
The Potential Revolution of Cancer Treatment with CRISPR Technology

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
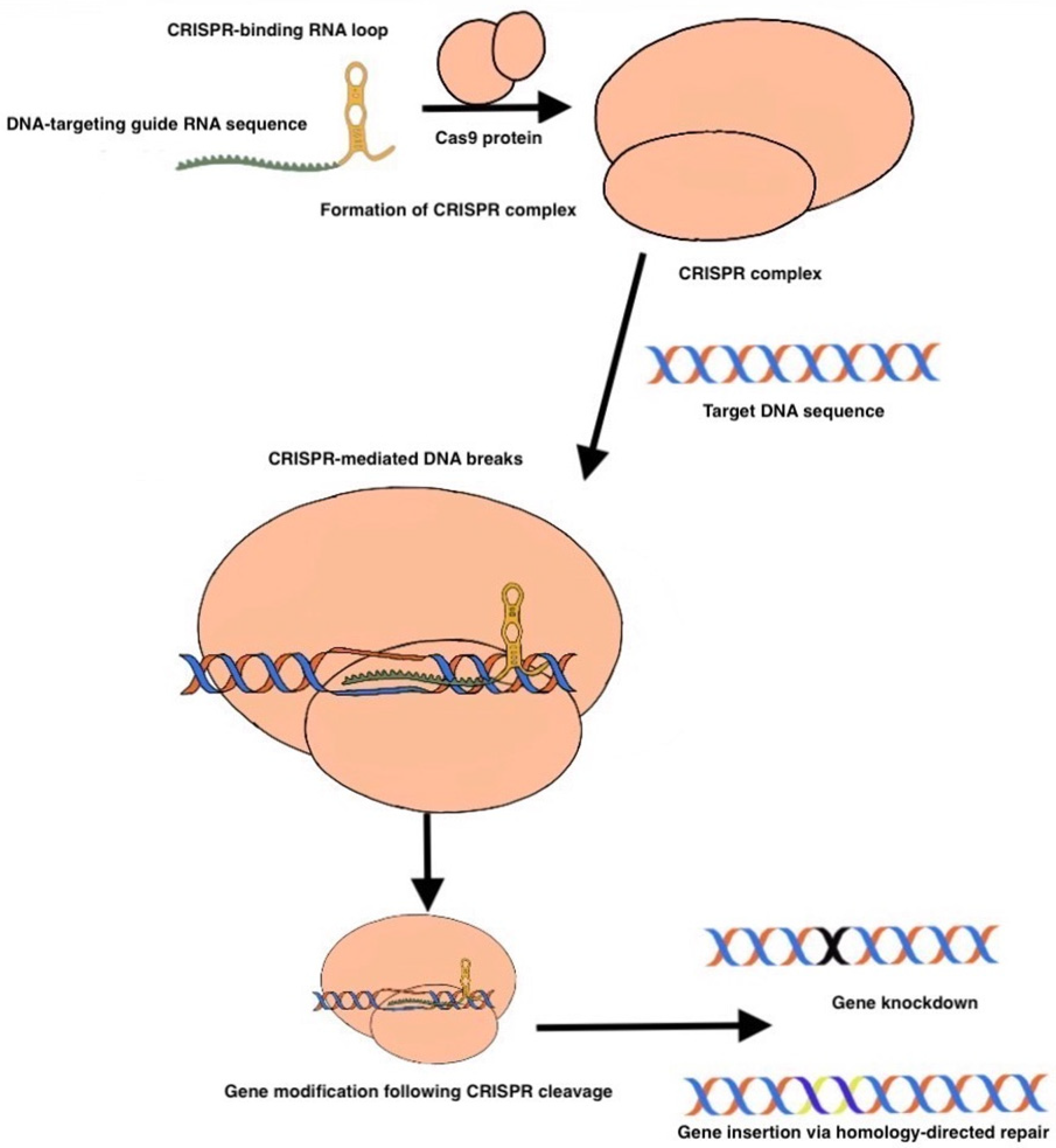
2. CRISPR
3. CRISPR Preclinical Use
4. CRISPR in Clinical Practice
5. CRISPR in Oncology—Preclinical Use
6. CRISPR Use in Cancer Prevention
7. CRISPR use for Clinical Cancer Treatment
8. CRISPR Use Limitations
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.M.; Hwu, W.-J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohrt, H.E.; Tumeh, P.C.; Benson, D.; Bhardwaj, N.; Brody, J.; Formenti, S.; Fox, B.A.; Galon, J.; June, C.H.; Kalos, M.; et al. Immunodynamics: A cancer immunotherapy trials network review of immune monitoring in immuno-oncology clinical trials. J. ImmunoTherapy Cancer 2016, 4, 15. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict Clinical Outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [Green Version]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Anagnostou, V.; Yarchoan, M.; Hansen, A.R.; Wang, H.; Verde, F.; Sharon, E.; Collyar, D.; Chow, L.Q.M.; Forde, P.M. Immuno-oncology Trial Endpoints: Capturing Clinically Meaningful Activity. Clin. Cancer Res. 2017, 23, 4959–4969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milton, C.K.; Self, A.J.; Clarke, P.A.; Banerji, U.; Piccioni, F.; Root, D.E.; Whittaker, S.R. A Genome-scale CRISPR Screen Identifies the ERBB and mTOR Signaling Networks as Key Determinants of Response to PI3K Inhibition in Pancreatic Cancer. Mol. Cancer 2020, 19, 1423–1435. [Google Scholar] [CrossRef]
- Qi, J.; Ouyang, Z. Targeting CDK4/6 for Anticancer Therapy. Biomedicines 2022, 10, 685. [Google Scholar] [CrossRef]
- Tsai, M.-L.; Lee, C.-H.; Huang, L.-C.; Chen, Y.-H.; Liu, W.-N.; Lin, C.-Y.; Hsu, K.-W.; Lee, A.-W.; Lin, C.-L. CRISPR-mediated knockout of VEGFR2/KDR inhibits cell growth in a squamous thyroid cancer cell line. FEBS OpenBio 2022, 12, 993–1005. [Google Scholar] [CrossRef]
- Zhang, C.; Quan, R.; Wang, J. Development and application of CRISPR/Cas9 technologies in genomic editing. Hum. Mol. Genet. 2018, 27, R79–R88. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA—Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrangou, R.; Doudna, J.A. Applications of CRISPR technologies in research and beyond. Nat. Biotechnol. 2016, 34, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; La Russa, M.; Qi, L.S. CRISPR/Cas9 in Genome Editing and Beyond. Annu. Rev. Biochem. 2016, 85, 227–264. [Google Scholar] [CrossRef] [Green Version]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., 3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [Green Version]
