
Oncolytic BHV-1 Is Sufficient to Induce Immunogenic Cell Death and Synergizes with Low-Dose Chemotherapy to Dampen Immunosuppressive T Regulatory Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Oncolytic Virus
2.3. Drug Preparation
2.4. Tumor Regression Studies in Mice Bearing B16-C10 Tumors
2.5. Histology and Image Analysis
2.6. Gold Standard ICD Immunization Assay
2.7. Tumor-Specific Activation of Circulating CD8+ T Cells
2.8. Re-Challenge Experiment
2.9. Flow Cytometry Analysis
2.10. Cytokine Analysis
2.11. Clariom S Assay Transcriptional Profiling
2.12. Statistical Analysis
3. Results
3.1. Use of B16-C10 Syngeneic Melanoma Model for Pre-Clinical Analysis of oBHV
3.2. oBHV Alone Is Sufficient to Induce ICD and Tumor-Specific CD8+ T Cell Activation
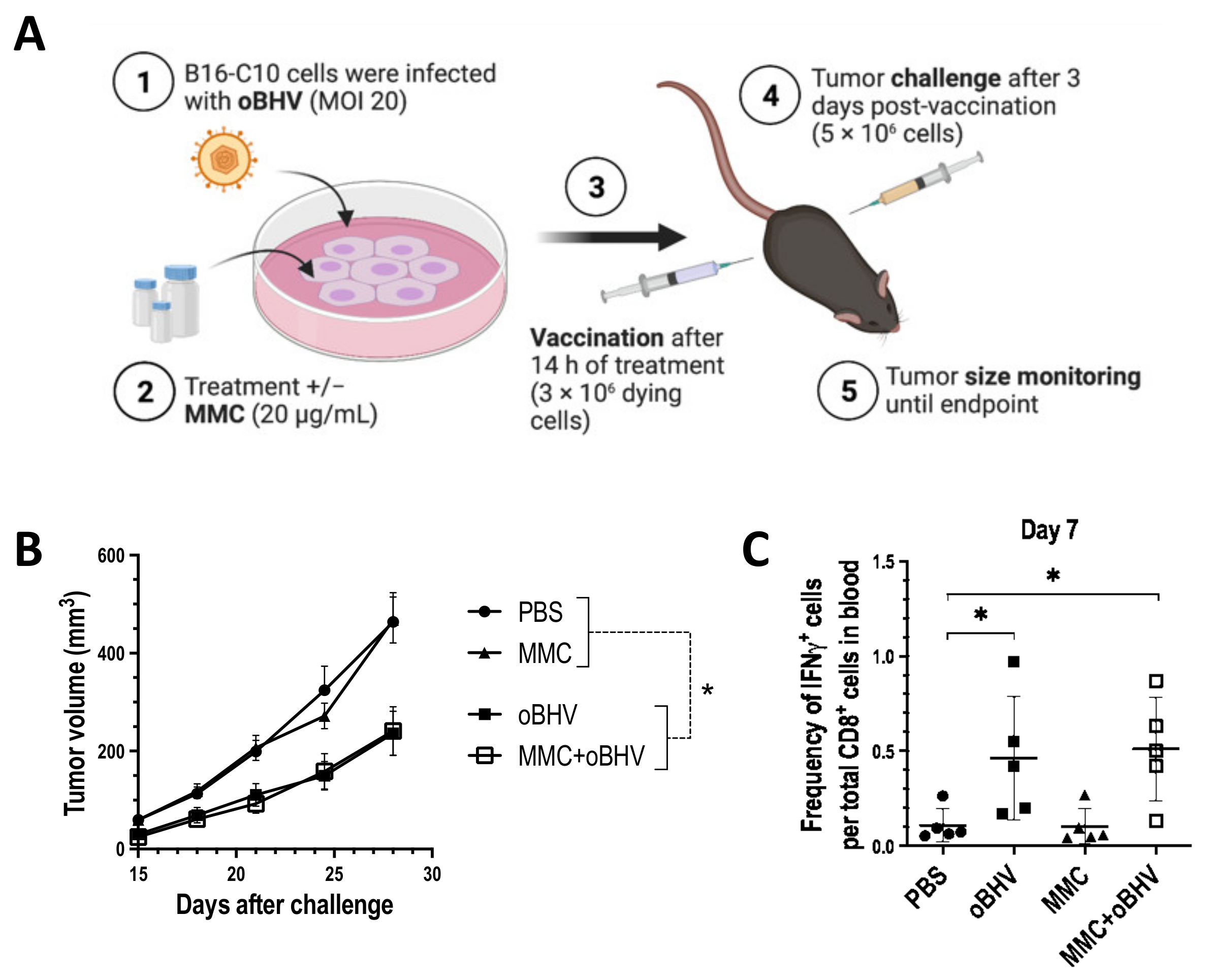
3.3. Low-Dose MMC Synergizes with oBHV to Sensitize Tumors to ICI and Induce Long-Term Protective Immunity
3.4. Therapeutic Efficacy of Triple Combination Correlates with Early Tumor Infiltration of CD8+ T Cells and Reduced Tumor Infiltration of Higly Suppresssive PD-1+ Treg Cells
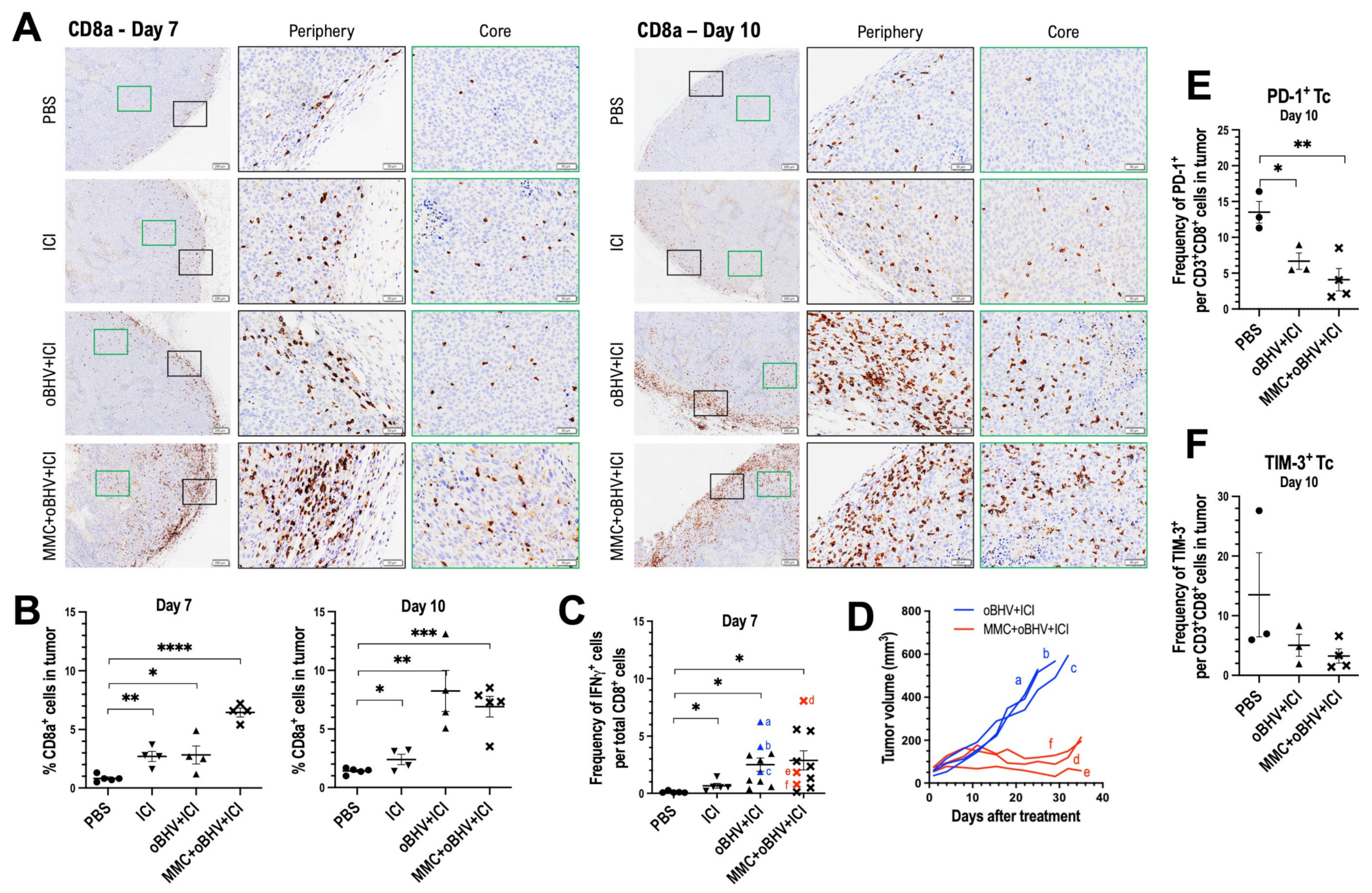

3.5. Addition of MMC to oBHV Induces Early Release of Anti-Tumor Cytokines and Alters the Tumor Microenvironment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lemay, C.G.; Keller, B.A.; Edge, R.E.; Abei, M.; Bell, J.C. Oncolytic Viruses: The Best is Yet to Come. Curr. Cancer Drug Targets 2018, 18, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Cuddington, B.P.; Dyer, A.L.; Workenhe, S.T.; Mossman, K.L. Oncolytic bovine herpesvirus type 1 infects and kills breast tumor cells and breast cancer-initiating cells irrespective of tumor subtype. Cancer Gene 2013, 20, 282–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, W.; Hu, P.; Wu, J.; Wang, J.; Li, J.; Lei, L.; Liu, R. The oncolytic herpes simplex virus vector G47 effectively targets breast cancer stem cells. Oncol. Rep. 2013, 29, 1108–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Workenhe, S.T.; Mossman, K.L. Oncolytic virotherapy and immunogenic cancer cell death: Sharpening the sword for improved cancer treatment strategies. Mol. Ther. 2014, 22, 251–256. [Google Scholar] [CrossRef] [Green Version]
- Workenhe, S.T.; Verschoor, M.L.; Mossman, K.L. The role of oncolytic virus immunotherapies to subvert cancer immune evasion. Future Oncol. 2015, 11, 675–689. [Google Scholar] [CrossRef]
- Pol, J.G.; Levesque, S.; Workenhe, S.T.; Gujar, S.; Le Boeuf, F.; Clements, D.R.; Fahrner, J.E.; Fend, L.; Bell, J.C.; Mossman, K.; et al. Trial Watch: Oncolytic viro-immunotherapy of hematologic and solid tumors. Oncoimmunology 2018, 7, e1503032. [Google Scholar] [CrossRef] [Green Version]
