
Inhibition of RNA Polymerase III Augments the Anti-Cancer Properties of TNFα


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. MTT Assay
2.3. Clonogenic Cell Survival Assay
2.4. Stable Cell Lines
2.5. Protein Extracts and Western Blotting
2.6. Fractionation
2.7. RNA Isolation and cDNA Synthesis
2.8. Quantitative PCR
2.9. Scratch Wound Assay
2.10. Cell Proliferation Assay
2.11. Propidium Iodide Exclusion Assay
2.12. Cell Death Assay Using IncuCyte
2.13. NF-κB Induction Reporter Assay
2.14. Confocal and High-Content Microscopy
3. Results
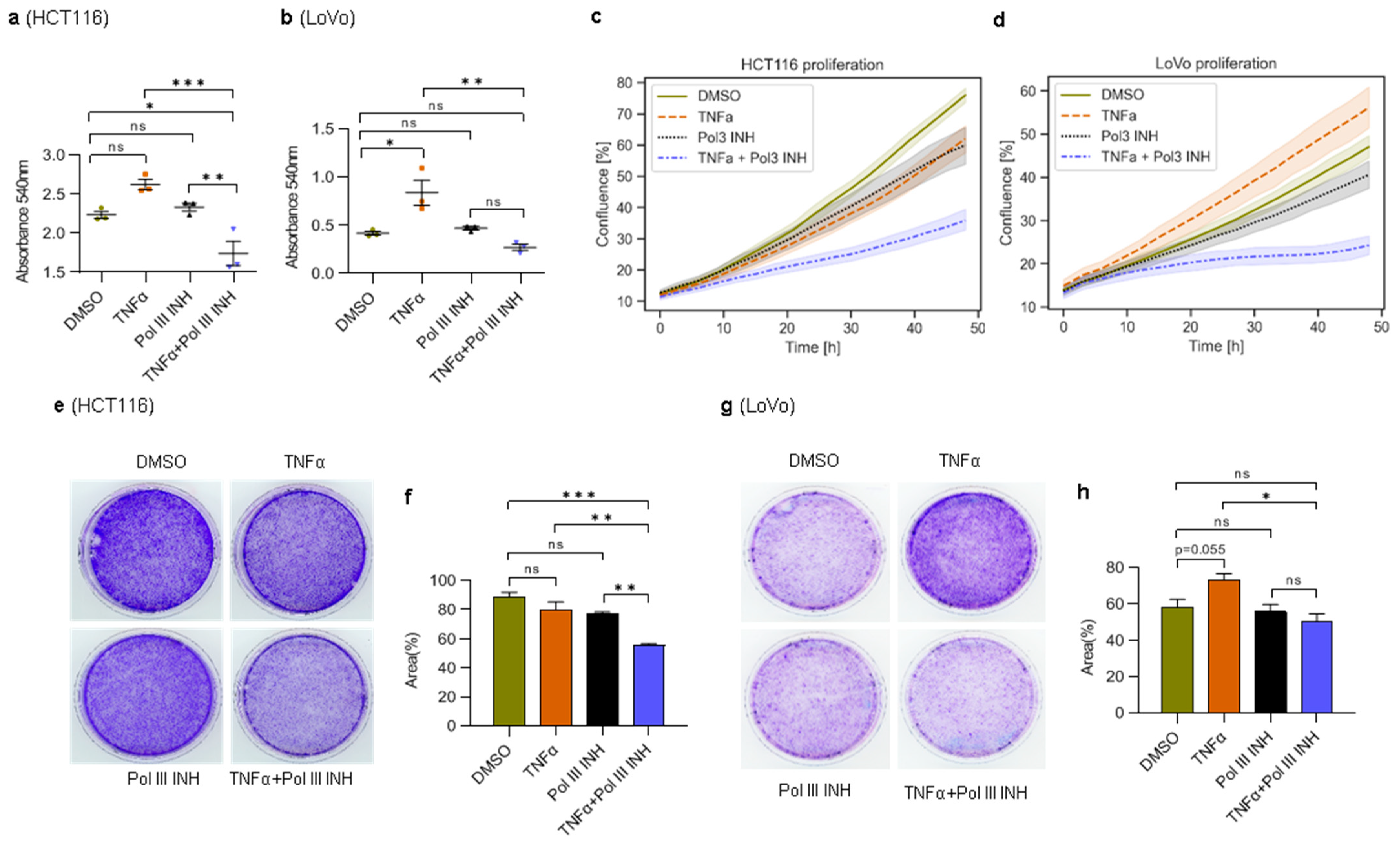
3.1. Inhibition of RNA Polymerase III Augments TNFα-Induced Cytotoxic and Cytostatic Effects in CRC Cells
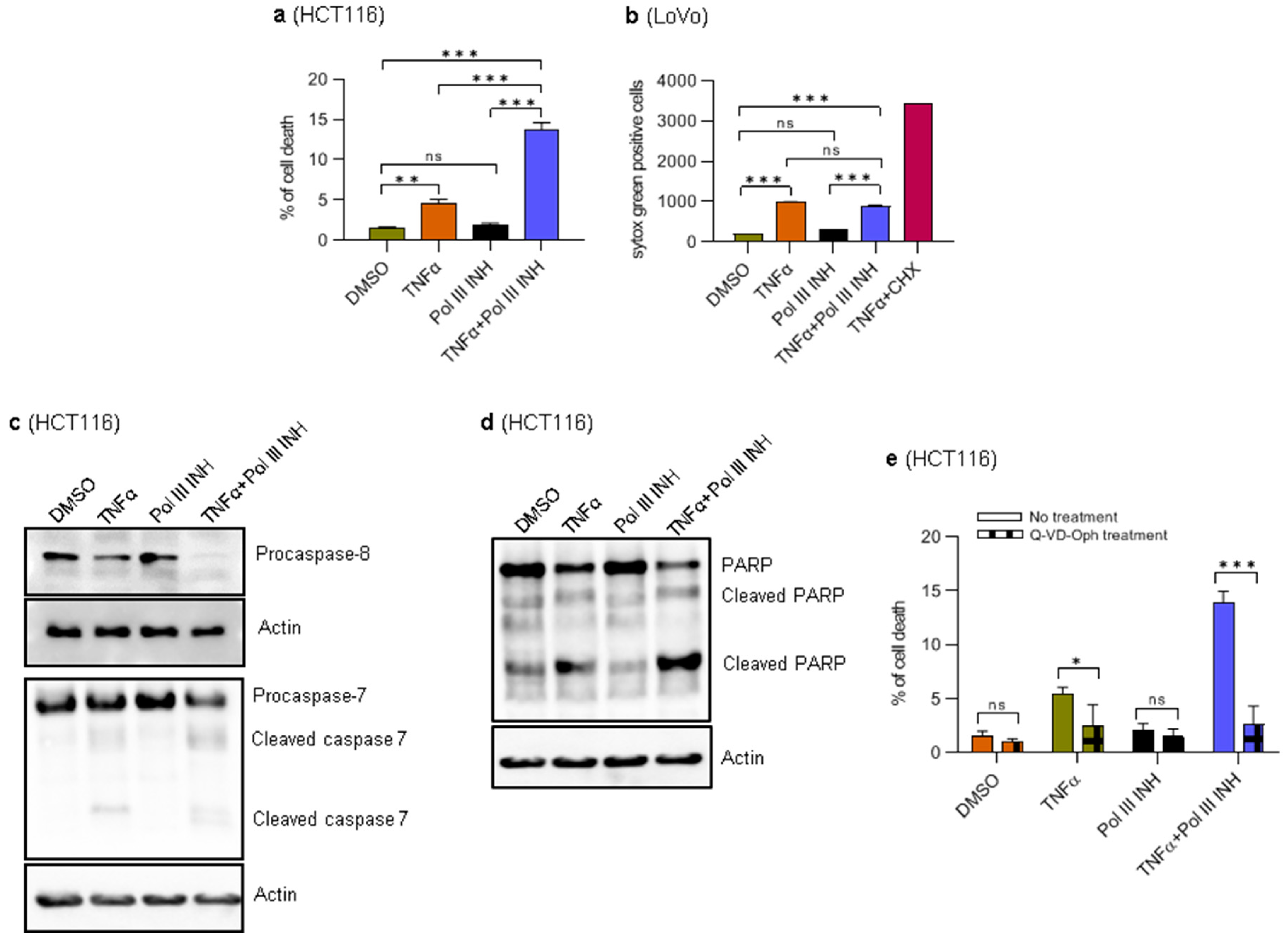
3.2. Combination of RNA Polymerase III Inhibitor with TNFα Induces Apoptosis in HCT116 Cells
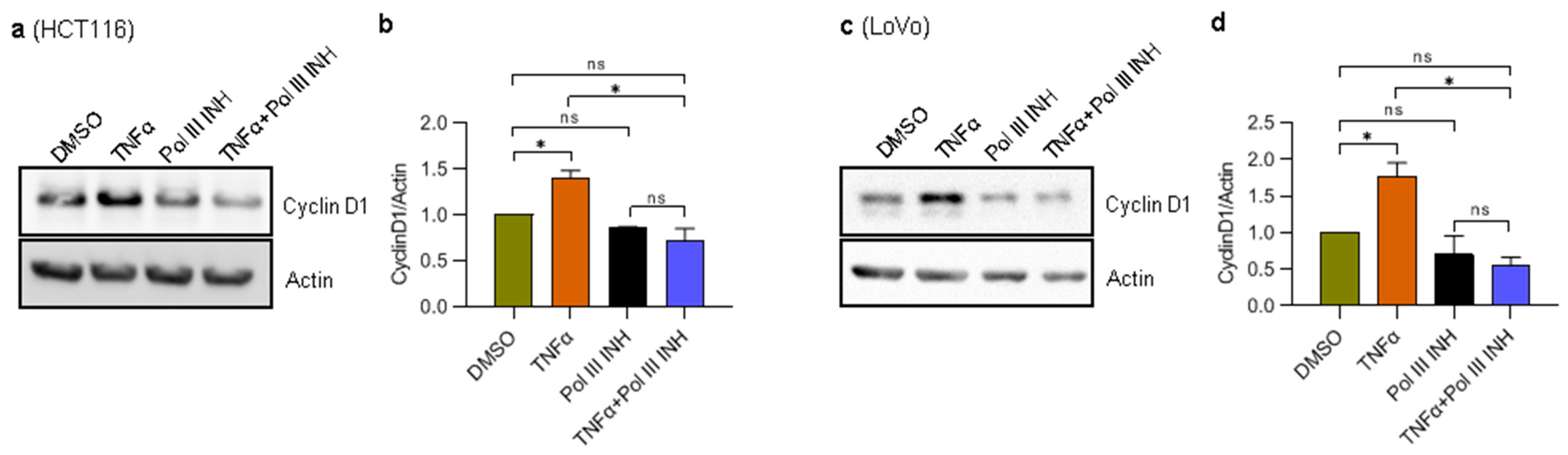
3.3. RNA Polymerase III Inhibition Affects the Levels of TNFα-Induced Cell Cycle Progression Markers in CRC Cells
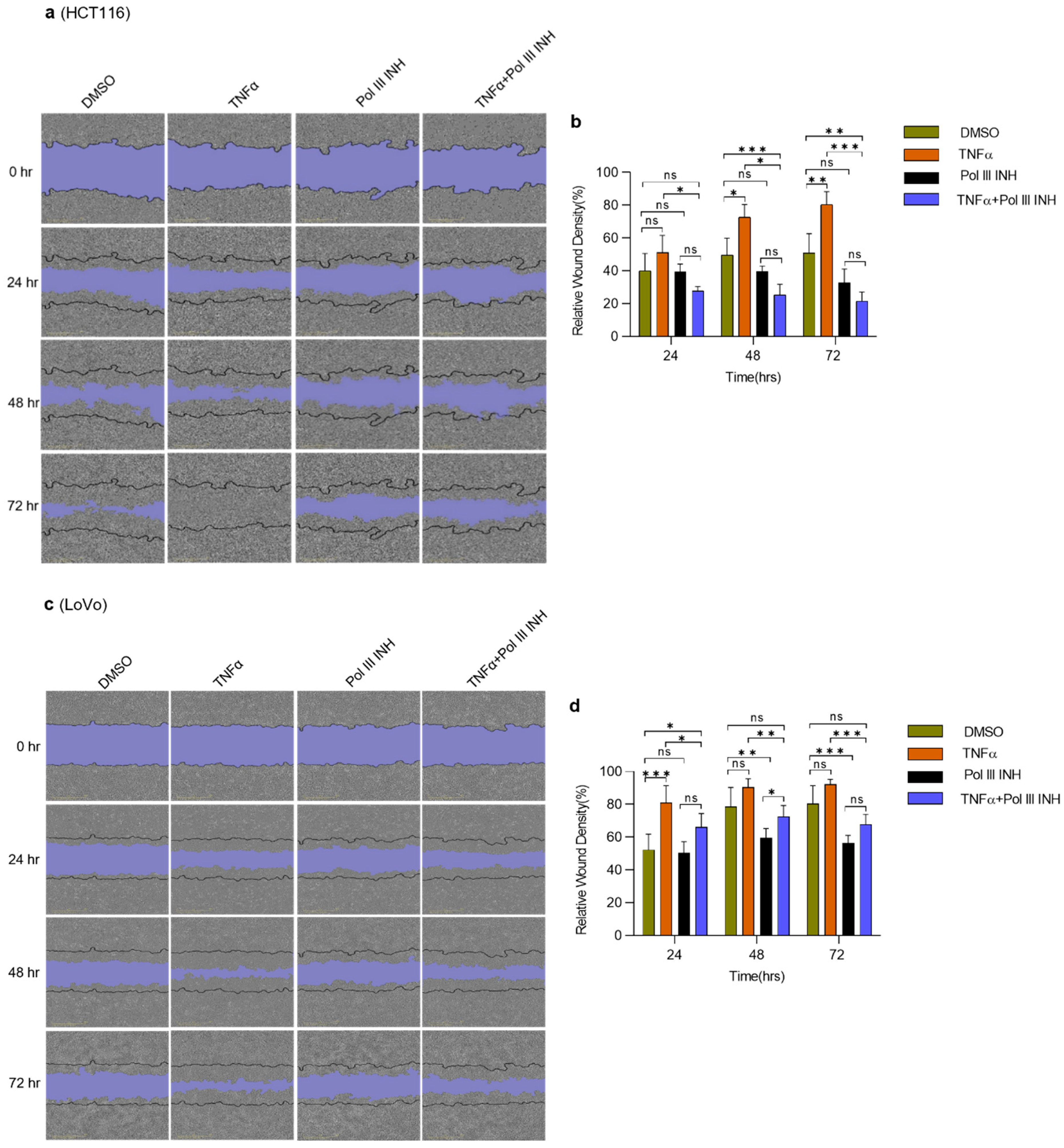
3.4. Inhibition of Pol III Decreases TNFα-Induced Migration in CRC Cells
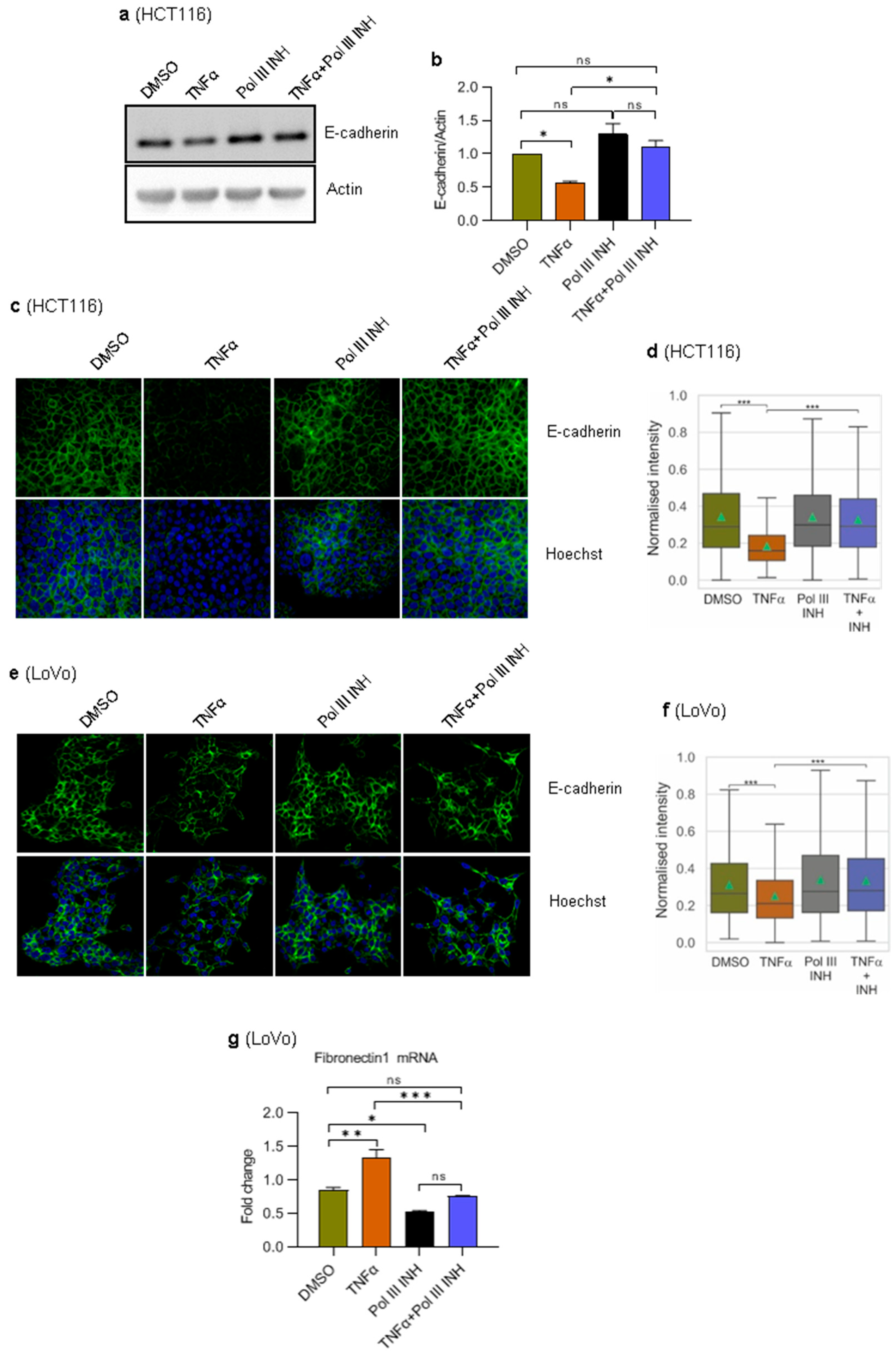
3.5. RNA Polymerase III Inhibition Blocks TNFα-Induced Suppression of E-Cadherin Expression in HCT116 Cells
3.6. Elevated Expression of Initiator Methionine tRNA Does Not Contribute to the Increased Proliferation and Migration of HCT116 Cells
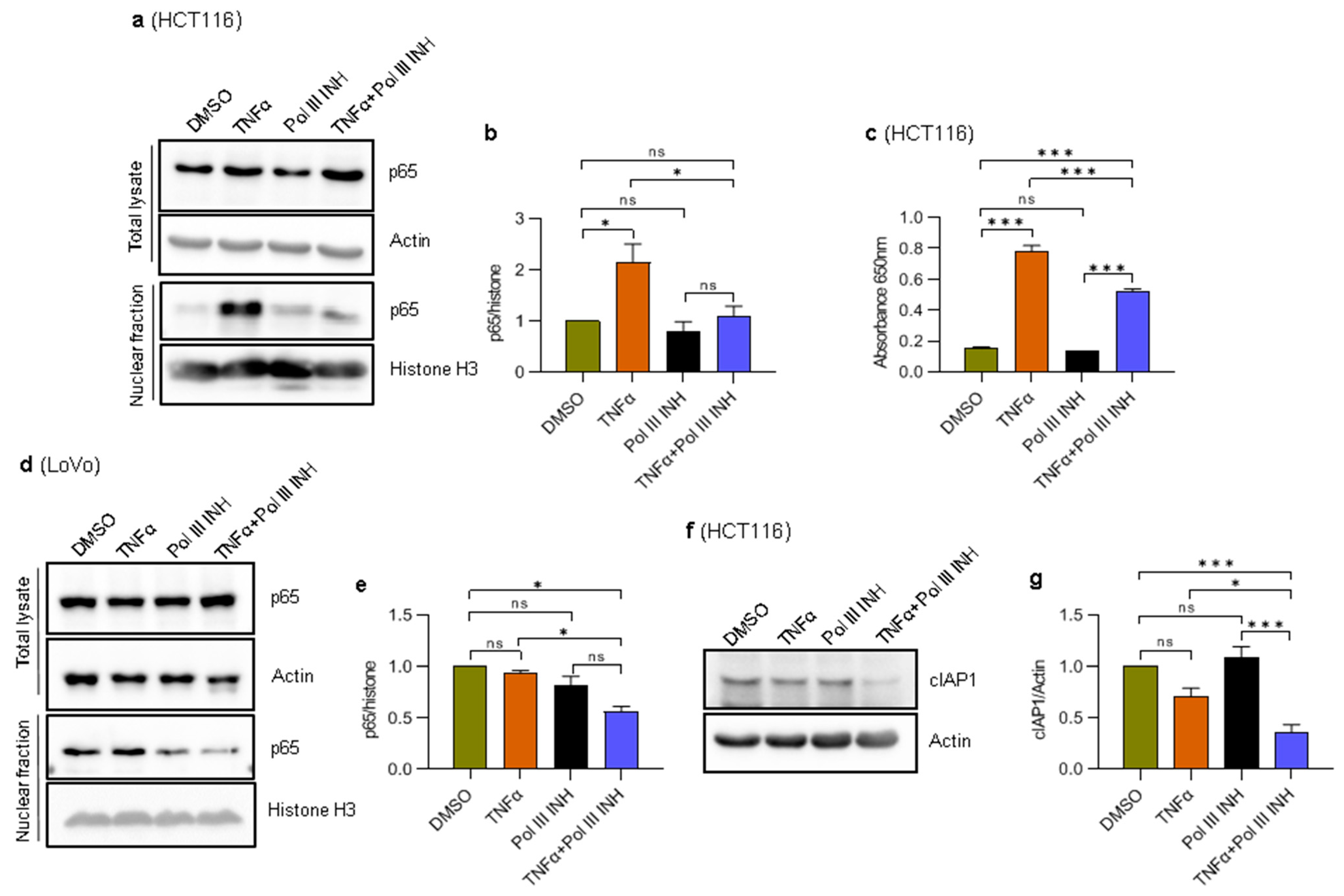
