

Emerging Therapies in CLL in the Era of Precision Medicine

{kind=link}
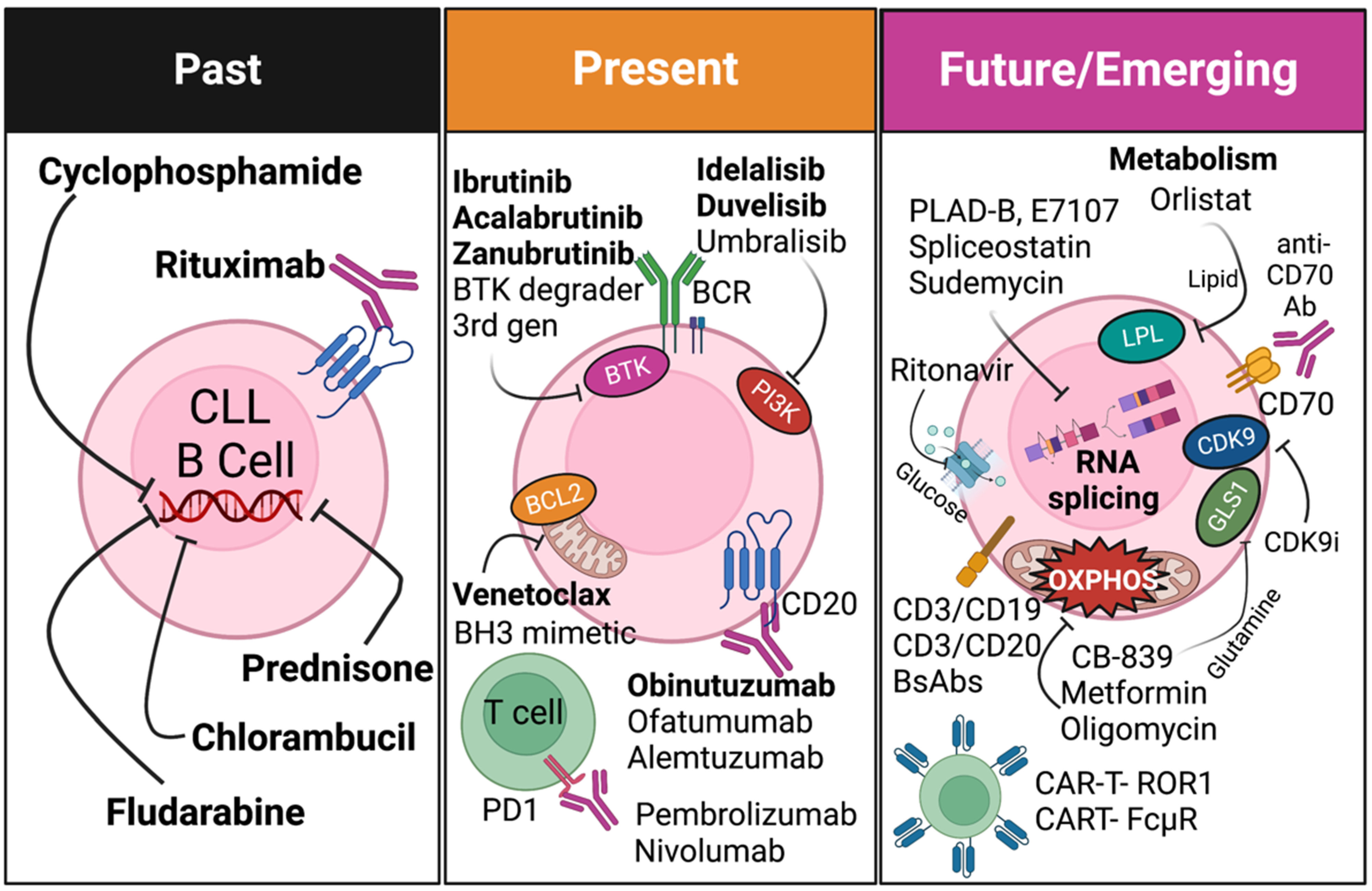{kind=link}
Simple Summary
Abstract
1. Introduction
2. Chemoimmunotherapy
2.1. Fludarabine, Cyclophosphamide, and Rituximab (FCR)
2.2. Monoclonal Antibody Treatment in CLL
3. Targeted Therapies
3.1. Targeting B-Cell Receptor (BCR) Signaling in CLL
3.2. Dasatinib and Ibrutinib
3.3. PI3 Kinase Inhibitors
3.3.1. Idelalisib (p110-PI3Kδ Inhibitor)
3.3.2. Duvelisib
3.3.3. Umbralisib
3.4. Targeting BCL2
4. Immunomodulatory Agents
4.1. Immune-Checkpoint Inhibitors
4.2. Bispecific Antibodies
4.3. Bi- or Tri-Specific Killer Engagers
4.4. CAR T-Cells
5. Emerging Novel Targets for CLL Treatment
5.1. Targeting RNA Splicing Dysregulation in CLL
5.2. Targeting Metabolism in CLL
5.2.1. Mitochondrial Metabolism
5.2.2. Glucose Metabolism
5.2.3. Glutamine Metabolism
5.2.4. Lipid Metabolism
6. Future Therapies
6.1. Combination Therapies
6.1.1. Targeted Therapy Coupled with Other Novel Chemotherapy
6.1.2. Novel Agents in Combination with Anti-CD20 Antibodies
6.1.3. Targeted Therapy Coupled with an Immunomodulatory Agent
6.2. Allogenic Transplant
6.3. Recent Targets and Trials in CLL Discussed in ASH2022
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- DeSantis, C.E.; Miller, K.D.; Dale, W.; Mohile, S.G.; Cohen, H.J.; Leach, C.R.; Sauer, A.G.; Jemal, A.; Siegel, R.L. Cancer statistics for adults aged 85 years and older, 2019. CA Cancer J. Clin. 2019, 69, 452–467. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Quinquenel, A.; Fornecker, L.-M.; Letestu, R.; Ysebaert, L.; Fleury, C.; Lazarian, G.; Dilhuydy, M.-S.; Nollet, D.; Guieze, R.; Feugier, P.; et al. Prevalence of BTK and PLCG2 mutations in a real-life CLL cohort still on ibrutinib after 3 years: A FILO group study. Blood 2019, 134, 641–644. [Google Scholar] [CrossRef]
- Landau, D.A.; Tausch, E.; Taylor-Weiner, A.N.; Stewart, C.; Reiter, J.G.; Bahlo, J.; Kluth, S.; Bozic, I.; Lawrence, M.; Böttcher, S.; et al. Mutations driving CLL and their evolution in progression and relapse. Nature 2015, 526, 525–530. [Google Scholar] [CrossRef]
- Puente, X.S.; Beà, S.; Valdés-Mas, R.; Villamor, N.; Gutiérrez-Abril, J.; Martín-Subero, J.I.; Munar, M.; Rubio-Pérez, C.; Jares, P.; Aymerich, M.; et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015, 526, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Knisbacher, B.A.; Lin, Z.; Hahn, C.K.; Nadeu, F.; Duran-Ferrer, M.; Stevenson, K.E.; Tausch, E.; Delgado, J.; Barbera-Mourelle, A.; Taylor-Weiner, A.; et al. Molecular map of chronic lymphocytic leukemia and its impact on outcome. Nat. Genet. 2022, 54, 1664–1674. [Google Scholar] [CrossRef]
- Puente, X.S.; Pinyol, M.; Quesada, V.; Conde, L.; Ordóñez, G.R.; Villamor, N.; Escaramis, G.; Jares, P.; Beà, S.; González-Díaz, M.; et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 2011, 475, 101–105. [Google Scholar] [CrossRef]
- Jeromin, S.; Weissmann, S.; Haferlach, C.; Dicker, F.; Bayer, K.; Grossmann, V.; Alpermann, T.; Roller, A.; Kohlmann, A.; Kern, W.; et al. SF3B1 mutations correlated to cytogenetics and mutations in NOTCH1, FBXW7, MYD88, XPO1 and TP53 in 1160 untreated CLL patients. Leukemia 2014, 28, 108–117. [Google Scholar] [CrossRef]
- Chiaretti, S.; Marinelli, M.; Del Giudice, I.; Bonina, S.; Piciocchi, A.; Messina, M.; Vignetti, M.; Rossi, D.; Di Maio, V.; Mauro, F.R.; et al. NOTCH1, SF3B1, BIRC3 and TP53 mutations in patients with chronic lymphocytic leukemia undergoing first-line treatment: Correlation with biological parameters and response to treatment. Leuk. Lymphoma 2014, 55, 2785–2792. [Google Scholar] [CrossRef]
- Rossi, D.; Rasi, S.; Fabbri, G.; Spina, V.; Fangazio, M.; Forconi, F.; Marasca, R.; Laurenti, L.; Bruscaggin, A.; Cerri, M.; et al. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood 2012, 119, 521–529. [Google Scholar] [CrossRef]
- Wang, L.; Lawrence, M.S.; Wan, Y.; Stojanov, P.; Sougnez, C.; Stevenson, K.; Werner, L.; Sivachenko, A.; DeLuca, D.S.; Zhang, L.; et al. SF3B1 and Other Novel Cancer Genes in Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2011, 365, 2497–2506. [Google Scholar] [CrossRef] [PubMed]
- Landau, D.A.; Carter, S.L.; Stojanov, P.; McKenna, A.; Stevenson, K.; Lawrence, M.S.; Sougnez, C.; Stewart, C.; Sivachenko, A.; Wang, L.; et al. Evolution and Impact of Subclonal Mutations in Chronic Lymphocytic Leukemia. Cell 2013, 152, 714–726. [Google Scholar] [CrossRef]
- Baliakas, P.; Hadzidimitriou, A.; Sutton, L.-A.; Rossi, D.; Minga, E.; Villamor, N.; Larrayoz, M.; Kminkova, J.; Agathangelidis, A.; Davis, Z.; et al. Recurrent mutations refine prognosis in chronic lymphocytic leukemia. Leukemia 2015, 29, 329–336. [Google Scholar] [CrossRef]
- Rossi, D.; Rasi, S.; Spina, V.; Bruscaggin, A.; Monti, S.; Ciardullo, C.; Deambrogi, C.; Khiabanian, H.; Serra, R.; Bertoni, F.; et al. Integrated mutational and cytogenetic analysis identifies new prognostic subgroups in chronic lymphocytic leukemia. Blood 2013, 121, 1403–1412. [Google Scholar] [CrossRef] [PubMed]
- Shuai, S.; Suzuki, H.; Diaz-Navarro, A.; Nadeu, F.; Kumar, S.A.; Gutierrez-Fernandez, A.; Delgado, J.; Pinyol, M.; López-Otín, C.; Puente, X.S.; et al. The U1 spliceosomal RNA is recurrently mutated in multiple cancers. Nature 2019, 574, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Ten Hacken, E.; Valentin, R.; Regis, F.F.D.; Sun, J.; Yin, S.; Werner, L.; Deng, J.; Gruber, M.; Wong, J.; Zheng, M.; et al. Splicing modulation sensitizes chronic lymphocytic leukemia cells to venetoclax by remodeling mitochondrial apoptotic dependencies. JCI Insight 2018, 3, e121438. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Simon-Molas, H.; Cretenet, G.; Valle-Argos, B.; Smith, L.D.; Forconi, F.; Schomakers, B.V.; van Weeghel, M.; Bryant, D.J.; van Bruggen, J.A.C.; et al. Characterization of metabolic alterations of chronic lymphocytic leukemia in the lymph node microenvironment. Blood 2022, 140, 630–643. [Google Scholar] [CrossRef]
