Simple Summary
Obesity is associated with an increased risk of colorectal cancer (CRC). Recent studies suggest that gut dysbiosis, i.e., abnormal perturbations in the gut microbiome (the highly diverse and complex community of microorganisms inhabiting our gastrointestinal tract) may play a crucial role in this obesity–CRC link. This microbiome imbalance can lead to alterations in the metabolism of the microbiome that can promote cancer development. Therefore, understanding the role of obesity and associated gut dysbiosis can help in identifying novel strategies for the prevention and treatment of CRC.
Abstract
The complexity and variety of gut microbiomes within and among individuals have been extensively studied in recent years in connection to human health and diseases. Our growing understanding of the bidirectional communication between metabolic diseases and the gut microbiome has also highlighted the significance of gut microbiome dysbiosis in the genesis and development of obesity-related cancers. Therefore, it is crucial to comprehend the possible role of the gut microbiota in the crosstalk between obesity and colorectal cancer (CRC). Through the induction of gut microbial dysbiosis, gut epithelial barrier impairment, metabolomic dysregulation, chronic inflammation, or dysregulation in energy harvesting, obesity may promote the development of colorectal tumors. It is well known that strategies for cancer prevention and treatment are most effective when combined with a healthy diet, physical activity, and active lifestyle choices. Recent studies also suggest that an improved understanding of the complex linkages between the gut microbiome and various cancers as well as metabolic diseases can potentially improve cancer treatments and overall outcomes. In this context, we herein review and summarize the clinical and experimental evidence supporting the functional role of the gut microbiome in the pathogenesis and progression of CRC concerning obesity and its metabolic correlates, which may pave the way for the development of novel prognostic tools for CRC prevention. Therapeutic approaches for restoring the microbiome homeostasis in conjunction with cancer treatments are also discussed herein.
1. Introduction
Colorectal cancer (CRC) is the third most common cancer, with an estimated one million deaths related to it each year, making it the second most frequent cause of cancer-related mortality worldwide [1]. According to the IARC (International Agency for Research on Cancer), this number is projected to increase by 56% between 2020 and 2040 [1]. An estimated 69% increase in disease-related fatalities is predicted, resulting in around 1.6 million deaths worldwide in 2040 [1]. The incidence of cancer is likely to keep increasing due to the rise in risk factors, particularly obesity and metabolic syndromes. Excess energy consumption and a lack of physical activity have been related to a sharp rise in the prevalence of obesity. Obesity is indicated by a BMI of ≥30 kg/m2, and almost all developed and developing countries are now experiencing a sharp increase in the incidence of overweight (BMI ≥ 25 kg /m2) and obesity, which affects up to 70% of adults in industrialized nations and is more common in women and urban regions [2]. Particularly in recent decades, obesity has escalated into a global epidemic and public health crisis, and its prevalence is still increasing at a greater frequency. According to the most recent NCD (Non-Communicable Diseases) Risk Factor Collaboration data, about 2 billion adults (39% of the world’s adult population) were predicted to be overweight in 2016, with 671 million (12% of the world’s adult population) obese, a threefold increase in obesity since 1975 [3]. If this trend continues, one billion adults could be obese by 2025 [4]. Obesity is linked to an increased risk of premature death and is a major contributor to the global burden of noncommunicable diseases, including cancer [4,5]. An analysis of the population-attributable percentage reveals that obesity is linked to 11.9% of cancer cases in men and 13.1% in women [2]. Numerous epidemiological studies demonstrate that adult obesity increases the risk of colon cancer by 1.2 to 2 times, with obesity thought to be responsible for between 14% and 35% of all cases of this cancer [6,7,8]. Ectopic fat tissue, which forms due to excessive or aberrant fat tissue deposition, that exceeds the epigenetically and genetically determined adipose tissue stores can increase the risk of developing CRC due to its effect on metabolic pathways and inflammatory processes [9]. Adipose tissue is distributed throughout the body in different compartments, each with a distinct role depending on the bodily location. Visceral adipose tissue, found mainly in the abdominal cavity around organs such as the liver, pancreas, and intestines, has been linked to an increased risk of developing CRC in several studies [10,11,12]. Unlike overall obesity, as measured by BMI, visceral obesity (VO) may be a more significant risk factor for CRC due to its metabolic implications. Visceral fat produces hormones such as adipokines, cytokines, and reactive species that promote inflammation and insulin resistance, both of which are associated with CRC development [13,14]. Additionally, obesity presents worse health effects when paired with sarcopenia, which is defined as the loss of skeletal muscle mass and strength. Sarcopenic obesity (SO) is caused by a number of endocrine-hormonal, metabolic, and lifestyle factors that, in turn, influence pathophysiological factors that may aid in the development of cancer [15,16]. Cancer also involves inflammatory components, such as the imbalanced production of inflammatory cytokines and reactive oxygen species, which are linked to both obesity and sarcopenia. Oncogenesis and SO are mediated by factors including extracellular matrix remodelling, type-2 diabetes, gut microbiota dysbiosis, immune system dysfunction, imbalance of adipokines and myokines, and insulin resistance [15,16,17]. Together, these phenomena correlate with the incidence of CRC development and progression. In addition to increasing the risk of CRC, both VO and SO have a negative impact on CRC prognosis and may have negative clinical implications in CRC patients such as higher recurrence rates, higher risk of dose-limiting toxicity or chemotherapy toxicity, surgical complications, delayed recovery after surgery, physical disability, shorter survival, and a greater risk of developing metastasis [18,19,20,21,22,23]. These implications might serve as a valuable point of reference for the prognostic significance of SO and VO. According to a dose-response meta-analysis, a body weight rise of 10 kg was associated with an approximately 8% higher risk of CRC [24]. Obese people in their youth have a higher risk of acquiring CRC than adults. As expected, a reduction in body weight through bariatric surgery results in a 27% lower incidence of CRC [24]. To understand the cellular and molecular processes at play in the obesity–cancer relationship is a vital step towards improving cancer prevention and treatment approaches. The relationship between obesity and cancer is a complicated one, with various factors influencing the development of the disease. Obesity-induced abnormal lipid metabolism, altered levels of adipokines and hormones, dysbiosis of the gut microbiota, chronic inflammation, and altered bile acid balance may all contribute to the intricate metabolic regulation of CRC carcinogenesis. The gut microbiome is constantly adapting to its human hosts, and changes in the last few decades are thought to be linked to an increase in obesity. Since the gut microbiota is known to play a significant part in regulating host metabolism, any alteration in the composition of the gut microbiota or the levels of its metabolites can affect a variety of physiological processes, including inflammation, energy metabolism, cell proliferation, and apoptosis, which are important factors in CRC development and progression. In these contexts, this review contemplates and discusses the role of obesity and gut microbiome dysbiosis in CRC etiology and pathophysiology, highlighting the fundamental biological mechanisms that underlie this association as well as recent advances that offer novel insights into pathogenetic mechanisms. In addition, potential therapeutic intervention approaches that aim to regulate the gut microbiome and microbial metabolic pathways that promote the rise of obesity and associated CRC are also discussed.
