

Pyruvate Dehydrogenase Kinase 4 Deficiency Increases Tumorigenesis in a Murine Model of Bladder Cancer
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods and Materials
2.1. Mice
2.2. BBN Protocol
2.3. Immunohistochemistry
2.4. Human Tissue and TCGA Analysis
2.5. qPCR
2.6. Statistics
3. Results
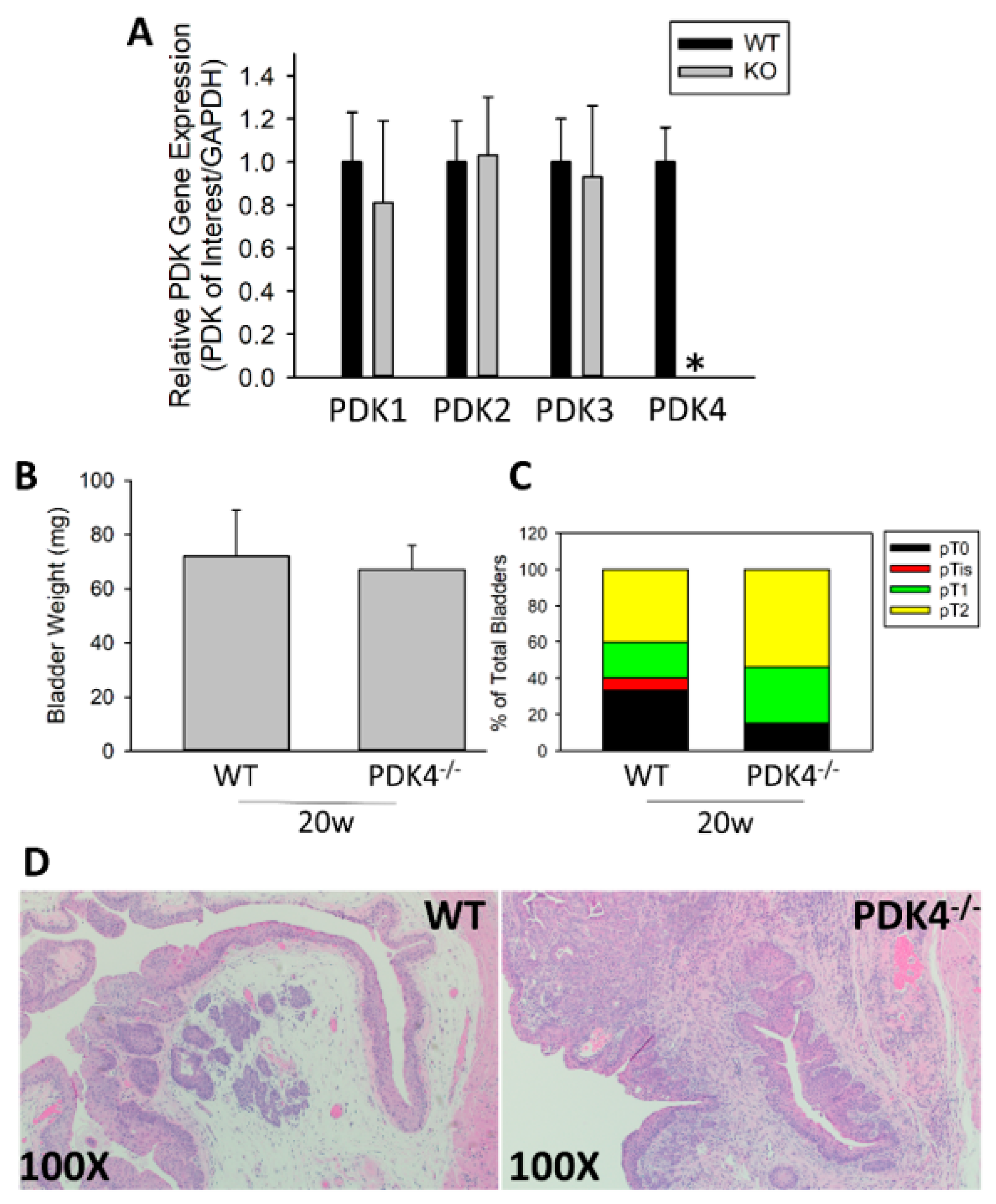
3.1. PDK4−/− Animals Have Larger, Higher-Stage Tumors after BBN Treatment at Later Time Points of Tumor Formation
3.2. Increased Proliferative Potential of PDK4−/− Tumors
3.3. PDK4 Expression Is Suppressed in Murine and Human BCa
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meeks, J.J.; Al-Ahmadie, H.; Faltas, B.M.; Taylor, J.A., 3rd; Flaig, T.W.; DeGraff, D.J.; Christensen, E.; Woolbright, B.L.; McConkey, D.J.; Dyrskjot, L. Genomic heterogeneity in bladder cancer: Challenges and possible solutions to improve outcomes. Nat. Rev. Urol. 2020, 17, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Kamat, A.M.; Hahn, N.M.; Efstathiou, J.A.; Lerner, S.P.; Malmstrom, P.U.; Choi, W.; Guo, C.C.; Lotan, Y.; Kassouf, W. Bladder cancer. Lancet 2016, 388, 2796–2810. [Google Scholar] [CrossRef]
- Huo, J.; Ray-Zack, M.D.; Shan, Y.; Chamie, K.; Boorjian, S.A.; Kerr, P.; Jana, B.; Freedland, S.J.; Kamat, A.M.; Mehta, H.B.; et al. Discerning Patterns and Quality of Neoadjuvant Chemotherapy Use Among Patients with Muscle-invasive Bladder Cancer. Eur. Urol. Oncol. 2019, 2, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Damish, A.W.; Frazier, Z.; Liu, D.; Reznichenko, E.; Kamburov, A.; Bell, A.; Zhao, H.; Jordan, E.J.; Gao, S.P.; et al. ERCC2 Helicase Domain Mutations Confer Nucleotide Excision Repair Deficiency and Drive Cisplatin Sensitivity in Muscle-Invasive Bladder Cancer. Clin. Cancer Res. 2019, 25, 977–988. [Google Scholar] [CrossRef] [Green Version]
- Teo, M.Y.; Bambury, R.M.; Zabor, E.C.; Jordan, E.; Al-Ahmadie, H.; Boyd, M.E.; Bouvier, N.; Mullane, S.A.; Cha, E.K.; Roper, N.; et al. DNA Damage Response and Repair Gene Alterations Are Associated with Improved Survival in Patients with Platinum-Treated Advanced Urothelial Carcinoma. Clin. Cancer Res. 2017, 23, 3610–3618. [Google Scholar] [CrossRef] [Green Version]