- Locy, H.; de Mey, S.; de Mey, W.; De Ridder, M.; Thielemans, K.; Maenhout, S.K. Immunomodulation of the Tumor Microenvironment: Turn Foe Into Friend. Front. Immunol. 2018, 9, 2909. [Google Scholar] [CrossRef] [Green Version]
- Raja, J.; Ludwig, J.M.; Gettinger, S.N.; Schalper, K.A.; Kim, H.S. Oncolytic virus immunotherapy: Future prospects for oncology. J. Immunother. Cancer 2018, 6, 140. [Google Scholar] [CrossRef]
- Gujar, S.; Pol, J.G.; Kroemer, G. Heating it up: Oncolytic viruses make tumors ‘hot’ and suitable for checkpoint blockade immunotherapies. Oncoimmunology 2018, 7, e1442169. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Guo, Z.S.; Liu, Z.; Bartlett, D.L. Oncolytic Immunotherapy: Dying the Right Way is a Key to Eliciting Potent Antitumor Immunity. Front. Oncol. 2014, 4, 74. [Google Scholar] [CrossRef] [Green Version]
- Filley, A.C.; Dey, M. Immune System, Friend or Foe of Oncolytic Virotherapy? Front. Oncol. 2017, 7, 106. [Google Scholar] [CrossRef] [Green Version]
- Workenhe, S.T.; Pol, J.G.; Lichty, B.D.; Cummings, D.T.; Mossman, K.L. Combining oncolytic HSV-1 with immunogenic cell death-inducing drug mitoxantrone breaks cancer immune tolerance and improves therapeutic efficacy. Cancer Immunol. Res. 2013, 1, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Workenhe, S.T.; Nguyen, A.; Bakhshinyan, D.; Wei, J.; Hare, D.N.; MacNeill, K.L.; Wan, Y.; Oberst, A.; Bramson, J.L.; Nasir, J.A.; et al. De novo necroptosis creates an inflammatory environment mediating tumor susceptibility to immune checkpoint inhibitors. Commun. Biol. 2020, 3, 645. [Google Scholar] [CrossRef]
- Vito, A.; Salem, O.; El-Sayes, N.; MacFawn, I.P.; Portillo, A.L.; Milne, K.; Harrington, D.; Ashkar, A.A.; Wan, Y.; Workenhe, S.T.; et al. Immune checkpoint blockade in triple negative breast cancer influenced by B cells through myeloid-derived suppressor cells. Commun. Biol. 2021, 4, 859. [Google Scholar] [CrossRef]
- El-Sayes, N.; Vito, A.; Salem, O.; Workenhe, S.T.; Wan, Y.; Mossman, K. A Combination of Chemotherapy and Oncolytic Virotherapy Sensitizes Colorectal Adenocarcinoma to Immune Checkpoint Inhibitors in a cDC1-Dependent Manner. Int. J. Mol. Sci. 2022, 23, 1754. [Google Scholar] [CrossRef]
- Warner, S.G.; O’Leary, M.P.; Fong, Y. Therapeutic oncolytic viruses: Clinical advances and future directions. Curr. Opin. Oncol. 2017, 29, 359–365. [Google Scholar] [CrossRef]