2. Obesity and Gut Microbiome Dysbiosis: An Intricate but Established Association
Our awareness of the gut microbiota as a factor that directly impacts our health or illness status has increased during the past decade. It has been specifically connected to the development of obesity. In fact, the makeup of gut bacteria seems to influence obesity [25]. The gut microbiome is an intricate and complex system that consists of an estimated 100 trillion microorganisms and contains nearly 200 times more genes than the human genome. This enormous variety of bacteria can be considered an independent “organ” [26]. Firmicutes and Bacteroides make up 90% of the bacteria in the gut of healthy adults, with Actinobacteria (mostly Bifidobacterium), Proteobacteria, Fusobacteria, and Verrucomicrobia making up the remaining 10% or less [27]. The gut microbiota’s composition varies significantly among individuals and even alters over time. The co-evolution of humans and their microbiota has resulted in biomolecular networks between them because of their mutual dependence on one another for survival. In this condition, bacterial populations are dynamic and susceptible to changes in the host’s environment and body [26]. To sustain its high population levels, the intestinal microbiota relies on food that is not digested by the human body, dead cells that act as nutrients, and the mucus secreted by the gut [28]. These microbes perform key functions such as producing short chain fatty acids (SCFAs), biodegrading polysaccharides, enriching specific lipopolysaccharide (LPS), and synthesizing essential amino acids and vitamins. The gut microbiota also generates a wide range of physiologically active molecules with beneficial properties, such as anti-inflammatory, antioxidant, and analgesic effects, as well as potential harm including carcinogens, neurotoxins, and immunotoxins. These substances can enter the bloodstream and alter gene expression, impacting metabolic and immunological processes [29,30]. Thus, a balanced gut flora is essential for maintaining metabolic balance and energy levels. Additionally, dynamic equilibrium allows the microbiota to resist disruption and return to its healthy state, such as after antibiotic therapy. Research has also uncovered a connection between gut microbiota composition and weight, as it can influence nutrient absorption and energy expenditure. Unbalanced gut microbial populations can lead to an increased intake of calories, increased storage of body fat, and metabolic issues resulting in obesity [31,32] (Table 1). It has also been linked to changes in hormones that regulate metabolism and appetite, leading to a greater risk of obesity. Through its impact on adiposity and glucose metabolism, the gut microbiota plays a significant role in the start, progression, and comorbidities of obesity [33]. Additionally, dysregulated autophagy activity, AMPK and PGC-1α signaling, and gut dysbiosis may be greatly implicated in the pathological link between SOB and related complications [17]. In addition, the gut microbiota can also play a role in regulating the immune system. This can lead to increased inflammation, which is associated with obesity. Recent research has shown that changes in the gut microbiota (at phylum or species level) as well as intestinal inflammation are related to weight gain and the metabolic effects of obesity [31]. Compared to healthy individuals, obese patients’ gut microbiota are less diverse [34]. The ratio of Firmicutes to Bacteriodetes, the Enterobacteriaceae species, and the Bacteroidales genera including Bacteroides spp., Lactobacillus spp., Enterococcus spp., and Bifidobacterium spp. are all upregulated in obesity, while Clostridia, including Clostridium leptum, and Enterobacter spp., are downregulated [35,36,37,38,39,40,41,42]. Sterile mice models lacking gut flora are protected from obesity, even after feeding a high-fat diet (HFD) [43]. Additionally, lean germ-free mice were injected with the intestinal microbiota of obese mice or women. These mice subsequently gained body fat and had metabolic problems linked to obesity [44,45]. Fecal microbiota transplantation (FMT) from lean to obese subjects improves insulin sensitivity in the recipients while also causing acute changes in the gut microbiota in humans [46]. Gut microbiota changes that may promote obesity and its associated comorbidities include appetite regulation via the gut–brain axis, various hormones, and neuromodulators, increasing host energy absorption by altering gene expression and levels of SCFAs, regulating fat storage through lipoprotein lipase and transcription factors, chronic inflammation brought on by LPS and inflammatory gene expression, disrupting the circadian rhythm by affecting the circadian transcription factors, epigenetic modifications, increasing fat accumulation by decreasing liver fatty acid oxidation by suppressing the adenosine monophosphate kinase (AMPk), and increasing the synthesis of toxic metabolites such as secondary bile acids (SBAs) [32,34]. These alterations are closely related to visceral fat accumulation; therefore, targeting specific microbial species and pathways closely associated with visceral fat accumulation might lead to new therapies for obesity and associated disorders [47]. Due to the richness and complexity of the gut microbiota, further research is still needed to determine the precise mechanism by which it causes obesity.

Table 1.
Studies demonstrating the role of the gut microbiota and its metabolites in obesity and CRC models.
3. Potential Mechanisms Linking Obesity, Microbiome, and CRC
Under normal circumstances, the gut microbiota is essential for maintaining the normal physiological function of the digestive tract, promoting food digestion and absorption, and regulating immunity. Dysbiosis is characterized by changed microbiota composition, bacterial bioactivity, and distribution in various regions of the human body. Dysbiosis occurs when the gut microbiota is influenced by multiple factors, including the diet, environment, and host genes. These changes can lead to diseases such as cancer, inflammatory bowel disease (IBD), cardiovascular disease, type 2 diabetes, and mental disorders. Despite accumulating evidence that there is a favorable correlation between obesity and CRC, the underlying molecular pathways remain poorly understood. The intricate metabolic regulation of CRC carcinogenesis may be significantly influenced by obesity-induced dysbiosis of the gut microbiota. Pharmacological interventions, such as taking antibiotics, host illnesses such as infectious diarrhea, and dietary factors, such as high-fat and low-fiber diets, can all lead to microbial dysbiosis, which alters the gut microbiota and its functioning and may trigger abnormal host immune responses. Human obesity, which is characterized by the decline in gut microbial diversity, is connected to the dysregulation of metabolism which leads to the development of additional disorders [71,72,73].
Intriguingly, the colon has one million times more bacteria than the small intestine, and the former has about 12 times more malignancies than the latter, suggesting that the gut microbiota may play a role in colorectal carcinogenesis [74]. Although the mechanisms underlying dysbiosis and changes in microbial richness are poorly understood, it is unclear whether dysbiosis is a cause or a result of CRC. A dysbiotic microbial community with pro-carcinogenic features can remodel the microbiome as a whole to drive pro-inflammatory reactions and epithelial cell transformation, which leads to cancer. In fact, the CRC microenvironment is characterized by host-derived immunological and inflammatory responses that may have an impact on microbial regulation, change the composition of the microbiota, and encourage the growth of particular bacteria that may have cancer-causing effects [75]. With the emergence of “keystone pathogens” that have significant impacts on bacterial composition and consequently promote dysbiosis, dysbiosis in CRC may thus result in the selection of microbiota composition via a tumor-linked microenvironment [53]. The altered gut microbiota of colon adenoma patients suggests dysbiosis may contribute to the early stages of CRC development [76,77]. According to another study, mice fed an HFD were more likely to develop CRC due to obesity-induced intestinal dysbiosis. They discovered that CRC formation and progression might be accelerated by simply transplanting the feces of obese Kras (G12Dint) mice to mice of normal weight [78]. The question of whether (and how) changes or disruptions in the microbiota contribute to the association between obesity and cancer risk has received little attention in the literature. Dysbiosis associated with obesity may result in physiological changes that increase the risk of cancer [48]. Numerous investigations have demonstrated that certain bacterial taxa associated with obesity may contribute to the pathogenesis of CRC [79,80,81,82] (Table 1). There is currently no major universal obesity-related intestinal microbiota profile linked to the development of CRC because of the interpersonal variability that drives CRC, including genetic factors, behavioral characteristics, and diet. The CRC “driver-passenger” model posits that symbiotic “driver” bacteria initiate tissue malignancy through cellular DNA damage, and colorectal tumorigenesis is then facilitated by transformations in the intestinal microenvironment that give an advantage to “passenger” opportunistic pathogens such as Fusobacterium spp., Roseburia spp., and Streptococcus bovis [83]. In terms of a particular group of colonizing gut bacteria, it has been found that resident bacteria are essential for maintaining gut homeostasis. Patients with CRC have been observed to possess an altered quantitative relationship between the bacterial composition of the lumen and mucosa, characterized by a decreased rate of colonization of Faecalibacterium, Bifidobacterium, and Blautia, and a prevalence of Mogibacterium, Porphyromonas spp., and mucin-degrading species [84]. Moreover, the bacterium Clostridium bolteae, associated with inflammation, dyslipidemia, and insulin resistance, has been linked to the presence of oncogenic human polyomaviruses. The most commonly observed oncogenic viruses in the guts of both obese and cancer patients were BK polyomavirus and Merkel cell polyomavirus, implying that an obese microbiome could provide an opportunity for tumor-driving/driver bacteria and viruses to bring about cell transformation [85].
