
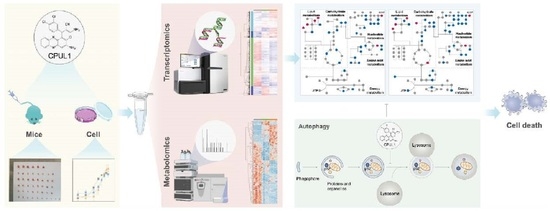
A Novel Phenazine Analog, CPUL1, Suppresses Autophagic Flux and Proliferation in Hepatocellular Carcinoma: Insight from Integrated Transcriptomic and Metabolomic Analysis

 , and
, and
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
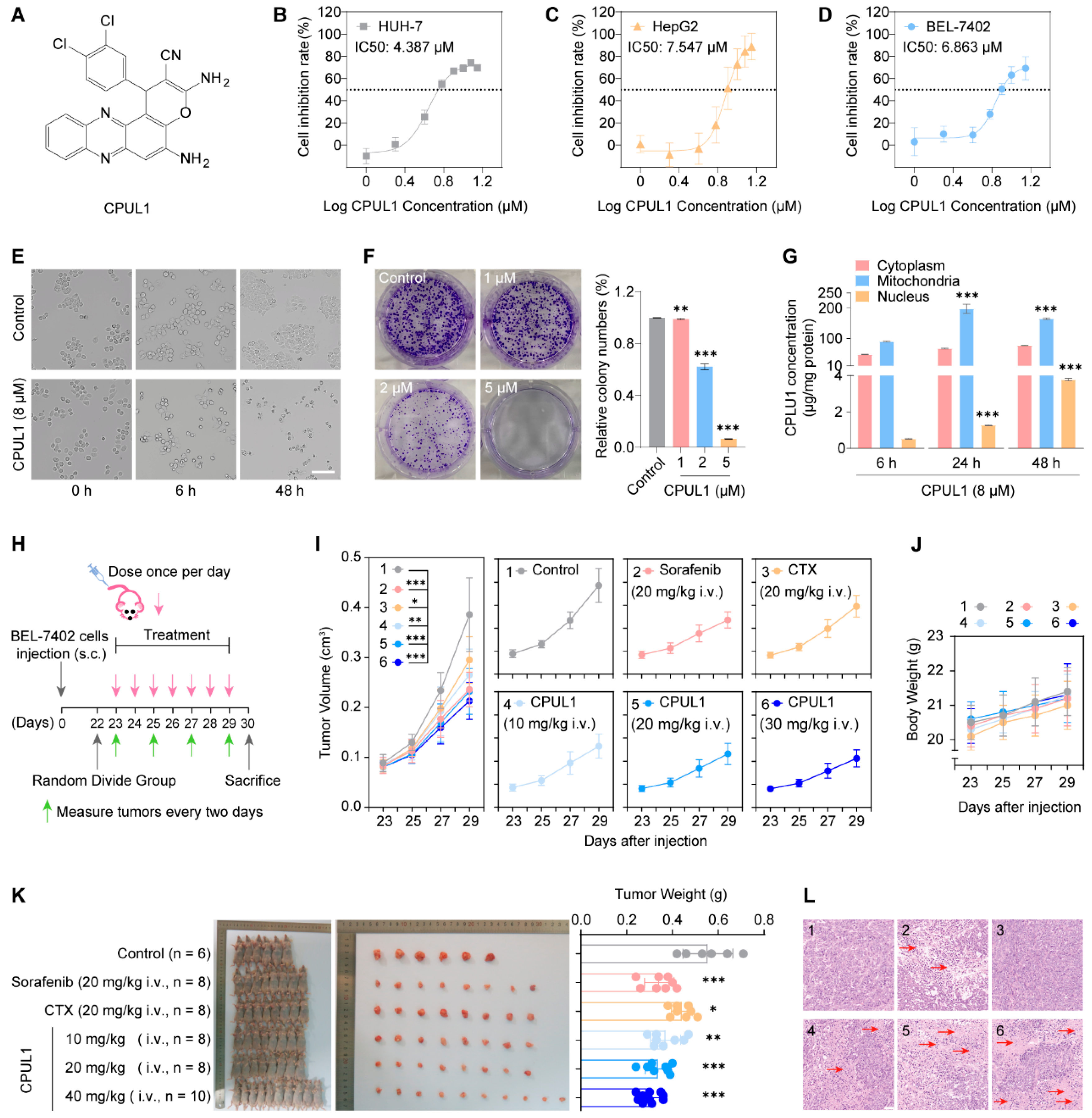
2.2. Cell Viability Assay
2.3. Cell Colony Formation Assay
2.4. Subcellular Localization of CPUL1
2.5. Antitumor Activity In Vivo
2.6. RNA Sequencing (RNA-Seq) Analysis
2.7. Metabolomics Analysis
2.8. Integrated Analysis of Transcriptomics and Metabolomics
2.9. Western Blotting Analysis
2.10. Transmission Electron Microscopy (TEM)
2.11. Immunofluorescence
2.12. Lyso-Tracker Red Staining
2.13. Statistical Analysis
3. Results
3.1. CPUL1 Exerted Antitumor Effects against HCC In Vitro
3.2. CPUL1 Exerted Antitumor Effects against HCC In Vivo
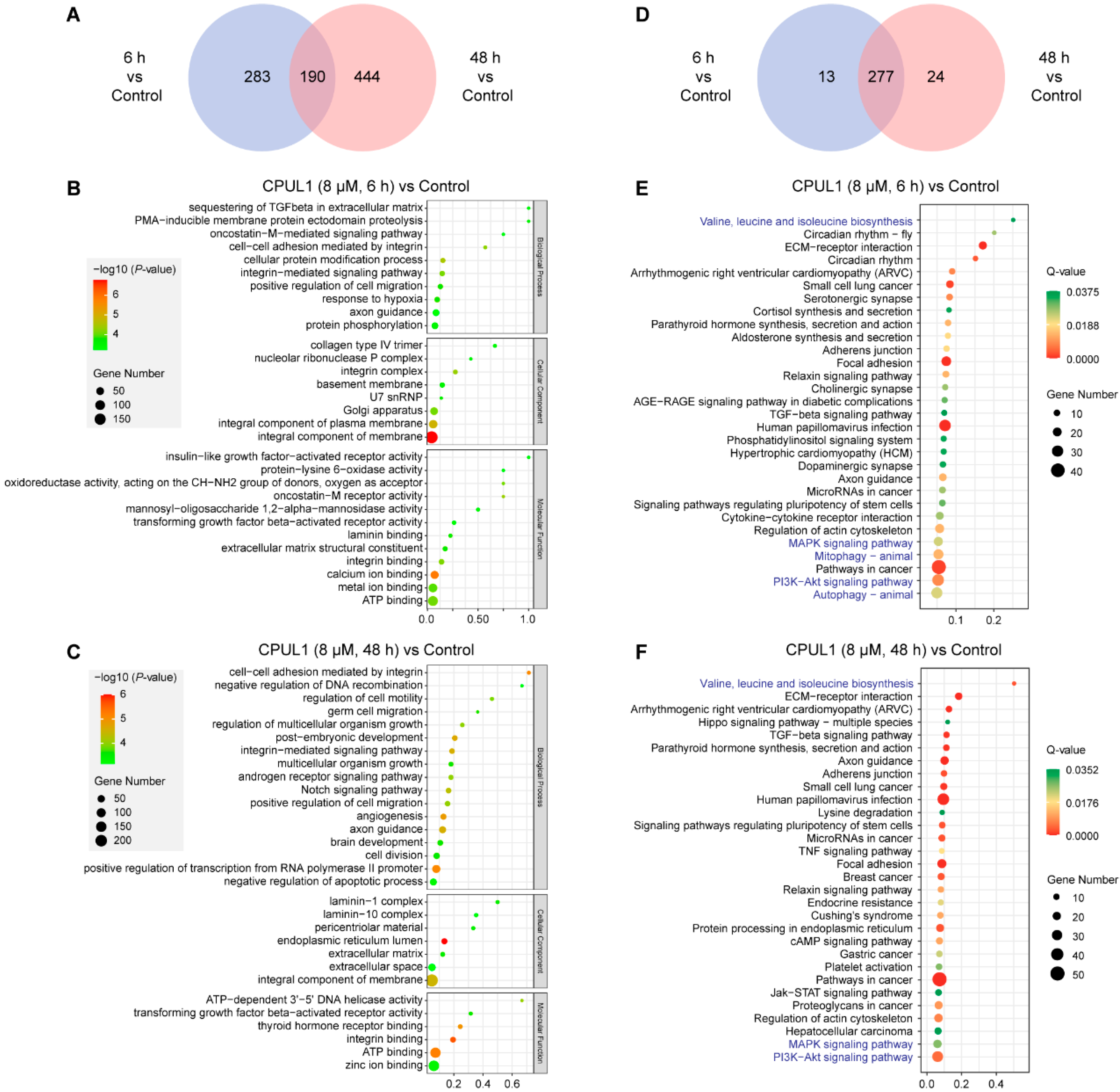
3.3. Transcriptomics Analysis on CPUL1-Treated BEL-7402 Cells
3.3.1. GO and KEGG Enrichment Analysis
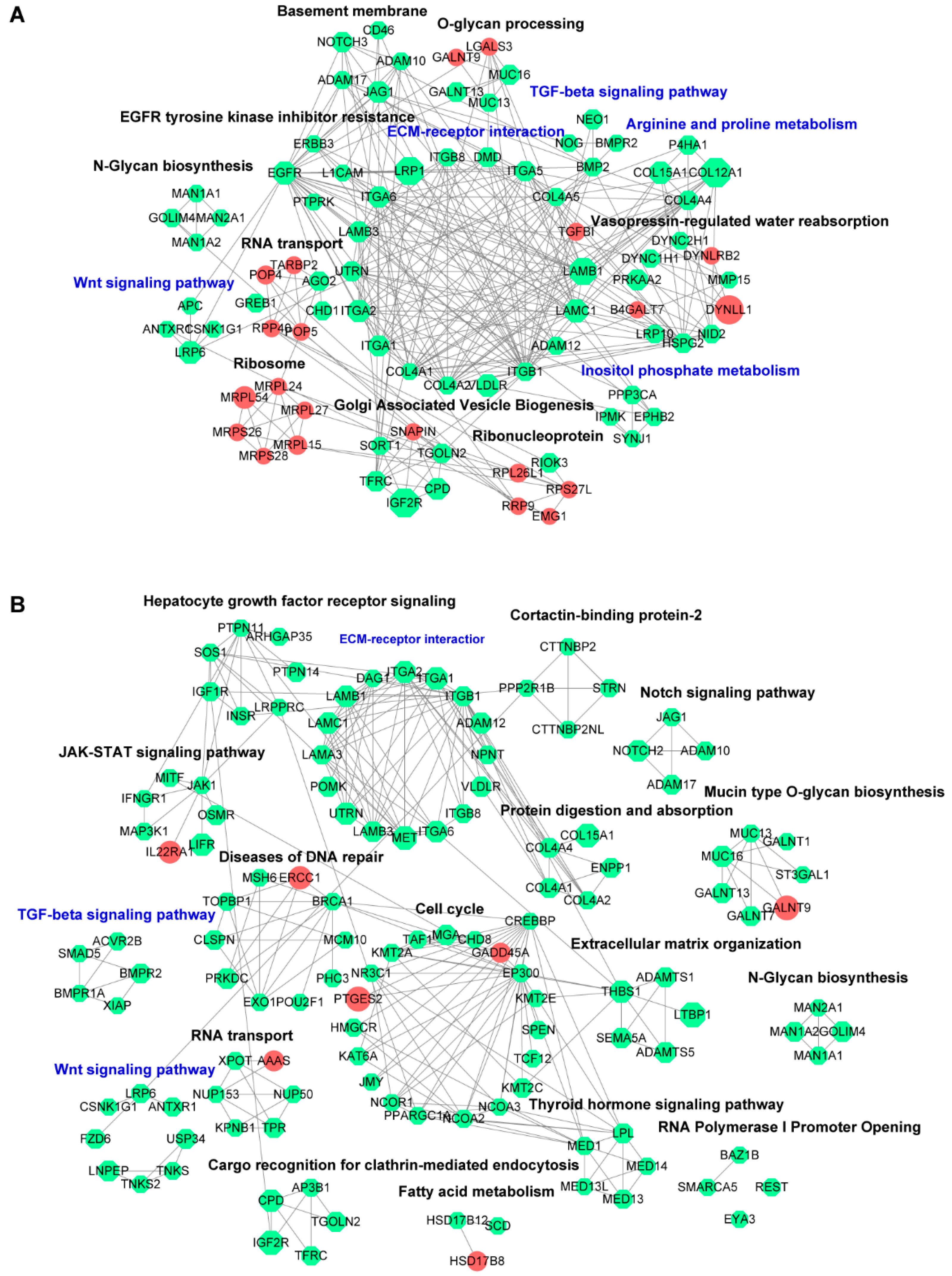
3.3.2. PPI Network Analysis
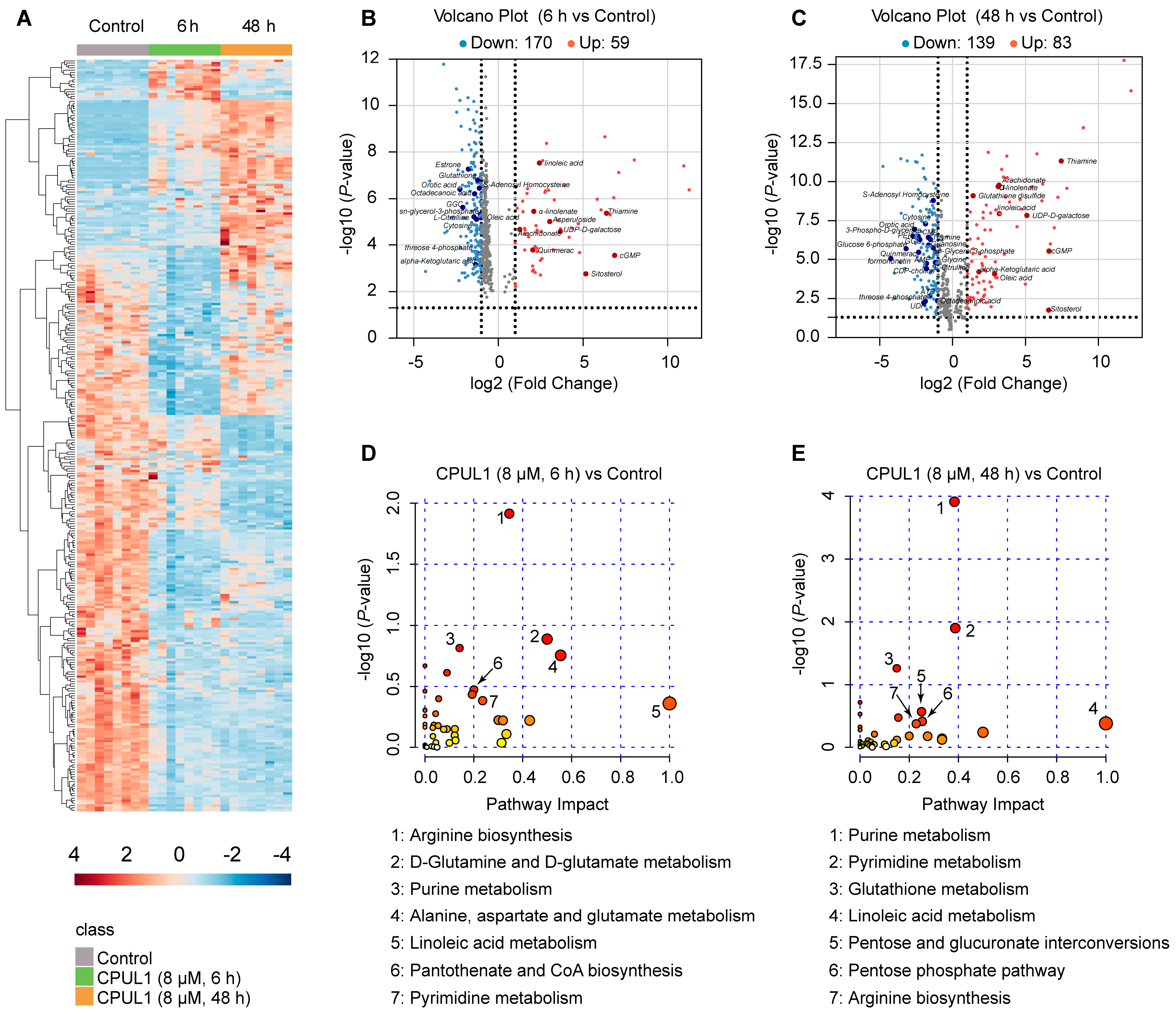
3.4. Non-Targeted Metabolomics Revealed Metabolic Perturbation Networks by CPUL1
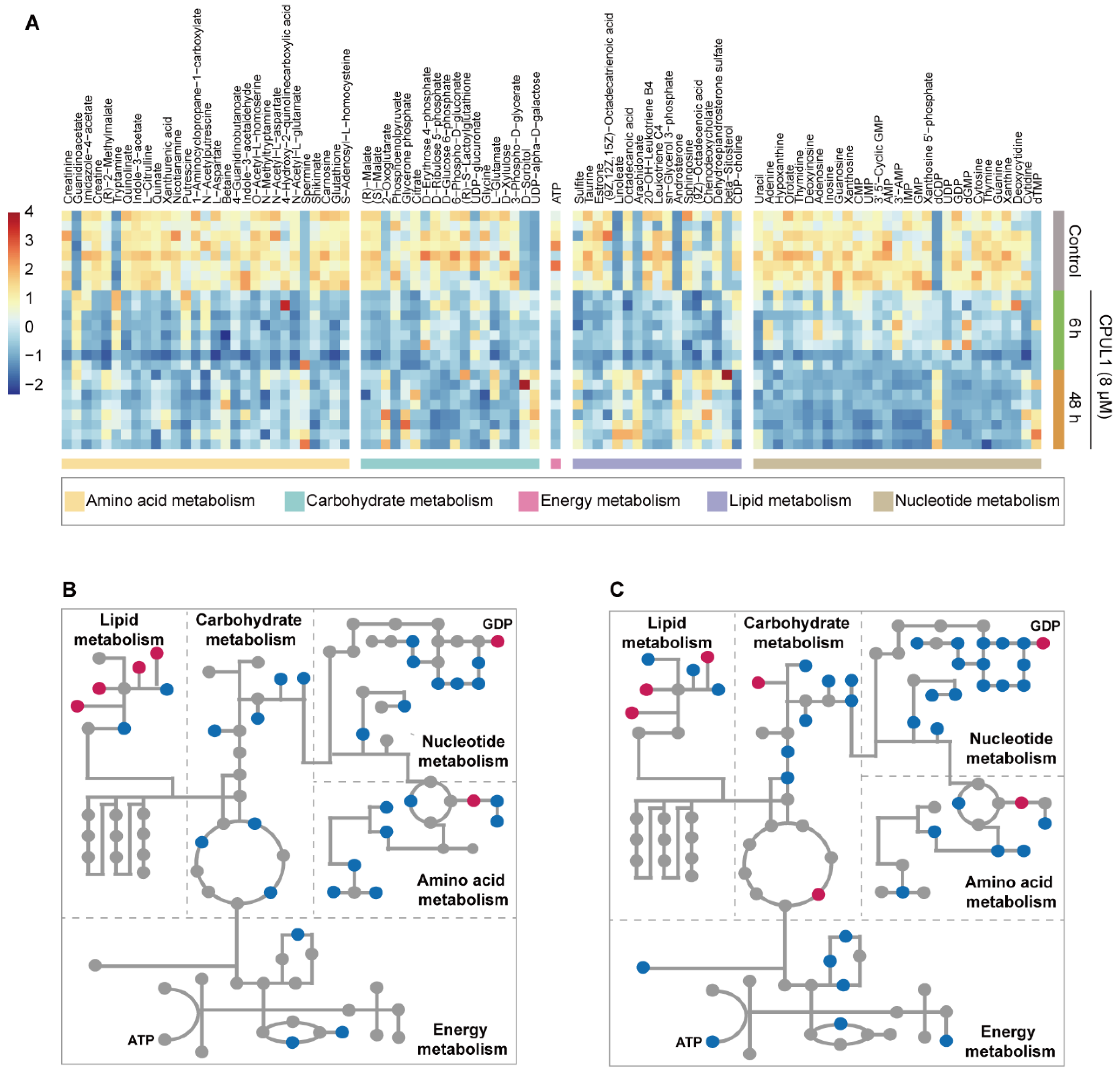
3.5. Integrative Omics Analysis Highlighted a Temporal Variegation of Metabolic Responses
3.6. CPUL1 Therapy Exacerbated Metabolic Dysregulation and Cellular Malnutrition in HCCs
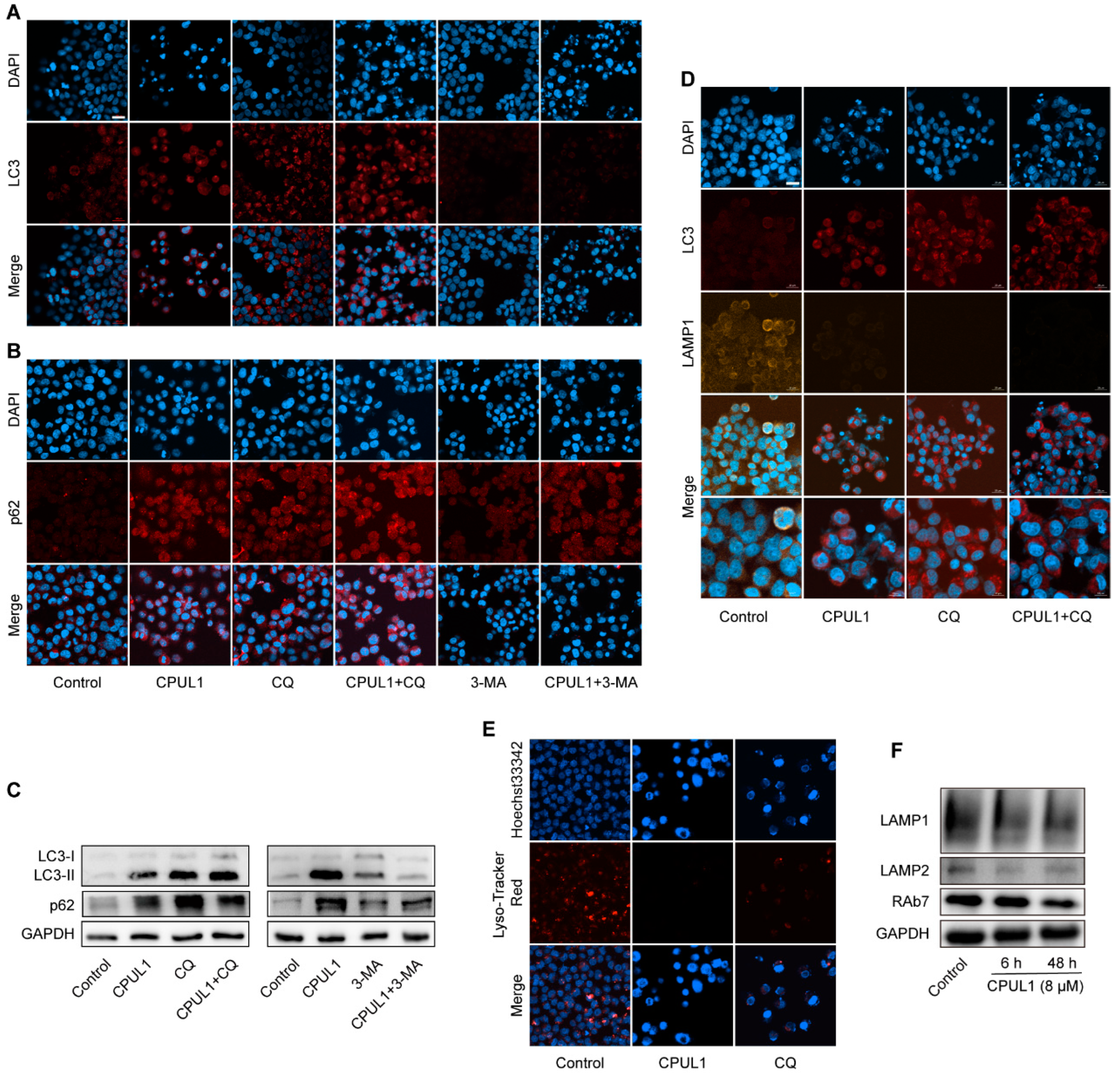
3.7. The Blockage of Autophagic Flux by CPUL1 Might Implicate Metabolism Perturbations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Morse, M.A.; Sun, W.; Kim, R.; He, A.R.; Abada, P.B.; Mynderse, M.; Finn, R.S. The Role of Angiogenesis in Hepatocellular Carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 912–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Lu, Z.; Zhao, X. Tumorigenesis, diagnosis, and therapeutic potential of exosomes in liver cancer. J. Hematol. Oncol. 2019, 12, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguayo, A.; Patt, Y.Z. Liver cancer. Clin. Liver Dis. 2001, 5, 479–507. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Morizane, C.; Ueno, M.; Okusaka, T.; Ishii, H.; Furuse, J. Chemotherapy for hepatocellular carcinoma: Current status and future perspectives. Jpn. J. Clin. Oncol. 2018, 48, 103–114. [Google Scholar] [CrossRef] [Green Version]
- Laursen, J.B.; Nielsen, J. Phenazine natural products: Biosynthesis, synthetic analogues, and biological activity. Chem. Rev. 2004, 104, 1663–1686. [Google Scholar] [CrossRef]
