. Cell Rep. 2014, 7, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, T.; Schuster, S.; Bonhoeffer, S. Cooperation and Competition in the Evolution of ATP-Producing Pathways. Science 2001, 292, 504–507. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does It Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Wei, J.; Huang, K.; Chen, Z.; Hu, M.; Bai, Y.; Lin, S.; Du, H. Characterization of Glycolysis-Associated Molecules in the Tumor Microenvironment Revealed by Pan-Cancer Tissues and Lung Cancer Single Cell Data. Cancers 2020, 12, 1788. [Google Scholar] [CrossRef]
- Mitchell, K.G.; Amini, B.; Wang, Y.; Carter, B.W.; Godoy, M.C.B.; Parra, E.R.; Behrens, C.; Villalobos, P.; Reuben, A.; Lee, J.J.; et al. 18 F-Fluorodeoxyglucose Positron Emission Tomography Correlates with Tumor Immunometabolic Phenotypes in Resected Lung Cancer. Cancer Immunol. Immunother. 2020, 69, 1519–1534. [Google Scholar] [CrossRef]
- Mathews, E.H.; Liebenberg, L.; Pelzer, R. High-Glycolytic Cancers and Their Interplay with the Body’s Glucose Demand and Supply Cycle. Med. Hypotheses 2011, 76, 157–165. [Google Scholar] [CrossRef]
- Graham, N.A.; Minasyan, A.; Lomova, A.; Cass, A.; Balanis, N.G.; Friedman, M.; Chan, S.; Zhao, S.; Delgado, A.; Go, J.; et al. Recurrent Patterns of DNA Copy Number Alterations in Tumors Reflect Metabolic Selection Pressures. Mol. Syst. Biol. 2017, 13, 914. [Google Scholar] [CrossRef]
- MacKeigan, J.P.; Murphy, L.O.; Blenis, J. Sensitized RNAi Screen of Human Kinases and Phosphatases Identifies New Regulators of Apoptosis and Chemoresistance. Nat. Cell Biol. 2005, 7, 591–600. [Google Scholar] [CrossRef]
- Ghosh, A.; Barman, S. Application of Euclidean Distance Measurement and Principal Component Analysis for Gene Identification. Gene 2016, 583, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Jaskowiak, P.A.; Costa, I.G.; Campello, R.J.G.B. Clustering of RNA-Seq Samples: Comparison Study on Cancer Data. Methods 2018, 132, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Hastie, T.; Friedman, J.; Tibshirani, R. The Elements of Statistical Learning; Springer: New York, NY, USA, 2001. [Google Scholar] [CrossRef]
- Rousseeuw, P.J. Silhouettes: A Graphical Aid to the Interpretation and Validation of Cluster Analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Huang, J.Z. Sparse Principal Component Analysis via Regularized Low Rank Matrix Approximation. J. Multivar. Anal. 2008, 99, 1015–1034. [Google Scholar] [CrossRef]
- Rohart, F.; Gautier, B.; Singh, A.; Lê Cao, K.A. MixOmics: An R Package for ‘omics Feature Selection and Multiple Data Integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 Facilitates Sensitive and Confident Localization of the Targets of Focal Somatic Copy-Number Alteration in Human Cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The Landscape of Somatic Copy-Number Alteration across Human Cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex Heatmaps Reveal Patterns and Correlations in Multidimensional Genomic Data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Ge, S.X.; Jung, D.; Jung, D.; Yao, R. ShinyGO: A Graphical Gene-Set Enrichment Tool for Animals and Plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]
- Castelli, S.; Ciccarone, F.; Tavian, D.; Ciriolo, M.R. ROS-Dependent HIF1α Activation under Forced Lipid Catabolism Entails Glycolysis and Mitophagy as Mediators of Higher Proliferation Rate in Cervical Cancer Cells. J. Exp. Clin. Cancer Res. 2021, 40, 94. [Google Scholar] [CrossRef]
- Shiratori, R.; Furuichi, K.; Yamaguchi, M.; Miyazaki, N.; Aoki, H.; Chibana, H.; Ito, K.; Aoki, S. Glycolytic Suppression Dramatically Changes the Intracellular Metabolic Profile of Multiple Cancer Cell Lines in a Mitochondrial Metabolism-Dependent Manner. Sci. Rep. 2019, 9, 18699. [Google Scholar] [CrossRef]