- Horvath, P.; Barrangou, R. CRISPR/Cas, the Immune System of Bacteria and Archaea. Science 2010, 327, 167–170. [Google Scholar] [CrossRef] [Green Version]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Jiang, F.; Doudna, J.A. CRISPR–Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.H.; Tsai, Y.T.; Justus, S.; Lee, T.T.; Zhang, L.; Lin, C.S.; Bassuk, A.G.; Mahajan, V.B.; Tsang, S.H. CRISPR Repair Reveals Causative Mutation in a Preclinical Model of Retinitis Pigmentosa. Mol. Ther. 2016, 24, 1388–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, L.; Zhao, C.; Song, J.; Ma, L.; Ruan, J.; Xia, X.; Chen, Y.E.; Zhang, J.; Ma, P.X.; Xu, J. CRISPR/Cas9-Mediated TERT Disruption in Cancer Cells. Int. J. Mol. Sci. 2020, 21, 653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Qian, X.; Wang, B.; Xia, Y.; Zheng, Y.; Du, L.; Xu, D.; Xing, D.; DePinho, R.A.; Lu, Z. Programmable base editing of mutated TERT promoter inhibits brain tumour growth. Nat. Cell Biol. 2020, 22, 282–288. [Google Scholar] [CrossRef]
- Torres-Ruiz, R.; Rodriguez-Perales, S. CRISPR-Cas9: A Revolutionary Tool for Cancer Modelling. Int. J. Mol. Sci. 2015, 16, 22151–22168. [Google Scholar] [CrossRef] [Green Version]
- Hazafa, A.; Mumtaz, M.; Farooq, M.F.; Bilal, S.; Chaudhry, S.N.; Firdous, M.; Naeem, H.; Ullah, M.O.; Yameen, M.; Mukhtiar, M.S.; et al. CRISPR/Cas9: A powerful genome editing technique for the treatment of cancer cells with present challenges and future directions. Life Sci. 2020, 263, 118525. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sanjana, N.E.; Zheng, K.; Shalem, O.; Lee, K.; Shi, X.; Scott, D.A.; Song, J.; Pan, J.Q.; Weissleder, R.; et al. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell 2015, 160, 1246–1260. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Shrager, J.B. CRISPR/Cas-mediated genome editing to treat EGFR-mutant lung cancer: A personalized molecular surgical therapy. EMBO Mol. Med. 2016, 8, 83–85. [Google Scholar] [CrossRef]