- Siefker-Radtke, A.O.; Cho, D.C.; Diab, A.; Sznol, M.; Bilen, M.A.; Balar, A.V.; Grignani, G.; Puente, E.; Tang, L.; Chien, D.; et al. Bempegaldesleukin plus Nivolumab in First-line Metastatic Urothelial Carcinoma: Results from PIVOT-02. Eur. Urol. 2022, 82, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Woolbright, B.L.; Rajendran, G.; Harris, R.A.; Taylor, J.A., 3rd. Metabolic Flexibility in Cancer: Targeting the Pyruvate Dehydrogenase Kinase:Pyruvate Dehydrogenase Axis. Mol. Cancer Ther. 2019, 18, 1673–1681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holness, M.J.; Sugden, M.C. Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem. Soc. Trans. 2003, 31 Pt 6, 1143–1151. [Google Scholar] [CrossRef]
- Stacpoole, P.W. Therapeutic Targeting of the Pyruvate Dehydrogenase Complex/Pyruvate Dehydrogenase Kinase (PDC/PDK) Axis in Cancer. J. Natl. Cancer Inst. 2017, 109, djx071. [Google Scholar] [CrossRef] [Green Version]
- Fuller, S.J.; Randle, P.J. Reversible phosphorylation of pyruvate dehydrogenase in rat skeletal-muscle mitochondria. Effects of starvation and diabetes. Biochem. J. 1984, 219, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Sugden, M.C.; Holness, M.J. Mechanisms underlying regulation of the expression and activities of the mammalian pyruvate dehydrogenase kinases. Arch. Physiol. Biochem. 2006, 112, 139–149. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Luengo, A.; Li, Z.; Gui, D.Y.; Sullivan, L.B.; Zagorulya, M.; Do, B.T.; Ferreira, R.; Naamati, A.; Ali, A.; Lewis, C.A.; et al. Increased demand for NAD(+) relative to ATP drives aerobic glycolysis. Mol. Cell 2021, 81, 691–707.e6. [Google Scholar] [CrossRef]
- Brough, P.A.; Aherne, W.; Barril, X.; Borgognoni, J.; Boxall, K.; Cansfield, J.E.; Cheung, K.M.; Collins, I.; Davies, N.G.; Drysdale, M.J.; et al. 4,5-diarylisoxazole Hsp90 chaperone inhibitors: Potential therapeutic agents for the treatment of cancer. J. Med. Chem. 2008, 51, 196–218. [Google Scholar] [CrossRef]
- Shen, Y.C.; Ou, D.L.; Hsu, C.; Lin, K.L.; Chang, C.Y.; Lin, C.Y.; Liu, S.H.; Cheng, A.L. Activating oxidative phosphorylation by a pyruvate dehydrogenase kinase inhibitor overcomes sorafenib resistance of hepatocellular carcinoma. Br. J. Cancer 2013, 108, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Morrell, J.A.; Orme, J.; Butlin, R.J.; Roche, T.E.; Mayers, R.M.; Kilgour, E. AZD7545 is a selective inhibitor of pyruvate dehydrogenase kinase 2. Biochem. Soc. Trans. 2003, 31 Pt 6, 1168–1170. [Google Scholar] [CrossRef]
- Tso, S.C.; Lou, M.; Wu, C.Y.; Gui, W.J.; Chuang, J.L.; Morlock, L.K.; Williams, N.S.; Wynn, R.M.; Qi, X.; Chuang, D.T. Development of Dihydroxyphenyl Sulfonylisoindoline Derivatives as Liver-Targeting Pyruvate Dehydrogenase Kinase Inhibitors. J. Med. Chem. 2017, 60, 1142–1150. [Google Scholar] [CrossRef]