3.7. Inhibition of RNA Polymerase III Blocks TNFα-Induced NF-κB Activation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Bogaert, J.; Prenen, H. Molecular genetics of colorectal cancer. Annu. Gastroenterol. Q. Publ. Hell. Soc. Gastroenterol. 2014, 27, 9–14. [Google Scholar]
- Long, A.G.; Lundsmith, E.T.; Hamilton, K.E. Inflammation and Colorectal Cancer. Curr. Colorectal. Cancer Rep. 2017, 13, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- West, N.R.; McCuaig, S.; Franchini, F.; Powrie, F. Emerging cytokine networks in colorectal cancer. Nat. Rev. Immunol. 2015, 15, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Tumour necrosis factor and cancer. Nat. Rev. Cancer. 2009, 9, 361–371. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Gupta, S.C.; Kim, J.H. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood 2012, 119, 651–665. [Google Scholar] [CrossRef] [Green Version]
- Verhoef, C.; de Wilt, J.H.; Grunhagen, D.J.; van Geel, A.N.; ten Hagen, T.L.; Eggermont, A.M. Isolated limb perfusion with melphalan and TNF-alpha in the treatment of extremity sarcoma. Curr. Treat Options Oncol. 2007, 8, 417–427. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, B.B. Signalling pathways of the TNF superfamily: A double-edged sword. Nat. Rev. Immunol. 2003, 3, 745–756. [Google Scholar] [CrossRef]
- Wajant, H.; Siegmund, D. TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front. Cell Dev. Biol. 2019, 7, 91. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, K.; Karin, M. NF-kappaB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Perkins, N.D. The diverse and complex roles of NF-kappaB subunits in cancer. Nat. Rev. Cancer 2012, 12, 121–132. [Google Scholar] [CrossRef]