- Tili, E.; Michaille, J.-J.; Luo, Z.; Volinia, S.; Rassenti, L.Z.; Kipps, T.J.; Croce, C.M. The down-regulation of miR-125b in chronic lymphocytic leukemias leads to metabolic adaptation of cells to a transformed state. Blood 2012, 120, 2631–2638. [Google Scholar] [CrossRef]
- Vangapandu, H.V.; Ayres, M.L.; Bristow, C.A.; Wierda, W.G.; Keating, M.J.; Balakrishnan, K.; Stellrecht, C.M.; Gandhi, V. The Stromal Microenvironment Modulates Mitochondrial Oxidative Phosphorylation in Chronic Lymphocytic Leukemia Cells. Neoplasia 2017, 19, 762–771. [Google Scholar] [CrossRef]
- Sharma, S.; Rai, K.R. Chronic lymphocytic leukemia (CLL) treatment: So many choices, such great options. Cancer 2019, 125, 1432–1440. [Google Scholar] [CrossRef]
- Eichhorst, B.F.; Busch, R.; Hopfinger, G.; Pasold, R.; Hensel, M.; Steinbrecher, C.; Siehl, S.; Jäger, U.; Bergmann, M.; Stilgenbauer, S.; et al. Fludarabine plus cyclophosphamide versus fludarabine alone in first-line therapy of younger patients with chronic lymphocytic leukemia. Blood 2006, 107, 885–891. [Google Scholar] [CrossRef]
- Hallek, M.; Fischer, K.; Fingerle-Rowson, G.; Fink, A.; Busch, R.; Mayer, J.; Hensel, M.; Hopfinger, G.; Hess, G.; von Grünhagen, U.; et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: A randomised, open-label, phase 3 trial. Lancet 2010, 376, 1164–1174. [Google Scholar] [CrossRef] [PubMed]
- Eichhorst, B.F.; Busch, R.; Stilgenbauer, S.; Stauch, M.; Bergmann, M.A.; Ritgen, M.; Kranzhöfer, N.; Rohrberg, R.; Söling, U.; Burkhard, O.; et al. First-line therapy with fludarabine compared with chlorambucil does not result in a major benefit for elderly patients with advanced chronic lymphocytic leukemia. Blood 2009, 114, 3382–3391. [Google Scholar] [CrossRef]
- Tam, C.S.; O’Brien, S.; Wierda, W.; Kantarjian, H.; Wen, S.; Do, K.-A.; Thomas, D.A.; Cortes, J.; Lerner, S.; Keating, M.J. Long-term results of the fludarabine, cyclophosphamide, and rituximab regimen as initial therapy of chronic lymphocytic leukemia. Blood 2008, 112, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.A.; Tam, C.S.; O’Brien, S.M.; Wierda, W.G.; Stingo, F.C.; Plunkett, W.; Smith, S.C.; Kantarjian, H.M.; Freireich, E.J.; Keating, M.J. Fludarabine, cyclophosphamide, and rituximab treatment achieves long-term disease-free survival in IGHV-mutated chronic lymphocytic leukemia. Blood 2016, 127, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Shanafelt, T.D.; Wang, X.V.; Hanson, C.A.; Paietta, E.M.; O’Brien, S.; Barrientos, J.; Jelinek, D.F.; Braggio, E.; Leis, J.F.; Zhang, C.C.; et al. Long-term outcomes for ibrutinib-rituximab and chemoimmunotherapy in CLL: Updated results of the E1912 trial. Blood 2022, 140, 112–120. [Google Scholar] [CrossRef]
- Eichhorst, B.; Fink, A.-M.; Bahlo, J.; Busch, R.; Kovacs, G.; Maurer, C.; Lange, E.; Köppler, H.; Kiehl, M.; Sökler, M.; et al. First-line chemoimmunotherapy with bendamustine and rituximab versus fludarabine, cyclophosphamide, and rituximab in patients with advanced chronic lymphocytic leukaemia (CLL10): An international, open-label, randomised, phase 3, non-inferiority trial. Lancet Oncol. 2016, 17, 928–942. [Google Scholar] [CrossRef]
- Hagenbeek, A.; Gadeberg, O.; Johnson, P.; Møller Pedersen, L.; Walewski, J.; Hellmann, A.; Link, B.K.; Robak, T.; Wojtukiewicz, M.; Pfreundschuh, M.; et al. First clinical use of ofatumumab, a novel fully human anti-CD20 monoclonal antibody in relapsed or refractory follicular lymphoma: Results of a phase 1/2 trial. Blood 2008, 111, 5486–5495. [Google Scholar] [CrossRef]
- Van Oers, M.H.; Kuliczkowski, K.; Smolej, L.; Petrini, M.; Offner, F.; Grosicki, S.; Levin, M.D.; Gupta, I.; Phillips, J.; Williams, V.; et al. Ofatumumab maintenance versus observation in relapsed chronic lymphocytic leukaemia (PROLONG): An open-label, multicentre, randomised phase 3 study. Lancet Oncol. 2015, 16, 1370–1379. [Google Scholar] [CrossRef]
- Wierda, W.G.; Kipps, T.J.; Dürig, J.; Griskevicius, L.; Stilgenbauer, S.; Mayer, J.; Smolej, L.; Hess, G.; Griniute, R.; Hernandez-Ilizaliturri, F.J.; et al. Chemoimmunotherapy with O-FC in previously untreated patients with chronic lymphocytic leukemia. Blood 2011, 117, 6450–6458. [Google Scholar] [CrossRef]
- Tedeschi, A.; Rossi, D.; Motta, M.; Quaresmini, G.; Rossi, M.; Coscia, M.; Anastasia, A.; Rossini, F.; Cortelezzi, A.; Nador, G.; et al. A phase II multi-center trial of pentostatin plus cyclophosphamide with ofatumumab in older previously untreated chronic lymphocytic leukemia patients. Haematologica 2015, 100, e501–e504. [Google Scholar] [CrossRef] [PubMed]
- Hillmen, P.; Skotnicki, A.B.; Robak, T.; Jaksic, B.; Dmoszynska, A.; Wu, J.; Sirard, C.; Mayer, J. Alemtuzumab Compared with Chlorambucil As First-Line Therapy for Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2007, 25, 5616–5623. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.A.; Keating, M.J.; O’Brien, S.; Wang, X.; Ferrajoli, A.; Faderl, S.; Burger, J.; Koller, C.; Estrov, Z.; Badoux, X.; et al. Frontline chemoimmunotherapy with fludarabine, cyclophosphamide, alemtuzumab, and rituximab for high-risk chronic lymphocytic leukemia. Blood 2011, 118, 2062–2068. [Google Scholar] [CrossRef]
- Cartron, G.; de Guibert, S.; Dilhuydy, M.S.; Morschhauser, F.; Leblond, V.; Dupuis, J.; Mahe, B.; Bouabdallah, R.; Lei, G.; Wenger, M.; et al. Obinutuzumab (GA101) in relapsed/refractory chronic lymphocytic leukemia: Final data from the phase 1/2 GAUGUIN study. Blood 2014, 124, 2196–2202. [Google Scholar] [CrossRef]