- Ott, P.A.; Hodi, F.S. Talimogene Laherparepvec for the Treatment of Advanced Melanoma. Clin. Cancer Res. 2016, 22, 3127–3131. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Quintanilla, J.; Seah, I.; Chua, M.; Shah, K. Oncolytic viruses: Overcoming translational challenges. J. Clin. Investig. 2019, 130, 1407–1418. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.H.; Lemoine, N.R.; Wang, Y. Oncolytic Viruses for Cancer Therapy: Overcoming the Obstacles. Viruses 2010, 2, 78–106. [Google Scholar] [CrossRef]
- Rodrigues, R.; Cuddington, B.; Mossman, K. Bovine herpesvirus type 1 as a novel oncolytic virus. Cancer Gene 2010, 17, 344–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuddington, B.P.; Mossman, K.L. Permissiveness of human cancer cells to oncolytic bovine herpesvirus 1 is mediated in part by KRAS activity. J. Virol. 2014, 88, 6885–6895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuddington, B.P.; Mossman, K.L. Oncolytic bovine herpesvirus type 1 as a broad spectrum cancer therapeutic. Curr. Opin. Virol. 2015, 13, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Hushur, O.; Takashima, Y.; Matsumoto, Y.; Otsuka, H. Restriction of bovine herpesvirus 1 (BHV-1) growth in non-permissive cells beyond the expression of immediate early genes. J. Vet. Med. Sci. 2004, 66, 453–455. [Google Scholar] [CrossRef] [Green Version]
- Campadelli-Fiume, G.; Cocchi, F.; Menotti, L.; Lopez, M. The novel receptors that mediate the entry of herpes simplex viruses and animal alphaherpesviruses into cells. Rev. Med. Virol. 2000, 10, 305–319. [Google Scholar] [CrossRef]
- Cuddington, B.; Verschoor, M.; Mossman, K. Handling of the cotton rat in studies for the pre-clinical evaluation of oncolytic viruses. J. Vis. Exp. 2014, 93, e52232. [Google Scholar] [CrossRef] [Green Version]
- Cuddington, B.P.; Verschoor, M.; Ashkar, A.; Mossman, K.L. Enhanced efficacy with azacytidine and oncolytic BHV-1 in a tolerized cotton rat model of breast adenocarcinoma. Mol. Oncolytics 2015, 2, 15004. [Google Scholar] [CrossRef]
- Miller, C.G.; Krummenacher, C.; Eisenberg, R.J.; Cohen, G.H.; Fraser, N.W. Development of a syngenic murine B16 cell line-derived melanoma susceptible to destruction by neuroattenuated HSV-1. Mol. Ther. 2001, 3, 160–168. [Google Scholar] [CrossRef]
- Spear, P.G.; Eisenberg, R.J.; Cohen, G.H. Three classes of cell surface receptors for alphaherpesvirus entry. Virology 2000, 275, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Geraghty, R.J.; Krummenacher, C.; Cohen, G.H.; Eisenberg, R.J.; Spear, P.G. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 1998, 280, 1618–1620. [Google Scholar] [CrossRef]