- Baldwin, A.S., Jr. The NF-kappa B and I kappa B proteins: New discoveries and insights. Annu. Rev. Immunol. 1996, 14, 649–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, S.J.; Lennon, S.V.; Bonham, A.M.; Cotter, T.G. Induction of apoptosis (programmed cell death) in human leukemic HL-60 cells by inhibition of RNA or protein synthesis. J. Immunol. 1990, 145, 1859–1867. [Google Scholar] [CrossRef]
- Graczyk, D.; White, R.J.; Ryan, K.M. Involvement of RNA Polymerase III in Immune Responses. Mol. Cell Biol. 2015, 35, 1848–1859. [Google Scholar] [CrossRef] [Green Version]
- White, R.J. RNA polymerases I and III, growth control and cancer. Nat. Rev. Mol. Cell Biol. 2005, 6, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.A.; Dubeau, L.; Johnson, D.L. Enhanced RNA polymerase III-dependent transcription is required for oncogenic transformation. J. Biol. Chem. 2008, 283, 19184–19191. [Google Scholar] [CrossRef] [Green Version]
- Petrie, J.L.; Swan, C.; Ingram, R.M.; Frame, F.M.; Collins, A.T.; Dumay-Odelot, H.; Teichmann, M.; Maitland, N.J.; White, R.J. Effects on prostate cancer cells of targeting RNA polymerase III. Nucleic Acids Res. 2019, 47, 3937–3956. [Google Scholar] [CrossRef] [Green Version]
- Grewal, S.S. Why should cancer biologists care about tRNAs? tRNA synthesis, mRNA translation and the control of growth. Biochim. Biophys. Acta 2015, 1849, 898–907. [Google Scholar] [CrossRef]