- Labuschagne, C.F.; Cheung, E.C.; Blagih, J.; Domart, M.-C.; Vousden Correspondence, K.H.; Vousden, K.H. Cell Clustering Promotes a Metabolic Switch That Supports Metastatic Colonization In Brief Cell Metabolism Article Cell Clustering Promotes a Metabolic Switch That Supports Metastatic Colonization. Cell Metab. 2019, 30, 720–734. [Google Scholar] [CrossRef] [PubMed]
- Gohil, V.M.; Sheth, S.A.; Nilsson, R.; Wojtovich, A.P.; Lee, J.H.; Perocchi, F.; Chen, W.; Clish, C.B.; Ayata, C.; Brookes, P.S.; et al. Nutrient-Sensitized Screening for Drugs That Shift Energy Metabolism from Mitochondrial Respiration to Glycolysis. Nat. Biotechnol. 2010, 28, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Christofk, H.R.; vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 Splice Isoform of Pyruvate Kinase Is Important for Cancer Metabolism and Tumour Growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, S.; Boschek, C.B.; Hugo, F.; Eigenbrodt, E. Pyruvate Kinase Type M2 and Its Role in Tumor Growth and Spreading. Semin. Cancer Biol. 2005, 15, 300–308. [Google Scholar] [CrossRef]
- Yuneva, M.O.; Fan, T.W.M.; Allen, T.D.; Higashi, R.M.; Ferraris, D.v.; Tsukamoto, T.; Matés, J.M.; Alonso, F.J.; Wang, C.; Seo, Y.; et al. The Metabolic Profile of Tumors Depends on Both the Responsible Genetic Lesion and Tissue Type. Cell Metab. 2012, 15, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Lanning, N.J.; Castle, J.P.; Singh, S.J.; Leon, A.N.; Tovar, E.A.; Sanghera, A.; MacKeigan, J.P.; Filipp, F.V.; Graveel, C.R. Metabolic Profiling of Triple-Negative Breast Cancer Cells Reveals Metabolic Vulnerabilities. Cancer Metab. 2017, 5, 6. [Google Scholar] [CrossRef]
- Magalhães, R.S.S.; Boechat, F.C.; Brasil, A.A.; Neto, J.R.M.; Ribeiro, G.D.; Paranhos, L.H.; Neves de Souza, N.; Vieira, T.; Outeiro, T.F.; Neves, B.C.; et al. Hexokinase 2: The Preferential Target of Trehalose-6-Phosphate over Hexokinase 1. J. Cell. Biochem. 2022, 123, 1808–1816. [Google Scholar] [CrossRef] [PubMed]
- Shibata, S.; Sogabe, S.; Miwa, M.; Fujimoto, T.; Takakura, N.; Naotsuka, A.; Kitamura, S.; Kawamoto, T.; Soga, T. Identification of the First Highly Selective Inhibitor of Human Lactate Dehydrogenase B. Sci. Rep. 2021, 11, 21353. [Google Scholar] [CrossRef] [PubMed]
- Heydasch, U.; Kessler, R.; Warnke, J.P.; Eschrich, K.; Scholz, N.; Bigl, M. Functional Diversity of PFKFB3 Splice Variants in Glioblastomas. PLoS ONE 2021, 16, e0241092. [Google Scholar] [CrossRef]
- Cadoux-Hudson, T.; Schofield, C.J.; McCullagh, J.S.O. Isocitrate Dehydrogenase Gene Variants in Cancer and Their Clinical Significance. Biochem. Soc. Trans. 2021, 49, 2561–2572. [Google Scholar] [CrossRef]
- Kobak, D.; Berens, P. The Art of Using T-SNE for Single-Cell Transcriptomics. Nat. Commun. 2019, 10, 5416. [Google Scholar] [CrossRef]
- Becht, E.; McInnes, L.; Healy, J.; Dutertre, C.A.; Kwok, I.W.H.; Ng, L.G.; Ginhoux, F.; Newell, E.W. Dimensionality Reduction for Visualizing Single-Cell Data Using UMAP. Nat. Biotechnol. 2018, 37, 38–47. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chacon-Barahona, J.A.; MacKeigan, J.P.; Lanning, N.J. Unique Metabolic Contexts Sensitize Cancer Cells and Discriminate between Glycolytic Tumor Types. Cancers 2023, 15, 1158. https://doi.org/10.3390/cancers15041158
Chacon-Barahona JA, MacKeigan JP, Lanning NJ. Unique Metabolic Contexts Sensitize Cancer Cells and Discriminate between Glycolytic Tumor Types. Cancers. 2023; 15(4):1158. https://doi.org/10.3390/cancers15041158
Chicago/Turabian StyleChacon-Barahona, Jonathan A., Jeffrey P. MacKeigan, and Nathan J. Lanning. 2023. "Unique Metabolic Contexts Sensitize Cancer Cells and Discriminate between Glycolytic Tumor Types" Cancers 15, no. 4: 1158. https://doi.org/10.3390/cancers15041158
APA StyleChacon-Barahona, J. A., MacKeigan, J. P., & Lanning, N. J. (2023). Unique Metabolic Contexts Sensitize Cancer Cells and Discriminate between Glycolytic Tumor Types. Cancers, 15(4), 1158. https://doi.org/10.3390/cancers15041158