- Workman, A.; Jones, C. Productive infection and bICP0 early promoter activity of bovine herpesvirus 1 are stimulated by E2F1. J. Virol. 2010, 84, 6308–6317. [Google Scholar] [CrossRef] [Green Version]
- Keil, G.M.; Höhle, C.; Giesow, K.; König, P. Engineering glycoprotein B of bovine herpesvirus 1 to function as transporter for secreted proteins: A new protein expression approach. J. Virol. 2005, 79, 791–799. [Google Scholar] [CrossRef] [Green Version]
- Abu Eid, R.; Razavi, G.S.E.; Mkrtichyan, M.; Janik, J.; Khleif, S.N. Old-School Chemotherapy in Immunotherapeutic Combination in Cancer, A Low-cost Drug Repurposed. Cancer Immunol. Res. 2016, 4, 377–382. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell 2015, 28, 690–714. [Google Scholar] [CrossRef] [Green Version]
- Landreneau, J.P.; Shurin, M.R.; Agassandian, M.V.; Keskinov, A.A.; Ma, Y.; Shurin, G.V. Immunological Mechanisms of Low and Ultra-Low Dose Cancer Chemotherapy. Cancer Microenviron. 2015, 8, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Panaretakis, T.; Tufi, R.; Joza, N.; van Endert, P.; Ghiringhelli, F.; Apetoh, L.; Chaput, N.; Flament, C.; et al. Ecto-calreticulin in immunogenic chemotherapy. Immunol. Rev. 2007, 220, 22–34. [Google Scholar] [CrossRef]
- Simpson, G.R.; Horvath, A.; Annels, N.E.; Pencavel, T.; Metcalf, S.; Seth, R.; Peschard, P.; Price, T.; Coffin, R.S.; Mostafid, H.; et al. Combination of a fusogenic glycoprotein, pro-drug activation and oncolytic HSV as an intravesical therapy for superficial bladder cancer. Br. J. Cancer 2012, 106, 496–507. [Google Scholar] [CrossRef] [Green Version]
- Mullerad, M.; Bochner, B.H.; Adusumilli, P.S.; Bhargava, A.; Kikuchi, E.; Hui-Ni, C.; Kattan, M.W.; Chou, T.C.; Fong, Y. Herpes simplex virus based gene therapy enhances the efficacy of mitomycin C for the treatment of human bladder transitional cell carcinoma. J. Urol. 2005, 174, 741–746. [Google Scholar] [CrossRef] [Green Version]
- Bennett, J.J.; Adusumilli, P.; Petrowsky, H.; Burt, B.M.; Roberts, G.; Delman, K.A.; Zager, J.S.; Chou, T.C.; Fong, Y. Up-regulation of GADD34 mediates the synergistic anticancer activity of mitomycin C and a γ134.5 deleted oncolytic herpes virus (G207). FASEB J. 2004, 18, 1001–1003. [Google Scholar] [CrossRef] [PubMed]