- Goede, V.; Fischer, K.; Busch, R.; Engelke, A.; Eichhorst, B.; Wendtner, C.M.; Chagorova, T.; de la Serna, J.; Dilhuydy, M.-S.; Illmer, T.; et al. Obinutuzumab plus Chlorambucil in Patients with CLL and Coexisting Conditions. N. Engl. J. Med. 2014, 370, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Al-Sawaf, O.; Gentile, B.; Devine, J.; Zhang, C.; Sail, K.; Tandon, M.; Fink, A.M.; Kutsch, N.; Wendtner, C.M.; Eichhorst, B.; et al. Health-related quality of life with fixed-duration venetoclax-obinutuzumab for previously untreated chronic lymphocytic leukemia: Results from the randomized, phase 3 CLL14 trial. Am. J. Hematol. 2021, 96, 1112–1119. [Google Scholar] [CrossRef]
- Tanaka, S.; Baba, Y. B Cell Receptor Signaling. Adv. Exp. Med. Biol. 2020, 1254, 23–36. [Google Scholar]
- Danilov, A.V. Targeted Therapy in Chronic Lymphocytic Leukemia: Past, Present, and Future. Clin. Ther. 2013, 35, 1258–1270. [Google Scholar] [CrossRef]
- Kwak, K.; Akkaya, M.; Pierce, S.K. B cell signaling in context. Nat. Immunol. 2019, 20, 963–969. [Google Scholar] [CrossRef]
- Woyach, J.A.; Johnson, A.J.; Byrd, J.C. The B-cell receptor signaling pathway as a therapeutic target in CLL. Blood 2012, 120, 1175–1184. [Google Scholar] [CrossRef]
- Stevenson, F.K.; Caligaris-Cappio, F. Chronic lymphocytic leukemia: Revelations from the B-cell receptor. Blood 2004, 103, 4389–4395. [Google Scholar] [CrossRef] [PubMed]
- Minden, M.D.V.; Übelhart, R.; Schneider, D.; Wossning, T.; Bach, M.P.; Buchner, M.; Hofmann, D.; Surova, E.; Follo, M.; Köhler, F.; et al. Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature 2012, 489, 309–312. [Google Scholar] [CrossRef]
- Burger, J.A.; Chiorazzi, N. B cell receptor signaling in chronic lymphocytic leukemia. Trends Immunol. 2013, 34, 592–601. [Google Scholar] [CrossRef]
- Ten Hacken, E.; Gounari, M.; Ghia, P.; Burger, J.A. The importance of B cell receptor isotypes and stereotypes in chronic lymphocytic leukemia. Leukemia 2019, 33, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Gobessi, S.; Laurenti, L.; Longo, P.G.; Sica, S.; Leone, G.; Efremov, D.G. ZAP-70 enhances B-cell–receptor signaling despite absent or inefficient tyrosine kinase activation in chronic lymphocytic leukemia and lymphoma B cells. Blood 2007, 109, 2032–2039. [Google Scholar] [CrossRef]
- Kipps, T.J.; Stevenson, F.K.; Wu, C.J.; Croce, C.M.; Packham, G.; Wierda, W.G.; O’brien, S.; Gribben, J.; Rai, K. Chronic lymphocytic leukaemia. Nat. Rev. Dis. Primers 2017, 3, 16096. [Google Scholar] [CrossRef] [PubMed]
- Kipps, T.J.; Stevenson, F.K.; Wu, C.J.; Croce, C.M.; Packham, G.; Wierda, W.G.; O’brien, S.; Gribben, J.; Rai, K. CD38 expression distinguishes two groups of B-cell chronic lymphocytic leukemias with different responses to anti-IgM antibodies and propensity to apoptosis. Blood 1996, 88, 1365–1374. [Google Scholar]
- Deaglio, S.; Capobianco, A.; Bergui, L.; Dürig, J.; Morabito, F.; Dührsen, U.; Malavasi, F. CD38 is a signaling molecule in B-cell chronic lymphocytic leukemia cells. Blood 2003, 102, 2146–2155. [Google Scholar] [CrossRef]
- Poggi, A.; Prevosto, C.; Catellani, S.; Rocco, I.; Garuti, A.; Zocchi, M.R. Engagement of CD31 delivers an activating signal that contributes to the survival of chronic lymphocytic leukaemia cells. Br. J. Haematol. 2010, 151, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Amrein, L.; Soulières, D.; Johnston, J.B.; Aloyz, R. p53 and autophagy contribute to dasatinib resistance in primary CLL lymphocytes. Leuk. Res. 2011, 35, 99–102. [Google Scholar] [CrossRef]
- Mccaig, A.M.; Cosimo, E.; Leach, M.T.; Michie, A.M. Dasatinib inhibits B cell receptor signalling in chronic lymphocytic leukaemia but novel combination approaches are required to overcome additional pro-survival microenvironmental signals. Br. J. Haematol. 2011, 153, 199–211. [Google Scholar] [CrossRef]
- Amrein, L.; Hernandez, T.A.; Ferrario, C.; Johnston, J.; Gibson, S.B.; Panasci, L.; Aloyz, R. Dasatinib sensitizes primary chronic lymphocytic leukaemia lymphocytes to chlorambucil and fludarabine in vitro. Br. J. Haematol. 2008, 143, 698–706. [Google Scholar] [CrossRef]
- Giannopoulos, K.; Karczmarczyk, A.; Karp, M.; Bojarska-Junak, A.; Kosior, K.; Kowal, M.; Tomczak, W.; Hus, M.; Machnicki, M.; Stokłosa, T. In vivo, ex vivo and in vitro dasatinib activity in chronic lymphocytic leukemia. Oncol. Lett. 2021, 21, 285. [Google Scholar] [CrossRef]
- Byrd, J.C.; Hillmen, P.; O’Brien, S.; Barrientos, J.C.; Reddy, N.M.; Coutre, S.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; Barr, P.M.; et al. Long-term follow-up of the RESONATE phase 3 trial of ibrutinib vs. ofatumumab. Blood 2019, 133, 2031–2042. [Google Scholar] [CrossRef]
- Tedeschi, A.; Barr, P.M.; Robak, T.; Owen, C.; Ghia, P.; Bairey, O.; Hillmen, P.; Bartlett, N.L.; Li, J.; Simpson, D.; et al. Results from the International, Randomized Phase 3 Study of Ibrutinib Versus Chlorambucil in Patients 65 Years and Older with Treatment-Naïve CLL/SLL (RESONATE-2TM). Blood 2015, 126, 495. [Google Scholar] [CrossRef]
- O’Brien, S.; Jones, J.A.; Coutre, S.; Mato, A.R.; Hillmen, M.P.; Tam, C.; Osterborg, A.; Siddiqi, T.; Thirman, M.J.; Furman, R.R.; et al. Efficacy and Safety of Ibrutinib in Patients with Relapsed or Refractory Chronic Lymphocytic Leukemia or Small Lymphocytic Leukemia with 17p Deletion: Results from the Phase II RESONATE™-17 Trial. Blood 2014, 124, 327. [Google Scholar] [CrossRef]
- Thompson, P.A.; O’Brien, S.M.; Wierda, W.G.; Ferrajoli, A.; Stingo, F.C.; Smith, S.C.; Burger, J.A.; Estrov, Z.; Jain, N.; Kantarjian, H.M.; et al. Complex karyotype is a stronger predictor than del(17p) for an inferior outcome in relapsed or refractory chronic lymphocytic leukemia patients treated with ibrutinib-based regimens. Cancer 2015, 121, 3612–3621. [Google Scholar] [CrossRef] [PubMed]
- Ghia, P.; Pluta, A.; Wach, M.; Lysak, D.; Kozak, T.; Šimkovič, M.; Kaplan, P.; Kraychok, I.; Illes, A.; DE LA Serna, J.; et al. ASCEND: Phase III, Randomized Trial of Acalabrutinib Versus Idelalisib Plus Rituximab or Bendamustine Plus Rituximab in Relapsed or Refractory Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2020, 38, 2849–2861. [Google Scholar] [CrossRef] [PubMed]
