

Neurofibromatosis Type 1: Pediatric Aspects and Review of Genotype–Phenotype Correlations
 , ,
, ,  ,
,  and
and
Abstract
:Simple Summary
Abstract
1. Background
2. Clinical Features of NF1 in Children and Adolescents
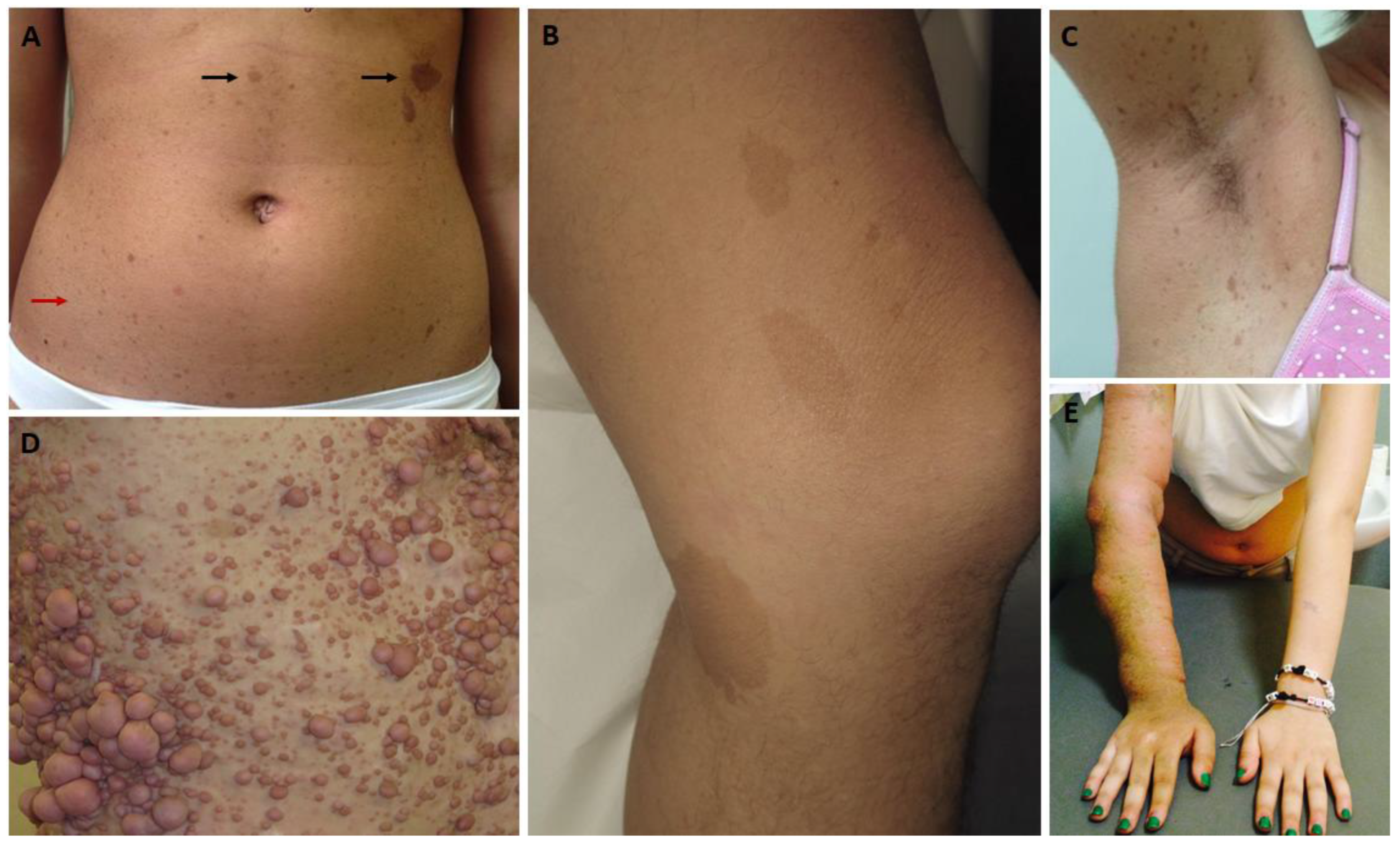
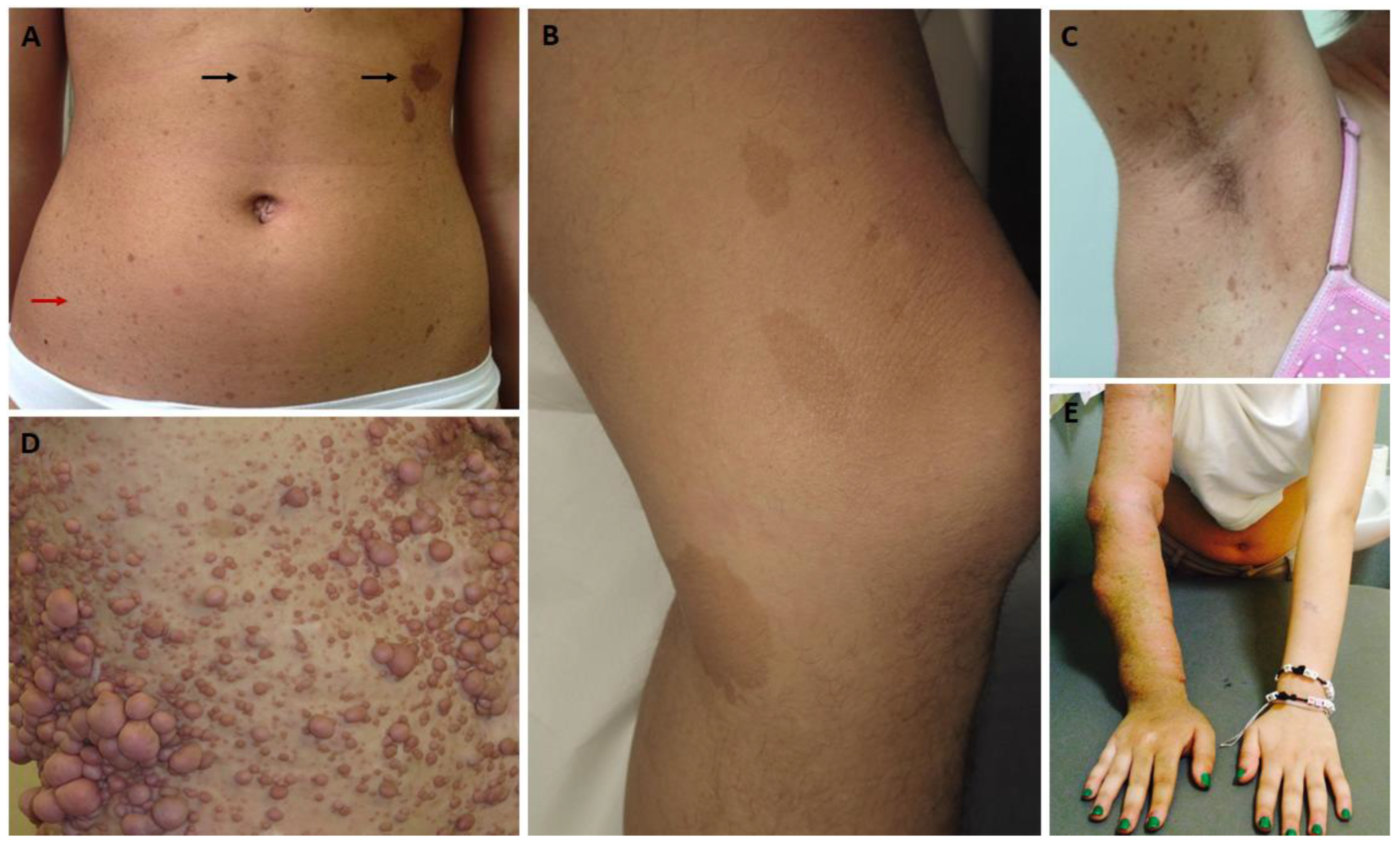
2.1. Cutaneous Findings
2.2. Eye Findings
2.3. Tumors
2.4. Other Complications Affecting the Nervous System
2.5. Vascular Diseases
2.6. Cardiovascular Involvement
2.7. Skeletal Manifestations
2.8. Miscellaneous
3. Follow-Up and Management
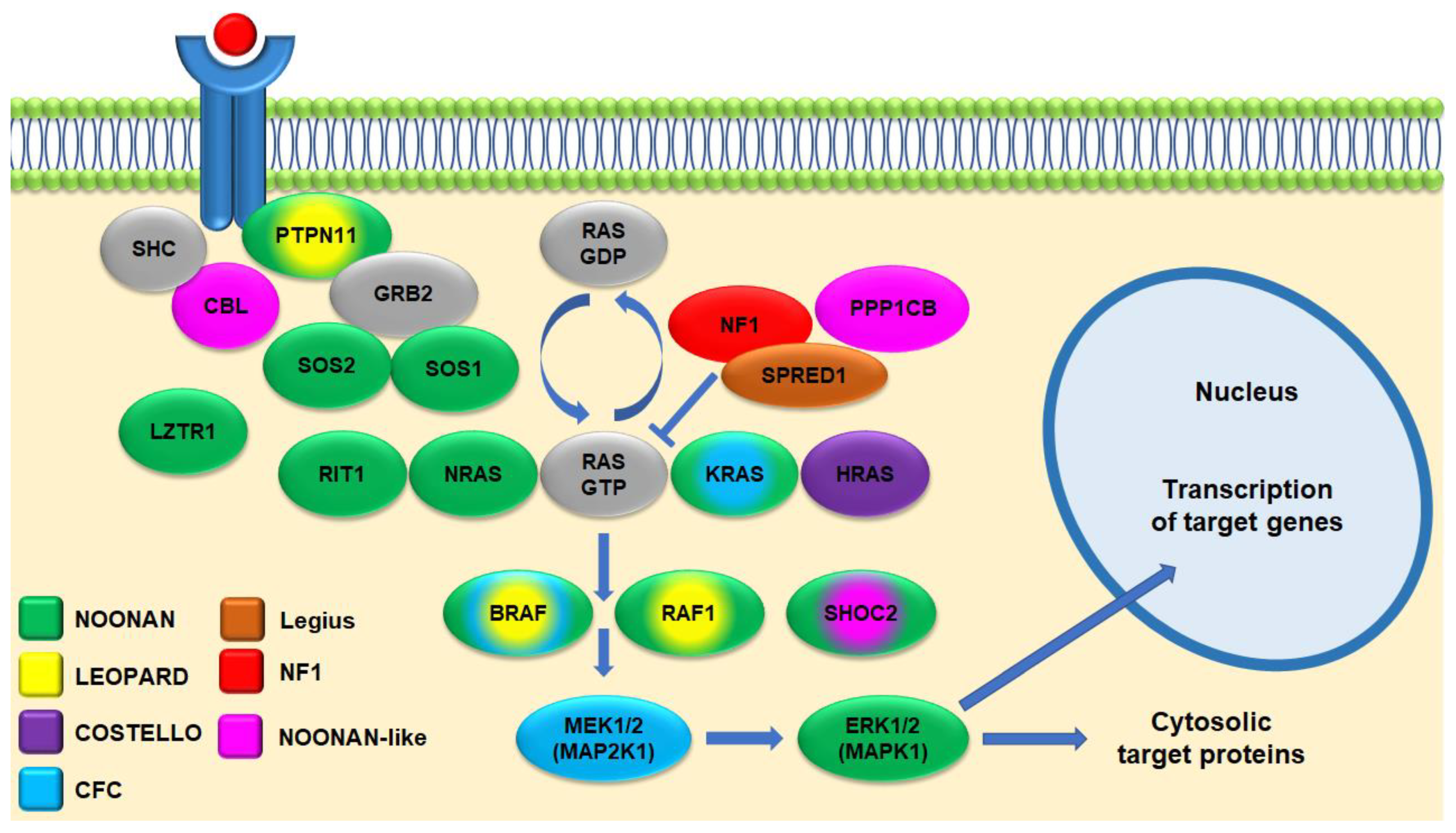
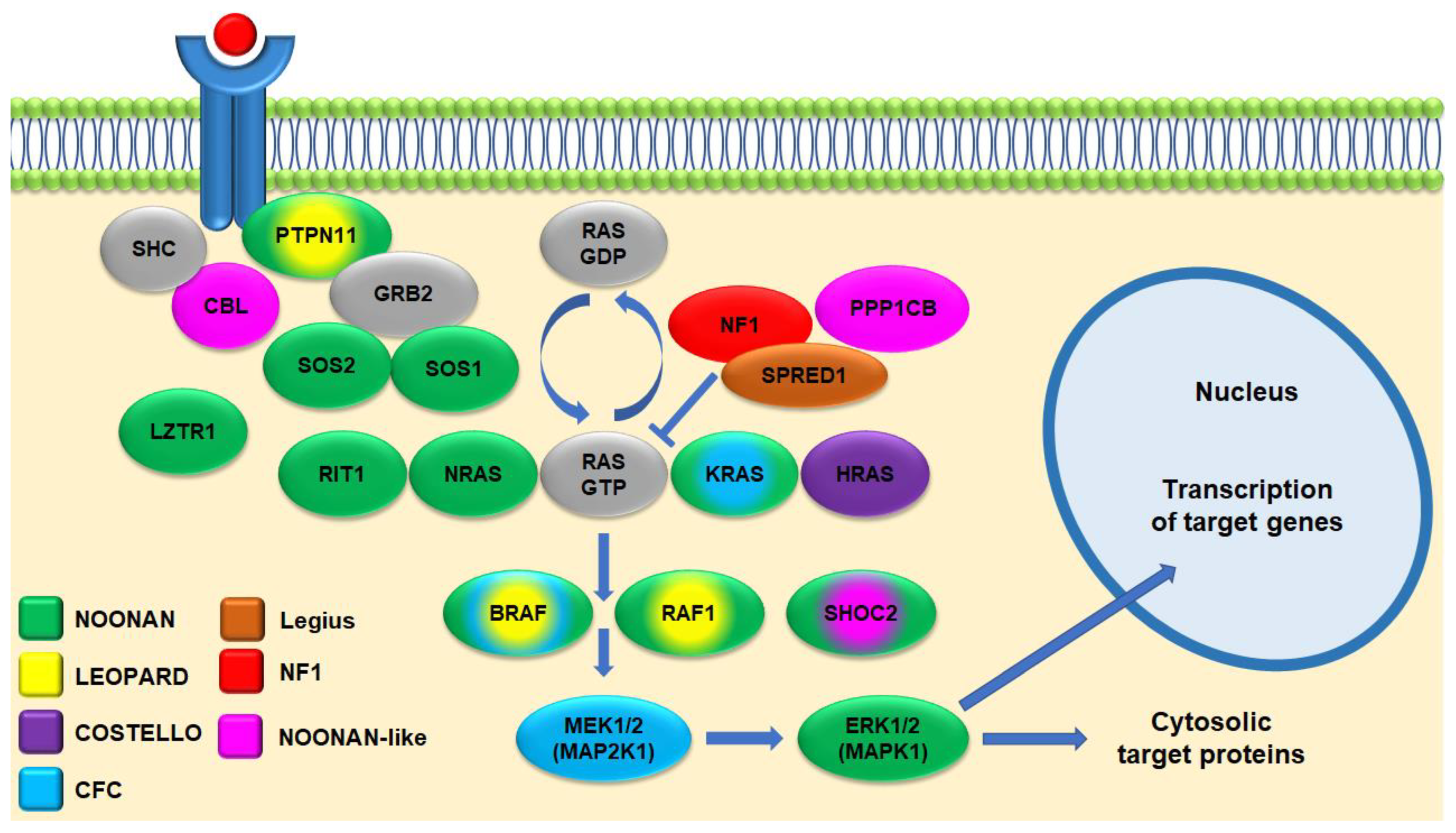
4. Genetics
4.1. Genotype–Phenotype Correlations
4.2. Association with Other Genetic Disorders
4.3. Molecular Diagnosis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Clementi, M.; Barbujani, G.; Turolla, L.; Tenconi, R. Neurofibromatosis-1: A maximum likelihood estimation of mutation rate. Hum. Genet. 1990, 84, 116–118. [Google Scholar] [CrossRef]
- Friedman, J.M. Epidemiology of neurofibromatosis type 1. Am. J. Med. Genet. 1999, 89, 1–6. [Google Scholar] [CrossRef]
- Kallionpaa, R.A.; Uusitalo, E.; Leppavirta, J.; Poyhonen, M.; Peltonen, S.; Peltonen, J. Prevalence of neurofibromatosis type 1 in the Finnish population. Genet. Med. 2018, 20, 1082–1086. [Google Scholar] [CrossRef]
- Lammert, M.; Friedman, J.M.; Kluwe, L.; Mautner, V.F. Prevalence of neurofibromatosis 1 in German children at elementary school enrollment. Arch. Dermatol. 2005, 141, 71–74. [Google Scholar] [CrossRef]
- Friedman, J.M. Neurofibromatosis 1. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Tidyman, W.E.; Rauen, K.A. The RASopathies: Developmental syndromes of Ras/MAPK pathway dysregulation. Curr. Opin. Genet. Dev. 2009, 19, 230–236. [Google Scholar] [CrossRef]
- Rauen, K.A. The RASopathies. Annu. Rev. Genom. Hum. Genet. 2013, 14, 355–369. [Google Scholar] [CrossRef]
- Santoro, C.; Giugliano, T.; Melone, M.A.B.; Cirillo, M.; Schettino, C.; Bernardo, P.; Cirillo, G.; Perrotta, S.; Piluso, G. Multiple spinal nerve enlargement and SOS1 mutation: Further evidence of overlap between neurofibromatosis type 1 and Noonan phenotype. Clin. Genet. 2018, 93, 138–143. [Google Scholar] [CrossRef]
- Santoro, C.; Pacileo, G.; Limongelli, G.; Scianguetta, S.; Giugliano, T.; Piluso, G.; Ragione, F.D.; Cirillo, M.; Mirone, G.; Perrotta, S. LEOPARD syndrome: Clinical dilemmas in differential diagnosis of RASopathies. BMC Med. Genet. 2014, 15, 44. [Google Scholar] [CrossRef]
- National Institutes of Health. National Institutes of Health Consensus Development Conference Statement: Neurofibromatosis. Neurofibromatosis 1988, 1, 172–178. [Google Scholar]
- Legius, E.; Messiaen, L.; Wolkenstein, P.; Pancza, P.; Avery, R.A.; Berman, Y.; Blakeley, J.; Babovic-Vuksanovic, D.; Cunha, K.S.; Ferner, R.; et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: An international consensus recommendation. Genet. Med. 2021, 23, 1506–1513. [Google Scholar] [CrossRef]
- Brems, H.; Chmara, M.; Sahbatou, M.; Denayer, E.; Taniguchi, K.; Kato, R.; Somers, R.; Messiaen, L.; De Schepper, S.; Fryns, J.P.; et al. Germline loss-of-function mutations in SPRED1 cause a neurofibromatosis 1-like phenotype. Nat. Genet. 2007, 39, 1120–1126. [Google Scholar] [CrossRef]
- Bergqvist, C.; Servy, A.; Valeyrie-Allanore, L.; Ferkal, S.; Combemale, P.; Wolkenstein, P.; Network, N.F.F. Neurofibromatosis 1 French national guidelines based on an extensive literature review since 1966. Orphanet J. Rare Dis. 2020, 15, 37. [Google Scholar] [CrossRef]
- Lalor, L.; Davies, O.M.T.; Basel, D.; Siegel, D.H. Cafe au lait spots: When and how to pursue their genetic origins. Clin. Dermatol. 2020, 38, 421–431. [Google Scholar] [CrossRef]
- Ozarslan, B.; Russo, T.; Argenziano, G.; Santoro, C.; Piccolo, V. Cutaneous Findings in Neurofibromatosis Type 1. Cancers 2021, 13, 463. [Google Scholar] [CrossRef]
- Miraglia, E.; Moliterni, E.; Iacovino, C.; Roberti, V.; Laghi, A.; Moramarco, A.; Giustini, S. Cutaneous manifestations in neurofibromatosis type 1. La Clin. Ter. 2020, 171, e371–e377. [Google Scholar] [CrossRef]
- Marque, M.; Roubertie, A.; Jaussent, A.; Carneiro, M.; Meunier, L.; Guillot, B.; Pinson, L.; Pinson, S.; Bessis, D. Nevus anemicus in neurofibromatosis type 1: A potential new diagnostic criterion. J. Am. Acad. Dermatol. 2013, 69, 768–775. [Google Scholar] [CrossRef]
- Vaassen, P.; Rosenbaum, T. Nevus Anemicus As an Additional Diagnostic Marker of Neurofibromatosis Type 1 in Childhood. Neuropediatrics 2016, 47, 190–193. [Google Scholar] [CrossRef]
- Carton, C.; Evans, D.G.; Blanco, I.; Friedrich, R.E.; Ferner, R.E.; Farschtschi, S.; Salvador, H.; Azizi, A.A.; Mautner, V.; Rohl, C.; et al. ERN GENTURIS tumour surveillance guidelines for individuals with neurofibromatosis type 1. EClinicalMedicine 2023, 56, 101818. [Google Scholar] [CrossRef]
- Liy-Wong, C.; Mohammed, J.; Carleton, A.; Pope, E.; Parkin, P.; Lara-Corrales, I. The relationship between neurofibromatosis type 1, juvenile xanthogranuloma, and malignancy: A retrospective case-control study. J. Am. Acad. Dermatol. 2017, 76, 1084–1087. [Google Scholar] [CrossRef]
- Senthilkumar, V.A.; Tripathy, K. Lisch Nodules. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Cosmo, E.; Frizziero, L.; Miglionico, G.; De Biasi, C.S.; Bruno, M.; Trevisson, E.; Gabbiato, I.; Midena, G.; Parrozzani, R. Choroidal Abnormalities in Pediatric NF1: A Cohort Natural History Study. Cancers 2022, 14, 1423. [Google Scholar] [CrossRef]
