
CD22 Exon 12 Deletion as an Independent Predictor of Poor Treatment Outcomes in B-ALL


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Comparative Analysis of the Expression Levels of CD22 Exons 11−14 in Primary Leukemia Cells from Newly Diagnosed Pediatric Patients with B-All and Normal Hematopoietic Cells from Non-Leukemic Controls Using a Human Genome Expression Microarray Platform for Transcriptome Profiling
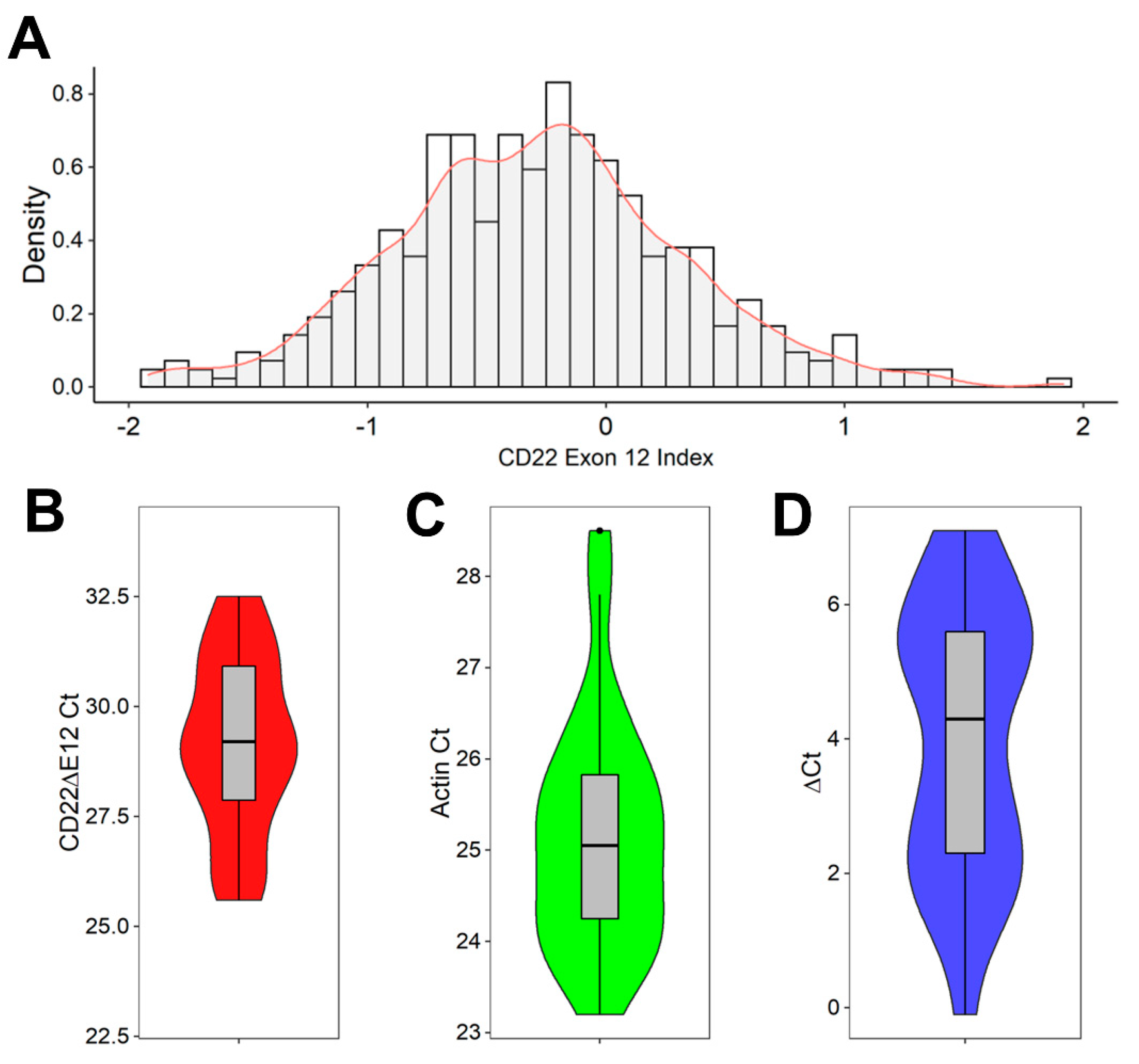
2.2. Detection of CD22ΔE12 mRNA in B-ALL Leukemia Samples via Real-Time Quantitative RT-PCR
2.3. Data Normalization for Exon-Level CD22 Gene Expression Data Derived from Primary B-ALL Cells
2.4. Hierarchical Clustering Analysis to Identify CD22E12low B-ALL Patients
2.5. Normalization of Gene Level RNAseq Data Derived from Primary B-ALL Cells
2.6. Gene Set Enrichment Analysis (GSEA) for Evaluation of Reactome Pathways in B-ALL Patients with Low CD22E12 Expression
2.7. Analysis of Treatment Outcomes according to RNAseq-Based CD22E12 mRNA Expression Levels
2.8. Multivariate and Univariate Cox Regression Models to Test for the Independent Effect of CD22E12low Status
3. Results
3.1. Interpatient Heterogeneity in Microarray-Based CD22E12 and qRT-PCR-Based CD22ΔE12 mRNA Expression Levels among Newly Diagnosed B-ALL Patients
3.2. Interpatient Heterogeneity in Selective Reduction of RNAseq-Based CD22E12 Expression Levels among B-ALL Patients
3.3. Presenting Features of CD22E12low B-ALL Patients
3.4. Gene Set Enrichment Analysis of Reactome Pathways in CD22E12low B-ALL Patients
3.5. Clinical Prognostic Significance of the Interpatient Heterogeneity in Selective Reduction of RNAseq-Based CD22 Exon 12 Expression Levels among B-ALL Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Crocker, P.R.; Redelinghuys, P. Siglecs as positive and negative regulators of the immune system. Biochem. Soc. Trans. 2008, 36, 1467–1471. [Google Scholar] [CrossRef] [PubMed]
- Tsubata, T. Inhibitory B cell co-receptors and autoimmune diseases. Immunol. Med. 2019, 42, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Tedder, T.F.; Poe, J.C.; Haas, K.M. CD22: A Multifunctional Receptor That Regulates B Lymphocyte Survival and Signal Transduction. Adv. Immunol. 2005, 88, 1–50. [Google Scholar] [CrossRef] [PubMed]
- Nitschke, L. The role of CD22 and other inhibitory co-receptors in B-cell activation. Curr. Opin. Immunol. 2005, 17, 290–297. [Google Scholar] [CrossRef]