- Takeda, H.; Kataoka, S.; Nakayama, M.; Ali, M.A.E.; Oshima, H.; Yamamoto, D.; Park, J.W.; Takegami, Y.; An, T.; Jenkins, N.A.; et al. CRISPR-Cas9-mediated gene knockout in intestinal tumor organoids provides functional validation for colorectal cancer driver genes. Proc. Natl. Acad. Sci. USA 2019, 116, 15635–15644. [Google Scholar] [CrossRef] [Green Version]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R.; et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017, 547, 413–418. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Shi, L.; Zhao, Z.; Du, P.; Ye, X.; Li, D.; Cai, Z.; Han, J.; Cai, J. Disruption of CTLA-4 expression on peripheral blood CD8 + T cell enhances anti-tumor efficacy in bladder cancer. Cancer Chemother. Pharmacol. 2019, 83, 911–920. [Google Scholar] [CrossRef]
- Zhang, X.; Cheng, C.; Sun, W.; Wang, H. Engineering T Cells Using CRISPR/Cas9 for Cancer Therapy. Methods Mol. Biol. 2020, 2115, 419–433. [Google Scholar] [CrossRef]
- U.S. Food & Drug Administration. FDA Approves Novel Gene Therapy to Treat Patients with a Rare Form of Inherited Vision Loss. 2017. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-novel-gene-therapy-treat-patients-rare-form-inherited-vision-loss (accessed on 26 January 2023).
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Day, J.W.; Finkel, R.S.; Chiriboga, C.A.; Connolly, A.M.; Crawford, T.O.; Darras, B.T.; Iannaccone, S.T.; Kuntz, N.L.; Peña, L.D.M.; Shieh, P.B.; et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy in patients with two copies of SMN2 (STR1VE): An open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol. 2021, 20, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Jaeger, M.; Walker, A.; Wei, D.; Leiker, K.; Weitao, T. Break Breast Cancer Addiction by CRISPR/Cas9 Genome Editing. J. Cancer 2018, 9, 219–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Salinas, F.; Rojo, R.; Martinez Amador, C.; Herrera-Gamboa, J.; Trevino, V. Transcriptomic and cellular analyses of CRISPR/Cas9-mediated edition of FASN show inhibition of aggressive characteristics in breast cancer cells. Biochem. Biophys. Res. Commun. 2020, 529, 321–327. [Google Scholar] [CrossRef]
- Faraoni, I.; Graziani, G. Role of BRCA Mutations in Cancer Treatment with Poly(ADP-ribose) Polymerase (PARP) Inhibitors. Cancers 2018, 10, 487. [Google Scholar] [CrossRef] [Green Version]
- National Cancer Institute (US). 2002. Available online: https://www.ncbi.nlm.nih.gov/books/NBK126744/ (accessed on 26 January 2023).
- Petrucelli, N.; Daly, M.B.; Pal, T. BRCA1- and BRCA2-Associated Hereditary Breast and Ovarian Cancer. 1998 Sep 4 [Updated 2022 May 26]. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Eds.; University of Washington: Seattle, WA, USA, 1993–2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1247/?report=reader (accessed on 26 January 2023).