- Sharman, J.P.; Egyed, M.; Jurczak, W.; Skarbnik, A.; Pagel, J.M.; Flinn, I.W.; Kamdar, M.; Munir, T.; Walewska, R.; Corbett, G.; et al. Acalabrutinib with or without obinutuzumab versus chlorambucil and obinutuzmab for treatment-naive chronic lymphocytic leukaemia (ELEVATE TN): A randomised, controlled, phase 3 trial. Lancet 2020, 395, 1278–1291. [Google Scholar] [CrossRef]
- Byrd, J.C.; Hillmen, P.; Ghia, P.; Kater, A.P.; Chanan-Khan, A.; Furman, R.R.; O’Brien, S.; Yenerel, M.N.; Illés, A.; Kay, N.; et al. Acalabrutinib Versus Ibrutinib in Previously Treated Chronic Lymphocytic Leukemia: Results of the First Randomized Phase III Trial. J. Clin. Oncol. 2021, 39, 3441–3452. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.S.; Robak, T.; Ghia, P.; Kahl, B.S.; Walker, P.; Janowski, W.; Simpson, D.; Shadman, M.; Ganly, P.S.; Laurenti, L.; et al. Zanubrutinib monotherapy for patients with treatment-naïve chronic lymphocytic leukemia and 17p deletion. Haematologica 2020, 106, 2354–2363. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; Hillmen, P.; Eichhorst, B.; Lamanna, N.; Brien, S.O.; . Tam, C.S.; Qui, L.; Kazmierczak, M.; Zhou, K.; Šimkovič, M.; et al. CLL-115 First Interim Analysis of ALPINE Study: Results of a Phase 3 Randomized Study of Zanubrutinib vs. Ibrutinib in Patients With Relapsed/Refractory (R/R) Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma (CLL/SLL). Clin. Lymphoma Myeloma Leuk. 2022, 22 (Suppl. 2), S266. [Google Scholar] [CrossRef]
- Tam, C.S.; Trotman, J.; Opat, S.; Burger, J.A.; Cull, G.; Gottlieb, D.; Harrup, R.; Johnston, P.B.; Marlton, P.; Munoz, J.; et al. Phase 1 study of the selective BTK inhibitor zanubrutinib in B-cell malignancies and safety and efficacy evaluation in CLL. Blood 2019, 134, 851–859. [Google Scholar] [CrossRef]
- Woyach, J.A.; Ruppert, A.S.; Guinn, D.; Lehman, A.; Blachly, J.S.; Lozanski, A.; Heerema, N.A.; Zhao, W.; Coleman, J.; Jones, D.; et al. BTK(C481S)-Mediated Resistance to Ibrutinib in Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2017, 35, 1437–1443. [Google Scholar] [CrossRef] [PubMed]
- Ahn, I.E.; Underbayev, C.; Albitar, A.; Herman, S.E.M.; Tian, X.; Maric, I.; Arthur, D.C.; Wake, L.; Pittaluga, S.; Yuan, C.M.; et al. Clonal evolution leading to ibrutinib resistance in chronic lymphocytic leukemia. Blood 2017, 129, 1469–1479. [Google Scholar] [CrossRef]
- Mato, A.R.; Shah, N.N.; Jurczak, W.; Cheah, C.Y.; Pagel, J.M.; Woyach, J.A.; Fakhri, B.; Eyre, T.A.; Lamanna, N.; Patel, M.R.; et al. Pirtobrutinib in relapsed or refractory B-cell malignancies (BRUIN): A phase 1/2 study. Lancet 2021, 397, 892–901. [Google Scholar] [CrossRef]
- Jayappa, K.D.; Portell, C.A.; Gordon, V.L.; Capaldo, B.J.; Bekiranov, S.; Axelrod, M.J.; Brett, L.K.; Wulfkuhle, J.D.; Gallagher, R.I.; Petricoin, E.F.; et al. Microenvironmental agonists generate de novo phenotypic resistance to combined ibrutinib plus venetoclax in CLL and MCL. Blood Adv. 2017, 1, 933–946. [Google Scholar] [CrossRef]
- Zhao, X.; Lwin, T.; Silva, A.; Shah, B.; Tao, J.; Fang, B.; Zhang, L.; Fu, K.; Bi, C.; Li, J.; et al. Unification of de novo and acquired ibrutinib resistance in mantle cell lymphoma. Nat. Commun. 2017, 8, 14920. [Google Scholar] [CrossRef]
- Furman, R.R.; Sharman, J.P.; Coutre, S.E.; Cheson, B.D.; Pagel, J.M.; Hillmen, P.; Barrientos, J.C.; Zelenetz, A.D.; Kipps, T.J.; Flinn, I.; et al. Idelalisib and Rituximab in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2014, 370, 997–1007. [Google Scholar] [CrossRef]
- Morabito, F.; Gentile, M.; Seymour, J.F.; Polliack, A. Ibrutinib, idelalisib and obinutuzumab for the treatment of patients with chronic lymphocytic leukemia: Three new arrows aiming at the target. Leuk. Lymphoma 2015, 56, 3250–3256. [Google Scholar] [CrossRef]
- O’Brien, S.M.; Lamanna, N.; Kipps, T.J.; Flinn, I.; Zelenetz, A.D.; Burger, J.A.; Keating, M.; Mitra, S.; Holes, L.; Yu, A.S.; et al. A phase 2 study of idelalisib plus rituximab in treatment-naïve older patients with chronic lymphocytic leukemia. Blood 2015, 126, 2686–2694. [Google Scholar] [CrossRef]
- O’Brien, S.; Patel, M.; Kahl, B.S.; Horwitz, S.M.; Foss, F.M.; Porcu, P.; Jones, J.; Burger, J.; Jain, N.; Allen, K.; et al. Duvelisib, an oral dual PI3K-δ,γ inhibitor, shows clinical and pharmacodynamic activity in chronic lymphocytic leukemia and small lymphocytic lymphoma in a phase 1 study. Am. J. Hematol. 2018, 93, 1318–1326. [Google Scholar] [CrossRef] [PubMed]
- Flinn, I.W.; Hillmen, P.; Montillo, M.; Nagy, Z.; Illés, A.; Etienne, G.; Delgado, J.; Kuss, B.J.; Tam, C.S.; Gasztonyi, Z.; et al. The phase 3 DUO trial: Duvelisib vs. ofatumumab in relapsed and refractory CLL/SLL. Blood 2018, 132, 2446–2455. [Google Scholar] [CrossRef]
- Barr, P.M.; Ma, S.; Zent, C.S.; Baran, M.A.M.; Bui, A.; Meacham, P.J.; Morrison, R.A.; Holkovic, R.K.; Liesveld, J.L.; Mulford, D.A.; et al. A Phase 1/2 Study of Umbralisib, Ublituximab, and Venetoclax (U2-Ven) in Patients with Relapsed or Refractory Chronic Lymphocytic Leukemia (CLL). Blood 2020, 136, 41–42. [Google Scholar] [CrossRef]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Seymour, J.F.; Ma, S.; Brander, D.M.; Choi, M.Y.; Barrientos, J.; Davids, M.S.; Anderson, M.A.; Beaven, A.W.; Rosen, S.T.; Tam, C.S.; et al. Venetoclax plus rituximab in relapsed or refractory chronic lymphocytic leukaemia: A phase 1b study. Lancet Oncol. 2017, 18, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Kater, A.P.; Seymour, J.F.; Hillmen, P.; Eichhorst, B.; Langerak, A.W.; Owen, C.; Verdugo, M.; Wu, J.; Punnoose, E.A.; Jiang, Y.; et al. Fixed Duration of Venetoclax-Rituximab in Relapsed/Refractory Chronic Lymphocytic Leukemia Eradicates Minimal Residual Disease and Prolongs Survival: Post-Treatment Follow-Up of the MURANO Phase III Study. J. Clin. Oncol. 2019, 37, 269–277. [Google Scholar] [CrossRef]