- Cornall, R.J.; Cyster, J.G.; Hibbs, M.L.; Dunn, A.R.; Otipoby, K.L.; A Clark, E.; Goodnow, C.C. Polygenic Autoimmune Traits: Lyn, CD22, and SHP-1 Are Limiting Elements of a Biochemical Pathway Regulating BCR Signaling and Selection. Immunity 1998, 8, 497–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasioli, J.; Paust, S.; Thomas, M.L. Definition of the Sites of Interaction between the Protein Tyrosine Phosphatase SHP-1 and CD22. J. Biol. Chem. 1999, 274, 2303–2307. [Google Scholar] [CrossRef] [Green Version]
- Uckun, F.M.; Goodman, P.; Ma, H.; Dibirdik, I.; Qazi, S. CD22 EXON 12 deletion as a pathogenic mechanism of human B-precursor leukemia. Proc. Natl. Acad. Sci. USA 2010, 107, 16852–16857. [Google Scholar] [CrossRef] [Green Version]
- Uckun, F.M.; Ma, H.; Cheng, J.; Myers, D.E.; Qazi, S. CD22ΔE12 as a molecular target for RNAi therapy. Br. J. Haematol. 2015, 169, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Uckun, F.M.; Qazi, S. Identification and targeting of CD22ΔE12 as a molecular RNAi target to overcome drug resistance in high-risk B-lineage leukemias and lymphomas. Cancer Drug Resist 2018, 1, 30–47. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Qazi, S.; Ozer, Z.; Gaynon, P.; Reaman, G.H.; Uckun, F.M. CD22 Exon 12 deletion is a characteristic genetic defect of therapy-refractory clones in paediatric acute lymphoblastic leukaemia. Br. J. Haematol. 2011, 156, 89–98. [Google Scholar] [CrossRef]
- Uckun, F.M.; Mitchell, L.G.; Qazi, S.; Liu, Y.; Zheng, N.; Myers, D.E.; Song, Z.; Ma, H.; Cheng, J. Development of Polypeptide-based Nanoparticles for Non-viral Delivery of CD22 RNA Trans-splicing Molecule as a New Precision Medicine Candidate Against B-lineage ALL. Ebiomedicine 2015, 2, 649–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uckun, F.M.; Qazi, S.; Ma, H.; Reaman, G.H.; Mitchell, L.G. CD22ΔE12 as a molecular target for corrective repair using RNAtrans-splicing: Anti-leukemic activity of a rationally designed RNAtrans-splicing molecule. Integr. Biol. 2015, 7, 237–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uckun, F.M.; Qazi, S.; Ma, H.; Yin, L.; Cheng, J. A rationally designed nanoparticle for RNA interference therapy in B-lineage lymphoid malignancies. Ebiomedicine 2014, 1, 141–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, W.P.; Larsen, E.L.; Devidas, M.; Linda, S.B.; Blach, L.; Carroll, A.J.; Carroll, W.L.; Pullen, D.J.; Shuster, J.; Willman, C.L.; et al. Augmented therapy improves outcome for pediatric high risk acute lymphocytic leukemia: Results of Children’s Oncology Group trial P9906. Pediatr. Blood Cancer 2011, 57, 569–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, E.C.; Devidas, M.; Chen, S.; Salzer, W.L.; Raetz, E.A.; Loh, M.L.; Mattano, L.A., Jr.; Cole, C.; Eicher, A.; Haugan, M.; et al. Dexamethasone and High-Dose Methotrexate Improve Outcome for Children and Young Adults With High-Risk B-Acute Lymphoblastic Leukemia: A Report From Children’s Oncology Group Study AALL0232. J. Clin. Oncol. 2016, 34, 2380–2388. [Google Scholar] [CrossRef] [PubMed]