- Viola, F.; Villani, E.; Natacci, F.; Selicorni, A.; Melloni, G.; Vezzola, D.; Barteselli, G.; Mapelli, C.; Pirondini, C.; Ratiglia, R. Choroidal abnormalities detected by near-infrared reflectance imaging as a new diagnostic criterion for neurofibromatosis 1. Ophthalmology 2012, 119, 369–375. [Google Scholar] [CrossRef]
- Parrozzani, R.; Clementi, M.; Frizziero, L.; Miglionico, G.; Perrini, P.; Cavarzeran, F.; Kotsafti, O.; Comacchio, F.; Trevisson, E.; Convento, E.; et al. In Vivo Detection of Choroidal Abnormalities Related to NF1: Feasibility and Comparison With Standard NIH Diagnostic Criteria in Pediatric Patients. Investig. Ophthalmol. Vis. Sci. 2015, 56, 6036–6042. [Google Scholar] [CrossRef]
- Vagge, A.; Camicione, P.; Capris, C.; Sburlati, C.; Panarello, S.; Calevo, M.G.; Traverso, C.E.; Capris, P. Choroidal abnormalities in neurofibromatosis type 1 detected by near-infrared reflectance imaging in paediatric population. Acta Ophthalmol. 2015, 93, e667–e671. [Google Scholar] [CrossRef]
- Yasunari, T.; Shiraki, K.; Hattori, H.; Miki, T. Frequency of choroidal abnormalities in neurofibromatosis type 1. Lancet 2000, 356, 988–992. [Google Scholar] [CrossRef]
- Campen, C.J.; Gutmann, D.H. Optic Pathway Gliomas in Neurofibromatosis Type 1. J. Child Neurol. 2018, 33, 73–81. [Google Scholar] [CrossRef]
- Shofty, B.; Ben Sira, L.; Constantini, S. Neurofibromatosis 1-associated optic pathway gliomas. Childs Nerv. Syst. 2020, 36, 2351–2361. [Google Scholar] [CrossRef]
- Barkovich, M.J.; Xu, D.; Desikan, R.S.; Williams, C.; Barkovich, A.J. Pediatric neuro MRI: Tricks to minimize sedation. Pediatr. Radiol. 2018, 48, 50–55. [Google Scholar] [CrossRef]
- Azizi, A.A.; Walker, D.A.; Liu, J.F.; Sehested, A.; Jaspan, T.; Pemp, B.; Simmons, I.; Ferner, R.; Grill, J.; Hargrave, D.; et al. NF1 optic pathway glioma. Analysing risk factors for visual outcome and indications to treat. Neuro Oncol. 2020, 23, 100–111. [Google Scholar] [CrossRef]
- Thirunavu, V.M.; Mohammad, L.M.; Kandula, V.; Beestrum, M.; Lam, S.K. Vision Outcomes for Pediatric Patients With Optic Pathway Gliomas Associated With Neurofibromatosis Type I: A Systematic Review of the Clinical Evidence. J. Pediatr. Hematol. Oncol. 2021, 43, 135–143. [Google Scholar] [CrossRef]
- Santoro, C.; Picariello, S.; Palladino, F.; Spennato, P.; Melis, D.; Roth, J.; Cirillo, M.; Quaglietta, L.; D′Amico, A.; Gaudino, G.; et al. Retrospective Multicentric Study on Non-Optic CNS Tumors in Children and Adolescents with Neurofibromatosis Type 1. Cancers 2020, 12, 1426. [Google Scholar] [CrossRef]
- Cambiaso, P.; Galassi, S.; Palmiero, M.; Mastronuzzi, A.; Del Bufalo, F.; Capolino, R.; Cacchione, A.; Buonuomo, P.S.; Gonfiantini, M.V.; Bartuli, A.; et al. Growth hormone excess in children with neurofibromatosis type-1 and optic glioma. Am. J. Med. Genet. A 2017, 173, 2353–2358. [Google Scholar] [CrossRef]
- Hannah-Shmouni, F.; Stratakis, C.A. Growth hormone excess in neurofibromatosis 1. Genet. Med. 2019, 21, 1254–1255. [Google Scholar] [CrossRef] [PubMed]
- Santoro, C.; Perrotta, S.; Picariello, S.; Scilipoti, M.; Cirillo, M.; Quaglietta, L.; Cinalli, G.; Cioffi, D.; Di Iorgi, N.; Maghnie, M.; et al. Pretreatment Endocrine Disorders Due to Optic Pathway Gliomas in Pediatric Neurofibromatosis Type 1: Multicenter Study. J. Clin. Endocrinol. Metab. 2020, 105, e2214–e2221. [Google Scholar] [CrossRef]
- Guillamo, J.S.; Creange, A.; Kalifa, C.; Grill, J.; Rodriguez, D.; Doz, F.; Barbarot, S.; Zerah, M.; Sanson, M.; Bastuji-Garin, S.; et al. Prognostic factors of CNS tumours in Neurofibromatosis 1 (NF1): A retrospective study of 104 patients. Brain 2003, 126, 152–160. [Google Scholar] [CrossRef]
- Landry, J.P.; Schertz, K.L.; Chiang, Y.J.; Bhalla, A.D.; Yi, M.; Keung, E.Z.; Scally, C.P.; Feig, B.W.; Hunt, K.K.; Roland, C.L.; et al. Comparison of Cancer Prevalence in Patients With Neurofibromatosis Type 1 at an Academic Cancer Center vs in the General Population From 1985 to 2020. JAMA Netw. Open 2021, 4, e210945. [Google Scholar] [CrossRef]
- Shofty, B.; Barzilai, O.; Khashan, M.; Lidar, Z.; Constantini, S. Spinal manifestations of Neurofibromatosis type 1. Childs Nerv. Syst. 2020, 36, 2401–2408. [Google Scholar] [CrossRef]
- Khajavi, M.; Khoshsirat, S.; Ahangarnazari, L.; Majdinasab, N. A brief report of plexiform neurofibroma. Curr. Probl. Cancer 2018, 42, 256–260. [Google Scholar] [CrossRef]
- Waggoner, D.J.; Towbin, J.; Gottesman, G.; Gutmann, D.H. Clinic-based study of plexiform neurofibromas in neurofibromatosis 1. Am. J. Med. Genet. 2000, 92, 132–135. [Google Scholar] [CrossRef]
- Klesse, L.J.; Jordan, J.T.; Radtke, H.B.; Rosser, T.; Schorry, E.; Ullrich, N.; Viskochil, D.; Knight, P.; Plotkin, S.R.; Yohay, K. The Use of MEK Inhibitors in Neurofibromatosis Type 1-Associated Tumors and Management of Toxicities. Oncologist 2020, 25, e1109–e1116. [Google Scholar] [CrossRef]
- de Blank, P.M.K.; Gross, A.M.; Akshintala, S.; Blakeley, J.O.; Bollag, G.; Cannon, A.; Dombi, E.; Fangusaro, J.; Gelb, B.D.; Hargrave, D.; et al. MEK inhibitors for neurofibromatosis type 1 manifestations: Clinical evidence and consensus. Neuro Oncol. 2022, 24, 1845–1856. [Google Scholar] [CrossRef]