- Toyoizumi, T.; Mick, R.; Abbas, A.E.; Kang, E.H.; Kaiser, L.R.; Molnar-Kimber, K.L. Combined therapy with chemotherapeutic agents and herpes simplex virus type 1 ICP34.5 mutant (HSV-1716) in human non-small cell lung cancer. Hum. Gene 1999, 10, 3013–3029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Workenhe, S.T.; Simmons, G.; Pol, J.G.; Lichty, B.D.; Halford, W.P.; Mossman, K.L. Immunogenic HSV-mediated oncolysis shapes the antitumor immune response and contributes to therapeutic efficacy. Mol. Ther. 2014, 22, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Senovilla, L.; Vitale, I.; Vacchelli, E.; Adjemian, S.; Agostinis, P.; Apetoh, L.; Aranda, F.; Barnaba, V.; Bloy, N.; et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014, 3, e955691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Vloten, J.P.; Workenhe, S.T.; Wootton, S.K.; Mossman, K.L.; Bridle, B.W. Critical Interactions between Immunogenic Cancer Cell Death, Oncolytic Viruses, and the Immune System Define the Rational Design of Combination Immunotherapies. J. Immunol. 2018, 200, 450–458. [Google Scholar] [CrossRef] [Green Version]
- Davola, M.E.; Mossman, K.L. Oncolytic viruses: How “lytic” must they be for therapeutic efficacy? Oncoimmunology 2019, 8, e1581528. [Google Scholar] [CrossRef] [Green Version]
- Briukhovetska, D.; Dörr, J.; Endres, S.; Libby, P.; Dinarello, C.A.; Kobold, S. Interleukins in cancer: From biology to therapy. Nat. Rev. Cancer 2021, 21, 481–499. [Google Scholar] [CrossRef]
- Pascual-García, M.; Bonfill-Teixidor, E.; Planas-Rigol, E.; Rubio-Perez, C.; Iurlaro, R.; Arias, A.; Cuartas, I.; Sala-Hojman, A.; Escudero, L.; Martínez-Ricarte, F.; et al. LIF regulates CXCL9 in tumor-associated macrophages and prevents CD8(+) T cell tumor-infiltration impairing anti-PD1 therapy. Nat. Commun. 2019, 10, 2416. [Google Scholar] [CrossRef] [Green Version]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Rodríguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castañón, E.; Melero, I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef] [Green Version]
- Adams, R.; Moser, B.; Karagiannis, S.N.; Lacy, K.E. Chemokine Pathways in Cutaneous Melanoma: Their Modulation by Cancer and Exploitation by the Clinician. Cancers 2021, 13, 5625. [Google Scholar] [CrossRef]
- Karagiannidis, I.; Salataj, E.; Said Abu Egal, E.; Beswick, E.J. G-CSF in tumors: Aggressiveness, tumor microenvironment and immune cell regulation. Cytokine 2021, 142, 155479. [Google Scholar] [CrossRef]
- Kamran, N.; Li, Y.; Sierra, M.; Alghamri, M.S.; Kadiyala, P.; Appelman, H.D.; Edwards, M.; Lowenstein, P.R.; Castro, M.G. Melanoma induced immunosuppression is mediated by hematopoietic dysregulation. Oncoimmunology 2018, 7, e1408750. [Google Scholar] [CrossRef] [Green Version]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Pol, J.G.; Workenhe, S.T.; Konda, P.; Gujar, S.; Kroemer, G. Cytokines in oncolytic virotherapy. Cytokine Growth Factor Rev. 2020, 56, 4–27. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. ImmunoTher. Cancer 2020, 8, e000337. [Google Scholar] [CrossRef] [Green Version]