Bacteria can contribute to carcinogenesis in multiple ways. Fusobacterium nucleatum has been shown to promote CRC progression through miRNA-mediated stimulation of Toll-like receptor (TLR)2 / TLR4 signaling and the suppression of apoptosis [86]. Peptostreptococcus produces metabolites that increase acid production, create a hypoxic tumor microenvironment, and promote bacterial colonization, all of which have pro-carcinogenic effects. Genotoxic bacteria including Fusobacterium nucleatum, Bacteroides fragilis, and certain strains of Escherichia coli carrying the polyketide synthase genomic island produce toxins such as cytolethal distending toxin, colibactin, hydroxyl radicals, or typhoid toxin, and induce DNA damage in IECs, which may initiate CRC development [87,88,89,90]. More recently, it has been demonstrated that these microbes instigate the development of CRC by forming biofilms. A polymicrobial community-based gut microbial biofilm development takes place in the inner layer of the colon [91]. Redistribution of colonic epithelial cell E-cadherin, increased gut epithelial permeability, impaired intestinal barrier function, elevated IL-6 production, and STAT3 activation are all caused by biofilm [91] (Figure 1). Along with the enhanced polyamine metabolism in colonic tissues, these microbial biofilms promote an inflammatory and pro-oncogenic condition that results in dysbiosis, onco-transformation, and the growth of tumors [91,92]. Secretion of secretory proteins known as secretomes or metabolites known as metabolomes make some bacteria carcinogenic via inducing the interactions between receptors in the host immune system and cancer cells [87,88]. The secretome is now referred to as the entire collection of proteins that are secreted into the extracellular space by a cell, tissue, organ, or organism at any given time and under any given set of circumstances via known and unknown secretory mechanisms involving constitutive and regulated secretory organelles [93]. The secretome encompasses functionally varied classes of molecules such as cytokines, chemokines, hormones, digestive enzymes, antibodies, extracellular proteinases, morphogens, toxins, and antimicrobial peptides, and can account for up to 30% of an organism’s proteome [94]. Several of these proteins are involved in a wide range of important biological activities, including cell adhesion, cell migration, cell–cell communication, differentiation, proliferation, morphogenesis, survival and defense, bacterial virulence factors, and immunological responses [94]. Proteins secreted by pathogens, in particular, mediate interactions with the host as these are present or active at the interface between the pathogen and the host cells [95]. Metabolomes are made up of several metabolic by-products of gut microbiota metabolism and oncometabolites implicated in carcinogenesis. Tumor metabolites such as succinate, lactate, D-2- or L-2-hydroxyglutarate, and fumarate accumulate in the cancerous cells after their metabolism. Furthermore, some metabolites generated by the microbiota, such as butyrate, can inhibit pro-inflammatory genes and tumor growth, while lactic acid can provide fuel for cancer cells and encourage their spread. Gut microbiota dysbiosis can contribute to the development and progression of CRC due to the wide-reaching implications of this imbalance, such as the generation of a chronic inflammatory state or immunological response, alterations to stem cell dynamics [96,97], the production of toxic and genotoxic chemicals, and the disruption of host metabolism.
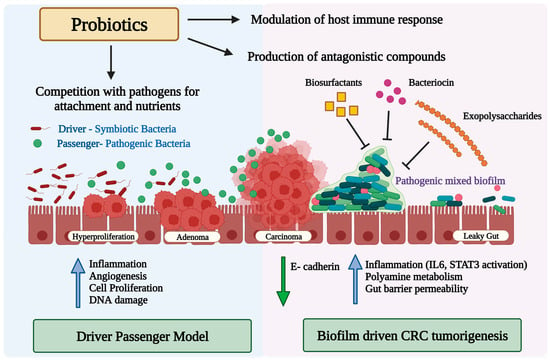
Figure 1.
Diagram illustrating the potential role of the gut microbiota and microbial biofilm in colorectal cancer carcinogenesis, and the potential mode of action of probiotics against pathogenic bacteria alone or in biofilms. Upward blue arrows and downward green arrows show upregulation and downregulation, respectively.
After CRC surgery, recurrence incidence can be as high as 40% during the first three years, with local recurrence happening in 1–23% of cases [98]. Several studies have suggested a link between obesity, gut dysbiosis, and CRC recurrence after resection [99,100,101,102], wherein chronic inflammation is believed to play a role in this link. Obesity is associated with chronic low-grade inflammation, which can lead to alterations in the gut microbiota. Additionally, anastomotic leakage (AL) and surgical site infections could result from the altered postoperative composition of the intestinal microbiota, which could also increase the virulence and number of pathogens and decrease the number of beneficial microorganisms [101]. These alterations can then lead to an increased risk of CRC recurrence after resection. Collagenolytic bacteria such as E. faecalis and P. aeruginosa, which comprise the prominent examples of this pathobiome, particularly in AL [103], have the ability to cause inflammation by activating MMP-9. There is evidence that patients with CRC who have high MMP-9 levels have tumor invasion and poorer prognosis [103]. High F. nucleatum levels were linked to CRC recurrence, possibly due to chemotherapy resistance from autophagy pathway stimulation [104]. Further research is needed to fully understand the mechanisms underlying relationships and to develop effective interventions to prevent/reduce the risk of CRC recurrence in obese individuals with gut dysbiosis.
4. Obesity-Associated Alterations in the Gut Microbiome That May Instigate CRC
CRC is triggered by a dysbiosis of the gut microbiota induced by obesity in several ways. The main processes are as follows: (1) intestinal microbiota dysbiosis can cause low-grade chronic colonic inflammation, which promotes colorectal tumorigenesis; (2) intestinal microbial metabolism in the obese state can contribute to the formation of toxic metabolites that can be carcinogenic; (3) disturbed gut microbiota can increase energy harnessing and nutrient availability, leading to metabolic disturbances (e.g., insulin resistance and altered adipokine function) that promote tumor growth (Figure 2).
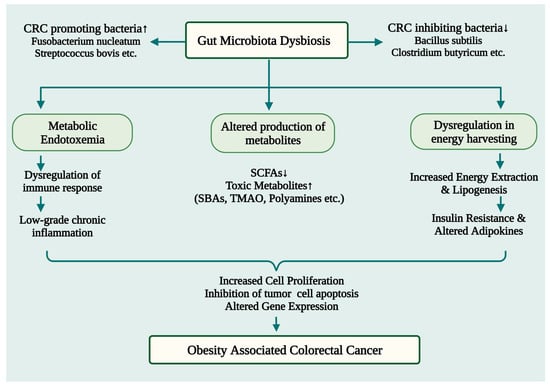
Figure 2.
A schematic illustration depicting the potential ways by which obesity-induced gut microbiome dysbiosis may lead to CRC development. SBAs: Secondary Bile Acids; TMAO: trimethylamine-N-oxide; SCFAs: Short chain fatty acids; CRC: Colorectal Cancer. ↑ Upregulation; ↓ Downregulation.
4.1. Obesity and CRC: Is Gut Dysbiosis-Induced Inflammation the Missing Link?
Inflammation is the physiological homeostatic response of the body’s innate system to infectious and non-infectious pathogens that alters vascular and immunological cell activity. The immune response to pathogens can lead to inflammation, which is a normal part of the resolution process. However, inflammation may contribute to the growth and spread of tumors if it continues unabated.
Intestinal inflammation and DNA damage of the intestinal cells due to the introduction of genotoxins have been suggested as the potential mechanisms linking gut microbial dysbiosis with carcinogenesis [105]. Obesity and CRC pathogenesis are both characterized by chronic low-grade inflammation [106]. Dysbiosis upsets the balance between pro- and anti-inflammatory cytokines, affecting intestinal homeostasis and accelerating the disease’s progression. Obesity and IBD-related chronic inflammation are risk factors for CRC. Inflammation has the potential to damage DNA and eventually lead to the development of cancer by producing reactive oxygen and nitrogen species that cause oxidative stress and increase cell turnover. Both sporadic and colitis-associated CRC is known to be largely influenced by inflammation and inflammatory cytokines [107]. HFD-fed mice have increased colonic TNF-α expression, which is a powerful inducer of IL-6 that is known to contribute to the promotion of CRC [108]. Colonic inflammation, which is linked to the development of CRC, has been demonstrated in numerous studies to be significantly influenced by intestinal microbiota [109,110,111] (Table 1). Furthermore, modifications in the intestinal barrier’s permeability brought on by obesity may also have an impact on the development of CRC. The combination of pre-existing adipose tissue inflammation and metabolic endotoxemia, which results from obesity-related gut barrier dysfunction, increases systemic inflammation, which encourages tumor growth and aids in the production of pro-inflammatory cytokines. The hypothesis that dietary dysbiosis alone is sufficient to promote inflammation is further supported by the fact that the transfer of gut microbiota from HFD mice to germ-free mice increased the stimulation of the NfkB1 inflammatory pathway [112]. These studies suggest that inflammation due to obesity-mediated intestinal dysbiosis and intestinal barrier dysfunction play a role in the increased risk of CRC in obese subjects. Therefore, it is important to consider the potential contribution of these factors to effectively reduce the risk of CRC in obese individuals.