- Borowitz, M.J.; Wood, B.L.; Devidas, M.; Loh, M.L.; Raetz, E.A.; Salzer, W.L.; Nachman, J.B.; Carroll, A.J.; Heerema, N.A.; Gastier-Foster, J.M.; et al. Prognostic significance of minimal residual disease in high risk B-ALL: A report from Children’s Oncology Group study AALL0232. Blood 2015, 126, 964–971. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.J.; Devidas, M.; Chen, Z.; Salzer, W.L.; Raetz, E.A.; Rabin, K.R.; Heerema, N.A.; Carroll, A.J.; Gastier-Foster, J.M.; Borowitz, M.J.; et al. Outcomes in adolescent and young adult patients (16 to 30 years) compared to younger patients treated for high-risk B-lymphoblastic leukemia: Report from Children’s Oncology Group Study AALL0232. Leukemia 2022, 36, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Maloney, K.W.; Devidas, M.; Wang, C.; Mattano, L.A.; Friedmann, A.M.; Buckley, P.; Borowitz, M.J.; Carroll, A.J.; Gastier-Foster, J.M.; Heerema, N.A.; et al. Outcome in Children With Standard-Risk B-Cell Acute Lymphoblastic Leukemia: Results of Children’s Oncology Group Trial AALL0331. J. Clin. Oncol. 2020, 38, 602–612. [Google Scholar] [CrossRef]
- Mattano, L.A., Jr.; Devidas, M.; Maloney, K.W.; Wang, C.; Friedmann, A.M.; Buckley, P.; Borowitz, M.J.; Carroll, A.J.; Gastier-Foster, J.M.; Heerema, N.A.; et al. Favorable Trisomies and ETV6-RUNX1 Predict Cure in Low-Risk B-Cell Acute Lymphoblastic Leukemia: Results From Children’s Oncology Group Trial AALL0331. J. Clin. Oncol. 2021, 39, 1540–1552. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Tran, T.H.; Langlois, S.; Meloche, C.; Caron, M.; Saint-Onge, P.; Rouette, A.; Bataille, A.R.; Jimenez-Cortes, C.; Sontag, T.; Bittencourt, H.; et al. Whole-transcriptome analysis in acute lymphoblastic leukemia: A report from the DFCI ALL Consortium Protocol 16-001. Blood Adv. 2022, 6, 1329–1341. [Google Scholar] [CrossRef] [PubMed]
- Eshibona, N.; Giwa, A.; Rossouw, S.C.; Gamieldien, J.; Christoffels, A.; Bendou, H. Upregulation of FHL1, SPNS3, and MPZL2 predicts poor prognosis in pediatric acute myeloid leukemia patients with FLT3-ITD mutation. Leuk. Lymphoma 2022, 63, 1897–1906. [Google Scholar] [CrossRef] [PubMed]