- Greene, J.T.; Brian, B.F.t.; Senevirathne, S.E.; Freedman, T.S. Regulation of myeloid-cell activation. Curr. Opin. Immunol. 2021, 73, 34–42. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, M.M.; de la Puente, P. Leukemia Inhibitory Factor: An Important Cytokine in Pathologies and Cancer. Biomolecules 2022, 12, 217. [Google Scholar] [CrossRef]
- Kusmartsev, S.; Gabrilovich, D.I. Effect of tumor-derived cytokines and growth factors on differentiation and immune suppressive features of myeloid cells in cancer. Cancer Metastasis Rev. 2006, 25, 323–331. [Google Scholar] [CrossRef]
- Kumar, A.H.; Martin, K.; Turner, E.C.; Buneker, C.K.; Dorgham, K.; Deterre, P.; Caplice, N.M. Role of CX3CR1 receptor in monocyte/macrophage driven neovascularization. PLoS ONE 2013, 8, e57230. [Google Scholar] [CrossRef] [Green Version]
- Marchica, V.; Toscani, D.; Corcione, A.; Bolzoni, M.; Storti, P.; Vescovini, R.; Ferretti, E.; Dalla Palma, B.; Vicario, E.; Accardi, F.; et al. Bone Marrow CX3CL1/Fractalkine is a New Player of the Pro-Angiogenic Microenvironment in Multiple Myeloma Patients. Cancers 2019, 11, 321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Zhao, Q.; Kong, L.Y.; Wang, J.; Yan, J.; Xia, X.; Jia, Z.; Heimberger, A.B.; Li, S. Regulation of tumor immune suppression and cancer cell survival by CXCL1/2 elevation in glioblastoma multiforme. Sci. Adv. 2021, 7, eabc2511. [Google Scholar] [CrossRef] [PubMed]
- Sumida, K.; Ohno, Y.; Ohtake, J.; Kaneumi, S.; Kishikawa, T.; Takahashi, N.; Taketomi, A.; Kitamura, H. IL-11 induces differentiation of myeloid-derived suppressor cells through activation of STAT3 signalling pathway. Sci. Rep. 2015, 5, 13650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magidey-Klein, K.; Cooper, T.J.; Kveler, K.; Normand, R.; Zhang, T.; Timaner, M.; Raviv, Z.; James, B.P.; Gazit, R.; Ronai, Z.e.A.; et al. IL-6 contributes to metastatic switch via the differentiation of monocytic-dendritic progenitors into prometastatic immune cells. J. ImmunoTher. Cancer 2021, 9, e002856. [Google Scholar] [CrossRef]
- Ding, X.; Yuan, W.; Yang, H.; Liu, C.; Li, S.; Zhu, L. β-Catenin-Specific Inhibitor, iCRT14, Promotes BoHV-1 Infection-Induced DNA Damage in Human A549 Lung Adenocarcinoma Cells by Enhancing Viral Protein Expression. Int. J. Mol. Sci. 2022, 23, 2328. [Google Scholar] [CrossRef]