Our increasing comprehension that changes in the microbiota precede obesity and the findings that altered microbiota-pattern recognition receptor (PRR) communications promote inflammation implicate inflammation as a possible key mechanism by which the microbiota modulates the association between cancer and obesity. Numerous studies have shown that eating foods high in saturated fat affects the quality and load of the microbiota, which negatively affects GI health [113,114]. Along with increased hyperplasia of visceral fat, epithelial permeability, and endotoxemia, animals that are predisposed to developing insulin resistance in response to a HFD also have altered microbiota (for example, decreased abundance of Enterococcus spp., Clostridium leptum, and Nitrospira spp. [115]. While permitting some nutrients and electrolytes to penetrate through, the intestinal epithelia play a crucial part in preserving barrier integrity. Numerous studies utilizing both murine and human models have linked obesity to higher endotoxemia, which undermined the epithelial barrier and increased luminal LPS permeability because tight junction protein (ZO-1) expression was downregulated [116,117,118,119,120]. In animal models, intestinal barrier leakage elevated endotoxemia, and stimulation of pro-inflammatory IL-23/IL-17 signaling that promotes tumor growth are all associated with the development of CRC [121,122,123]. The gut’s luminal LPS absorption is assumed to be caused by either LPS leakage via the gut epithelia’s tight junctions or LPS absorption via lipid-rich chylomicrons. These pathways can cooperate to generate metabolic endotoxemia and are not mutually exclusive [124]. The gut barrier works as a complex system of interconnected components to prevent the gut microbiota from colonizing epithelial cells and to limit the entry of pathogen-associated molecular patterns (PAMPs) such as LPS [125]. Obesity-induced microbial imbalances in the gut can be caused by a thinner mucous membrane, uneven distribution and localization of tight junction proteins (TJP), and an aberrant immune response involving immunoglobulin A (IgA) and antimicrobial peptides. These defects combine to allow LPS leakage, which then triggers the TLR4 myeloid differentiation primary response protein (MYD88) reaction and NF-κB, culminating in the generation of an inflammatory response [37,125,126]. Leaky gut, increased levels of the pro-inflammatory cytokine IL-1β, and the harmful bacterial metabolite trimethylamine-N-oxide (TMAO) have been seen in OB-CRC patients, which may be related to their unique intestinal microbial profile marked by an excess of opportunistic pathogens and insufficiency in butyrate-producing bacteria [48]. Inflammatory signals can activate several kinases, including c-Jun N-terminal kinases (JNK) and IkappaB kinase (IKK), that regulate transcription factors, most notably NF-κB, and inhibit insulin action. Interaction between LPS and TLR4 activates a downstream signal cascade that causes Iκβ to become phosphorylated, which then leads to the relocation of NF-κB and initiates the transcription of proinflammatory genes. Inflammation can influence many of the key molecular and cellular processes needed for tumorigenesis, such as the interplay between external and internal signals, which can affect genomic instability, cell proliferation, survival, and abnormal differentiation. An example of this is the activation of the STAT family members (especially STAT3) that often occurs in tumor development in different tissues and links to CRC’s oncogenic process [127]. STAT3 boosts the expression of anti-apoptotic genes, which leads to cell growth and survival by promoting cyclin D family members and the proto-oncogene Myc. Many studies have shown that inflammatory processes activate the NK-κB-IL-6-STAT3 signaling cascade, resulting in initiating or advancing of cancer. Therefore, the changes to the microbiota associated with obesity may cause cancer progression by activating these inflammatory pathways [127].
4.2. Gut Microbial Metabolites as Key Regulators of Obesity and Related CRC
The link between obesity and cancer is complex and the precise mechanism by which the gut microbiota may be involved is not yet fully understood. However, it is known that the microbiota can affect nutrient metabolism and the production of metabolites that may raise cancer risk. Specifically, the gut microbiota can transform dietary environmental toxins and chemicals into obesogenic and diabetogenic substances, which can contribute to the development of gastrointestinal tumors.
Additionally, the microbiota produces a wide range of metabolites from ingested food and host metabolic products, including SCFAs and SBAs, and some of these compounds may become toxic or carcinogenic when exposed to certain physiological conditions. Research is ongoing to better understand the role of gut microbiota in the development of cancer and the prevention of obesity-related cancers.
The gut microbiome plays an important role in the health of the digestive system and in the prevention of CRC. Gut bacteria, such as Clostridium cluster IV and Faecalibacterium, produce tumor suppressor metabolites, such as SCFAs, through the fermentation of dietary fiber [128]. SCFAs, such as butyrate, acetate, propionate, and valerate, regulate immune responses and help to maintain a healthy immune system. SCFAs can also enhance intestinal barrier performance by promoting connexin expression and mucus formation, as well as reducing the amount of oxygen in the intestinal cavity. Consequently, SCFAs are effective in preventing the development of CRC [109,128,129] (Table 1). Colon cancer is a serious disease that can have a wide range of effects on the body. Recent research has demonstrated potential anti-carcinogenic properties in cellular and animal models when exposed to SCFAs [130,131,132,133]. Notably, butyrate, acetate, and propionate all have positive effects on health, with butyrate being the most effective. Butyrate has anti-inflammatory and anticancer properties, which are believed to be mediated through cell metabolism, microbiota homeostasis, immunological control, and gene epigenetic modification [128]. In particular, butyrate serves as the primary source of energy for colonocytes and controls epithelial proliferation. Furthermore, it prevents histone deacetylase activity in colonocytes and immune cells, which reduces the production of pro-inflammatory cytokines and causes CRC cells to undergo apoptosis [134]. SCFAs can also significantly lower fecal pH in the colon, thus inhibiting pathogen growth and DNA damage, promoting apoptosis, and limiting cancer cell proliferation. Furthermore, butyrate and propionate shape the mucosal immune system by controlling the differentiation of colonic regulatory T cells and suppressing colonic inflammation and carcinogenesis [109,129,135]. Overall, SCFAs have the potential to be a powerful tool in the fight against cancer, with butyrate being the primary one.
An HFD- or obesity-related modification in the gut microbiota results in a decrease in bacteria that produce SCFAs, mostly butyrate-producing bacteria, and an elevation in pathogenic bacteria, which affects the synthesis and absorption of SCFAs. Pathobiont overgrowth impairs barrier function, induces inflammation, and downregulates the receptor and transporter for SCFAs, as seen in IBD patients and in vivo IBD models [136,137,138,139]. A major cause of CRC is the chronic inflammatory condition associated with IBD. Due to this, individuals with advanced colorectal adenoma have a higher chance of developing CRC when their SCFA levels are lowered [140]. According to clinical case studies, there were lower levels of fecal SCFAs in CRC patients compared to control groups. This is likely due to fewer SCFAs-producing organisms such as Lachnospiraceae, Roseburia spp., and Bifidobacterium spp. [52,141]. Furthermore, compared to nearby mucosa, metabolomic investigations have shown considerable abnormalities of SCFA metabolism in CRC [142]. SCFAs have been shown to inhibit colitis and the accompanying carcinogenesis by binding to the receptor Gpr109a [135]. Activation of this receptor has been demonstrated to cause an increase in IL-18 production in intestinal epithelial cells, which in turn can stimulate the repair of mucosal tissue. This is done by controlling the production and availability of IL-22 [143]. In murine models, the absence of IL-18 has been linked to dysbiosis of the gut microbiota, a disruption of the inflammatory response, and a disruption of homeostasis and mucosal repair [144,145], all of which can lead to an increased risk of CRC carcinogenesis [143,146,147].
Given the importance of SCFAs, numerous epidemiological studies have revealed that people with diets low in SCFAs or low fecal SCFA levels have an increased risk of developing inflammatory diseases and cancer, particularly breast and gastrointestinal cancers [148,149]. It is now believed that high-fiber diets can have anti-cancer effects due to the formation of SCFAs such as butyrate through the action of the microbiota [150]. Although SCFAs, can shield the host from diet-induced obesity, excessive SCFAs may provide extra energy to the host that may promote obesity. Thus, SCFAs might act as a double-edged sword and this aspect needs to be further explored.
Recent research has suggested that bacteria, including Fusobacterium nucleaturm, Helicobacter pylori, Streptococcus bovis, Clostridium septicum, and Bacteroides fragilis, are involved in the carcinogenesis of CRC [151]. The gut microbiota can contribute to carcinogenesis in a variety of ways. Risk factors such as host genetics, obesity, and an HFD can lead to an alteration of the gut microbiota, resulting in reduced mucus layer thickness, high intestinal permeability, and the translocation of commensal microbiota and its metabolites. Maleficent bacteria can produce large amounts of pathogen-associated molecular patterns (PAMPs) such as LPS that can be recognized by the TLR4 of macrophages and dendritic cells, which then release pro-inflammatory cytokines (IL-1β, TNF-α, IL-23). Other unfavorable metabolites produced by the gut microbiota, such as SBAs, hydrogen sulfide (H2S), TMAO, and N-nitroso compounds (NOCs), may also change the ecological makeup and metabolic activity of intestinal microorganisms, leading to an increase in sensitivity to carcinogenic stimuli. These metabolites may lead to DNA damage and low-grade inflammation, activate tumorigenic signaling pathways, and control tumor immunity, leading to an increased risk of CRC [152]. Furthermore, these metabolites may directly affect IECs after crossing the mucosal barrier, induce immunological responses in the intestinal stroma, release pro-inflammatory signals such as TNF and IL-17, or cause immunosuppression in the tumor microenvironment (TME). NOCs may also contribute to DNA damage through DNA adducts and DNA alkylation, as well as increased production of ROS [152].