- Tam, C.S.; Allan, J.N.; Siddiqi, T.; Kipps, T.J.; Jacobs, R.W.; Opat, S.; Barr, P.M.; Tedeschi, A.; Trentin, L.; Bannerji, R.; et al. Fixed-duration ibrutinib plus venetoclax for first-line treatment of CLL: Primary analysis of the CAPTIVATE FD cohort. Blood 2022, 139, 3278–3289. [Google Scholar] [CrossRef]
- Bagnara, D.; Kaufman, M.S.; Calissano, C.; Marsilio, S.; Patten, P.; Simone, R.; Chum, P.; Yan, X.-J.; Allen, S.; Kolitz, J.E.; et al. A novel adoptive transfer model of chronic lymphocytic leukemia suggests a key role for T lymphocytes in the disease. Blood 2011, 117, 5463–5472. [Google Scholar] [CrossRef]
- Patten, P.; Ferrer, G.; Chen, S.-S.; Simone, R.; Marsilio, S.; Yan, X.-J.; Gitto, Z.; Yuan, C.; Kolitz, J.E.; Barrientos, J.; et al. Chronic lymphocytic leukemia cells diversify and differentiate in vivo via a nonclassical Th1-dependent, Bcl-6–deficient process. J. Clin. Investig. 2016, 1, e86288. [Google Scholar] [CrossRef]
- Griggio, V.; Perutelli, F.; Salvetti, C.; Boccellato, E.; Boccadoro, M.; Vitale, C.; Coscia, M. Immune Dysfunctions and Immune-Based Therapeutic Interventions in Chronic Lymphocytic Leukemia. Front. Immunol. 2020, 11, 594556. [Google Scholar] [CrossRef]
- Hallek, M. Chronic lymphocytic leukemia: 2020 update on diagnosis, risk stratification and treatment. Am. J. Hematol. 2019, 94, 1266–1287. [Google Scholar] [CrossRef]
- Younes, A.; Brody, J.; Carpio, C.; Lopez-Guillermo, A.; Ben-Yehuda, D.; Ferhanoglu, B.; Nagler, A.; Ozcan, M.; Avivi, I.; Bosch, F.; et al. Safety and activity of ibrutinib in combination with nivolumab in patients with relapsed non-Hodgkin lymphoma or chronic lymphocytic leukaemia: A phase 1/2a study. Lancet Haematol. 2019, 6, e67–e78. [Google Scholar] [CrossRef] [PubMed]
- Mato, A.R.; Svoboda, J.; Prak, E.T.L.; Schuster, S.J.; Tsao, P.; Dorsey, C.; Becker, P.S.; Brander, D.M.; Nasta, S.D.; Landsburg, D.J.; et al. Phase I/II Study of Umbralisib (TGR-1202) in Combination with Ublituximab (TG-1101) and Pembrolizumab in Patients with Relapsed/Refractory CLL and Richter’s Transformation. Blood 2018, 132 (Suppl. 1), 297. [Google Scholar] [CrossRef]
- Robinson, H.R.; Qi, J.; Cook, E.M.; Nichols, C.; Dadashian, E.L.; Underbayev, C.; Herman, S.E.; Saba, N.S.; Keyvanfar, K.; Sun, C.; et al. A CD19/CD3 bispecific antibody for effective immunotherapy of chronic lymphocytic leukemia in the ibrutinib era. Blood 2018, 132, 521–532. [Google Scholar] [CrossRef]
- Wong, R.; Pepper, C.; Brennan, P.; Nagorsen, D.; Man, S.; Fegan, C. Blinatumomab induces autologous T-cell killing of chronic lymphocytic leukemia cells. Haematologica 2013, 98, 1930–1938. [Google Scholar] [CrossRef] [PubMed]
- Martens, A.W.; Janssen, S.R.; Derks, I.A.; Adams III, H.C.; Izhak, L.; van Kampen, R.; Tonino, S.H.; Eldering, E.; van der Windt, G.J.; Kater, A.P. CD3xCD19 DART molecule treatment induces non-apoptotic killing and is efficient against high-risk chemotherapy and venetoclax-resistant chronic lymphocytic leukemia cells. J. Immunother. Cancer 2020, 8, e000218. [Google Scholar] [CrossRef]
- De Weerdt, I.; Lameris, R.; Scheffer, G.L.; Vree, J.; de Boer, R.; Stam, A.G.; van de Ven, R.; Levin, M.D.; Pals, S.T.; Roovers, R.C.; et al. A Bispecific Antibody Antagonizes Prosurvival CD40 Signaling and Promotes Vgamma9Vdelta2 T cell-Mediated Antitumor Responses in Human B-cell Malignancies. Cancer Immunol. Res. 2021, 9, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Vyas, M.; Schneider, A.C.; Shatnyeva, O.; Reiners, K.S.; Tawadros, S.; Kloess, S.; Köhl, U.; Hallek, M.; Hansen, H.P.; Pogge von Strandmann, E. Mono- and dual-targeting triplebodies activate natural killer cells and have anti-tumor activity in vitro and in vivo against chronic lymphocytic leukemia. Oncoimmunology 2016, 5, e1211220. [Google Scholar] [CrossRef]
- Gleason, M.K.; Verneris, M.R.; Todhunter, D.A.; Zhang, B.; McCullar, V.; Zhou, S.X.; Panoskaltsis-Mortari, A.; Weiner, L.M.; Vallera, D.A.; Miller, J.S. Bispecific and Trispecific Killer Cell Engagers Directly Activate Human NK Cells through CD16 Signaling and Induce Cytotoxicity and Cytokine Production. Mol. Cancer Ther. 2012, 11, 2674–2684. [Google Scholar] [CrossRef]
- Figueroa, J.A.; Reidy, A.; Mirandola, L.; Trotter, K.; Suvorava, N.; Figueroa, A.; Konala, V.M.; Aulakh, A.; Littlefield, L.; Grizzi, F.; et al. Chimeric Antigen Receptor Engineering: A Right Step in the Evolution of Adoptive Cellular Immunotherapy. Int. Rev. Immunol. 2015, 34, 154–187. [Google Scholar] [CrossRef]
- Dotti, G.; Gottschalk, S.; Savoldo, B.; Brenner, M.K. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol. Rev. 2013, 257, 107–126. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, A.G.; Johnson, A.J.; Lee, A.M.; Görgün, G.; Le Dieu, R.; Blum, W.; Byrd, J.C.; Gribben, J.G. Chronic lymphocytic leukemia T cells show impaired immunological synapse formation that can be reversed with an immunomodulating drug. J. Clin. Investig. 2008, 118, 2427–2437. [Google Scholar] [CrossRef]
- Riches, J.C.; Davies, J.K.; McClanahan, F.; Fatah, R.; Iqbal, S.; Agrawal, S.; Ramsay, A.G.; Gribben, J.G. T cells from CLL patients exhibit features of T-cell exhaustion but retain capacity for cytokine production. Blood 2013, 121, 1612–1621. [Google Scholar] [CrossRef] [PubMed]
- Turtle, C.J.; Hay, K.; Hanafi, L.-A.; Li, D.; Cherian, S.; Chen, X.; Wood, B.; Lozanski, A.; Byrd, J.C.; Heimfeld, S.; et al. Durable Molecular Remissions in Chronic Lymphocytic Leukemia Treated with CD19-Specific Chimeric Antigen Receptor–Modified T Cells After Failure of Ibrutinib. J. Clin. Oncol. 2017, 35, 3010–3020. [Google Scholar] [CrossRef]
- Porter, D.L.; Hwang, W.-T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef]
- Siddiqi, T.; Soumerai, J.D.; Dorritie, K.A.; Stephens, D.M.; Riedell, P.A.; Arnason, J.E.; Kipps, T.J.; Gillenwater, H.H.; Gong, L.; Yang, L.; et al. Phase 1 TRANSCEND CLL 004 study of lisocabtagene maraleucel in patients with relapsed/refractory CLL or SLL. Blood 2022, 139, 1794–1806. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, R.; Shou, P.; Ahn, S.; Sun, C.; West, J.; Savoldo, B.; Dotti, G. CAR T cells Targeting Human Immunoglobulin Light Chains Eradicate Mature B-cell Malignancies While Sparing a Subset of Normal B Cells. Clin. Cancer Res. 2021, 27, 5951–5960. [Google Scholar] [CrossRef] [PubMed]