- Evans, D.G.; Baser, M.E.; McGaughran, J.; Sharif, S.; Howard, E.; Moran, A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J. Med. Genet. 2002, 39, 311–314. [Google Scholar] [CrossRef] [PubMed]
- King, A.A.; Debaun, M.R.; Riccardi, V.M.; Gutmann, D.H. Malignant peripheral nerve sheath tumors in neurofibromatosis 1. Am. J. Med. Genet. 2000, 93, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Huson, S.M.; Compston, D.A.; Clark, P.; Harper, P.S. A genetic study of von Recklinghausen neurofibromatosis in south east Wales. I. Prevalence, fitness, mutation rate, and effect of parental transmission on severity. J. Med. Genet. 1989, 26, 704–711. [Google Scholar] [CrossRef] [PubMed]
- Durbin, A.D.; Ki, D.H.; He, S.; Look, A.T. Malignant Peripheral Nerve Sheath Tumors. Adv. Exp. Med. Biol. 2016, 916, 495–530. [Google Scholar] [CrossRef]
- Farid, M.; Demicco, E.G.; Garcia, R.; Ahn, L.; Merola, P.R.; Cioffi, A.; Maki, R.G. Malignant peripheral nerve sheath tumors. Oncologist 2014, 19, 193–201. [Google Scholar] [CrossRef]
- Ducatman, B.S.; Scheithauer, B.W.; Piepgras, D.G.; Reiman, H.M.; Ilstrup, D.M. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer 1986, 57, 2006–2021. [Google Scholar] [CrossRef]
- Ferraz-Filho, J.R.; Jose da Rocha, A.; Muniz, M.P.; Souza, A.S.; Goloni-Bertollo, E.M.; Pavarino-Bertelli, E.C. Unidentified bright objects in neurofibromatosis type 1: Conventional MRI in the follow-up and correlation of microstructural lesions on diffusion tensor images. Eur. J. Paediatr. Neurol. 2012, 16, 42–47. [Google Scholar] [CrossRef]
- Lopes Ferraz Filho, J.R.; Munis, M.P.; Soares Souza, A.; Sanches, R.A.; Goloni-Bertollo, E.M.; Pavarino-Bertelli, E.C. Unidentified bright objects on brain MRI in children as a diagnostic criterion for neurofibromatosis type 1. Pediatr. Radiol. 2008, 38, 305–310. [Google Scholar] [CrossRef]
- D′Amico, A.; Mazio, F.; Ugga, L.; Cuocolo, R.; Cirillo, M.; Santoro, C.; Perrotta, S.; Melis, D.; Brunetti, A. Medullary unidentified bright objects in Neurofibromatosis type 1: A case series. BMC Pediatr. 2018, 18, 91. [Google Scholar] [CrossRef]
- Al-Farsi, F.A.H.; Al-Alyani, O.B.S.; Al-Kumzari, A.; Al-Saadi, T. Systemic Review and Meta-analysis of the Intellectual Integrity of Children with Neurofibromatosis Type 1. World Neurosurg. 2022, 157, 69–74. [Google Scholar] [CrossRef]
- Cutting, L.E.; Levine, T.M. Cognitive profile of children with neurofibromatosis and reading disabilities. Child Neuropsychol. 2010, 16, 417–432. [Google Scholar] [CrossRef] [PubMed]
- Torres Nupan, M.M.; Velez Van Meerbeke, A.; Lopez Cabra, C.A.; Herrera Gomez, P.M. Cognitive and Behavioral Disorders in Children with Neurofibromatosis Type 1. Front. Pediatr. 2017, 5, 227. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.C.; Gutmann, D.H.; Morris, S.M. Neurodevelopmental disorders in children with neurofibromatosis type 1. Dev. Med. Child Neurol. 2017, 59, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Lehtonen, A.; Howie, E.; Trump, D.; Huson, S.M. Behaviour in children with neurofibromatosis type 1: Cognition, executive function, attention, emotion, and social competence. Dev. Med. Child Neurol. 2013, 55, 111–125. [Google Scholar] [CrossRef]
- Bernardo, P.; Cinalli, G.; Santoro, C. Epilepsy in NF1: A systematic review of the literature. Childs Nerv. Syst. 2020, 36, 2333–2350. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, P.; Santoro, C.; Rubino, A.; Mirone, G.; Cinalli, G. Epilepsy surgery in neurofibromatosis type 1: An overlooked therapeutic approach. Childs Nerv. Syst. 2020, 36, 2909–2910. [Google Scholar] [CrossRef]
- Nix, J.S.; Blakeley, J.; Rodriguez, F.J. An update on the central nervous system manifestations of neurofibromatosis type 1. Acta Neuropathol. 2020, 139, 625–641. [Google Scholar] [CrossRef]
- Roth, J.; Constantini, S.; Cinalli, G. Neurofibromatosis type 1-related hydrocephalus: Causes and treatment considerations. Childs Nerv. Syst. 2020, 36, 2385–2390. [Google Scholar] [CrossRef]
- Roth, J.; Ber, R.; Wisoff, J.H.; Hidalgo, E.T.; Limbrick, D.D.; Berger, D.S.; Thomale, U.W.; Schulz, M.; Cinalli, G.; Santoro, C.; et al. Endoscopic Third Ventriculostomy in Patients with Neurofibromatosis Type 1: A Multicenter International Experience. World Neurosurg. 2017, 107, 623–629. [Google Scholar] [CrossRef]
- Pozetti, M.; Belsuzarri, T.A.; Belsuzarri, N.C.; Seixas, N.B.; Araujo, J.F. Neurofibromatosis type 1 and Chiari type 1 malformation: A case report and literature review of a rare association. Surg. Neurol. Int. 2016, 7, S469–S472. [Google Scholar] [CrossRef]
- Raborn, J.; McCafferty, B.J.; Gunn, A.J.; Moawad, S.; Mahmoud, K.; Aal, A.K.A.; Saddekni, S. Endovascular Management of Neurofibromatosis Type I-Associated Vasculopathy: A Case Series and Brief Review of the Literature. Vasc. Endovasc. Surg. 2020, 54, 182–190. [Google Scholar] [CrossRef]
- Fossali, E.; Signorini, E.; Intermite, R.C.; Casalini, E.; Lovaria, A.; Maninetti, M.M.; Rossi, L.N. Renovascular disease and hypertension in children with neurofibromatosis. Pediatr. Nephrol. 2000, 14, 806–810. [Google Scholar] [CrossRef]