Obesity or an HFD may cause the liver to produce more bile acids, which in turn causes more bile acid to accumulate in the gut. The gut microbiota produces 7-dehydroxylase, which turns primary bile acids into SBAs. SBAs are mostly composed of deoxycholic acid (DCA), lithocholic acid (LCA), and a trace quantity of ursodeoxycholic acid (UDCA), and levels of these acids have been linked to CRC, particularly DCA, which is thought to be a carcinogen [153]. The development and progression of CRC is a multistep, multigene, multi-pathway process involving gene mutations, DNA mismatch repair gene inactivation, chromosomal instability, and multiple abnormalities in cell signaling pathways that is influenced by a variety of factors [153]. SBAs, especially DCA, are considered to be genotoxic because they can cause DNA damage in intestinal mucosal epithelial cells [128] and facilitate intestinal tumorigenesis and progression [154]. DCA induces genomic instability through several mechanisms, including mitochondrial and endoplasmic reticulum damage, oxidative DNA damage, chromosomal aneuploid mutations, and increased micronuclei [155]. Additionally, it suppresses CRC cell apoptosis while encouraging CRC cell proliferation [156,157]. To stimulate CRC growth, SBAs also interact with a number of intracellular transduction networks, including the PKC-p38 MAPK signaling pathway, the EGFR-ERK1/2 signaling system, and the Wnt/β-catenin signaling circuit [153]. Furthermore, the propensity of bile acids to dissolve the phospholipid bilayer to provide antimicrobial qualities results in alterations to the gut flora and an increase in pathogens. Therefore, an HFD can enhance the level of SBAs in CRC patients, which can further affect colonic epithelial cell proliferation and apoptosis, erode the gut barrier, modify the intestinal microbiota, and promote the occurrence of CRC [153]. Therefore, it is essential to be aware of the potential risks associated with the presence of these molecules to reduce the risk of CRC.
The above-mentioned well-known compounds as well as lesser-known ones including polyamine, ammonia, heterocyclic amines (HCAs), and lactate are all examples of the varied reservoir of metabolites from intestinal microbiota that play a role in the development and promotion of CRC [152]. When meat is cooked at a high temperature, a family of mutagenic substances known as HCAs is generated by the reaction of creatine (or creatinine), amino acids, and sugars. The HCAs are transformed into genotoxic metabolites after being consumed and absorbed [158,159]. Numerous studies have postulated HCA as an oncogenic agent for CRC due to the correlation between greater HCA levels and a higher risk of CRC [160,161,162]. According to reports, HCAs can cause oxidative base damage, strand breakage, microsatellite instability, frameshift mutations, and microsatellite instability [163]. More specifically, mutations in the colon can be brought on by HCA-induced DNA adducts, which are regarded to be the main executor of DNA-damaging and carcinogenic properties [164].
Products of protein fermentation such as polyamines and ammonia are among the gut microbiota-derived compounds having cancer-promoting effects [165]. To fulfil their enormous metabolic needs, most cancers have a significantly elevated need for polyamines as they are essential for cell growth and differentiation [166]. Through strictly controlled production, breakdown, absorption, and output routes, intracellular polyamine levels are kept at a constant level. Interestingly, a recent metagenomic study discovered a connection between the altered polyamine metabolism and the microbiota linked to CRC, indicating that these metabolites may be crucial to the onset and progression of CRC [167]. Colonocytes that are continually exposed to free ammonia may play a role in the development of CRC [152]. Serum ammonia levels are elevated in CRC patients [168]. High ammonia concentrations cause T cells to undergo metabolic reprogramming, which reduces proliferation and enhances exhaustion. The ammonia-related gene profile correlates with aberrant T-cell responses, poor patient prognosis, and inability to respond to immunotherapy. Ammonia removal leads to the reactivation of T cells and lowers the risk of CRC [168].
Another metabolite, lactate, can serve an immunosuppressive effect in the TME and promote tumor formation by attracting and stimulating the function of immunosuppressive molecules and cells [169]. It has been found to boost angiogenesis and provide oxygen and glucose to cancer cells, which in turn facilitates the growth, invasion, and migration of colon cells [169]. The lactate metabolized by cancerous cells exacerbates CRC progression by prompting macrophages to exhibit M2-type polarization and the production of high-mobility group box 1 (HMGB1), which is essential in maintaining nucleosome structure and controlling the transcription of many genes [170]. In addition, the accumulation of lactate in the TME creates an acidic environment that impairs the cytotoxic and effector capabilities of T cells, thus enabling tumors to escape the immune system and reducing the effectiveness of certain chemotherapy drugs, all of which have a detrimental influence on CRC prognosis [152].
Although the pathogenic effects of numerous gut microbiota metabolites on CRC have been widely reported, their precise mechanisms are still not fully understood. In addition, there is an ongoing debate regarding the dual nature of some metabolites, as they have both anti-carcinogenic and pro-tumorigenic properties. These properties may vary depending on the concentration of the metabolites in the lumen, interactions with certain other metabolites, the length of colonic stasis, and the stage of tumor development. To gain a better understanding of the role of metabolites in CRC progression, further research is necessary.
4.3. Obesity-Induced Dysregulation in Energy Homeostasis Metabolism Links Gut Microbiome and CRC
A lifestyle that is related to energy imbalance, excess body fat, a poor diet, inactivity, and sedentary behavior are risk factors for CRC. These factors are particularly prone to clustering and share similar pathways in colon carcinogenesis, such as chronic systemic inflammation and insulin resistance.
Within the ecosystems of human bodies, cancer cells and microbes coevolve, and both are dependent on external nutrients for life and reproduction. This implies that our diet—and specifically, whether we have a surplus of energy and nutrients—can influence how quickly both cancerous and microbial cells develop. Furthermore, the multiplication and survival of cancer cells and microorganisms can be influenced by one another through the synthesis of certain metabolites. Under ideal circumstances, the growth and proliferation of microbes and the host’s somatic cells are limited by the availability of resources such as protein, carbohydrates, and fat, as well as by somatic cell cycle regulators. Most human cells prefer glucose as a fuel source, and from this source, cellular energy in the form of ATP is used to power cellular operations. This oxygen-dependent route shifts to less effective mechanisms such as fermentation in the event of cancer. This metabolic malfunction, also known as Warburg metabolism, produces lactate, which damages the extracellular environment and promotes metastasis and invasion into new tissues [171]. The breakdown of xenobiotics, such as drugs and environmental pollutants, digestion of complex carbohydrates, and the synthesis of necessary amino acids and beneficial fatty acids are all key aspects of human metabolism that are influenced by interactions between bacteria and human cells. As cancer progresses, these metabolic connections between bacteria and human cells may change from ones that promote health to ones that harm it as microbes start interacting with cancerous cells instead of healthy human cells. It has been discovered that microbial dysbiosis aids in the development of gastrointestinal cancer. In addition, it is believed that this dysregulated energy homeostasis and risk of CRC are related to the metabolic effects of obesity. Obese people tend to have a higher calorie intake, which can cause an elevation in the circulating fatty acid and glucose levels in the body. This can further upset the delicate balance of energy homeostasis and possibly lead to additional disturbance of the gut microbiota. Gut microbial metabolism may act as a mediator in this relationship between excessive energy consumption and CRC. Studies on the effects of caloric restriction imply that improved cancer outcomes result from increased gut microbial diversity and a consequent decrease in inflammation [172,173,174]. Improved gut barrier integrity, which reduces the translocation of microbially derived inflammatory markers such as LPS, as well as advantageous shifts in microbial abundance (such as an increase in Lachnospiraceae abundance), which have been seen in obese women on very low-calorie diets (800 kcal/day), may be the mechanisms underlying this [172]. The gut microbiota may control energy storage by supplying lipogenic substrates (monosaccharides, SCFAs) to the liver and upregulating the activity of the lipoprotein lipase (LPL) enzyme as a result of suppressing the fasting-induced adipose factor in the gut [175].