- Hetzel, S.; Mattei, A.L.; Kretzmer, H.; Qu, C.; Chen, X.; Fan, Y.; Wu, G.; Roberts, K.G.; Luger, S.; Litzow, M.; et al. Acute lymphoblastic leukemia displays a distinct highly methylated genome. Nat. Cancer 2022, 3, 768–782. [Google Scholar] [CrossRef] [PubMed]
- Huber, W.; von Heydebreck, A.; Sueltmann, H.; Poustka, A.; Vingron, M. “Variance Stabilization Applied to Microarray Data Calibration and to the Quantification of Differential Expression. Bioinformatics 2002, 18 (Suppl. 1), S96–S104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickrell, J.K.; Marioni, J.C.; Pai, A.A.; Degner, J.F.; Engelhardt, B.E.; Nkadori, E.; Veyrieras, J.-B.; Stephens, M.; Gilad, Y.; Pritchard, J.K. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature 2010, 464, 768–772. [Google Scholar] [CrossRef] [Green Version]
- Korotkevich, G.; Sukhov, V.; Sergushichev, A. Fast gene set enrichment analysis. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Black, K.L.; Naqvi, A.; Asnani, M.; Hayer, K.; Yang, S.; Gillespie, E.; Bagashev, A.; Pillai, V.; Tasian, S.K.; Gazzara, M.R.; et al. Aberrant splicing in B-cell acute lymphoblastic leukemia. Nucleic Acids Res. 2018, 46, 11357–11369. [Google Scholar] [CrossRef] [Green Version]
- Venables, J.P. Aberrant and Alternative Splicing in Cancer. Cancer Res. 2004, 64, 7647–7654. [Google Scholar] [CrossRef] [Green Version]
- Hui, J.; Hung, L.-H.; Heiner, M.; Schreiner, S.; Neumüller, N.; Reither, G.; A Haas, S.; Bindereif, A. Intronic CA-repeat and CA-rich elements: A new class of regulators of mammalian alternative splicing. EMBO J. 2005, 24, 1988–1998. [Google Scholar] [CrossRef] [Green Version]
- Venables, J.P.; Koh, C.-S.; Froehlich, U.; Lapointe, E.; Couture, S.; Inkel, L.; Bramard, A.; Paquet, E.R.; Watier, V.; Durand, M.; et al. Multiple and Specific mRNA Processing Targets for the Major Human hnRNP Proteins. Mol. Cell. Biol. 2008, 28, 6033–6043. [Google Scholar] [CrossRef] [Green Version]
- Hung, L.-H.; Heiner, M.; Hui, J.; Schreiner, S.; Benes, V.; Bindereif, A. Diverse roles of hnRNP L in mammalian mRNA processing: A combined microarray and RNAi analysis. Rna 2008, 14, 284–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, S.; Gillespie, E.; Naqvi, A.S.; Hayer, K.E.; Ang, Z.; Torres-Diz, M.; Quesnel-Vallières, M.; Hottman, D.A.; Bagashev, A.; Chukinas, J.; et al. Modulation of CD22 Protein Expression in Childhood Leukemia by Pervasive Splicing Aberrations: Implications for CD22-Directed Immunotherapies. Blood Cancer Discov. 