- Kaas, B.; Huisman, T.A.; Tekes, A.; Bergner, A.; Blakeley, J.O.; Jordan, L.C. Spectrum and prevalence of vasculopathy in pediatric neurofibromatosis type 1. J. Child Neurol. 2013, 28, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Lama, G.; Graziano, L.; Calabrese, E.; Grassia, C.; Rambaldi, P.F.; Cioce, F.; Tedesco, M.A.; Di Salvo, G.; Esposito-Salsano, M. Blood pressure and cardiovascular involvement in children with neurofibromatosis type1. Pediatr. Nephrol. 2004, 19, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Sivasubramanian, R.; Meyers, K.E. Hypertension in Children and Adolescents with Turner Syndrome (TS), Neurofibromatosis 1 (NF1), and Williams Syndrome (WS). Curr. Hypertens. Rep. 2021, 23, 18. [Google Scholar] [CrossRef] [PubMed]
- Duat-Rodriguez, A.; Carceller Lechon, F.; Lopez Pino, M.A.; Rodriguez Fernandez, C.; Gonzalez-Gutierrez-Solana, L. Neurofibromatosis type 1 associated with moyamoya syndrome in children. Pediatr. Neurol. 2014, 50, 96–98. [Google Scholar] [CrossRef] [PubMed]
- D′Amico, A.; Ugga, L.; Cocozza, S.; Giorgio, S.; Cicala, D.; Santoro, C.; Melis, D.; Cinalli, G.; Brunetti, A.; Pappata, S. Multimodal evaluation of the cerebrovascular reserve in Neurofibromatosis type 1 patients with Moyamoya syndrome. Neurol. Sci. 2020, 42, 655–663. [Google Scholar] [CrossRef]
- Scott, R.M.; Smith, E.R. Moyamoya disease and moyamoya syndrome. N. Engl. J. Med. 2009, 360, 1226–1237. [Google Scholar] [CrossRef] [PubMed]
- Mirone, G.; Cicala, D.; Meucci, C.; d′Amico, A.; Santoro, C.; Muto, M.; Cinalli, G. Multiple Burr-Hole Surgery for the Treatment of Moyamoya Disease and Quasi-Moyamoya Disease in Children: Preliminary Surgical and Imaging Results. World Neurosurg. 2019, 127, e843–e855. [Google Scholar] [CrossRef] [PubMed]
- Santoro, C.; Di Rocco, F.; Kossorotoff, M.; Zerah, M.; Boddaert, N.; Calmon, R.; Vidaud, D.; Cirillo, M.; Cinalli, G.; Mirone, G.; et al. Moyamoya syndrome in children with neurofibromatosis type 1: Italian-French experience. Am. J. Med. Genet. A 2017, 173, 1521–1530. [Google Scholar] [CrossRef]
- Phi, J.H.; Choi, J.W.; Seong, M.W.; Kim, T.; Moon, Y.J.; Lee, J.; Koh, E.J.; Ryu, S.K.; Kang, T.H.; Bang, J.S.; et al. Association between moyamoya syndrome and the RNF213 c.14576G>A variant in patients with neurofibromatosis Type 1. J. Neurosurg. Pediatr. 2016, 17, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Santoro, C.; Giugliano, T.; Kraemer, M.; Torella, A.; Schwitalla, J.C.; Cirillo, M.; Melis, D.; Berlit, P.; Nigro, V.; Perrotta, S.; et al. Whole exome sequencing identifies MRVI1 as a susceptibility gene for moyamoya syndrome in neurofibromatosis type 1. PLoS ONE 2018, 13, e0200446. [Google Scholar] [CrossRef] [PubMed]
- Santoro, C.; Mirone, G.; Zanobio, M.; Ranucci, G.; D′Amico, A.; Cicala, D.; Iascone, M.; Bernardo, P.; Piccolo, V.; Ronchi, A.; et al. Mystery(n) Phenotypic Presentation in Europeans: Report of Three Further Novel Missense RNF213 Variants Leading to Severe Syndromic Forms of Moyamoya Angiopathy and Literature Review. Int. J. Mol. Sci. 2022, 23, 8952. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.E.; Birch, P.H.; Korf, B.R.; Tenconi, R.; Niimura, M.; Poyhonen, M.; Armfield Uhas, K.; Sigorini, M.; Virdis, R.; Romano, C.; et al. Cardiovascular malformations and other cardiovascular abnormalities in neurofibromatosis 1. Am. J. Med. Genet. 2000, 95, 108–117. [Google Scholar] [CrossRef]
- Jutant, E.M.; Girerd, B.; Jais, X.; Savale, L.; O′Connell, C.; Perros, F.; Sitbon, O.; Humbert, M.; Montani, D. Pulmonary hypertension associated with neurofibromatosis type 1. Eur. Respir. Rev. 2018, 27, 180053. [Google Scholar] [CrossRef]
- Pinna, V.; Daniele, P.; Calcagni, G.; Mariniello, L.; Criscione, R.; Giardina, C.; Lepri, F.R.; Hozhabri, H.; Alberico, A.; Cavone, S.; et al. Prevalence, Type, and Molecular Spectrum of NF1 Mutations in Patients with Neurofibromatosis Type 1 and Congenital Heart Disease. Genes 2019, 10, 675. [Google Scholar] [CrossRef]
- Bird, A.R.; Kent, P.; Moores, P.P.; Elliott, T. Haemoglobin M-Hyde Park associated with polyagglutinable red blood cells in a South African family. Br. J. Haematol. 1988, 68, 459–464. [Google Scholar] [CrossRef]
- Virdis, R.; Balestrazzi, P.; Zampolli, M.; Donadio, A.; Street, M.; Lorenzetti, E. Hypertension in children with neurofibromatosis. J. Hum. Hypertens. 1994, 8, 395–397. [Google Scholar]
- Friedman, J.M.; Arbiser, J.; Epstein, J.A.; Gutmann, D.H.; Huot, S.J.; Lin, A.E.; McManus, B.; Korf, B.R. Cardiovascular disease in neurofibromatosis 1: Report of the NF1 Cardiovascular Task Force. Genet. Med. 2002, 4, 105–111. [Google Scholar] [CrossRef]
- Evans, D.G.; Howard, E.; Giblin, C.; Clancy, T.; Spencer, H.; Huson, S.M.; Lalloo, F. Birth incidence and prevalence of tumor-prone syndromes: Estimates from a UK family genetic register service. Am. J. Med. Genet. A 2010, 152A, 327–332. [Google Scholar] [CrossRef]
- Al-Sharefi, A.; Javaid, U.; Perros, P.; Ealing, J.; Truran, P.; Nag, S.; Kamaruddin, S.; Abouglila, K.; Cains, F.; Lewis, L.; et al. Clinical Presentation and Outcomes of Phaeochromocytomas/Paragangliomas in Neurofibromatosis Type 1. Eur. Endocrinol. 2019, 15, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Clementi, M.; Milani, S.; Mammi, I.; Boni, S.; Monciotti, C.; Tenconi, R. Neurofibromatosis type 1 growth charts. Am. J. Med. Genet. 1999, 87, 317–323. [Google Scholar] [CrossRef]
- Soucy, E.A.; van Oppen, D.; Nejedly, N.L.; Gao, F.; Gutmann, D.H.; Hollander, A.S. Height assessments in children with neurofibromatosis type 1. J. Child Neurol. 2013, 28, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Durrani, A.A.; Crawford, A.H.; Chouhdry, S.N.; Saifuddin, A.; Morley, T.R. Modulation of spinal deformities in patients with neurofibromatosis type 1. Spine (Phila Pa 1976) 2000, 25, 69–75. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Ferner, R.E.; Listernick, R.H.; Korf, B.R.; Wolters, P.L.; Johnson, K.J. Neurofibromatosis type 1. Nat. Rev. Dis. Prim. 2017, 3, 17004. [Google Scholar] [CrossRef]