These results imply that gut microbial dysbiosis may promote energy harnessing or disrupt the storage of energy in the host by altering various metabolic hormones, such as insulin, adiponectin, and leptin, thereby resulting in an unfavorable energy balance and predisposition to obesity [175]. Alterations in energy balance-responsive hormones could increase the likelihood of CRC and other cancers. Numerous pro-tumorigenic effects of insulin resistance include increased levels of systemic TNFα, increased NF-κΒ activation, activation of the mTOR pathway, and increased proliferative/survival signals mediated by IGF-1 [176]. Obesity-induced dysregulation in energy homeostasis metabolism, which links gut microbiota to CRC, is a significant topic of research. The processes underlying this correlation are currently being researched; nevertheless, knowing the mechanisms that underpin this connection could provide insights into potential therapeutic approaches. Dietary therapies, for example, aimed at restoring energy homeostasis balance, may be effective in lowering the risk of developing CRC.
5. Potential Applications of the Gut Microbiome in Clinical Practice
The gut microbiome has a variety of possible roles in the management of CRC; for instance, it may be used as a biomarker for screening, prognosis, and/or prediction, as well as a modifiable element influencing the efficacy of systemic CRC treatment. Due to their greater prevalence in fecal samples from patients with adenomas and CRC, certain bacterial species, such as F. nucleatum, can be employed as screening markers [177,178]. Other screening markers, such as the metabolic metabolites and genotoxic byproducts of some strains, may be used to identify and screen CRC in its initial stages. A patient’s clinical outcome, responsiveness to treatment, and potential adverse effects may all be predicted by the gut microbiota, which can also function as a predictive and/or prognostic biomarker. For instance, in a study, larger levels of F. nucleatum in CRC tissue were linked to worse clinical outcomes, such as shorter survival periods and a poorer prognosis [179]. Even though only a few studies using various metabolomic approaches have demonstrated the diagnostic value of metabolites such SCFAs [180,181], feces metabolome screening may be another possible non-invasive tool for creating a specific metabolic fingerprint to identify CRC [182]. The notion that metabolic changes can be used as indicators of an early precancerous environment in screening processes has not been tested and verified on a large scale yet, but this could be a valuable approach.
5.1. Modulation of Gut Microbiome
One of the primary processes behind the association between obesity and cancer is gut microbial dysbiosis, and effective strategies to repair the gut microbiota potentially present novel treatments that may reduce the risk of CRC associated with obesity. By modifying the gut flora in high-risk individuals, CRC can be prevented, and chemotherapy and immunotherapy treatments can be more effective. Probiotics, prebiotics, postbiotics, FMT, and bacteriophage therapy are all methods for positively modifying the gut microbiome. These biotherapies balance bacterial populations and encourage their beneficial metabolic activity to create a healthy gut environment (Figure 3).
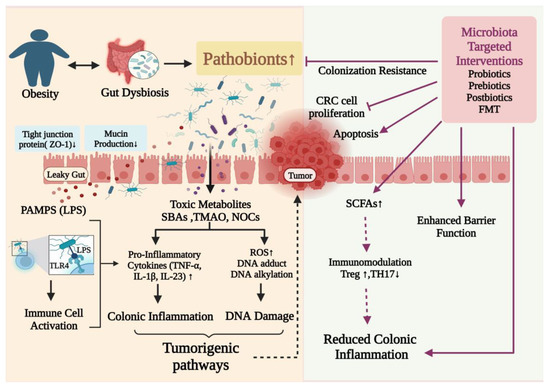
Figure 3.
Putative mechanisms of action of gut microbiome–targeted therapeutic interventions in the treatment of obesity and associated CRC. PAMPS: Pathogen-associated molecular patterns; LPS: lipopolysaccharides; ROS: reactive oxygen species; TNFα: tumor necrosis factor alpha; SBAs: secondary bile acids; TMAO: trimethylamine-N-oxide; NOCs: N-nitroso compounds. ↑ Upregulation; ↓ Downregulation.
5.1.1. Probiotics
Probiotics are microbes, including bacteria, yeasts, and molds, that, when administered in adequate quantities, can improve the host’s health. The most popular bacterial genera utilized as probiotics include Lactobacillus, Bifidobacterium, Bacillus, Streptococcus, and Enterococcus [183]. Probiotics may have an impact on the gut in the prevention and treatment of CRC through mechanisms such as immunomodulation by promoting homeostatic immune responses, supporting the expansion of Treg cell-mediated anti-inflammatory responses, modulating the release of pro-inflammatory cytokines, supporting production of anticarcinogenic and antimicrobial compounds, enhancing the host’s antioxidant system, degrading carcinogens, inhibiting the colonization of pathogenic bacteria, improving the function of the gut barrier by upregulating the synthesis of mucin and tight junction proteins, and encouraging apoptosis in CRC cells [184].
The administration of probiotics has a strong preventive effect against CRC, according to several investigations using chemically-induced animal models. Faecalibacterium prausnitzii, a putative probiotic, inhibits the NF-κB pathway in intestinal epithelial cells and prevents colitis in animal models by producing hydrophobic microbial anti-inflammatory compounds [185]. Treatment with a probiotic combination of L. acidophilus, L. plantarum, and Bifidobacterium longum enhanced the number of cell junction proteins, enhancing the integrity of the intestinal mucosal barrier in CRC patients [186]. Patients who had undergone resection experienced less colorectal tumor atypia while taking the probiotic orally [187]. Single-genus and multi-strain probiotics (L. fermentum NCIMB 5221 and L. acidophilus ATCC 314 in vitro and in vivo study) [188], single-strain probiotics (L. acidophilus or L. rhamnosus GG; L. plantarum; L. casei BL23 in vivo study) [189,190,191] and multigenus and multistrain probiotics (VSL#3 containing, B. longum, B. breve, B. infantis, L. casei, L. acidophilus, L. plantarum, L. bulgaricus, and S. thermophilus along with balsalazide in vivo study) [192] are some of the probiotic strains that have been described as adjuvant treatment agents for CRC management. Intervention of B. lactis and L. acidophilus patients with CRC showed higher levels of butyrate-producing bacteria such as Clostridiales spp. and Faecalibacterium and lower levels of CRC-associated taxa including Peptostreptococcus and Fusobacterium [193]. Diverse probiotic supplements have been demonstrated to considerably minimize postoperative complications, improve postoperative quality of life in CRC patients [194,195,196,197] and gastrointestinal function, and reduce the frequency of diarrhea [198]. In addition, permeability of horseradish peroxidase, lactulose-mannitol ratio, bacterial translocation, enteropathogenic bacterial load, and infection incidence were all significantly decreased, while tight junction protein expression, transepithelial resistance, and serum zonulin levels were all improved in CRC patients on taking probiotics [186,199]. Probiotics can also work through a variety of methods to inhibit biofilm formation and biofilm pathogen survival. Among these processes are competition with pathogens, the manipulation of host immunological responses, and the synthesis of antagonistic substances such as exopolysaccharides, bacteriocins, and biosurfactants that have antibiofilm action [91,200,201]. These antagonistic substances have been demonstrated to hinder the adhesion and development of biofilms as well as the thinning of mature biofilms.
The positive effects of probiotic supplementation depend on the host’s physiology, the severity of the condition, the strain, the dosage, the length of the intervention, other dietary supplements, etc. Probiotic supplements strengthened the immune system and intestinal health in CRC patients, boosted antimicrobial defense, and neutralized carcinogenic chemicals. However, not all probiotic treatments have noticeable advantages for CRC patients’ health. To enhance the functionality of enhancing therapeutic efficacy, the dosage of probiotics provided to patients in the future should be evaluated through preclinical and clinical experiments. To determine the precise mechanism and the potential of probiotics in CRC prevention, more research is highly advised.
5.1.2. Prebiotics
Prebiotics are selectively fermented substances that elicit specific changes in the makeup and/or activities of the host’s gut microbiota. The polyunsaturated fatty acids (PUFAs), polyphenols, and carbohydrates such as inulin, fructans, fructooligosaccharides (FOS), galactooligosaccharides (GOS), and xylooligosaccharides (XOS) possess prebiotic properties. Prebiotics function through a variety of mechanisms, including selective induction of the growth or activity of beneficial intestinal bacteria, fermentation via gut microbiota, direct uptake by the intestine and anti-inflammatory activity, and preventing pathogen colonization by interacting with them.