- Moore, J.D.; Staniszewska, A.; Shaw, T.; D’Alessandro, J.; Davis, B.; Surgenor, A.; Baker, L.; Matassova, N.; Murray, J.; Macias, A.; et al. VER-246608, a novel pan-isoform ATP competitive inhibitor of pyruvate dehydrogenase kinase, disrupts Warburg metabolism and induces context-dependent cytostasis in cancer cells. Oncotarget 2014, 5, 12862–12876. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.J.; Lin, T.Y.; Lin, X.L.; Liu, Y.; Zhao, W.T.; Li, X.Y.; Lian, M.; Chen, H.W.; Li, Y.L.; Zhang, X.L.; et al. Loss of PDK4 expression promotes proliferation, tumorigenicity, motility and invasion of hepatocellular carcinoma cells. J. Cancer 2020, 11, 4397–4405. [Google Scholar] [CrossRef]
- Yang, C.; Wang, S.; Ruan, H.; Li, B.; Cheng, Z.; He, J.; Zuo, Q.; Yu, C.; Wang, H.; Lv, Y.; et al. Downregulation of PDK4 Increases Lipogenesis and Associates with Poor Prognosis in Hepatocellular Carcinoma. J. Cancer 2019, 10, 918–926. [Google Scholar] [CrossRef]
- Li, G.; Li, M.; Hu, J.; Lei, R.; Xiong, H.; Ji, H.; Yin, H.; Wei, Q.; Hu, G. The microRNA-182-PDK4 axis regulates lung tumorigenesis by modulating pyruvate dehydrogenase and lipogenesis. Oncogene 2017, 36, 989–998. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Choudhary, D.; Mikhalyuk, A.; Trammel, C.; Shanmugam, S.; Abbott, E.; Pilbeam, C.C.; Taylor, J.A., 3rd. The Role of Pyruvate Dehydrogenase Kinase-4 (PDK4) in Bladder Cancer and Chemoresistance. Mol. Cancer Ther. 2018, 17, 2004–2012. [Google Scholar] [CrossRef] [Green Version]
- Nagao, M.; Suzuki, E.; Yasuo, K.; Yahagi, T.; Seino, Y. Mutagenicity of N-butyl-N-(4-hydroxybutyl)nitrosamine, a bladder carcinogen, and related compounds. Cancer Res. 1977, 37, 399–407. [Google Scholar]
- Akagi, G.; Akagi, A.; Kimura, M.; Otsuka, H. Comparison of bladder tumors induced in rats and mice with N-butyl-N-(4-hydroxybutyl)-nitrosoamine. Gan 1973, 64, 331–336. [Google Scholar]
- Chandrashekar, D.S.; Karthikeyan, S.K.; Korla, P.K.; Patel, H.; Shovon, A.R.; Athar, M.; Netto, G.J.; Qin, Z.S.; Kumar, S.; Manne, U.; et al. UALCAN: An update to the integrated cancer data analysis platform. Neoplasia 2022, 25, 18–27. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [Green Version]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repecka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Fantini, D.; Meeks, J.J. The BBN model: A mouse bladder cancer model featuring basal-subtype gene expression and MLL3/MLL4 genetic disruption. Oncoscience 2018, 5, 172–173. [Google Scholar] [CrossRef]