- Guha, T.; Malkin, D. Inherited TP53 Mutations and the Li-Fraumeni Syndrome. Cold Spring Harb. Perspect. Med. 2017, 7, a026187. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Mao, A.; Xu, M.; Weng, Q.; Mao, J.; Ji, J. CRISPR-Cas9 for cancer therapy: Opportunities and challenges. Cancer Lett. 2019, 447, 48–55. [Google Scholar] [CrossRef]
- Mercuri, E.; Muntoni, F.; Baranello, G.; Masson, R.; Boespflug-Tanguy, O.; Bruno, C.; Corti, S.; Daron, A.; Deconinck, N.; Servais, L.; et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy type 1 (STR1VE-EU): An open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol. 2021, 20, 832–841. [Google Scholar] [CrossRef]
- Ribeil, J.A.; Hacein-Bey-Abina, S.; Payen, E.; Magnani, A.; Semeraro, M.; Magrin, E.; Caccavelli, L.; Neven, B.; Bourget, P.; El Nemer, W.; et al. Gene Therapy in a Patient with Sickle Cell Disease. N. Engl. J. Med. 2017, 376, 848–855. [Google Scholar] [CrossRef]
- Kanter, J.; Walters, M.C.; Krishnamurti, L.; Mapara, M.Y.; Kwiatkowski, J.L.; Rifkin-Zenenberg, S.; Aygun, B.; Kasow, K.A.; Pierciey, F.J.; Bonner, M.; et al. Biologic and Clinical Efficacy of LentiGlobin for Sickle Cell Disease. N. Engl. J. Med. 2021, 386, 617–628. [Google Scholar] [CrossRef]
- Thompson, A.A.; Walters, M.C.; Kwiatkowski, J.; Rasko, J.E.J.; Ribeil, J.A.; Hongeng, S.; Magrin, E.; Schiller, G.J.; Payen, E.; Semeraro, M.; et al. Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia. N. Engl. J. Med. 2018, 378, 1479–1493. [Google Scholar] [CrossRef] [PubMed]
- Frangoul, H.; Altshuler, D.; Cappellini, M.D.; Chen, Y.S.; Domm, J.; Eustace, B.K.; Foell, J.; de la Fuente, J.; Grupp, S.; Handgretinger, R.; et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 2021, 384, 252–260. [Google Scholar] [CrossRef]
- Kontomanolis, E.N.; Koutras, A.; Syllaios, A.; Schizas, D.; Mastoraki, A.; Garmpis, N.; Diakosavvas, M.; Angelou, K.; Tsatsaris, G.; Pagkalos, A.; et al. Role of Oncogenes and Tumor-suppressor Genes in Carcinogenesis: A Review. Anticancer Res. 2020, 40, 6009–6015. [Google Scholar] [CrossRef] [PubMed]
- CT Coronary Angiography (CTCA). Does the DNA Editing Tool CRISPR Have a Future in Cancer Treatment? In Cancer Treatment Centers of America; 2021; Available online: https://www.cancercenter.com/community/blog/2021/04/cancer-crispr (accessed on 26 January 2023).
- Cancer.Net. The Genetics of Cancer. 2018. Available online: https://www.cancer.net/navigating-cancer-care/cancer-basics/genetics/genetics-cancer (accessed on 26 January 2023).
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef]
- Tian, X.; Gu, T.; Patel, S.; Bode, A.M.; Lee, M.-H.; Dong, Z. CRISPR/Cas9—An evolving biological tool kit for cancer biology and oncology. NPJ Precis. Oncol. 2019, 3, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, E.; Khan, A. Erratic journey of CRISPR/Cas9 in oncology from bench-work to successful-clinical therapy. Cancer Treat. Res. Commun. 2021, 27, 100289. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; He, Y.; He, W.; Wu, G.; Zhou, X.; Sheng, Q.; Zhong, W.; Lu, Y.; Ding, Y.; Lu, Q.; et al. Exhausted CD8+ T Cells in the Tumor Immune Microenvironment: New Pathways to Therapy. Front. Immunol. 2020, 11, 622509. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Dimitri, A.; Herbst, F.; Fraietta, J.A. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol. Cancer 2022, 21, 78. [Google Scholar] [CrossRef]
- Cheung, A.H.; Chow, C.; Zhang, J.; Zhou, Y.; Huang, T.; Ng, K.C.; Or, T.C.; Yao, Y.Y.; Dong, Y.; Fung, J.M.; et al. Specific targeting of point mutations in EGFR L858R-positive lung cancer by CRISPR/Cas9. Lab. Investig. 2018, 98, 968–976. [Google Scholar] [CrossRef] [Green Version]