Numerous studies in animal models have demonstrated that supplementing different amounts of FOS and GOS to diets may alter the composition of gut microbes and encourage the growth of advantageous bacteria such as Lactobacillus, Bifidobacterium, Akkermansia, Ruminococcus, Roseburia, and Faecalibacterium [202,203,204]. This microbiota regulation can considerably reduce body weight and total fat while improving gut barrier performance [33]. Furthermore, better gut barrier integrity improves glucose tolerance and insulin resistance in mouse models [205]. Recent studies on animal models that were genetically predisposed to CRC or were exposed to induction carcinogens such as azoxymethane/dextran sodium sulfate (AOM/DSS) found that different prebiotics, including GOS derived from lactulose, inulin, and phenolic compounds such as anthocyanins and ellagic acid, had inhibitory effects on the development of CRC. Notably, the use of FOS allows researchers to observe the beneficial effects of prebiotics on the development of CRC in a variety of human colon cell lines [206]. Zheng et al. [207] formulated spores-dex, i.e., prebiotics-encapsulated probiotic spores containing C. butyricum (as a probiotic) and chemically modified dextran (as a prebiotic) and evaluated their anti-cancer effects in colon tumor models [207]. As a result of C. butyricum fermenting dextran inside the lesions, SCFAs with anti-cancer potential were produced. Additionally, they also boost the number of SCFA-producing bacteria, such as Eubacterium and Roseburia, considerably slowing the growth of tumors. SCFA-producing bacteria help to inhibit tumors by creating a tumor-suppressing microenvironment in the gut [207]. Furthermore, SCFAs have been shown to induce CRC apoptosis, modulate oxidative stress, improve the epithelial barrier, and suppress inflammation. The novel combination of prebiotics GOS and inulin exhibited stronger preventive activity against CRC by inhibiting aberrant crypt foci formation and biomarkers of colon cancer in the murine models [208]. Prebiotics can be added to the diet to regulate and enrich the intestinal microbiota, with a focus on biologically active substances found in foods of plant origin that can prevent or attenuate the development of CRC.
5.1.3. Postbiotics
Postbiotics are bioactive compounds produced during fermentation carried out by probiotic live cells in the intestine and offer various health benefits to the host. These may include inactivated microbial cells, microbial cell fractions, cell metabolites, functional proteins, and others. Considering that postbiotics are found in the conditioned/supernatants media of bacterial culture, they are comparatively safer than live microbes. They function as anti-tumor agents by specifically suppressing tumor cells while protecting the intestinal epithelium by preventing apoptosis of epithelial cells and boosting IgA production. In colon cancer cells, Saccharomyces boulardii-derived postbiotics reduced cell viability, inhibited the first (G0/G1) phase of cell division, affected the nucleus of the treated cells, and induced apoptosis by decreasing the expression of RelA and Bcl-XL genes and elevating the expression of Caspas3 and PTEN genes [209]. Postbiotics modify the makeup of the gut microbiota and the function of the immune system, in addition to enhancing the efficacy of CRC treatment and minimizing its side effects in CRC patients because of their anti-inflammatory, anti-proliferative, anti-oxidant, and anti-cancer capabilities [209,210]. Postbiotics can reportedly be used as promising drugs for both preventative and adjuvant therapy strategies in CRC patients without having any noticeable negative side effects. This is owing to their particular economic (low production costs) clinical (safe origin), and technical (stability) qualities.
5.1.4. Fecal Microbiota Transplantation (FMT)
FMT is a fascinating and revolutionary biotherapy that entails introducing the intestinal tract of a person with an illness to the fecal liquid from a healthy donor to recreate a healthy microbiome and manage a range of health problems. FMT has been approved for the treatment of C. difficile infection (CDI) [211] but has also shown promise in the treatment of obesity and associated disorders. It has been shown to restore microbial balance in unbalanced communities and can be used to manage multiple gastrointestinal ailments [212]. According to research by Rosshart et al., transplanting the feces of wild mice into laboratory mice can improve resistance to CRC induced by mutagen/inflammatory drugs while also promoting fitness [213]. Intriguingly, Wong et al. showed both conventional and germ-free mice fed the carcinogen azoxymethane produced more tumors and had lower microbial abundance when exposed to fecal microbiota from CRC patients [214]. Additionally, the animals increased the frequency of Th1 and Th17 cells and elevated C-X-C motif chemokine receptor 1 (CXCR1), CXCR2, IL-22, IL-17A, and IL-23 [214]. Metagenomic and metabolomics data indicate that FMT and anti-PD-1 therapy are synergistic in the treatment of CRC [215]. FMT increases the effectiveness of anti-cancer therapy, which may aid in the development of novel microbiota-based anti-cancer treatments. While FMT has been successful, there is a lack of control as the entire gut microbiota is transferred with the therapeutic microorganisms. It is thus essential to be mindful of the donor’s health and the composition of their gut microbiome for optimum results. The success of this method depends on having clear inclusion and exclusion criteria for donors.
5.1.5. Bacteriophage Therapy
Recent advances in scientific research have led to the discovery of various potential treatments for CRC. Bacteriophage therapy is among the most promising treatment options used for the modulation of gut microbiota and involves the use of viruses that infect and replicate in bacteria. This type of treatment has been shown to induce bacterial and metabolic changes in a gut microbiome model, as well as modulate the human immune system, suggesting it could have an important role in the treatment of disorders including obesity and cancer brought on by gut microbiota dysbiosis. The precise mechanism of action of phage therapy in cancer is not yet clear, but researchers believe it involves an immunological response triggered by the presence of the virus. In a co-culture system with HCT116 colon cancer cells, a recent study using the lytic bacteriophage EFA1 discovered that it could break E. faecalis biofilms and alter the growth-stimulatory effects of E. faecalis [216]. To fully comprehend phage therapy in cancer and its potential uses, more research on the bacteriophage EFA1 in these systems and in vivo models is suggested. A decrease in immunosuppressive myeloid-derived suppressor cells (MDSCs) at the tumor site has been linked to treatment with the bioinorganic hybrid bacteriophage M13@Ag. The host immune system may be stimulated, and CRC may be suppressed as a result of activating antigen-presenting cells. Additionally, studies have shown that M13@Ag in conjunction with either immunotherapy or chemotherapy may be able to prolong overall mice survival in an orthotopic CRC model [217]. Bacteriophages have the potential to provide significant benefits in cancer treatment, including their ability to kill carcinogenic bacteria, modulate the immune system, and introduce toxins to the TME. However, further study is needed to understand how exactly bacteriophages interact with tumor cells, bacteria, and the TME as a whole. This knowledge could help to optimize the use of bacteriophages to treat cancer and thus should be a priority in cancer research. Additionally, efforts must be made to ensure that the use of bacteriophages does not lead to the inadvertent introduction of new forms of bacteria that could have a deleterious effect on the TME. Overall, bacteriophage therapy seems like a viable alternative for treating CRC. The available data point to the therapy’s safety and efficacy, but additional study is required to fully grasp its potential and create more efficient therapies.
6. Conclusions and Prospects
It is crucial to comprehend the causes of CRC because the disease is widespread and has a high fatality rate. Through its impact on metabolic and immunological processes, the intestine’s microbial environment has a significant impact on human physiology. Disturbances in intestinal homeostasis and the gut microbiota brought on by obesity may favor the development of certain diseases, including CRC. Due to the strong and fundamental links between intestinal microbiota and these illnesses, identifying microbial profiles important in the pathogenesis of obesity and CRC may offer a helpful marker for the early detection or prediction of these diseases, and modulating the species composition of these communities may offer an appealing therapeutic alternative. A multifaceted strategy for public health is required to combat the global epidemic of obesity-related cancer risk and progression. Collaborative research projects combining different omics platforms (metabolomics, proteomics, lipidomics, and glycomics) may be able to reveal the main molecular pathways underlying this process, paving the way for the creation of microbiome-based preventative and treatment paradigms. It will, however, be important to distinguish between bacteria and metabolites that are good for health and those that are linked to disease. Furthermore, additional research into the metabolic interactions between cancer cells and the microbiome where the tumor is growing needs to be conducted. Finally, as the role of microorganisms becomes clearer, more research into the mechanisms of action of each microorganism in obesity and associated CRC carcinogenesis may be undertaken. Major microbial variations between healthy and malignant tissue as the disease advances can help identify dysbiosis indicators. Understanding dysbiosis may also help explain why some people with comparable clinical traits experience varying disease progression and therapeutic outcomes. The logical next steps are to identify the causes of these diseases and create better diagnostics and treatments for them. By altering the microbiota through dietary changes, prebiotics, probiotics, symbiotics, postbiotics, FMT, and bacteriophage therapy, it is hoped to move dysbiosis toward eubiosis. Although intriguing, microbiome research has its own set of issues. The study cohorts must be carefully planned, and all pertinent characteristics must be taken into account in statistical analyses, as the composition of the microbiota might vary with geography, age, eating habits, BMI, prescription medications, antibiotics, and pet ownership. Two other crucial factors for microbiome research are the preservation of the original microbiota and the prevention of contamination during sample collection and analysis. Future trials could incorporate a personalized, integrated approach that considers the unique clinical and pathological histories of every patient. This way, the positive effects of gut microbiota-targeted treatments can be maximized while any potential negative side effects may be avoided.