2022, 3, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Bourcier, J.; Abdel-Wahab, O. Splicing-Mediated Antigen Escape from Immunotherapy for B-cell Malignancies. Blood Cancer Discov. 2022, 3, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018, 24, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Brivio, E.; Locatelli, F.; Lopez-Yurda, M.; Malone, A.; Díaz-De-Heredia, C.; Bielorai, B.; Rossig, C.; van der Velden, V.H.J.; Ammerlaan, A.C.J.; Thano, A.; et al. A phase 1 study of inotuzumab ozogamicin in pediatric relapsed/refractory acute lymphoblastic leukemia (ITCC-059 study). Blood 2021, 137, 1582–1590. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | CD22E12low (N = 21) | All Others (N = 120) | p-Value | |||
|---|---|---|---|---|---|---|
| Mean/Median(Range) orN (% Evaluable) | Mean/Median(Range) or N (% Evaluable) | Mann-Whitney U Test or Fisher’s Exact | ||||
| Age (yrs) | ||||||
| Mean ± SEM/Median (Range) | 8.2 ± 1.2 | 7.6 (1.4–18.1) | 7.9 ± 0.5 | 6.4 (1.2–30) | 0.8 | |
| WBC (×109/L) | ||||||
| Mean ± SEM/Median (Range) | 45.5 ± 13.1 | 15.9 (1.3–214.5) | 76.8 ± 12 | 33 (1.1–1149) | 0.2 | |
| MRD at Day 29 | ||||||
| Mean ± SEM/Median (Range) | 0.07 ± 0.03 | 0 (0–0.57) | 0.9 ± 0.4 | 0 (0–26) | 0.4 | |
| MRD at End of Consolidation | ||||||
| Mean ± SEM/Median (Range) | 0 ± 0 (N = 3) | 0 (0–0) | 1.6 ± 1.4 (N = 23) | 0 (0–31.5) | 0.3 | |
| Age category | ||||||
| Adult (≥18 yrs) | 1/21 | (4.8%) | 4/120 | (3.3%) | 0.6 | |
| CNS Status at Diagnosis | ||||||
| CNS 2 + CNS 3 Induction Failure | 5/21 0/21 | (23.8%) (0%) | 23/120 2/119 | (19.2%) (1.7%) | 0.6 1.0 | |
| NCI Risk | ||||||
| High Risk | 11/21 | (52.4%) | 79/120 | (65.8%) | 0.3 | |
| Age risk | ||||||
| Poor (Age <2 yrs or ≥10 years) | 11/21 | (52.4%) | 53/120 | (44.2%) | 0.6 | |
| WBC category | ||||||
| ≥20 × 109/L MRD at Day 8 MRD > 0 | 10/21 7/7 | (47.6%) (100%) | 73/120 33/33 | (60.8%) (100%) | 0.3 1.0 | |
| MRD at Day 29 | ||||||
| MRD > 0 MRD at End of Consolidation MRD > 0 | 9/21 0/3 | (42.9%) (0%) | 64/120 8/23 | (53.3%) (34.8%) | 0.5 0.5 | |
| Cytogenetics (N = 106) | ||||||
| Pseudodiploid | 5/12 | (41.7%) | 38/94 | (40.4%) | 1.0 | |
| Pseudodiploid + Hypodiploid or Hyperdiploid with SCA | 7/12 | (58.3%) | 71/94 | (75.5%) | 0.3 | |
| Molecular Markers/FISH (N = 132) | ||||||
| BCR-ABL1 | 0/20 | (0%) | 5/112 | (4.5%) | 1.0 | |
| MLL/KMT2A rearranged | 0/20 | (0%) | 4/112 | (3.6%) | 1.0 | |
| TCF3-PBX1 | 3/20 | (15%) | 11/112 | (9.8%) | 0.4 | |
| Hyperdiploid with Trisomy of chromosomes 4 and 10 | 0/20 | (0%) | 10/112 | (8.9%) | 0.4 | |
| ETV6-RUNX1 | 0/20 | (0%) | 11/112 | (9.8%) | 0.2 | |