- Price-Whelan, A.; Dietrich, L.E.; Newman, D.K. Rethinking ‘secondary’ metabolism: Physiological roles for phenazine antibiotics. Nat. Chem. Biol. 2006, 2, 71–78. [Google Scholar] [CrossRef]
- Cimmino, A.; Evidente, A.; Mathieu, V.; Andolfi, A.; Lefranc, F.; Kornienko, A.; Kiss, R. Phenazines and cancer. Nat. Prod. Rep. 2012, 29, 487–501. [Google Scholar] [CrossRef]
- Mavrodi, D.V.; Blankenfeldt, W.; Thomashow, L.S. Phenazine compounds in fluorescent Pseudomonas spp. biosynthesis and regulation. Annu. Rev. Phytopathol. 2006, 44, 417–445. [Google Scholar] [CrossRef]
- Varsha, K.K.; Nishant, G.; Sneha, S.M.; Shilpa, G.; Devendra, L.; Priya, S.; Nampoothiri, K.M. Antifungal, anticancer and aminopeptidase inhibitory potential of a phenazine compound produced by lactococcus BSN307. Indian J. Microbiol. 2016, 56, 411–416. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Yan, Y.; Wang, L.; Wang, X.; Gao, J.; Xi, T.; Wang, Z.; Jiang, F. Design, facile synthesis and biological evaluations of novel pyrano[3,2-a]phenazine hybrid molecules as antitumor agents. Eur. J. Med. Chem. 2017, 127, 928–943. [Google Scholar] [CrossRef]
- Liao, J.; Wang, L.; Wu, Z.; Wang, Z.; Chen, J.; Zhong, Y.; Jiang, F.; Lu, Y. Identification of phenazine analogue as a novel scaffold for thioredoxin reductase I inhibitors against Hep G2 cancer cell lines. J. Enzym. Inhib. Med. Chem. 2019, 34, 1158–1163. [Google Scholar] [CrossRef]
- Pan, Y.; Lei, X.; Zhang, Y. Association predictions of genomics, proteinomics, transcriptomics, microbiome, metabolomics, pathomics, radiomics, drug, symptoms, environment factor, and disease networks: A comprehensive approach. Med. Res. Rev. 2022, 42, 441–461. [Google Scholar] [CrossRef]
- Hassan, M.A.; Al-Sakkaf, K.; Shait Mohammed, M.R.; Dallol, A.; Al-Maghrabi, J.; Aldahlawi, A.; Ashoor, S.; Maamra, M.; Ragoussis, J.; Wu, W.; et al. Integration of Transcriptome and Metabolome Provides Unique Insights to Pathways Associated With Obese Breast Cancer Patients. Front. Oncol. 2020, 10, 804. [Google Scholar] [CrossRef]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-omics Data Integration, Interpretation, and Its Application. Bioinform. Biol. Insights 2020, 14, 1177932219899051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, X.; Xu, S.; Zhong, Y.; Tu, T.; Xu, Y.; Li, X.; Wang, B.; Yang, F. High gene expression levels of VEGFA and CXCL8 