- Bensaude, O. Inhibiting eukaryotic transcription: Which compound to choose? How to evaluate its activity? Transcription 2011, 2, 103–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beg, A.A.; Baltimore, D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science 1996, 274, 782–784. [Google Scholar] [CrossRef] [PubMed]
- Van Antwerp, D.J.; Martin, S.J.; Kafri, T.; Green, D.R.; Verma, I.M. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science 1996, 274, 787–789. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Mayo, M.W.; Baldwin, A.S., Jr. TNF- and cancer therapy-induced apoptosis: Potentiation by inhibition of NF-kappaB. Science 1996, 274, 784–787. [Google Scholar] [CrossRef]
- Birch, J.; Clarke, C.J.; Campbell, A.D.; Campbell, K.; Mitchell, L.; Liko, D.; Kalna, G.; Strathdee, D.; Sansom, O.J.; Neilson, M.; et al. The initiator methionine tRNA drives cell migration and invasion leading to increased metastatic potential in melanoma. Biol. Open 2016, 5, 1371–1379. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Bose, P.; Leong-Quong, R.Y.; Fujita, D.J.; Riabowol, K. REAP: A two minute cell fractionation method. BMC Res. Notes 2010, 3, 294. [Google Scholar] [CrossRef] [Green Version]
- Pietras, Z.; Wojcik, M.A.; Borowski, L.S.; Szewczyk, M.; Kulinski, T.M.; Cysewski, D.; Stepien, P.P.; Dziembowski, A.; Szczesny, R.J. Dedicated surveillance mechanism controls G-quadruplex forming non-coding RNAs in human mitochondria. Nat. Commun. 2018, 9, 2558. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Du, F.; Wang, X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008, 133, 693–703. [Google Scholar] [CrossRef] [Green Version]