- Zhu, L.; Fu, X.; Yuan, C.; Jiang, X.; Zhang, G. Induction of Oxidative DNA Damage in Bovine Herpesvirus 1 Infected Bovine Kidney Cells (MDBK Cells) and Human Tumor Cells (A549 Cells and U2OS Cells). Viruses 2018, 10, 393. [Google Scholar] [CrossRef] [Green Version]
- Martins, I.; Wang, Y.; Michaud, M.; Ma, Y.; Sukkurwala, A.Q.; Shen, S.; Kepp, O.; Metivier, D.; Galluzzi, L.; Perfettini, J.L.; et al. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ. 2014, 21, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Wang, H.; Chavan, S.S.; Andersson, U. High Mobility Group Box Protein 1 (HMGB1): The Prototypical Endogenous Danger Molecule. Mol. Med. 2015, 21 (Suppl. S1), S6–S12. [Google Scholar] [CrossRef]
- Garg, A.D.; Galluzzi, L.; Apetoh, L.; Baert, T.; Birge, R.B.; Bravo-San Pedro, J.M.; Breckpot, K.; Brough, D.; Chaurio, R.; Cirone, M.; et al. Molecular and Translational Classifications of DAMPs in Immunogenic Cell Death. Front. Immunol. 2015, 6, 588. [Google Scholar] [CrossRef] [Green Version]
- Groeneveldt, C.; Kinderman, P.; van den Wollenberg, D.J.M.; van den Oever, R.L.; Middelburg, J.; Mustafa, D.A.M.; Hoeben, R.C.; van der Burg, S.H.; van Hall, T.; van Montfoort, N. Preconditioning of the tumor microenvironment with oncolytic reovirus converts CD3-bispecific antibody treatment into effective immunotherapy. J. ImmunoTher. Cancer 2020, 8, e001191. [Google Scholar] [CrossRef]
- Bhatt, D.K.; Chammas, R.; Daemen, T. Resistance Mechanisms Influencing Oncolytic Virotherapy, a Systematic Analysis. Vaccines 2021, 9, 1166. [Google Scholar] [CrossRef]
- Chen, F.Y.-S.; Chi, C.-W.; Shieh, H.-R.; Lin, C.-P.; Ko, C.-C.; Chung, Y.-C.; Lai, J.C.-Y.; Tai, H.-C.; Chen, Y.-J. Mitomycin C modulates tumor microenvironment and enhances radiosensitivity in rectal cancer. Ther. Radiol. Oncol. 2019, 3, 29. [Google Scholar] [CrossRef]
- Han, S.; Toker, A.; Liu, Z.Q.; Ohashi, P.S. Turning the Tide Against Regulatory T Cells. Front. Oncol. 2019, 9, 279. [Google Scholar] [CrossRef]
- Wang, D.; Quiros, J.; Mahuron, K.; Pai, C.C.; Ranzani, V.; Young, A.; Silveria, S.; Harwin, T.; Abnousian, A.; Pagani, M.; et al. Targeting EZH2 Reprograms Intratumoral Regulatory T Cells to Enhance Cancer Immunity. Cell Rep. 2018, 23, 3262–3274. [Google Scholar] [CrossRef]
- Paluskievicz, C.M.; Cao, X.; Abdi, R.; Zheng, P.; Liu, Y.; Bromberg, J.S. T Regulatory Cells and Priming the Suppressive Tumor Microenvironment. Front. Immunol. 2019, 10, 2453. [Google Scholar] [CrossRef] [Green Version]