- Heyman, B.M.; Tzachanis, D.; Kipps, T.J. Recent Advances in CAR T-Cell Therapy for Patients with Chronic Lymphocytic Leukemia. Cancers 2022, 14, 1715. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.S.; Johnstone, T.G.; Baturevych, A.; Hause, R.J.; Ragan, S.P.; Clouser, C.R.; Jones, J.C.; Ponce, R.; Krejsa, C.M.; Salmon, R.A.; et al. Antitumor Potency of an Anti-CD19 Chimeric Antigen Receptor T-Cell Therapy, Lisocabtagene Maraleucel in Combination with Ibrutinib or Acalabrutinib. J. Immunother. 2020, 43, 107–120. [Google Scholar] [CrossRef]
- Till, B.G.; Jensen, M.C.; Wang, J.; Qian, X.; Gopal, A.K.; Maloney, D.G.; Lindgren, C.G.; Lin, Y.; Pagel, J.M.; Budde, L.E.; et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: Pilot clinical trial results. Blood 2012, 119, 3940–3950. [Google Scholar] [CrossRef]
- Hudecek, M.; Schmitt, T.M.; Baskar, S.; Lupo-Stanghellini, M.T.; Nishida, T.; Yamamoto, T.N.; Bleakley, M.; Turtle, C.J.; Chang, W.-C.; Greisman, H.A.; et al. The B-cell tumor–associated antigen ROR1 can be targeted with T cells modified to express a ROR1-specific chimeric antigen receptor. Blood 2010, 116, 4532–4541. [Google Scholar] [CrossRef] [PubMed]
- Faitschuk, E.; Hombach, A.A.; Frenzel, L.P.; Wendtner, C.M.; Abken, H. Chimeric antigen receptor T cells targeting Fc mu receptor selectively eliminate CLL cells while sparing healthy B cells. Blood 2016, 128, 1711–1722. [Google Scholar] [CrossRef]
- Brody, E.; Abelson, J. The “Spliceosome”: Yeast Pre-Messenger RNA Associates with a 40 S Complex in a Splicing-Dependent Reaction. Science 1985, 228, 963–967. [Google Scholar] [CrossRef]
- Will, C.L.; Lührmann, R. Spliceosome Structure and Function. Cold Spring Harb. Perspect. Biol. 2011, 3, a003707. [Google Scholar] [CrossRef] [PubMed]
- Stanley, R.F.; Abdel-Wahab, O. Dysregulation and therapeutic targeting of RNA splicing in cancer. Nat. Cancer 2022, 3, 536–546. [Google Scholar] [CrossRef]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Graubert, T.; Shen, D.; Ding, L.; Okeyo-Owuor, T.; Lunn, C.L.; Shao, J.; Krysiak, K.; Harris, C.C.; Koboldt, D.C.; Larson, D.; et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat. Genet. 2011, 44, 53–57. [Google Scholar] [CrossRef]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.W.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.-B.; Murphy, M.E.; et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2015, 27, 617–630. [Google Scholar] [CrossRef]
- Pararajalingam, P.; Coyle, K.M.; Arthur, S.E.; Thomas, N.; Alcaide, M.; Meissner, B.; Boyle, M.; Qureshi, Q.; Grande, B.M.; Rushton, C.; et al. Coding and noncoding drivers of mantle cell lymphoma identified through exome and genome sequencing. Blood 2020, 136, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Kumar, S.A.; Shuai, S.; Diaz-Navarro, A.; Gutierrez-Fernandez, A.; De Antonellis, P.; Cavalli, F.M.G.; Juraschka, K.; Farooq, H.; Shibahara, I.; et al. Recurrent noncoding U1 snRNA mutations drive cryptic splicing in SHH medulloblastoma. Nature 2019, 574, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Jayasinghe, R.G.; Cao, S.; Gao, Q.; Wendl, M.C.; Vo, N.S.; Reynolds, S.M.; Zhao, Y.; Climente-González, H.; Chai, S.; Wang, F.; et al. Systematic Analysis of Splice-Site-Creating Mutations in Cancer. Cell Rep. 2018, 23, 270–281.e3. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.-W.; North, K.; Kim, E.; Jang, E.; Obeng, E.; Lu, S.X.; Liu, B.; Inoue, D.; Yoshimi, A.; Ki, M.; et al. Synthetic Lethal and Convergent Biological Effects of Cancer-Associated Spliceosomal Gene Mutations. Cancer Cell 2018, 34, 225–241.e8. [Google Scholar] [CrossRef]
- Chen, S.; Benbarche, S.; Abdel-Wahab, O. Splicing factor mutations in hematologic malignancies. Blood 2021, 138, 599–612. [Google Scholar] [CrossRef]
- Kotake, Y.; Sagane, K.; Owa, T.; Mimori-Kiyosue, Y.; Shimizu, H.; Uesugi, M.; Ishihama, Y.; Iwata, M.; Mizui, Y. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat. Chem. Biol. 2007, 3, 570–575. [Google Scholar] [CrossRef]
- Yokoi, A.; Kotake, Y.; Takahashi, K.; Kadowaki, T.; Matsumoto, Y.; Minoshima, Y.; Sugi, N.H.; Sagane, K.; Hamaguchi, M.; Iwata, M.; et al. Biological validation that SF3b is a target of the antitumor macrolide pladienolide. FEBS J. 2011, 278, 4870–4880. [Google Scholar] [CrossRef]
- Kaida, D.; Motoyoshi, H.; Tashiro, E.; Nojima, T.; Hagiwara, M.; Ishigami, K.; Watanabe, H.; Kitahara, T.; Yoshida, T.; Nakajima, H.; et al. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat. Chem. Biol. 2007, 3, 576–583. [Google Scholar] [CrossRef]
- Larrayoz, M.; Blakemore, S.J.; Dobson, R.C.; Blunt, M.D.; Rose-Zerilli, M.J.J.; Walewska, R.; Duncombe, A.; Oscier, D.; Koide, K.; Forconi, F.; et al. The SF3B1 inhibitor spliceostatin A (SSA) elicits apoptosis in chronic lymphocytic leukaemia cells through downregulation of Mcl-1. Leukemia 2016, 30, 351–360. [Google Scholar] [CrossRef]
- Xargay-Torrent, S.; López-Guerra, M.; Rosich, L.; Montraveta, A.; Roldán, J.; Rodríguez, V.; Villamor, N.; Aymerich, M.; Lagisetti, C.; Webb, T.R.; et al. The splicing modulator sudemycin induces a specific antitumor response and cooperates with ibrutinib in chronic lymphocytic leukemia. Oncotarget 2015, 6, 22734–22749. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Cannizzaro, E.; Meier-Abt, F.; Scheinost, S.; Bruch, P.M.; Giles, H.A.; Lütge, A.; Hüllein, J.; Wagner, L.; Giacopelli, B.; et al. Multi-omics reveals clinically relevant proliferative drive associated with mTOR-MYC-OXPHOS activity in chronic lymphocytic leukemia. Nat. Cancer 2021, 2, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Jitschin, R.; Hofmann, A.D.; Bruns, H.; Gießl, A.; Bricks, J.; Berger, J.; Saul, D.; Eckart, M.J.; Mackensen, A.; Mougiakakos, D. Mitochondrial metabolism contributes to oxidative stress and reveals therapeutic targets in chronic lymphocytic leukemia. Blood 2014, 123, 2663–2672. [Google Scholar] [CrossRef] [PubMed]