in the peritumoral brain zone are associated with the recurrence of glioblastoma: A bioinformatics analysis. Oncol. Lett. 2019, 18, 6171–6179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Zhang, S.; Yang, J.; Yi, P.; Xu, P.; Yi, M.; Peng, W. Integrated transcriptomic and metabolomic analyses to characterize the anti-cancer effects of (-)-epigallocatechin-3-gallate in human colon cancer cells. Toxicol. Appl. Pharmacol. 2020, 401, 115100. [Google Scholar] [CrossRef]
- Chen, Y.; Gao, Y.; Yi, X.; Zhang, J.; Chen, Z.; Wu, Y. Integration of Transcriptomics and Metabolomics Reveals the Antitumor Mechanism Underlying Shikonin in Colon Cancer. Front. Pharmacol. 2020, 11, 544647. [Google Scholar] [CrossRef]
- Yang, Y.; Misra, B.B.; Liang, L.; Bi, D.; Weng, W.; Wu, W.; Cai, S.; Qin, H.; Goel, A.; Li, X.; et al. Integrated microbiome and metabolome analysis reveals a novel interplay between commensal bacteria and metabolites in colorectal cancer. Theranostics 2019, 9, 4101–4114. [Google Scholar] [CrossRef]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef]
- Chazotte, B. Labeling lysosomes in live cells with LysoTracker. Cold Spring Harb. Protoc. 2011, 2011, pdb-prot5571. [Google Scholar] [CrossRef]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. 4), S11. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Chandrashekar, D.S.; Varambally, S.; Creighton, C.J. Pan-cancer molecular subtypes revealed by mass-spectrometry-based proteomic characterization of more than 500 human cancers. Nat. Commun. 2019, 10, 5679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef] [PubMed]
- Lieu, E.L.; Nguyen, T.; Rhyne, S.; Kim, J. Amino acids in cancer. Exp. Mol. Med. 2020, 52, 15–30. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Antioxidants, T.I.J.; Signaling, R. Autophagosome formation and molecular mechanism of autophagy. Antioxid. Redox Signal. 2011, 14, 2201–2214. [Google Scholar] [CrossRef]
- Sanchez-Martin, P.; Komatsu, M. p62/SQSTM1—Steering the cell through health and disease. J. Cell Sci. 2018, 131, jcs222836. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wang, Q.; Song, D.; Zen, R.; Zhang, L.; Wang, Y.; Yang, H.; Zhang, D.; Jia, J.; Zhang, J.; et al. Lysosomal dysfunction and autophagy blockade contribute to autophagy-related cancer suppressing peptide-induced cytotoxic death of cervical cancer cells through the AMPK/mTOR pathway. J. Exp. Clin. Cancer Res. CR 2020, 39, 197. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, Y.; Yuan, M.; Chen, J.; Xu, Y.; Jie, Q.; An, Z.; Liao, J.; Liu, Y.; Li, J.; et al. Development and validation of a liquid chromatography-tandem mass spectrometry method for CPUL1, a novel antitumor candidate compound, and its application to pharmacokinetic studies. J. Sep. Sci. 2022, 45, 4397–4406. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [Green Version]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saftig, P.; Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: Trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Lehallier, B.; Shokhirev, M.N.; Wyss-Coray, T.; Johnson, A.A. Data mining of human plasma proteins generates a multitude of highly predictive aging clocks that reflect different aspects of aging. Aging Cell 2020, 19, e13256. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K. Recent advances in medical management of hepatocellular carcinoma. Hepatol. Res. 2019, 49, 14–32. [Google Scholar] [CrossRef] [Green Version]
- Duan, S.; Gong, B.; Wang, P.; Huang, H.; Luo, L.; Liu, F. Novel prognostic biomarkers of gastric cancer based on gene expression microarray: COL12A1, GSTA3, FGA and FGG. Mol. Med. Rep. 2018, 18, 3727–3736. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Ma, X.; Ha, W. Identification of Potential Prognostic Biomarkers Associated With Macrophage M2 Infiltration in Gastric Cancer. Front. Genet. 2021, 12, 827444. [Google Scholar] [CrossRef]
- He, C.K.; Li, Z.B.; Yi, D.; Zhu, X.Y.; Liu, R.R.; Zhang, D.X.; Cao, Q.; Chen, Y.P. LncRNA FGD5-AS1 enhances the proliferation and stemness of hepatocellular carcinoma cells through targeting miR-223 and regulating the expression of ECT2 and FAT1. Hepatol. Res. 2022, 52, 614–629. [Google Scholar] [CrossRef]
- Chen, Z.G.; Saba, N.F.; Teng, Y. The diverse functions of FAT1 in cancer progression: Good, bad, or ugly? J. Exp. Clin. Cancer Res. CR 2022, 41, 248. [Google Scholar] [CrossRef]
- van Loon, K.; Yemelyanenko-Lyalenko, J.; Margadant, C.; Griffioen, A.W.; Huijbers, E.J.M. Role of fibrillin-2 in the control of TGF-beta activation in tumor angiogenesis and connective tissue disorders. Biochim. Et Biophys. Acta. Rev. Cancer 2020, 1873, 188354. [Google Scholar] [CrossRef]
- Zhang, N.; Shao, F.; Jia, W. Upregulation of microfibrillar-associated protein 2 is closely associated with tumor angiogenesis and poor prognosis in hepatocellular carcinoma. Oncol. Lett. 2021, 22, 739. [Google Scholar] [CrossRef]