- Fantini, D.; Glaser, A.P.; Rimar, K.J.; Wang, Y.; Schipma, M.; Varghese, N.; Rademaker, A.; Behdad, A.; Yellapa, A.; Yu, Y.; et al. A Carcinogen-induced mouse model recapitulates the molecular alterations of human muscle invasive bladder cancer. Oncogene 2018, 37, 1911–1925. [Google Scholar] [CrossRef]
- Jeon, J.H.; Thoudam, T.; Choi, E.J.; Kim, M.J.; Harris, R.A.; Lee, I.K. Loss of metabolic flexibility as a result of overexpression of pyruvate dehydrogenase kinases in muscle, liver and the immune system: Therapeutic targets in metabolic diseases. J. Diabetes Investig. 2021, 12, 21–31. [Google Scholar] [CrossRef]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007, 11, 37–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.H.; Chung, J.W.; Sung, E.; Yoon, B.H.; Jeon, M.; Park, S.; Chun, S.Y.; Lee, J.N.; Kim, B.S.; Kim, H.T.; et al. Anti-Metastatic Effect of Pyruvate Dehydrogenase Kinase 4 Inhibition in Bladder Cancer via the ERK, SRC, and JNK Pathways. Int. J. Mol. Sci. 2022, 23, 13240. [Google Scholar] [CrossRef] [PubMed]
- Atas, E.; Oberhuber, M.; Kenner, L. The Implications of PDK1-4 on Tumor Energy Metabolism, Aggressiveness and Therapy Resistance. Front. Oncol. 2020, 10, 583217. [Google Scholar] [CrossRef] [PubMed]
- Gerriets, V.A.; Kishton, R.J.; Nichols, A.G.; Macintyre, A.N.; Inoue, M.; Ilkayeva, O.; Winter, P.S.; Liu, X.; Priyadharshini, B.; Slawinska, M.E.; et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J. Clin. Investig. 2015, 125, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Podojil, J.R.; Glaser, A.P.; Baker, D.; Courtois, E.T.; Fantini, D.; Yu, Y.; Eaton, V.; Sivajothi, S.; Chiang, M.; Das, A.; et al. Antibody targeting of B7-H4 enhances the immune response in urothelial carcinoma. Oncoimmunology 2020, 9, 1744897. [Google Scholar] [CrossRef] [Green Version]
- Min, B.K.; Park, S.; Kang, H.J.; Kim, D.W.; Ham, H.J.; Ha, C.M.; Choi, B.J.; Lee, J.Y.; Oh, C.J.; Yoo, E.K.; et al. Pyruvate Dehydrogenase Kinase Is a Metabolic Checkpoint for Polarization of Macrophages to the M1 Phenotype. Front. Immunol. 2019, 10, 944. [Google Scholar] [CrossRef] [Green Version]
- Thoudam, T.; Chanda, D.; Sinam, I.S.; Kim, B.G.; Kim, M.J.; Oh, C.J.; Lee, J.Y.; Kim, M.J.; Park, S.Y.; Lee, S.Y.; et al. Noncanonical PDK4 action alters mitochondrial dynamics to affect the cellular respiratory status. Proc. Natl. Acad. Sci. USA 2022, 119, e2120157119. [Google Scholar] [CrossRef]
- Boulton, D.P.; Caino, M.C. Mitochondrial Fission and Fusion in Tumor Progression to Metastasis. Front. Cell Dev. Biol. 2022, 10, 849962. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 2013, 32, 4814–4824. [Google Scholar] [CrossRef]