- Stevenson, D.A.; Birch, P.H.; Friedman, J.M.; Viskochil, D.H.; Balestrazzi, P.; Boni, S.; Buske, A.; Korf, B.R.; Niimura, M.; Pivnick, E.K.; et al. Descriptive analysis of tibial pseudarthrosis in patients with neurofibromatosis 1. Am. J. Med. Genet. 1999, 84, 413–419. [Google Scholar] [CrossRef]
- Toro, G.; Santoro, C.; Ambrosio, D.; Landi, G.; Scilipoti, M.; Moretti, A.; Paoletta, M.; Liguori, S.; Schiavone Panni, A.; Picariello, S.; et al. Natural History of Scoliosis in Children with NF1: An Observation Study. Healthcare 2021, 9, 881. [Google Scholar] [CrossRef]
- Heerva, E.; Koffert, A.; Jokinen, E.; Kuorilehto, T.; Peltonen, S.; Aro, H.T.; Peltonen, J. A controlled register-based study of 460 neurofibromatosis 1 patients: Increased fracture risk in children and adults over 41 years of age. J. Bone Miner. Res. 2012, 27, 2333–2337. [Google Scholar] [CrossRef]
- Riccardi, C.; Perrone, L.; Napolitano, F.; Sampaolo, S.; Melone, M.A.B. Understanding the Biological Activities of Vitamin D in Type 1 Neurofibromatosis: New Insights into Disease Pathogenesis and Therapeutic Design. Cancers 2020, 12, 2965. [Google Scholar] [CrossRef]
- Tucker, T.; Birch, P.; Savoy, D.M.; Friedman, J.M. Increased dental caries in people with neurofibromatosis 1. Clin. Genet. 2007, 72, 524–527. [Google Scholar] [CrossRef]
- Wotjiuk, F.; Hyon, I.; Dajean-Trutaud, S.; Badran, Z.; Prud’homme, T. Dental Management of Neurofibromatosis Type 1: A Case Report and Literature Review. Int. J. Clin. Pediatr. Dent. 2019, 12, 577–581. [Google Scholar] [CrossRef]
- Karvonen, M.; Saari, A.; Hannila, M.L.; Lonnqvist, T.; Dunkel, L.; Sankilampi, U. Elevated head circumference-to-height ratio is an early and frequent feature in children with neurofibromatosis type 1. Horm. Res. Paediatr. 2013, 79, 97–102. [Google Scholar] [CrossRef]
- Virdis, R.; Street, M.E.; Bandello, M.A.; Tripodi, C.; Donadio, A.; Villani, A.R.; Cagozzi, L.; Garavelli, L.; Bernasconi, S. Growth and pubertal disorders in neurofibromatosis type 1. J. Pediatr. Endocrinol. Metab. 2003, 16 (Suppl. 2), 289–292. [Google Scholar]
- Riccardi, V.M.; Kleiner, B. Neurofibromatosis: A neoplastic birth defect with two age peaks of severe problems. Birth Defects Orig. Artic. Ser. 1977, 13, 131–138. [Google Scholar]
- Kehrer-Sawatzki, H.; Mautner, V.F.; Cooper, D.N. Emerging genotype-phenotype relationships in patients with large NF1 deletions. Hum. Genet. 2017, 136, 349–376. [Google Scholar] [CrossRef]
- Giugliano, T.; Santoro, C.; Torella, A.; Del Vecchio Blanco, F.; Grandone, A.; Onore, M.E.; Melone, M.A.B.; Straccia, G.; Melis, D.; Piccolo, V.; et al. Clinical and Genetic Findings in Children with Neurofibromatosis Type 1, Legius Syndrome, and Other Related Neurocutaneous Disorders. Genes 2019, 10, 580. [Google Scholar] [CrossRef]
- Sabbagh, A.; Pasmant, E.; Imbard, A.; Luscan, A.; Soares, M.; Blanche, H.; Laurendeau, I.; Ferkal, S.; Vidaud, M.; Pinson, S.; et al. NF1 molecular characterization and neurofibromatosis type I genotype-phenotype correlation: The French experience. Hum. Mutat. 2013, 34, 1510–1518. [Google Scholar] [CrossRef] [PubMed]
- Kehrer-Sawatzki, H.; Cooper, D.N. Classification of NF1 microdeletions and its importance for establishing genotype/phenotype correlations in patients with NF1 microdeletions. Hum. Genet. 2021, 140, 1635–1649. [Google Scholar] [CrossRef] [PubMed]
- Kehrer-Sawatzki, H.; Kluwe, L.; Sandig, C.; Kohn, M.; Wimmer, K.; Krammer, U.; Peyrl, A.; Jenne, D.E.; Hansmann, I.; Mautner, V.F. High frequency of mosaicism among patients with neurofibromatosis type 1 (NF1) with microdeletions caused by somatic recombination of the JJAZ1 gene. Am. J. Hum. Genet. 2004, 75, 410–423. [Google Scholar] [CrossRef] [PubMed]
- Bengesser, K.; Cooper, D.N.; Steinmann, K.; Kluwe, L.; Chuzhanova, N.A.; Wimmer, K.; Tatagiba, M.; Tinschert, S.; Mautner, V.F.; Kehrer-Sawatzki, H. A novel third type of recurrent NF1 microdeletion mediated by nonallelic homologous recombination between LRRC37B-containing low-copy repeats in 17q11.2. Hum. Mutat. 2010, 31, 742–751. [Google Scholar] [CrossRef]
- Buki, G.; Zsigmond, A.; Czako, M.; Szalai, R.; Antal, G.; Farkas, V.; Fekete, G.; Nagy, D.; Szell, M.; Tihanyi, M.; et al. Genotype-Phenotype Associations in Patients With Type-1, Type-2, and Atypical NF1 Microdeletions. Front. Genet. 2021, 12, 673025. [Google Scholar] [CrossRef]
- Kehrer-Sawatzki, H.; Wahllander, U.; Cooper, D.N.; Mautner, V.F. Atypical NF1 Microdeletions: Challenges and Opportunities for Genotype/Phenotype Correlations in Patients with Large NF1 Deletions. Genes 2021, 12, 1639. [Google Scholar] [CrossRef] [PubMed]
- Santoro, C.; Bernardo, P.; Coppola, A.; Pugliese, U.; Cirillo, M.; Giugliano, T.; Piluso, G.; Cinalli, G.; Striano, S.; Bravaccio, C.; et al. Seizures in children with neurofibromatosis type 1: Is neurofibromatosis type 1 enough? Ital. J. Pediatr. 2018, 44, 41. [Google Scholar] [CrossRef] [PubMed]