- Schmidt, D.R.; Patel, R.; Kirsch, D.G.; Lewis, C.A.; Vander Heiden, M.G.; Locasale, J.W. Metabolomics in cancer research and emerging applications in clinical oncology. CA Cancer J. Clin. 2021, 71, 333–358. [Google Scholar] [CrossRef]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef]
- Abla, H.; Sollazzo, M.; Gasparre, G.; Iommarini, L.; Porcelli, A.M. The multifaceted contribution of alpha-ketoglutarate to tumor progression: An opportunity to exploit? Semin. Cell Dev. Biol. 2020, 98, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Zhou, B.; Sun, R.Y.; Ai, Y.L.; Cheng, K.; Li, F.N.; Wang, B.R.; Liu, F.J.; Jiang, Z.H.; Wang, W.J.; et al. The metabolite alpha-KG induces GSDMC-dependent pyroptosis through death receptor 6-activated caspase-8. Cell Res. 2021, 31, 980–997. [Google Scholar] [CrossRef] [PubMed]
- Che, L.; Chi, W.; Qiao, Y.; Zhang, J.; Song, X.; Liu, Y.; Li, L.; Jia, J.; Pilo, M.G.; Wang, J.; et al. Cholesterol biosynthesis supports the growth of hepatocarcinoma lesions depleted of fatty acid synthase in mice and humans. Gut 2020, 69, 177–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satriano, L.; Lewinska, M.; Rodrigues, P.M.; Banales, J.M.; Andersen, J.B. Metabolic rearrangements in primary liver cancers: Cause and consequences. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 748–766. [Google Scholar] [CrossRef]
- Chen, D.; Zhang, Y.; Wang, W.; Chen, H.; Ling, T.; Yang, R.; Wang, Y.; Duan, C.; Liu, Y.; Guo, X.; et al. Identification and Characterization of Robust Hepatocellular Carcinoma Prognostic Subtypes Based on an Integrative Metabolite-Protein Interaction Network. Adv. Sci. 2021, 8, e2100311. [Google Scholar] [CrossRef]
- Qin, X.Y.; Su, T.; Yu, W.; Kojima, S. Lipid desaturation-associated endoplasmic reticulum stress regulates MYCN gene expression in hepatocellular carcinoma cells. Cell Death Dis. 2020, 11, 66. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, W.K.; Clemans, G.W.; Hutchinson, M.L. Essential fatty acids deficiency in humans. Prog. Lipid Res. 1980, 19, 187–215. [Google Scholar] [CrossRef]
- Qiu, J.F.; Zhang, K.L.; Zhang, X.J.; Hu, Y.J.; Li, P.; Shang, C.Z.; Wan, J.B. Abnormalities in Plasma Phospholipid Fatty Acid Profiles of Patients with Hepatocellular Carcinoma. Lipids 2015, 50, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Ning, Z.; Guo, X.; Liu, X.; Lu, C.; Wang, A.; Wang, X.; Wang, W.; Chen, H.; Qin, W.; Liu, X.; et al. USP22 regulates lipidome accumulation by stabilizing PPARgamma in hepatocellular carcinoma. Nat. Commun. 2022, 13, 2187. [Google Scholar] [CrossRef]