- Koo, T.; Yoon, A.R.; Cho, H.Y.; Bae, S.; Yun, C.O.; Kim, J.S. Selective disruption of an oncogenic mutant allele by CRISPR/Cas9 induces efficient tumor regression. Nucleic Acids Res. 2017, 45, 7897–7908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perumal, E.; So Youn, K.; Sun, S.; Seung-Hyun, J.; Suji, M.; Jieying, L.; Yeun-Jun, C. PTEN inactivation induces epithelial-mesenchymal transition and metastasis by intranuclear translocation of β-catenin and snail/slug in non-small cell lung carcinoma cells. Lung Cancer 2019, 130, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Yau, E.H.; Kummetha, I.R.; Lichinchi, G.; Tang, R.; Zhang, Y.; Rana, T.M. Genome-Wide CRISPR Screen for Essential Cell Growth Mediators in Mutant KRAS Colorectal Cancers. Cancer Res. 2017, 77, 6330–6339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, J.-Y.; Choi, Y.J.; Won, E.-J.; Hui, E.; Kim, H.-S.; Cho, Y.-S.; Yoon, T.-J. Gene editing particle system as a therapeutic approach for drug-resistant colorectal cancer. Nano Res. 2020, 13, 1576–1585. [Google Scholar] [CrossRef]
- Ebright, R.Y.; Lee, S.; Wittner, B.S.; Niederhoffer, K.L.; Nicholson, B.T.; Bardia, A.; Truesdell, S.; Wiley, D.F.; Wesley, B.; Li, S.; et al. Deregulation of ribosomal protein expression and translation promotes breast cancer metastasis. Science 2020, 367, 1468–1473. [Google Scholar] [CrossRef]
- Warner, M.; Wu, W.-F.; Montanholi, L.; Nalvarte, I.; Antonson, P.; Gustafsson, J.-A. Ventral prostate and mammary gland phenotype in mice with complete deletion of the ERβ gene. Proc. Natl. Acad. Sci. USA 2020, 117, 4902–4909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.; Fan, S.; Wen, C.; Du, X. CRISPR/Cas9 for cancer treatment: Technology, clinical applications and challenges. Brief. Funct. Genom. 2020, 19, 209–214. [Google Scholar] [CrossRef]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. CRISPR–Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef] [Green Version]
- Luther, D.C.; Lee, Y.W.; Nagaraj, H.; Scaletti, F.; Rotello, V.M. Delivery approaches for CRISPR/Cas9 therapeutics in vivo: Advances and challenges. Expert Opin. Drug Deliv. 2018, 15, 905–913. [Google Scholar] [CrossRef]
- Inturi, R.; Jemth, P. CRISPR/Cas9-based inactivation of human papillomavirus oncogenes E6 or E7 induces senescence in cervical cancer cells. Virology 2021, 562, 92–102. [Google Scholar] [CrossRef]
- Seeger, C.; Sohn, J.A. Complete Spectrum of CRISPR/Cas9-induced Mutations on HBV cccDNA. Mol. Ther. 2016, 24, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Moyo, B.; Bloom, K.; Scott, T.; Ely, A.; Arbuthnot, P. Advances with using CRISPR/Cas-mediated gene editing to treat infections with hepatitis B virus and hepatitis C virus. Virus Res. 2018, 244, 311–320. [Google Scholar] [CrossRef]
- Stone, D.; Long, K.R.; Loprieno, M.A.; De Silva Feelixge, H.S.; Kenkel, E.J.; Liley, R.M.; Rapp, S.; Roychoudhury, P.; Nguyen, T.; Stensland, L.; et al. CRISPR-Cas9 gene editing of hepatitis B virus in chronically infected humanized mice. Mol. Ther.-Methods Clin. Dev. 2021, 20, 258–275. [Google Scholar] [CrossRef]
- Yuen, K.-S.; Chan, C.-P.; Wong, N.-H.M.; Ho, C.-H.; Ho, T.-H.; Lei, T.; Deng, W.; Tsao, S.W.; Chen, H.; Kok, K.-H.; et al. CRISPR/Cas9-mediated genome editing of Epstein–Barr virus in human cells. J. Gen. Virol. 2015, 96, 626–636. [Google Scholar] [CrossRef] [Green Version]
- Huo, H.; Hu, G. CRISPR/Cas9-mediated LMP1 knockout inhibits Epstein-Barr virus infection and nasopharyngeal carcinoma cell growth. Infect. Agents Cancer 2019, 14, 30. [Google Scholar] [CrossRef]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Xue, J.; Deng, T.; Zhou, X.; Yu, K.; Deng, L.; Huang, M.; Yi, X.; Liang, M.; Wang, Y.; et al. Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat. Med. 2020, 26, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, N.; Feng, K.; Chen, M.; Zhang, Y.; Liu, Y.; Yang, Q.; Nie, J.; Tang, N.; Zhang, X.; et al. Phase I study of CAR-T cells with PD-1 and TCR disruption in mesothelin-positive solid tumors. Cell. Mol. Immunol. 2021, 18, 2188–2198. [Google Scholar] [CrossRef]
- Liao, Y.; Chen, L.; Feng, Y.; Shen, J.; Gao, Y.; Cote, G.; Choy, E.; Harmon, D.; Mankin, H.; Hornicek, F.; et al. Targeting programmed cell death ligand 1 by CRISPR/Cas9 in osteosarcoma cells. Oncotarget 2017, 8, 30276–30287. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov Identifier: NCT03057912. A Safety and Efficacy Study of TALEN and CRISPR/Cas9 in the Treatment of HPV-related Cervical Intraepithelial Neoplasia I. 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03057912?term=NCT03057912&draw=2&rank=1 (accessed on 26 January 2023).