- Koczkowska, M.; Callens, T.; Gomes, A.; Sharp, A.; Chen, Y.; Hicks, A.D.; Aylsworth, A.S.; Azizi, A.A.; Basel, D.G.; Bellus, G.; et al. Expanding the clinical phenotype of individuals with a 3-bp in-frame deletion of the NF1 gene (c.2970_2972del): An update of genotype–phenotype correlation. Genet. Med. 2019, 21, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Upadhyaya, M.; Huson, S.M.; Davies, M.; Thomas, N.; Chuzhanova, N.; Giovannini, S.; Evans, D.G.; Howard, E.; Kerr, B.; Griffiths, S.; et al. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): Evidence of a clinically significant NF1 genotype-phenotype correlation. Am. J. Hum. Genet. 2007, 80, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Santoro, C.; Maietta, A.; Giugliano, T.; Melis, D.; Perrotta, S.; Nigro, V.; Piluso, G. Arg(1809) substitution in neurofibromin: Further evidence of a genotype-phenotype correlation in neurofibromatosis type 1. Eur. J. Hum. Genet. 2015, 23, 1460–1461. [Google Scholar] [CrossRef]
- Rojnueangnit, K.; Xie, J.; Gomes, A.; Sharp, A.; Callens, T.; Chen, Y.; Liu, Y.; Cochran, M.; Abbott, M.A.; Atkin, J.; et al. High Incidence of Noonan Syndrome Features Including Short Stature and Pulmonic Stenosis in Patients carrying NF1 Missense Mutations Affecting p.Arg1809: Genotype-Phenotype Correlation. Hum. Mutat. 2015, 36, 1052–1063. [Google Scholar] [CrossRef]
- Pinna, V.; Lanari, V.; Daniele, P.; Consoli, F.; Agolini, E.; Margiotti, K.; Bottillo, I.; Torrente, I.; Bruselles, A.; Fusilli, C.; et al. p.Arg1809Cys substitution in neurofibromin is associated with a distinctive NF1 phenotype without neurofibromas. Eur. J. Hum. Genet. 2015, 23, 1068–1071. [Google Scholar] [CrossRef]
- Santoro, C.; Boccia, R.; Iovino, C.; Piluso, G.; Perrotta, S.; Simonelli, F. Patients carrying Arg1809 substitution with no choroidal abnormalities: A further proof of a "Quasi-Incomplete" NF1 phenotype. Eur. J. Hum. Genet. 2022, 31, 136–137. [Google Scholar] [CrossRef]
- Dunzendorfer-Matt, T.; Mercado, E.L.; Maly, K.; McCormick, F.; Scheffzek, K. The neurofibromin recruitment factor Spred1 binds to the GAP related domain without affecting Ras inactivation. Proc. Natl. Acad. Sci. USA 2016, 113, 7497–7502. [Google Scholar] [CrossRef]
- Koczkowska, M.; Chen, Y.; Callens, T.; Gomes, A.; Sharp, A.; Johnson, S.; Hsiao, M.C.; Chen, Z.; Balasubramanian, M.; Barnett, C.P.; et al. Genotype-Phenotype Correlation in NF1: Evidence for a More Severe Phenotype Associated with Missense Mutations Affecting NF1 Codons 844-848. Am. J. Hum. Genet. 2018, 102, 69–87. [Google Scholar] [CrossRef] [PubMed]
- Koczkowska, M.; Callens, T.; Chen, Y.; Gomes, A.; Hicks, A.D.; Sharp, A.; Johns, E.; Uhas, K.A.; Armstrong, L.; Bosanko, K.A.; et al. Clinical spectrum of individuals with pathogenic NF1 missense variants affecting p.Met1149, p.Arg1276, and p.Lys1423: Genotype-phenotype study in neurofibromatosis type 1. Hum. Mutat. 2020, 41, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shachar, S.; Constantini, S.; Hallevi, H.; Sach, E.K.; Upadhyaya, M.; Evans, G.D.; Huson, S.M. Increased rate of missense/in-frame mutations in individuals with NF1-related pulmonary stenosis: A novel genotype-phenotype correlation. Eur. J. Hum. Genet. 2013, 21, 535–539. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Bottillo, I.; Sarkozy, A.; Carta, C.; Neri, C.; Bellacchio, E.; Schirinzi, A.; Conti, E.; Zampino, G.; Battaglia, A.; et al. NF1 gene mutations represent the major molecular event underlying neurofibromatosis-Noonan syndrome. Am. J. Hum. Genet. 2005, 77, 1092–1101. [Google Scholar] [CrossRef]
- Watson, G.H. Pulmonary stenosis, cafe-au-lait spots, and dull intelligence. Arch. Dis. Child. 1967, 42, 303–307. [Google Scholar] [CrossRef]
- Sharafi, P.; Ayter, S. Possible modifier genes in the variation of neurofibromatosis type 1 clinical phenotypes. J. Neurogenet. 2018, 32, 65–77. [Google Scholar] [CrossRef]
- D′Amico, A.; Rosano, C.; Pannone, L.; Pinna, V.; Assunto, A.; Motta, M.; Ugga, L.; Daniele, P.; Mandile, R.; Mariniello, L.; et al. Clinical variability of neurofibromatosis 1: A modifying role of cooccurring PTPN11 variants and atypical brain MRI findings. Clin. Genet. 2021, 100, 563–572. [Google Scholar] [CrossRef]
- Thiel, C.; Wilken, M.; Zenker, M.; Sticht, H.; Fahsold, R.; Gusek-Schneider, G.C.; Rauch, A. Independent NF1 and PTPN11 mutations in a family with neurofibromatosis-Noonan syndrome. Am. J. Med. Genet. A 2009, 149A, 1263–1267. [Google Scholar] [CrossRef]
- Kawakami, I.; Katsuse, O.; Aoki, N.; Togo, T.; Suzuki, K.; Isojima, D.; Kondo, D.; Iseki, E.; Kosaka, K.; Akiyama, H.; et al. Autopsy case of concurrent Huntington’s disease and neurofibromatosis type 1. Psychogeriatrics 2014, 14, 81–86. [Google Scholar] [CrossRef]
- Martin, F.; Kana, V.; Mori, A.C.; Fischer, D.; Parkin, N.; Boltshauser, E.; Rushing, E.J.; Klein, A. Neurofibromatosis type 1 (NF1) with an unusually severe phenotype due to digeny for NF1 and ryanodine receptor 1 associated myopathy. Eur. J. Pediatr. 2014, 173, 1691–1694. [Google Scholar] [CrossRef]
- Campos, B.; Balmana, J.; Gardenyes, J.; Valenzuela, I.; Abad, O.; Fabregas, P.; Volpini, V.; Diez, O. Germline mutations in NF1 and BRCA1 in a family with neurofibromatosis type 1 and early-onset breast cancer. Breast Cancer Res. Treat. 2013, 139, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Ercolino, T.; Lai, R.; Giache, V.; Melchionda, S.; Carella, M.; Delitala, A.; Mannelli, M.; Fanciulli, G. Patient affected by neurofibromatosis type 1 and thyroid C-cell hyperplasia harboring pathogenic germ-line mutations in both NF1 and RET genes. Gene 2014, 536, 332–335. [Google Scholar] [CrossRef] [PubMed]