- Haberl, E.M.; Pohl, R.; Rein-Fischboeck, L.; Horing, M.; Krautbauer, S.; Liebisch, G.; Buechler, C. Accumulation of cholesterol, triglycerides and ceramides in hepatocellular carcinomas of diethylnitrosamine injected mice. Lipids Health Dis. 2021, 20, 135. [Google Scholar] [CrossRef] [PubMed]
- Cotte, A.K.; Cottet, V.; Aires, V.; Mouillot, T.; Rizk, M.; Vinault, S.; Binquet, C.; de Barros, J.P.; Hillon, P.; Delmas, D. Phospholipid profiles and hepatocellular carcinoma risk and prognosis in cirrhotic patients. Oncotarget 2019, 10, 2161–2172. [Google Scholar] [CrossRef] [PubMed]
- Hall, Z.; Chiarugi, D.; Charidemou, E.; Leslie, J.; Scott, E.; Pellegrinet, L.; Allison, M.; Mocciaro, G.; Anstee, Q.M.; Evan, G.I.; et al. Lipid Remodeling in Hepatocyte Proliferation and Hepatocellular Carcinoma. Hepatology 2021, 73, 1028–1044. [Google Scholar] [CrossRef]
- Mugabo, Y.; Zhao, S.; Seifried, A.; Gezzar, S.; Al-Mass, A.; Zhang, D.; Lamontagne, J.; Attane, C.; Poursharifi, P.; Iglesias, J.; et al. Identification of a mammalian glycerol-3-phosphate phosphatase: Role in metabolism and signaling in pancreatic beta-cells and hepatocytes. Proc. Natl. Acad. Sci. USA 2016, 113, E430–E439. [Google Scholar] [CrossRef] [Green Version]
- Gibellini, F.; Smith, T.K. The Kennedy pathway–De novo synthesis of phosphatidylethanolamine and phosphatidylcholine. IUBMB Life 2010, 62, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.; van der Meer, L.T.; van Leeuwen, F.N. Amino Acid Depletion Therapies: Starving Cancer Cells to Death. Trends Endocrinol. Metab. TEM 2021, 32, 367–381. [Google Scholar] [CrossRef]
- Bonifácio, V.D.B.; Pereira, S.A.; Serpa, J.; Vicente, J.B. Cysteine metabolic circuitries: Druggable targets in cancer. Br. J. Cancer 2021, 124, 862–879. [Google Scholar] [CrossRef]
- Jung, S.; Jeong, H.; Yu, S.W. Autophagy as a decisive process for cell death. Exp. Mol. Med. 2020, 52, 921–930. [Google Scholar] [CrossRef]
- Van Gool, B.; Dedieu, S.; Emonard, H.; Roebroek, A.J. The Matricellular Receptor LRP1 Forms an Interface for Signaling and Endocytosis in Modulation of the Extracellular Tumor Environment. Front. Pharmacol. 2015, 6, 271. [Google Scholar] [CrossRef] [Green Version]