| ETV6-RUNX1 + Trisomy of chromosomes 4 and 10 | 0/20 | (0%) | 21/112 | (18.8%) | 0.04 | |
| Reactome Pathway | Enrichment Score in CD22E12low B-ALL | Enrichment Score in CD22ΔE12-Tg Mice | ||
|---|---|---|---|---|
| NES | p-Value | NES | p-Value | |
| Reactomes Involved in Transcription | ||||
| mRNA 3’-end processing | 2.4 | 2.8 × 10−5 | 2.4 | 1.3 × 10−5 |
| RNA polymerase II transcription termination | 2.3 | 2.8 × 10−5 | 2.5 | 1.3 × 10−5 |
| Transport of mature mRNA derived from an intronless transcript | 2.1 | 2.7 × 10−5 | 2.4 | 1.4 × 10−5 |
| RNA polymerase II pretranscription events | 1.8 | 4.7 × 10−4 | 2.6 | 1.2 × 10−5 |
| RNA polymerase II transcription elongation | 1.7 | 1.6 × 10−3 | 2.4 | 1.3 × 10−5 |
| Transcriptional regulation by small RNAs | 1.5 | 9.9 × 10−3 | 2.4 | 1.3 × 10−5 |
| Positive epigenetic regulation of rRNA expression | 1.5 | 1.9 × 10−2 | 2.0 | 1.3 × 10−5 |
| Reactomes Involved in mRNA Processing | ||||
| Regulation of mRNA stability by proteins that bind AU-rich elements | 1.8 | 2.4 × 0−4 | 2.5 | 1.2 × 10−5 |
| mRNA splicing—minor pathway | 1.6 | 1.3 × 10−2 | 2.3 | 1.3 × 10−5 |
| tRNA processing in the nucleus | 1.5 | 2.3 × 10−2 | 2.5 | 1.3 × 10−5 |
| Metabolism of non-coding RNA | 1.5 | 3.0 × 10−2 | 2.6 | 1.3 × 10−5 |
| Reactomes Involved in mRNA Transport | ||||
| Transport of mature transcript to cytoplasm | 2.6 | 3.0 × 10−5 | 2.6 | 1.2 × 10−5 |
| Transport of mature mRNA derived from an intron-containing transcript | 2.5 | 2.9 × 10−5 | 2.6 | 1.3 × 10−5 |
| Transport of mature mRNAs’ intronless transcripts | 2.2 | 2.7 × 10−5 | 2.4 | 1.3 × 10−5 |
| Reactomes Involved in Translation | ||||
| Formation of a pool of free 40S subunits | 3.0 | 3.1 × 10−5 | 2.8 | 1.2 × 10−5 |
| Eukaryotic translation elongation | 3.0 | 3.0 × 10−5 | 2.8 | 1.2 × 10−5 |
| Peptide chain elongation | 2.9 | 3.0 × 10−5 | 2.7 | 1.2 × 10−5 |
| Eukaryotic translation termination | 2.6 | 3.0 × 10−5 | 2.7 | 1.2 × 10−5 |
| Ribosomal scanning and start codon recognition | 2.6 | 2.8 × 10−5 | 2.6 | 1.3 × 10−5 |
| Translation initiation complex formation | 2.6 | 2.8 × 10−5 | 2.6 | 1.3 × 10−5 |
| Activation of the mRNA upon binding of the cap-binding complex and eIFs and subsequent binding to 43S | 2.4 | 2.8 × 10−5 | 2.6 | 1.3 × 10−5 |
| Formation of the ternary complex and, subsequently, the 43S complex | 2.4 | 2.7 × 10−5 | 2.5 | 1.3 × 10−5 |
| Reactomes Involved in Post-Translational Protein Modification | ||||
| SUMOylation of RNA binding proteins | 2.2 | 2.7 × 10−5 | 2.4 | 1.3 × 10−5 |
| Synthesis of active ubiquitin: roles of E1/E2 enzymes | 2.2 | 2.5 × 10−5 | 2.0 | 1.8 × 10−4 |