- Zobor, D.; Kaufmann, D.H.; Weckerle, P.; Sauer, A.; Wissinger, B.; Wilhelm, H.; Kohl, S. Cone-rod dystrophy associated with amelogenesis imperfecta in a child with neurofibromatosis type 1. Ophthalmic Genet. 2012, 33, 34–38. [Google Scholar] [CrossRef]
- Santoro, C.; Malan, V.; Bertoli, M.; Boddaert, N.; Vidaud, D.; Lyonnet, S. Sporadic NF1 mutation associated with a de-novo 20q11.3 deletion explains the association of unusual facies, Moyamoya vasculopathy, and developmental delay, reported by Bertoli et al. in 2009. Clin. Dysmorphol. 2013, 22, 42–43. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, R.; Goss, L.; Romer, M.M.; Kalamchi, S. Down syndrome and neurofibromatosis: A case report. Spec. Care Dent. 2014, 34, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Santoro, C.; Giugliano, T.; Bernardo, P.; Palladino, F.; Torella, A.; Del Vecchio Blanco, F.; Onore, M.E.; Carotenuto, M.; Nigro, V.; Piluso, G. A novel RAB39B mutation and concurrent de novo NF1 mutation in a boy with neurofibromatosis type 1, intellectual disability, and autism: A case report. BMC Neurol. 2020, 20, 327. [Google Scholar] [CrossRef] [PubMed]
- Santoro, C.; Riccio, S.; Palladino, F.; Aliberti, F.; Carotenuto, M.; Zanobio, M.; Peduto, C.; Nigro, V.; Perrotta, S.; Piluso, G. A novel MEIS2 mutation explains the complex phenotype in a boy with a typical NF1 microdeletion syndrome. Eur. J. Med. Genet. 2021, 64, 104190. [Google Scholar] [CrossRef]
- Castellanos, E.; Rosas, I.; Negro, A.; Gel, B.; Alibes, A.; Baena, N.; Pineda, M.; Pi, G.; Pintos, G.; Salvador, H.; et al. Mutational spectrum by phenotype: Panel-based NGS testing of patients with clinical suspicion of RASopathy and children with multiple cafe-au-lait macules. Clin. Genet. 2020, 97, 264–275. [Google Scholar] [CrossRef]
- De Luca, A.; Bottillo, I.; Dasdia, M.C.; Morella, A.; Lanari, V.; Bernardini, L.; Divona, L.; Giustini, S.; Sinibaldi, L.; Novelli, A.; et al. Deletions of NF1 gene and exons detected by multiplex ligation-dependent probe amplification. J. Med. Genet. 2007, 44, 800–808. [Google Scholar] [CrossRef]
- Valero, M.C.; Martin, Y.; Hernandez-Imaz, E.; Marina Hernandez, A.; Melean, G.; Valero, A.M.; Javier Rodriguez-Alvarez, F.; Telleria, D.; Hernandez-Chico, C. A highly sensitive genetic protocol to detect NF1 mutations. J. Mol. Diagn. 2011, 13, 113–122. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
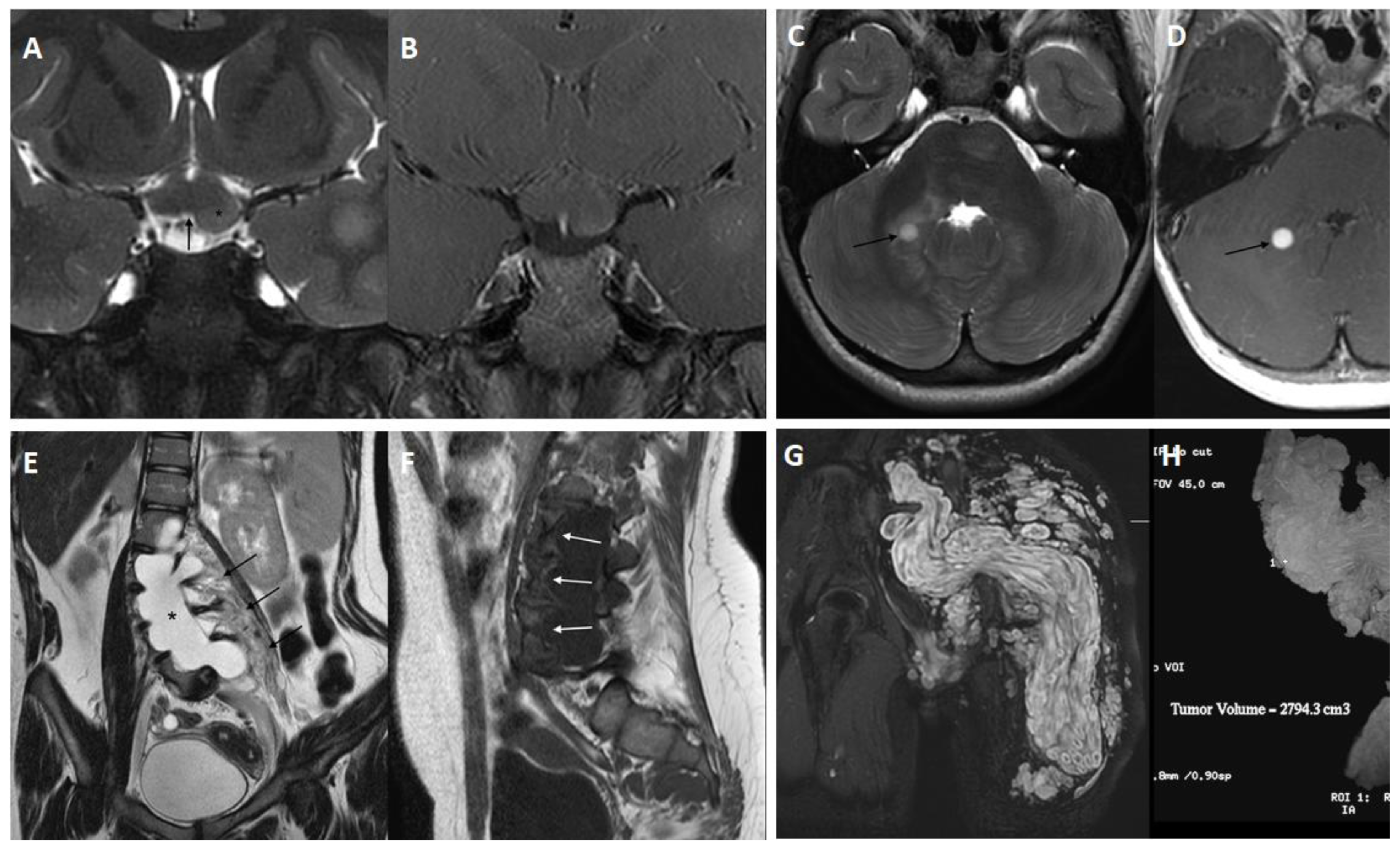{kind=link}
{kind=link}
{kind=link}
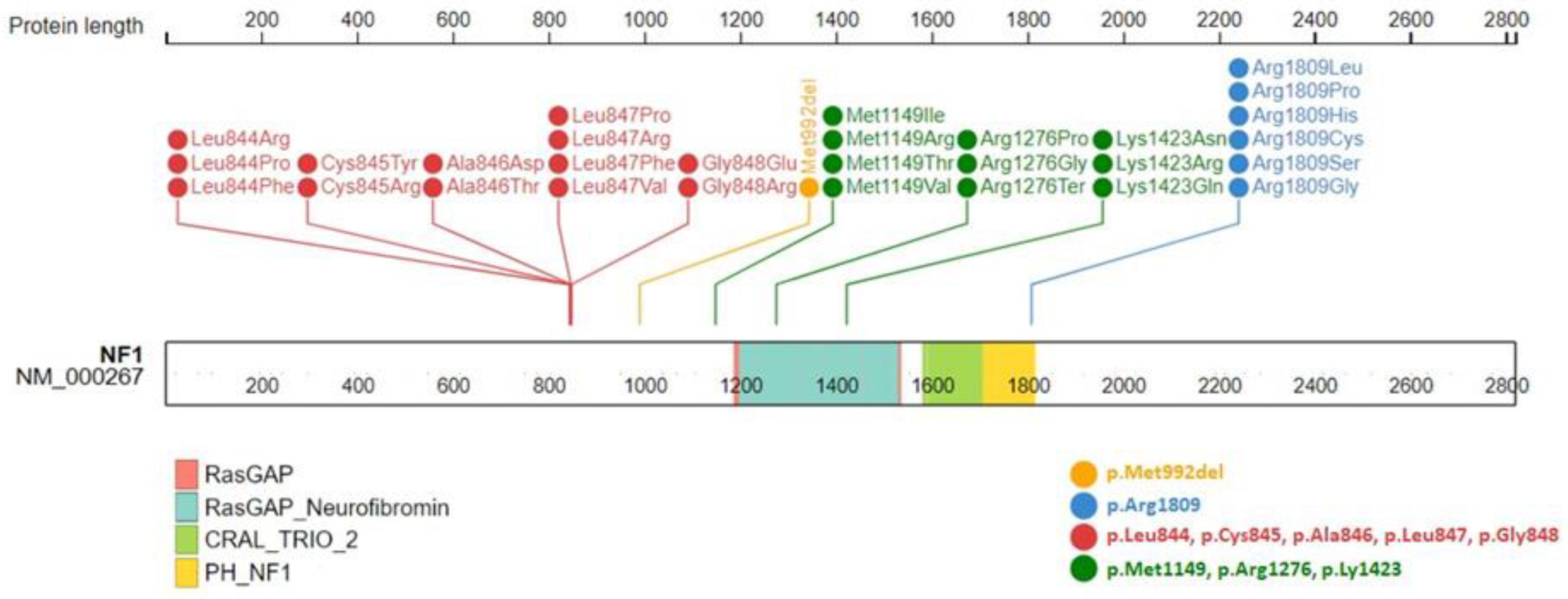{kind=link}
| Revised Diagnostic Criteria for Neurofibromatosis Type 1 (NF1) |
|---|
| A: The diagnostic criteria for NF1 are met in an individual who does not have a parent diagnosed with NF1 if two or more of the following are present: |
| • Six or more café-au-lait macules over 5 mm in greatest diameter in prepubertal individuals and over 15 mm in greatest diameter in post-pubertal individuals 1 |
| • Freckling in the axillary or inguinal region 2 |
| • Two or more neurofibromas of any type or one plexiform neurofibroma |
| • Optic pathway glioma (OPG) |
| • Two or more iris Lisch nodules identified by slit lamp examination or two or more choroidal abnormalities (CAs)—defined as bright, patchy nodules imaged by optical coherence tomography (OCT)/near-infrared reflectance (NIR) imaging |
| • A distinctive osseous lesion such as sphenoid dysplasia 3, anterolateral bowing of the tibia 4, or pseudarthrosis of a long bone 5 |
| • A heterozygous pathogenic NF1 variant with a variant allele fraction of 50% in apparently normal tissue such as white blood cells |
| B: A child of a parent who meets the diagnostic criteria specified in A merits a diagnosis of NF1 if one or more of the criteria in A are present |
| 1 If only café-au-lait macules and freckling are present, the diagnosis is most likely NF1 but exceptionally the person might have another diagnosis such as Legius syndrome. At least one of the two pigmentary findings (café-au-lait macules or freckling) should be bilateral. 2 If only café-au-lait spots and freckles are present, the diagnosis is probably NF1 but exceptionally the person may have another diagnosis such as Legius syndrome. At least one of the two pigment findings (café-au-lait spots or freckles) should be bilateral. 3 Sphenoid wing dysplasia is not a separate criterion in case of an ipsilateral orbital plexiform neurofibroma. 4 Congenital bowing is usually associated with thickening of the cortex of the long bone. 5 Pseudarthrosis is usually preceded by congenital curvature of a long bone and only rarely by thinning of the cortex. |
| NF1 Additional Features | |
|---|---|
| Dystrophic scoliosis | Typical and should be clearly defined |
| Juvenile xanthogranuloma | Present in up to 30% of very young patients Not reported in Legius syndrome |
| Anemic nevus | Present in up to 50% of children with NF1 and may allow for clinical diagnosis at young age, but may also be seen in other conditions and in the general population |
| Choroidal anomalies | Present in 60–70% of children with NF1 |
| Focal areas of signal intensity (FASI) | Previously known as unidentified bright objects (UBOs) Common neuroradiological findings in brain MRI and may disappear over time |