- Peeters, S.; De Kinderen, P.; Meester, J.A.N.; Verstraeten, A.; Loeys, B.L. The fibrillinopathies: New insights with focus on the paradigm of opposing phenotypes for both FBN1 and FBN2. Hum. Mutat. 2022, 43, 815–831. [Google Scholar] [CrossRef]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.; Qu, Y.; An, Y.; Hou, L.; Li, J.; Li, W.; Fan, G.; Song, B.L.; Li, E.; Zhang, L.; et al. Induction of senescence-associated secretory phenotype underlies the therapeutic efficacy of PRC2 inhibition in cancer. Cell Death Dis. 2022, 13, 155. [Google Scholar] [CrossRef] [PubMed]
- Irshad, K.; Srivastava, C.; Malik, N.; Arora, M.; Gupta, Y.; Goswami, S.; Sarkar, C.; Suri, V.; Mahajan, S.; Gupta, D.K.; et al. Upregulation of Atypical Cadherin FAT1 Promotes an Immunosuppressive Tumor Microenvironment via TGF-β. Front. Immunol. 2022, 13, 813888. [Google Scholar] [CrossRef] [PubMed]
- De Souza, A.T.; Hankins, G.R.; Washington, M.K.; Orton, T.C.; Jirtle, R.L. M6P/IGF2R gene is mutated in human hepatocellular carcinomas with loss of heterozygosity. Nat. Genet. 1995, 11, 447–449. [Google Scholar] [CrossRef]
- Murakami, R.; Mizuno, T. Proximal-distal sequence of development of the skeletal tissues in the penis of rat and the inductive effect of epithelium. J. Embryol. Exp. Morphol. 1986, 92, 133–143. [Google Scholar] [CrossRef]
- Hamlin, A.N.; Basford, J.E.; Jaeschke, A.; Hui, D.Y. LRP1 Protein Deficiency Exacerbates Palmitate-induced Steatosis and Toxicity in Hepatocytes. J. Biol. Chem. 2016, 291, 16610–16619. [Google Scholar] [CrossRef] [Green Version]
- Hamlin, A.N.; Chinnarasu, S.; Ding, Y.; Xian, X.; Herz, J.; Jaeschke, A.; Hui, D.Y. Low-density lipoprotein receptor-related protein-1 dysfunction synergizes with dietary cholesterol to accelerate steatohepatitis progression. J. Biol. Chem. 2018, 293, 9674–9684. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.; Zheng, L.; Xu, X.; Hao, Z.; Zhang, Y.; Lu, J.; Sun, Z.; Dai, J.; Shi, D.; Guo, B.; et al. Heterozygous LRP1 deficiency causes developmental dysplasia of the hip by impairing triradiate chondrocytes differentiation due to inhibition of autophagy. Proc. Natl. Acad. Sci. USA 2022, 119, e2203557119. [Google Scholar] [CrossRef] [PubMed]
- Tumbarello, D.A.; Waxse, B.J.; Arden, S.D.; Bright, N.A.; Kendrick-Jones, J.; Buss, F. Autophagy receptors link myosin VI to autophagosomes to mediate Tom1-dependent autophagosome maturation and fusion with the lysosome. Nat. Cell Biol. 2012, 14, 1024–1035. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petroni, G.; Cantley, L.C.; Santambrogio, L.; Formenti, S.C.; Galluzzi, L. Radiotherapy as a tool to elicit clinically actionable signalling pathways in cancer. Nat. Rev. Clin. Oncol. 2022, 19, 114–131. [Google Scholar] [CrossRef] [PubMed]