| SUMOylation of SUMOylation proteins | 2.1 | 7.7 × 10−5 | 2.3 | 1.4 × 10−5 |
| SUMOylation of DNA replication proteins | 2.0 | 1.1 × 10−4 | 2.4 | 1.3 × 10−5 |
| SUMOylation of ubiquitinylation proteins | 2.0 | 2.9 × 10−4 | 2.3 | 1.4 × 10−5 |
| Protein ubiquitination | 2.0 | 8.6 × 10−5 | 2.3 | 1.3 × 10−5 |
| SUMOylation of transcription cofactors | 1.9 | 3.2 × 10−4 | 1.9 | 7.9 × 10−5 |
| SUMOylation of chromatin organization proteins | 1.8 | 1.1 × 10−3 | 2.3 | 1.3 × 10−5 |
| SUMOylation of DNA damage response and repair proteins | 1.6 | 7.1 × 10−3 | 2.4 | 1.2 × 10−5 |
| Reactomes Involved in Signal Transduction | ||||
| RAF activation | 2.2 | 2.6 × 10−5 | 1.7 | 8.7 × 10−3 |
| MAP kinase activation | 1.9 | 2.5 × 10−4 | 1.5 | 1.5 × 10−2 |
| RHOBTB2 GTPase cycle | 1.9 | 2.1 × 10−3 | 1.8 | 2.7 × 10−3 |
| Regulation of RAS by GAPs | 1.9 | 2.6 × 10−4 | 2.0 | 1.2 × 10−5 |
| MAPK6/MAPK4 signaling | 1.8 | 4.5 × 10−4 | 2.1 | 1.2 × 10−5 |
| Reactomes Involved in Cell Cycle Pathway | ||||
| Postmitotic nuclear pore complex (NPC) reformation | 1.9 | 1.5 × 10−3 | 2.1 | 4.2 × 10−5 |
| Nuclear envelope (NE) reassembly | 1.8 | 9.7 × 10−4 | 2.2 | 1.2 × 10−5 |
| Mitotic telophase/cytokinesis | 1.8 | 1.2 × 10−2 | 1.9 | 1.3 × 10−5 |
| Regulation of apoptosis | 1.7 | 3.8 × 10−3 | 2.2 | 1.3 × 10−5 |
| Establishment of sister chromatid cohesion | 1.7 | 2.4 × 10−2 | 1.9 | 1.3 × 10−3 |
| Nuclear pore complex (NPC) disassembly | 1.7 | 7.9 × 10−3 | 2.3 | 1.4 × 10−5 |
| Nuclear envelope breakdown | 1.6 | 1.3 × 10−2 | 2.3 | 1.3 × 10−5 |
| Amplification of signal from unattached kinetochores via a MAD2 inhibitory signal | 1.5 | 1.3 × 10−2 | 2.7 | 1.2 × 10−5 |
| Amplification of signal from the kinetochores | 1.5 | 1.3 × 10−2 | 2.7 | 1.2 × 10−5 |
| APC/C-mediated degradation of cell cycle proteins | 1.5 | 1.6 × 10−2 | 2.4 | 1.2 × 10−5 |
| Regulation of mitotic cell cycle | 1.5 | 1.6 × 10−2 | 2.4 | 1.2 × 10−5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qazi, S.; Uckun, F.M. CD22 Exon 12 Deletion as an Independent Predictor of Poor Treatment Outcomes in B-ALL. Cancers 2023, 15, 1599. https://doi.org/10.3390/cancers15051599
Qazi S, Uckun FM. CD22 Exon 12 Deletion as an Independent Predictor of Poor Treatment Outcomes in B-ALL. Cancers. 2023; 15(5):1599. https://doi.org/10.3390/cancers15051599
Chicago/Turabian StyleQazi, Sanjive, and Fatih M. Uckun. 2023. "CD22 Exon 12 Deletion as an Independent Predictor of Poor Treatment Outcomes in B-ALL" Cancers 15, no. 5: 1599. https://doi.org/10.3390/cancers15051599