- Tummers, B.; Green, D.R. Caspase-8: Regulating life and death. Immunol. Rev. 2017, 277, 76–89. [Google Scholar] [CrossRef] [Green Version]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef] [Green Version]
- Caserta, T.M.; Smith, A.N.; Gultice, A.D.; Reedy, M.A.; Brown, T.L. Q-VD-OPh, a broad spectrum caspase inhibitor with potent antiapoptotic properties. Apoptosis 2003, 8, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Sugarman, B.J.; Aggarwal, B.B.; Hass, P.E.; Figari, I.S.; Palladino, M.A., Jr.; Shepard, H.M. Recombinant human tumor necrosis factor-alpha: Effects on proliferation of normal and transformed cells in vitro. Science 1985, 230, 943–945. [Google Scholar] [CrossRef]
- Rubio, M.F.; Werbajh, S.; Cafferata, E.G.; Quaglino, A.; Colo, G.P.; Nojek, I.M.; Kordon, E.C.; Nahmod, V.E.; Costas, M.A. TNF-alpha enhances estrogen-induced cell proliferation of estrogen-dependent breast tumor cells through a complex containing nuclear factor-kappa B. Oncogene 2006, 25, 1367–1377. [Google Scholar] [CrossRef] [Green Version]
- Qie, S.; Diehl, J.A. Cyclin D1, cancer progression, and opportunities in cancer treatment. J. Mol. Med. 2016, 94, 1313–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef] [PubMed]
- Li, C.W.; Xia, W.; Huo, L.; Lim, S.O.; Wu, Y.; Hsu, J.L.; Chao, C.H.; Yamaguchi, H.; Yang, N.K.; Ding, Q.; et al. Epithelial-mesenchymal transition induced by TNF-alpha requires NF-kappaB-mediated transcriptional upregulation of Twist1. Cancer Res. 2012, 72, 1290–1300. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Zhang, Z. TNF-alpha promotes colon cancer cell migration and invasion by upregulating TROP-2. Oncol. Lett. 2018, 15, 3820–3827. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- van Roy, F.; Berx, G. The cell-cell adhesion molecule E-cadherin. Cell Mol. Life Sci. 2008, 65, 3756–3788. [Google Scholar] [CrossRef]
- Kim, Y.J.; Kang, H.B.; Yim, H.S.; Kim, J.H.; Kim, J.W. NDRG2 positively regulates E-cadherin expression and prolongs overall survival in colon cancer patients. Oncol. Rep. 2013, 30, 1890–1898. [Google Scholar] [CrossRef] [Green Version]
- Sudo, T.; Iwaya, T.; Nishida, N.; Sawada, G.; Takahashi, Y.; Ishibashi, M.; Shibata, K.; Fujita, H.; Shirouzu, K.; Mori, M.; et al. Expression of mesenchymal markers vimentin and fibronectin: The clinical significance in esophageal squamous cell carcinoma. Annu. Surg. Oncol. 2013, 20 (Suppl. 3), S324–S335. [Google Scholar] [CrossRef]
- Pavon-Eternod, M.; Gomes, S.; Rosner, M.R.; Pan, T. Overexpression of initiator methionine tRNA leads to global reprogramming of tRNA expression and increased proliferation in human epithelial cells. RNA 2013, 19, 461–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guttridge, D.C.; Albanese, C.; Reuther, J.Y.; Pestell, R.G.; Baldwin, A.S., Jr. NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol. Cell Biol. 1999, 19, 5785–5799. [Google Scholar] [CrossRef] [Green Version]