- Mele, L.; Del Vecchio, V.; Liccardo, D.; Prisco, C.; Schwerdtfeger, M.; Robinson, N.; Desiderio, V.; Tirino, V.; Papaccio, G.; La Noce, M. The role of autophagy in resistance to targeted therapies. Cancer Treat. Rev. 2020, 88, 102043. [Google Scholar] [CrossRef]
- Shimizu, S.; Takehara, T.; Hikita, H.; Kodama, T.; Tsunematsu, H.; Miyagi, T.; Hosui, A.; Ishida, H.; Tatsumi, T.; Kanto, T.; et al. Inhibition of autophagy potentiates the antitumor effect of the multikinase inhibitor sorafenib in hepatocellular carcinoma. Int. J. Cancer 2012, 131, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, J.; Li, K.; Deng, L.; Wang, H. Combination of an Autophagy Inducer and an Autophagy Inhibitor: A Smarter Strategy Emerging in Cancer Therapy. Front. Pharmacol. 2020, 11, 408. [Google Scholar] [CrossRef] [Green Version]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Feng, D.; Lu, Y.; Zhang, Y.; Jiang, H.; Yuan, M.; Xu, Y.; Zou, J.; Zhu, Y.; Zhang, J.; et al. A Novel Phenazine Analog, CPUL1, Suppresses Autophagic Flux and Proliferation in Hepatocellular Carcinoma: Insight from Integrated Transcriptomic and Metabolomic Analysis. Cancers 2023, 15, 1607. https://doi.org/10.3390/cancers15051607
Chen J, Feng D, Lu Y, Zhang Y, Jiang H, Yuan M, Xu Y, Zou J, Zhu Y, Zhang J, et al. A Novel Phenazine Analog, CPUL1, Suppresses Autophagic Flux and Proliferation in Hepatocellular Carcinoma: Insight from Integrated Transcriptomic and Metabolomic Analysis. Cancers. 2023; 15(5):1607. https://doi.org/10.3390/cancers15051607
Chicago/Turabian StyleChen, Jiaqin, Dong Feng, Yuanyuan Lu, Yanjun Zhang, Hanxiang Jiang, Man Yuan, Yifan Xu, Jianjun Zou, Yubing Zhu, Jingjing Zhang, and et al. 2023. "A Novel Phenazine Analog, CPUL1, Suppresses Autophagic Flux and Proliferation in Hepatocellular Carcinoma: Insight from Integrated Transcriptomic and Metabolomic Analysis" Cancers 15, no. 5: 1607. https://doi.org/10.3390/cancers15051607
APA StyleChen, J., Feng, D., Lu, Y., Zhang, Y., Jiang, H., Yuan, M., Xu, Y., Zou, J., Zhu, Y., Zhang, J., Ge, C., & Wang, Y. (2023). A Novel Phenazine Analog, CPUL1, Suppresses Autophagic Flux and Proliferation in Hepatocellular Carcinoma: Insight from Integrated Transcriptomic and Metabolomic Analysis. Cancers, 15(5), 1607. https://doi.org/10.3390/cancers15051607