- Dyrskjot, L.; Zieger, K.; Kruhoffer, M.; Thykjaer, T.; Jensen, J.L.; Primdahl, H.; Aziz, N.; Marcussen, N.; Moller, K.; Orntoft, T.F. A molecular signature in superficial bladder carcinoma predicts clinical outcome. Clin. Cancer Res. 2005, 11, 4029–4036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyrskjot, L.; Kruhoffer, M.; Thykjaer, T.; Marcussen, N.; Jensen, J.L.; Moller, K.; Orntoft, T.F. Gene expression in the urinary bladder: A common carcinoma in situ gene expression signature exists disregarding histopathological classification. Cancer Res. 2004, 64, 4040–4048. [Google Scholar] [CrossRef] [Green Version]
- Choiniere, J.; Lin, M.J.; Wang, L.; Wu, J. Deficiency of pyruvate dehydrogenase kinase 4 sensitizes mouse liver to diethylnitrosamine and arsenic toxicity through inducing apoptosis. Liver Res. 2018, 2, 100–107. [Google Scholar] [CrossRef]
- Choiniere, J.; Wu, J.; Wang, L. Pyruvate Dehydrogenase Kinase 4 Deficiency Results in Expedited Cellular Proliferation through E2F1-Mediated Increase of Cyclins. Mol. Pharmacol. 2017, 91, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Wu, J.; Choiniere, J.; Yang, Z.; Huang, Y.; Bennett, J.; Wang, L. Arsenic silences hepatic PDK4 expression through activation of histone H3K9 methylatransferase G9a. Toxicol. Appl. Pharmacol. 2016, 304, 42–47. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wang, L.; Wang, L.; Feng, Z.; Peng, C. Single-Cell Sequencing of Hepatocellular Carcinoma Reveals Cell Interactions and Cell Heterogeneity in the Microenvironment. Int. J. Gen. Med. 2021, 14, 10141–10153. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woolbright, B.L.; Rajendran, G.; Abbott, E.; Martin, A.; Didde, R.; Dennis, K.; Harris, R.A.; Taylor, J.A., III. Pyruvate Dehydrogenase Kinase 4 Deficiency Increases Tumorigenesis in a Murine Model of Bladder Cancer. Cancers 2023, 15, 1654. https://doi.org/10.3390/cancers15061654
Woolbright BL, Rajendran G, Abbott E, Martin A, Didde R, Dennis K, Harris RA, Taylor JA III. Pyruvate Dehydrogenase Kinase 4 Deficiency Increases Tumorigenesis in a Murine Model of Bladder Cancer. Cancers. 2023; 15(6):1654. https://doi.org/10.3390/cancers15061654
Chicago/Turabian StyleWoolbright, Benjamin L., Ganeshkumar Rajendran, Erika Abbott, Austin Martin, Ryan Didde, Katie Dennis, Robert A. Harris, and John A. Taylor, III. 2023. "Pyruvate Dehydrogenase Kinase 4 Deficiency Increases Tumorigenesis in a Murine Model of Bladder Cancer" Cancers 15, no. 6: 1654. https://doi.org/10.3390/cancers15061654