- Hinz, M.; Krappmann, D.; Eichten, A.; Heder, A.; Scheidereit, C.; Strauss, M. NF-kappaB function in growth control: Regulation of cyclin D1 expression and G0/G1-to-S-phase transition. Mol. Cell Biol. 1999, 19, 2690–2698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karin, M.; Lin, A. NF-kappaB at the crossroads of life and death. Nat. Immunol. 2002, 3, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.C.; Lee, N.H.; Hsu, H.H.; Ho, T.J.; Tu, C.C.; Chen, R.J.; Lin, Y.M.; Viswanadha, V.P.; Kuo, W.W.; Huang, C.Y. Inhibition of NF-kappaB and metastasis in irinotecan (CPT-11)-resistant LoVo colon cancer cells by thymoquinone via JNK and p38. Environ. Toxicol. 2017, 32, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Gao, J.; Zhang, X.; Peng, Y.; Wei, W.; Xu, J.; Li, Z.; Wang, C.; Zhou, M.; Tian, X.; et al. Characterization of a small-molecule inhibitor targeting NEMO/IKKbeta to suppress colorectal cancer growth. Signal Transduct. Target. Ther. 2022, 7, 71. [Google Scholar] [CrossRef]
- Ioele, G.; Chieffallo, M.; Occhiuzzi, M.A.; De Luca, M.; Garofalo, A.; Ragno, G.; Grande, F. Anticancer Drugs: Recent Strategies to Improve Stability Profile, Pharmacokinetic and Pharmacodynamic Properties. Molecules 2022, 27, 5436. [Google Scholar] [CrossRef]
- Chua, H.L.; Bhat-Nakshatri, P.; Clare, S.E.; Morimiya, A.; Badve, S.; Nakshatri, H. NF-kappaB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: Potential involvement of ZEB-1 and ZEB-2. Oncogene 2007, 26, 711–724. [Google Scholar] [CrossRef] [Green Version]
- Brlek, P.; Bukovac, A.; Kafka, A.; Pecina-Slaus, N. TWIST1 upregulation affects E-cadherin expression in brain metastases. Clin. Transl. Oncol. 2021, 23, 1085–1095. [Google Scholar] [CrossRef]