- Rozovski, U.; Grgurevic, S.; Bueso-Ramos, C.; Harris, D.M.; Li, P.; Liu, Z.; Wu, J.Y.; Jain, P.; Wierda, W.; Burger, J.; et al. Aberrant LPL Expression, Driven by STAT3, Mediates Free Fatty Acid Metabolism in CLL Cells. Mol. Cancer Res. 2015, 13, 944–953. [Google Scholar] [CrossRef]
- Zelenetz, A.D. Chronic Lymphocytic Leukemia: Individualizing Treatment Approach. J. Natl. Compr. Cancer Netw. 2017, 15, 713–715. [Google Scholar] [CrossRef]
- Galicia-Vázquez, G.; Smith, S.; Aloyz, R. Del11q-positive CLL lymphocytes exhibit altered glutamine metabolism and differential response to GLS1 and glucose metabolism inhibition. Blood Cancer J. 2018, 8, 13. [Google Scholar] [CrossRef]
- Martinez Marignac, V.L.; Smith, S.; Toban, N.; Bazile, M.; Aloyz, R. Resistance to Dasatinib in primary chronic lymphocytic leukemia lymphocytes involves AMPK-mediated energetic re-programming. Oncotarget 2013, 4, 2550–2566. [Google Scholar] [CrossRef]
- Brown, J.R. Insulin receptor activation in deletion 11q chronic lymphocytic leukemia. Clin. Cancer Res. 2011, 17, 2605–2607. [Google Scholar] [CrossRef]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.-Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Fuchs, B.C.; Bode, B.P. Amino acid transporters ASCT2 and LAT1 in cancer: Partners in crime? Semin. Cancer Biol. 2005, 15, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Heintel, D.; Kienle, D.; Shehata, M.; Kröber, A.; Kroemer, E.; Schwarzinger, I.; Mitteregger, D.; Le, T.; Gleiß, A.; Mannhalter, C.; et al. High expression of lipoprotein lipase in poor risk B-cell chronic lymphocytic leukemia. Leukemia 2005, 19, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Mead, J.R.; Irvine, S.A.; Ramji, D.P. Lipoprotein lipase: Structure, function, regulation, and role in disease. J. Mol. Med. 2002, 80, 753–769. [Google Scholar] [CrossRef] [PubMed]
- Pallasch, C.P.; Schwamb, J.; Königs, S.; Schulz, A.; Debey, S.; Kofler, D.; Schultze, J.; Hallek, M.; Ultsch, A.; Wendtner, C.-M. Targeting lipid metabolism by the lipoprotein lipase inhibitor orlistat results in apoptosis of B-cell chronic lymphocytic leukemia cells. Leukemia 2008, 22, 585–592. [Google Scholar] [CrossRef]
- Galicia-Vázquez, G.; Aloyz, R. Ibrutinib Resistance Is Reduced by an Inhibitor of Fatty Acid Oxidation in Primary CLL Lymphocytes. Front. Oncol. 2018, 8, 411. [Google Scholar] [CrossRef]
- Dimier, N.; Delmar, P.; Ward, C.; Morariu-Zamfir, R.; Fingerle-Rowson, G.; Bahlo, J.; Fischer, K.; Eichhorst, B.; Goede, V.; Van Dongen, J.J.M.; et al. A model for predicting effect of treatment on progression-free survival using MRD as a surrogate end point in CLL. Blood 2018, 131, 955–962. [Google Scholar] [CrossRef]
- Böttcher, S.; Ritgen, M.; Fischer, K.; Stilgenbauer, S.; Busch, R.M.; Fingerle-Rowson, G.; Fink, A.M.; Bühler, A.; Zenz, T.; Wenger, M.K.; et al. Minimal Residual Disease Quantification Is an Independent Predictor of Progression-Free and Overall Survival in Chronic Lymphocytic Leukemia: A Multivariate Analysis From the Randomized GCLLSG CLL8 Trial. J. Clin. Oncol. 2012, 30, 980–988. [Google Scholar] [CrossRef]
- Kovacs, G.; Robrecht, S.; Fink, A.M.; Bahlo, J.; Cramer, P.; Von Tresckow, J.; Maurer, C.; Langerbeins, P.; Fingerle-Rowson, G.; Ritgen, M.; et al. Minimal Residual Disease Assessment Improves Prediction of Outcome in Patients with Chronic Lymphocytic Leukemia (CLL) Who Achieve Partial Response: Comprehensive Analysis of Two Phase III Studies of the German CLL Study Group. J. Clin. Oncol. 2016, 34, 3758–3765. [Google Scholar] [CrossRef]
- Chanan-Khan, A.; Cramer, P.; Demirkan, F.; Fraser, G.; Silva, R.S.; Grosicki, S.; Pristupa, A.; Janssens, A.; Mayer, J.; Bartlett, N.L.; et al. Ibrutinib combined with bendamustine and rituximab compared with placebo, bendamustine, and rituximab for previously treated chronic lymphocytic leukaemia or small lymphocytic lymphoma (HELIOS): A randomised, double-blind, phase 3 study. Lancet Oncol. 2016, 17, 200–211. [Google Scholar] [CrossRef]
- Fraser, G.; Cramer, P.; Demirkan, F.; Silva, R.S.; Grosicki, S.; Pristupa, A.; Janssens, A.; Mayer, J.; Bartlett, N.L.; Dilhuydy, M.-S.; et al. Updated results from the phase 3 HELIOS study of ibrutinib, bendamustine, and rituximab in relapsed chronic lymphocytic leukemia/small lymphocytic lymphoma. Leukemia 2019, 33, 969–980. [Google Scholar] [CrossRef]
- Zelenetz, A.D.; Barrientos, J.C.; Brown, J.R.; Coiffier, B.; Delgado, J.; Egyed, M.; Ghia, P.; Illés, Á.; Jurczak, W.; Marlton, P.; et al. Idelalisib or placebo in combination with bendamustine and rituximab in patients with relapsed or refractory chronic lymphocytic leukaemia: Interim results from a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2017, 18, 297–311. [Google Scholar] [CrossRef]
- Jain, N.; Thompson, P.; Burger, J.; Ferrajoli, A.; Takahashi, K.; Estrov, Z.; Borthakur, G.; Bose, P.; Kadia, T.; Pemmaraju, N.; et al. Ibrutinib, fludarabine, cyclophosphamide, and obinutuzumab (iFCG) regimen for chronic lymphocytic leukemia (CLL) with mutated IGHV and without TP53 aberrations. Leukemia 2021, 35, 3421–3429. [Google Scholar] [CrossRef] [PubMed]
- Woyach, J.A.; Ruppert, A.S.; Heerema, N.A.; Zhao, W.; Booth, A.M.; Ding, W.; Bartlett, N.L.; Brander, D.M.; Barr, P.M.; Rogers, K.A.; et al. Ibrutinib Regimens versus Chemoimmunotherapy in Older Patients with Untreated CLL. N. Engl. J. Med. 2018, 379, 2517–2528. [Google Scholar] [CrossRef] [PubMed]
- Moreno, C.; Greil, R.; Demirkan, F.; Tedeschi, A.; Anz, B.; Larratt, L.; Simkovic, M.; Samoilova, O.; Novak, J.; Ben-Yehuda, D.; et al. Ibrutinib plus obinutuzumab versus chlorambucil plus obinutuzumab in first-line treatment of chronic lymphocytic leukaemia (iLLUMINATE): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2018, 20, 43–56. [Google Scholar] [CrossRef]