- Foy, S.P.; Jacoby, K.; Bota, D.A.; Hunter, T.; Pan, Z.; Stawiski, E.; Ma, Y.; Lu, W.; Peng, S.; Wang, C.L.; et al. Non-viral precision T cell receptor replacement for personalized cell therapy. Nature 2022. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov Identifier: NCT04035434. A Safety and Efficacy Study Evaluating CTX110 in Subjects with Relapsed or Refractory B-Cell Malignancies (CARBON). 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT04035434 (accessed on 26 January 2023).
- ClinicalTrials.gov Identifier: NCT04244656. A Safety and Efficacy Study Evaluating CTX120 in Subjects with Relapsed or Refractory Multiple Myeloma. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT04244656 (accessed on 26 January 2023).
- ClinicalTrials.gov Identifier: NCT04438083. A Safety and Efficacy Study Evaluating CTX130 in Subjects with Relapsed or Refractory Renal Cell Carcinoma (COBALT-RCC). 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT04438083 (accessed on 26 January 2023).
- ClinicalTrials.gov Identifier: NCT04037566. CRISPR (HPK1) Edited CD19-specific CAR-T Cells (XYF19 CAR-T Cells) for CD19+ Leukemia or Lymphoma. 2019. Available online: https://clinicaltrials.gov/ct2/show/record/NCT04037566 (accessed on 26 January 2023).
- ClinicalTrials.gov Identifier: NCT05643742. A Safety and Efficacy Study Evaluating CTX112 in Subjects with Relapsed or Refractory B-Cell Malignancies. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT05643742?cond=NCT05643742&draw=2&rank=1 (accessed on 26 January 2023).
- ClinicalTrials.gov Identifier: NCT03044743. PD-1 Knockout EBV-CTLs for Advanced Stage Epstein-Barr Virus (EBV) Associated Malignancies. 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03044743?term=NCT03044743&draw=2&rank=1 (accessed on 26 January 2023).
- ClinicalTrials.gov Identifier: NCT03166878. A Study Evaluating UCART019 in Patients with Relapsed or Refractory CD19+ Leukemia and Lymphoma. 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03166878?term=NCT03166878&draw=2&rank=1 (accessed on 26 January 2023).
- ClinicalTrials.gov Identifier: NCT03398967. A Feasibility and Safety Study of Universal Dual Specificity CD19 and CD20 or CD22 CAR-T Cell Immunotherapy for Relapsed or Refractory Leukemia and Lymphoma. 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT03398967?term=NCT03398967&draw=2&rank=1 (accessed on 26 January 2023).
- ClinicalTrials.gov Identifier: NCT03081715. PD-1 Knockout Engineered T Cells for Advanced Esophageal Cancer. 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT03081715?term=NCT03081715&draw=2&rank=1 (accessed on 26 January 2023).