- Zhu, Q.Q.; Ma, C.; Wang, Q.; Song, Y.; Lv, T. The role of TWIST1 in epithelial-mesenchymal transition and cancers. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Schumacher, S.E.; Wu, W.H.; Tang, F.; Beroukhim, R.; Chan, T.A. Pan-Cancer Analysis Links PARK2 to BCL-XL-Dependent Control of Apoptosis. Neoplasia 2017, 19, 75–83. [Google Scholar] [CrossRef]
- Thomas, L.W.; Lam, C.; Edwards, S.W. Mcl-1; the molecular regulation of protein function. FEBS Lett. 2010, 584, 2981–2989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitts, S.; Laiho, M. Regulation of RNA Polymerase I Stability and Function. Cancers 2022, 14, 5776. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nair, H.G.; Jurkiewicz, A.; Graczyk, D. Inhibition of RNA Polymerase III Augments the Anti-Cancer Properties of TNFα. Cancers 2023, 15, 1495. https://doi.org/10.3390/cancers15051495
Nair HG, Jurkiewicz A, Graczyk D. Inhibition of RNA Polymerase III Augments the Anti-Cancer Properties of TNFα. Cancers. 2023; 15(5):1495. https://doi.org/10.3390/cancers15051495
Chicago/Turabian StyleNair, Hitha Gopalan, Aneta Jurkiewicz, and Damian Graczyk. 2023. "Inhibition of RNA Polymerase III Augments the Anti-Cancer Properties of TNFα" Cancers 15, no. 5: 1495. https://doi.org/10.3390/cancers15051495
APA StyleNair, H. G., Jurkiewicz, A., & Graczyk, D. (2023). Inhibition of RNA Polymerase III Augments the Anti-Cancer Properties of TNFα. Cancers, 15(5), 1495. https://doi.org/10.3390/cancers15051495