- Cramer, P.; von Tresckow, J.; Bahlo, J.; Robrecht, S.; Langerbeins, P.; Al-Sawaf, O.; Engelke, A.; Fink, A.-M.; Fischer, K.; Tausch, E.; et al. Bendamustine followed by obinutuzumab and venetoclax in chronic lymphocytic leukaemia (CLL2-BAG): Primary endpoint analysis of a multicentre, open-label, phase 2 trial. Lancet Oncol. 2019, 19, 1215–1228. [Google Scholar] [CrossRef]
- Hillmen, P.; Rawstron, A.C.; Brock, K.; Muñoz-Vicente, S.; Yates, F.J.; Bishop, R.; Boucher, R.; Macdonald, D.; Fegan, C.; McCaig, A.; et al. Ibrutinib Plus Venetoclax in Relapsed/Refractory Chronic Lymphocytic Leukemia: The CLARITY Study. J. Clin. Oncol. 2019, 37, 2722–2729. [Google Scholar] [CrossRef]
- Chanan-Khan, A.A.; Chitta, K.; Ersing, N.; Paulus, A.; Masood, A.; Sher, T.; Swaika, A.; Wallace, P.; Jr, T.L.M.; Wilding, G.; et al. Biological effects and clinical significance of lenalidomide-induced tumour flare reaction in patients with chronic lymphocytic leukaemia: In vivo evidence of immune activation and antitumour response. Br. J. Haematol. 2011, 155, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Chanan-Khan, A.; Miller, K.C.; Musial, L.; Lawrence, D.; Padmanabhan, S.; Takeshita, K.; Porter, C.W.; Goodrich, D.W.; Bernstein, Z.P.; Wallace, P.; et al. Clinical Efficacy of Lenalidomide in Patients with Relapsed or Refractory Chronic Lymphocytic Leukemia: Results of a Phase II Study. J. Clin. Oncol. 2006, 24, 5343–5349. [Google Scholar] [CrossRef] [PubMed]
- Moreno, C.; Villamor, N.; Colomer, D.; Esteve, J.; Martino, R.; Nomdedéu, J.; Bosch, F.; López-Guillermo, A.; Campo, E.; Sierra, J.; et al. Allogeneic Stem-Cell Transplantation May Overcome the Adverse Prognosis of Unmutated VH Gene in Patients with Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2005, 23, 3433–3438. [Google Scholar] [CrossRef]
- Gribben, J.G. How and when I do allogeneic transplant in CLL. Blood 2018, 132, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Sorror, M.L.; Storer, B.E.; Maloney, D.G.; Sandmaier, B.M.; Martin, P.J.; Storb, R. Outcomes after allogeneic hematopoietic cell transplantation with nonmyeloablative or myeloablative conditioning regimens for treatment of lymphoma and chronic lymphocytic leukemia. Blood 2008, 111, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Sorror, M.L.; Storer, B.E.; Sandmaier, B.M.; Maris, M.; Shizuru, J.; Maziarz, R.; Agura, E.; Chauncey, T.R.; Pulsipher, M.A.; McSweeney, P.A.; et al. Five-Year Follow-Up of Patients with Advanced Chronic Lymphocytic Leukemia Treated with Allogeneic Hematopoietic Cell Transplantation After Nonmyeloablative Conditioning. J. Clin. Oncol. 2008, 26, 4912–4920. [Google Scholar] [CrossRef]
- Khouri, I.F.; Wei, W.; Korbling, M.; Turturro, F.; Ahmed, S.; Alousi, A.; Anderlini, P.; Ciurea, S.; Jabbour, E.; Oran, B.; et al. BFR (bendamustine, fludarabine, and rituximab) allogeneic conditioning for chronic lymphocytic leukemia/lymphoma: Reduced myelosuppression and GVHD. Blood 2014, 124, 2306–2312. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shadman, M.; Maloney, D.G.; Storer, B.; Sandmaier, B.M.; Chauncey, T.R.; Andersen, N.S.; Niederwieser, D.; Shizuru, J.; Bruno, B.; Pulsipher, M.A.; et al. Rituximab-based allogeneic transplant for chronic lymphocytic leukemia with comparison to historical experience. Bone Marrow Transpl. 2020, 55, 172–181. [Google Scholar] [CrossRef]
- Diaz Rohena, D.; Slawin, B.; Ravikrishnan, J.; Liu, C.; Hu, W.; Zhang, P.; Thompson, P.A.; Wierda, W.G.; Jain, N.; Zheng, G.; et al. Targeting Venetoclax Resistant CLL Using a Protac-Based BCL-2/BCL-XL Degrader. Blood 2022, 140 (Suppl. 1), 497–498. [Google Scholar] [CrossRef]
- Bhagwat, N.; Ruggeri, B.; Zhang, Y.; Mosesson, Y.; Killick-Cole, C.; Jagannathan, V.; Scherle, P. PRT2527, a Novel Highly Selective Cyclin-Dependent Kinase 9 (CDK9) Inhibitor, Has Potent Anti-Leukemic Activity in Preclinical Primary Models of Human B.-ALL, T.-ALL, and CLL. Blood 2022, 140, 505–506. [Google Scholar] [CrossRef]
- Hu, X.; Zhang, Y.; Wang, H.; Han, Y.; Tian, Z.; Zhang, X.; Lu, L.; Wang, X. BRD9 Facilitates Oncogenic Nrf2 Pathway and Dampens Venetoclax Sensitivity By Remodeling Chromatin Accessibility in Chronic Lymphocytic Leukemia. Blood 2022, 140, 356–357. [Google Scholar] [CrossRef]
- Chen, S.-S.; Yan, X.-J.; Van Rompaey, L.; Zabrocki, P.; Chiorazzi, N. Anti-CD70 Monoclonal Antibodies Show Efficacy in Preclinical Models for Chronic Lymphoblastic Leukemia. Blood 2022, 140, 1803–1804. [Google Scholar] [CrossRef]
- Iyer, P.; Zhang, B.; Liu, T.; Jin, M.; Hart, K.; Song, J.Y.; Chan, W.C.; Siddiqi, T.; Danilov, A.V.; Rosen, S.T.; et al. Disrupting MGA-MYC Driven Metabolic Reprogramming in Richter’s Syndrome Pre-Clinical Models Via Novel Therapeutic Approaches. Blood 2022, 140 (Suppl. 1), 9842–9843. [Google Scholar] [CrossRef]
- Kater, A.P.; Ye, J.C.; Sandoval-Sus, J.; Bellido, M.; Christensen, J.H.; Mato, A.R.; Janssens, A.; Oki, T.; Hoehn, D.; Rios, M.; et al. Subcutaneous Epcoritamab in Patients with Richter’s Syndrome: Early Results from Phase 1b/2 Trial (EPCORE CLL-1). Blood 2022, 140 (Suppl. 1), 850–851. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iyer, P.; Wang, L. Emerging Therapies in CLL in the Era of Precision Medicine. Cancers 2023, 15, 1583. https://doi.org/10.3390/cancers15051583
Iyer P, Wang L. Emerging Therapies in CLL in the Era of Precision Medicine. Cancers. 2023; 15(5):1583. https://doi.org/10.3390/cancers15051583
Chicago/Turabian StyleIyer, Prajish, and Lili Wang. 2023. "Emerging Therapies in CLL in the Era of Precision Medicine" Cancers 15, no. 5: 1583. https://doi.org/10.3390/cancers15051583
APA StyleIyer, P., & Wang, L. (2023). Emerging Therapies in CLL in the Era of Precision Medicine. Cancers, 15(5), 1583. https://doi.org/10.3390/cancers15051583