- Kim, S.; Koo, T.; Jee, H.G.; Cho, H.Y.; Lee, G.; Lim, D.G.; Shin, H.S.; Kim, J.S. CRISPR RNAs trigger innate immune responses in human cells. Genome Res. 2018, 28, 367–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| Target Genes | Cancer Type | Related CRISPR Method | Reference |
|---|---|---|---|
| TERT | Glioblastoma | sgRNA and Cas9-fused adenine base editor | [27] |
| TP53 | Prostate cancer | sgRNA and Cas9-fused adenine base editor | [28,29] |
| PKC | Colon cancer | sgRNA and Cas9-fused adenine base editor | [28,29] |
| Genes on non-metastatic cancer cell line | Lung metastases | Evaluate gene phenotypes via knockdown | [30] |
| Colorectal cancer driver genes | Intestinal tumors | Evaluate gene phenotypes via knockdown | [32] |
| Novel gene involved in PD-1 resistance | Melanoma | Evaluate gene phenotypes via knockdown | [33] |
| CTLA-4 | Bladder cancer | Evaluate gene phenotypes via knockdown | [34] |
| EGFR | NSCLC | Blocked the tumor PTEN gene | [35] |
| KRAS, BRAF | Colorectal | Genome screening of novel pathways | [36,37] |
| RPL15 | Breast cancer metastasis | Genome screening of novel pathways | [38] |
| FASN | Breast cancer | Knockdown | [39] |
| PARP1 | Breast cancer | Genome screening of novel pathways | [40] |
| ERβ | Prostate cancer | Genome screening of novel pathways | [41] |
| Status | Name/Trial | Trade Name | Disease | Reference |
|---|---|---|---|---|
| 2017 FDA approved | Voretigene neparvovec | Luxterna | Retinal dystrophy | [43,44] |
| 2019 FDA approved | Onasemnogene abeparvovec | Zolgensma | SMA (Pediatric Patients, < 2 y/o) | [45,46] |
| Currently evaluated in clinical trials | Lovotibeglogene autotemcel | LentiGlobin BB305 | SCD, Thalassemia, TDT | [47] |
| Currently evaluated in clinical trials | CLIMB THAL-111, CLIMB SCD-111 | - | SCD, TDT | [48] |
| Clinical Trial | CANCER TYPE | Related CRISPR Method | Reference |
|---|---|---|---|
| Phase 1 | Refractory cancers | Delete two genes that encode endogenous TCR and a gene encoding PD-1 | [76] |
| Phase 1 | Advanced NSCLC | Edite PD-1 on T cells | [77] |
| Phase 1 | Mesothelin-positive solid tumors | Generate PD-1 and TCR deficient CAR-T cells specific to mesothelin | [78] |
| Phase 1 | Osteosarcoma | PD-L1 possible target for knockout | [79] |
| Phase 1 | Refractory solid cancers | Knockout two T cell receptor genes | [81] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stefanoudakis, D.; Kathuria-Prakash, N.; Sun, A.W.; Abel, M.; Drolen, C.E.; Ashbaugh, C.; Zhang, S.; Hui, G.; Tabatabaei, Y.A.; Zektser, Y.; et al. The Potential Revolution of Cancer Treatment with CRISPR Technology. Cancers 2023, 15, 1813. https://doi.org/10.3390/cancers15061813
Stefanoudakis D, Kathuria-Prakash N, Sun AW, Abel M, Drolen CE, Ashbaugh C, Zhang S, Hui G, Tabatabaei YA, Zektser Y, et al. The Potential Revolution of Cancer Treatment with CRISPR Technology. Cancers. 2023; 15(6):1813. https://doi.org/10.3390/cancers15061813
Chicago/Turabian StyleStefanoudakis, Dimitrios, Nikhita Kathuria-Prakash, Alexander W. Sun, Melissa Abel, Claire E. Drolen, Camille Ashbaugh, Shiliang Zhang, Gavin Hui, Yeganeh A. Tabatabaei, Yuliya Zektser, and et al. 2023. "The Potential Revolution of Cancer Treatment with CRISPR Technology" Cancers 15, no. 6: 1813. https://doi.org/10.3390/cancers15061813
APA StyleStefanoudakis, D., Kathuria-Prakash, N., Sun, A. W., Abel, M., Drolen, C. E., Ashbaugh, C., Zhang, S., Hui, G., Tabatabaei, Y. A., Zektser, Y., Lopez, L. P., Pantuck, A., & Drakaki, A. (2023). The Potential Revolution of Cancer Treatment with CRISPR Technology. Cancers, 15(6), 1813. https://doi.org/10.3390/cancers15061813