- Showalter, A.; Limaye, A.; Oyer, J.L.; Igarashi, R.; Kittipatarin, C.; Copik, A.J.; Khaled, A.R. Cytokines in immunogenic cell death: Applications for cancer immunotherapy. Cytokine 2017, 97, 123–132. [Google Scholar] [CrossRef]
- Donnelly, O.G.; Errington-Mais, F.; Steele, L.; Hadac, E.; Jennings, V.; Scott, K.; Peach, H.; Phillips, R.M.; Bond, J.; Pandha, H.; et al. Measles virus causes immunogenic cell death in human melanoma. Gene 2013, 20, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef]
- Lv, M.; Xu, Y.; Tang, R.; Ren, J.; Shen, S.; Chen, Y.; Liu, B.; Hou, Y.; Wang, T. miR141–CXCL1–CXCR2 Signaling–Induced Treg Recruitment Regulates Metastases and Survival of Non–Small Cell Lung Cancer. Mol. Cancer Ther. 2014, 13, 3152–3162. [Google Scholar] [CrossRef] [Green Version]
- Krombach, J.; Hennel, R.; Brix, N.; Orth, M.; Schoetz, U.; Ernst, A.; Schuster, J.; Zuchtriegel, G.; Reichel, C.A.; Bierschenk, S.; et al. Priming anti-tumor immunity by radiotherapy: Dying tumor cell-derived DAMPs trigger endothelial cell activation and recruitment of myeloid cells. OncoImmunology 2019, 8, e1523097. [Google Scholar] [CrossRef] [Green Version]
- Engblom, C.; Pfirschke, C.; Pittet, M.J. The role of myeloid cells in cancer therapies. Nat. Rev. Cancer 2016, 16, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Neophytou, C.M.; Pierides, C.; Christodoulou, M.I.; Costeas, P.; Kyriakou, T.C.; Papageorgis, P. The Role of Tumor-Associated Myeloid Cells in Modulating Cancer Therapy. Front. Oncol. 2020, 10, 899. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davola, M.E.; Cormier, O.; Vito, A.; El-Sayes, N.; Collins, S.; Salem, O.; Revill, S.; Ask, K.; Wan, Y.; Mossman, K. Oncolytic BHV-1 Is Sufficient to Induce Immunogenic Cell Death and Synergizes with Low-Dose Chemotherapy to Dampen Immunosuppressive T Regulatory Cells. Cancers 2023, 15, 1295. https://doi.org/10.3390/cancers15041295
Davola ME, Cormier O, Vito A, El-Sayes N, Collins S, Salem O, Revill S, Ask K, Wan Y, Mossman K. Oncolytic BHV-1 Is Sufficient to Induce Immunogenic Cell Death and Synergizes with Low-Dose Chemotherapy to Dampen Immunosuppressive T Regulatory Cells. Cancers. 2023; 15(4):1295. https://doi.org/10.3390/cancers15041295
Chicago/Turabian StyleDavola, Maria Eugenia, Olga Cormier, Alyssa Vito, Nader El-Sayes, Susan Collins, Omar Salem, Spencer Revill, Kjetil Ask, Yonghong Wan, and Karen Mossman. 2023. "Oncolytic BHV-1 Is Sufficient to Induce Immunogenic Cell Death and Synergizes with Low-Dose Chemotherapy to Dampen Immunosuppressive T Regulatory Cells" Cancers 15, no. 4: 1295. https://doi.org/10.3390/cancers15041295