| Screening for Major NF1 Complications | |||
|---|---|---|---|
| Sought Complications | Affected Patients | Screening Modality | |
| Dermatological manifestations | Subcutaneous, internal, and plexiform NF: malignant transformation? Esthetic or functional problems? | Children, adults | Clinical examination: Pain, neurological deficit, increase in size, functional and psychological repercussions. Additional examinations: optional Indications: suspicion of malignancy, preoperative, internal NF risk factor |
| Juvenile xanthogranuloma (JXG) | Children | Physical examination: If JXG present: palpation of ganglionic areas and complete blood count * | |
| Orthopedic manifestations | Bone dysplasia and pseudarthrosis of the long bones, fractures | Children, adults | Clinical examination: search for gibbosity, bone deformity. X-ray if abnormalities found on clinical examination. |
| Scoliosis | Children, adults | Physical examination Additional examinations (optional): Front and profile X-ray views of the spine if clinical abnormalities found (1st line) MRI should be reserved for forms with vertebral and/or costal dysplasia (expert consensus) Pulmonary function tests to evaluate the impact of severe scoliosis | |
| Bone mineralization disorder, osteoporosis | Children, adults | Consider bone densitometry scans based on clinical examination, vitamin D levels and X-ray results | |
| Endocrinological manifestations | Pubertal and growth disorders | Children | Follow pubertal development and the growth curve, measure head circumference |
| Cardiac and vascular manifestations | Essential and secondary hypertension | Children, adults | Physical examination: Blood pressure measurement at each consultation (at least annually), discuss the possibility of ambulatory measurement. Look for signs suggestive of pheochromocytoma. Additional examinations if high blood pressure. As a first-line examination: angio-CT scan of the renal arteries and abdominal CT. Plasma and/or urinary determination of metanephrines in adults. |
| Cardiac abnormalities | Children, adults | Clinical examination | |
| Hemorrhagic manifestations | Children, adults | Assess hemostasis before any surgical, dental, or obstetric procedure. | |
| Pain, psychological repercussions, quality of life | Children, adults | Clinical examination Offer psychological counseling, pain specialist referral | |
| Otolaryngologic manifestations | Deafness, neurinoma, phonatory disorder, laryngeal NF | Children, adults | Otolaryngologic examination with tuning fork |
| Neurological manifestation | OPG | Children | Interview: repeated falls leading to suspicion of decrease visual acuity or visual field amputation Neurological and ocular examination: strabismus, nystagmus, low visual acuity, neurological deficit, signs of intracranial hypertension. Early puberty, deviation from the growth curve, measurement of head circumference Ophthalmological screening at least once per year until the age of 13 years and then if signs appear. MRI of the optic and cerebral pathways is not systematic and should be performed only if suspicion of OPG |
| Epilepsy, hydrocephalus, intracranial, hypertension, stroke, headache | Children, adults | Neurological examination Cerebral MRI and electroencephalogram guided by the abnormalities detected on clinical examination | |
| Developmental delay, learning difficulties, behavioral problems | Children | Evaluation of psychomotor development and academic proficiency at each consultation Search for learning difficulties Comprehensive neuropsychomotor assessment before entering elementary school, support for school integration. | |
| Medullary and nerve compression, peripheral neuropathy, socio-professional integration | Adults | Clinical examination | |
| Cancers | MPNST (60% of cancers in NF1 patients) | Children, adults | Clinical examination: recent increase in size of plexiform NF, appearance of pain. Additional examinations if signs appear. If high NF1 score: screening for internal neurofibromas by imaging (preferably by MRI). |
| All other cancers | Children, adults | Clinical examination: asthenia, high blood pressure, intracranial hypertension symptoms, abdominal mass, bladder signs, appearance of mass, compressive syndrome Screening identical to that of the general population except for earlier breast screening (>40 years) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peduto, C.; Zanobio, M.; Nigro, V.; Perrotta, S.; Piluso, G.; Santoro, C. Neurofibromatosis Type 1: Pediatric Aspects and Review of Genotype–Phenotype Correlations. Cancers 2023, 15, 1217. https://doi.org/10.3390/cancers15041217
Peduto C, Zanobio M, Nigro V, Perrotta S, Piluso G, Santoro C. Neurofibromatosis Type 1: Pediatric Aspects and Review of Genotype–Phenotype Correlations. Cancers. 2023; 15(4):1217. https://doi.org/10.3390/cancers15041217
Chicago/Turabian StylePeduto, Cristina, Mariateresa Zanobio, Vincenzo Nigro, Silverio Perrotta, Giulio Piluso, and Claudia Santoro. 2023. "Neurofibromatosis Type 1: Pediatric Aspects and Review of Genotype–Phenotype Correlations" Cancers 15, no. 4: 1217. https://doi.org/10.3390/cancers15041217
APA StylePeduto, C., Zanobio, M., Nigro, V., Perrotta, S., Piluso, G., & Santoro, C. (2023). Neurofibromatosis Type 1: Pediatric Aspects and Review of Genotype–Phenotype Correlations. Cancers, 15(4), 1217. https://doi.org/10.3390/cancers15041217