Author Contributions
Writing—original draft preparation and conceptualization, S.S., R.N. and M.K. (Manoj Kumar); Review and editing, P.S., D.K.S. and M.K. (Manoj Kumawat); Visualization, V.V. and R.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors wish to thank all the colleagues and collaborators that provided suggestions and feedback about this work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- IARC (WHO). Colorectal Cancer Awareness Month 2022; WHO: Geneva, Switzerland, 2022. [Google Scholar]
- Avgerinos, K.I.; Spyrou, N.; Mantzoros, C.S.; Dalamaga, M. Obesity and cancer risk: Emerging biological mechanisms and perspectives. Metabolism 2019, 92, 121–135. [Google Scholar] [CrossRef]
- Abarca-Gómez, L.; Abdeen, Z.A.; Hamid, Z.A.; Abu-Rmeileh, N.M.; Acosta-Cazares, B.; Acuin, C.; Adams, R.J.; Aekplakorn, W.; Afsana, K.; Aguilar-Salinas, C.A.J. Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: A pooled analysis of 2416 population-based measurement studies in 128· 9 million children, adolescents, and adults. Lancet 2017, 390, 2627–2642. [Google Scholar] [CrossRef]
- Loos, R.J.F.; Yeo, G.S.H. The genetics of obesity: From discovery to biology. Nat. Rev. Genet. 2022, 23, 120–133. [Google Scholar] [CrossRef] [PubMed]
- GBD 2015 Obesity Collaborators. Health effects of overweight and obesity in 195 countries over 25 years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Giovannucci, E.; Ascherio, A.; Rimm, E.B.; Colditz, G.A.; Stampfer, M.J.; Willett, W.C. Physical activity, obesity, and risk for colon cancer and adenoma in men. Ann. Intern. Med. 1995, 122, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Engeland, A.; Tretli, S.; Austad, G.; Bjørge, T.J. Height and body mass index in relation to colorectal and gallbladder cancer in two million Norwegian men and women. Cancer Causes Control. 2005, 16, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Greathouse, K.L.; White, J.R.; Padgett, R.N.; Perrotta, B.G.; Jenkins, G.D.; Chia, N.; Chen, J. Gut microbiome meta-analysis reveals dysbiosis is independent of body mass index in predicting risk of obesity-associated CRC. BMJ Open Gastroenterol. 2019, 6, e000247. [Google Scholar] [CrossRef] [PubMed]
- Hesson, L.B. Gut microbiota and obesity-related gastrointestinal cancer: A focus on epigenetics. Trans. Gastrointest Cancer 2013, 2, 204–210. [Google Scholar]
- Akay, S.; Urkan, M.; Balyemez, U.; Erşen, M.; Taşar, M. Is visceral obesity associated with colorectal cancer? The first volumetric study using all CT slices. Diagn. Interv. Radiol. 2019, 25, 338–345. [Google Scholar] [CrossRef]
- Charette, N.; Vandeputte, C.; Ameye, L.; Bogaert, C.V.; Krygier, J.; Guiot, T.; Deleporte, A.; Delaunoit, T.; Geboes, K.; Van Laethem, J.L.; et al. Prognostic value of adipose tissue and muscle mass in advanced colorectal cancer: A post hoc analysis of two non-randomized phase II trials. BMC Cancer 2019, 19, 134. [Google Scholar] [CrossRef]
- Okamura, T.; Hashimoto, Y.; Hamaguchi, M.; Obora, A.; Kojima, T.; Fukui, M. Visceral Adiposity Index is a predictor of incident colorectal cancer: A population-based longitudinal study. BMJ Open Gastroenterol. 2020, 7, e000400. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.M. Subcutaneous and visceral adipose tissue: Structural and functional differences. Obes. Rev. Off. J. Int. Assoc. Study Obes. 2010, 11, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Chaplin, A.; Rodriguez, R.M.; Segura-Sampedro, J.J.; Ochogavía-Seguí, A.; Romaguera, D.; Barceló-Coblijn, G. Insights behind the Relationship between Colorectal Cancer and Obesity: Is Visceral Adipose Tissue the Missing Link? Int. J. Mol. Sci. 2022, 23, 13128. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.-Q.; Xiao, G.-l.; Fan, Y.-B.; He, M.; Lv, S.; Li, Y.-S. Sarcopenic obesity: Research advances in pathogenesis and diagnostic criteria. Aging Clin. Exp. Res. 2021, 33, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-H.; Choi, K.M. Sarcopenic obesity, insulin resistance, and their implications in cardiovascular and metabolic consequences. Int. J. Mol. Sci. 2020, 21, 494. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.Y.; Choi, H.M.; Yang, H.I.; Kim, K.S. Dysregulated Autophagy Mediates Sarcopenic Obesity and Its Complications via AMPK and PGC1α Signaling Pathways: Potential Involvement of Gut Dysbiosis as a Pathological Link. Int. J. Mol. Sci. 2020, 21, 6887. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Chang, S.Y.; Lim, J.S.; Park, S.J.; Park, J.J.; Cheon, J.H.; Kim, W.H.; Kim, T.I. Impact of Visceral Fat on Survival and Metastasis of Stage III Colorectal Cancer. Gut Liver 2022, 16, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Kuwada, K.; Kuroda, S.; Kikuchi, S.; Yoshida, R.; Nishizaki, M.; Kagawa, S.; Fujiwara, T. Clinical Impact of Sarcopenia on Gastric Cancer. J. Anticancer. Res. 2019, 39, 2241–2249. [Google Scholar] [CrossRef]
- Carneiro, I.P.; Mazurak, V.C.; Prado, C.M. Clinical Implications of Sarcopenic Obesity in Cancer. Curr. Oncol. Rep. 2016, 18, 62. [Google Scholar] [CrossRef]
- Chen, W.-Z.; Chen, X.-D.; Ma, L.-L.; Zhang, F.-M.; Lin, J.; Zhuang, C.-L.; Yu, Z.; Chen, X.-L.; Chen, X.-X. Impact of Visceral Obesity and Sarcopenia on Short-Term Outcomes After Colorectal Cancer Surgery. Dig. Dis. Sci. 2018, 63, 1620–1630. [Google Scholar] [CrossRef]
- Wang, P.; Wang, S.; Ma, Y.; Li, H.; Liu, Z.; Lin, G.; Li, X.; Yang, F.; Qiu, M. Sarcopenic obesity and therapeutic outcomes in gastrointestinal surgical oncology: A meta-analysis. Front. Nutr. 2022, 9, 921817. [Google Scholar] [CrossRef] [PubMed]
- Pedrazzani, C.; Conti, C.; Zamboni, G.A.; Chincarini, M.; Turri, G.; Valdegamberi, A.; Guglielmi, A. Impact of visceral obesity and sarcobesity on surgical outcomes and recovery after laparoscopic resection for colorectal cancer. Clin. Nutr. 2020, 39, 3763–3770. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Xi, Y.; Huang, Z.; Xu, P. Linking Obesity with Colorectal Cancer: Epidemiology and Mechanistic Insights. Cancers 2020, 12, 1408. [Google Scholar] [CrossRef]
- Baothman, O.A.; Zamzami, M.A.; Taher, I.; Abubaker, J.; Abu-Farha, M. The role of gut microbiota in the development of obesity and diabetes. Lipids Health Dis. 2016, 15, 1–8. [Google Scholar] [CrossRef]
- Singh, S.; Sharma, P.; Pal, N.; Kumawat, M.; Shubham, S.; Sarma, D.K.; Tiwari, R.R.; Kumar, M.; Nagpal, R. Impact of Environmental Pollutants on Gut Microbiome and Mental Health via the Gut-Brain Axis. Microorganisms 2022, 10, 1457. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.-M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- Gentile, C.L.; Weir, T.L. The gut microbiota at the intersection of diet and human health. Science 2018, 362, 776–780. [Google Scholar] [CrossRef]
- Miyamoto, J.; Igarashi, M.; Watanabe, K.; Karaki, S.-I.; Mukouyama, H.; Kishino, S.; Li, X.; Ichimura, A.; Irie, J.; Sugimoto, Y.; et al. Gut microbiota confers host resistance to obesity by metabolizing dietary polyunsaturated fatty acids. Nat. Commun. 2019, 10, 4007. [Google Scholar] [CrossRef]
- Canfora, E.E.; Meex, R.C.; Venema, K.; Blaak, E.E. Gut microbial metabolites in obesity, NAFLD and T2DM. Nat. Rev. Endocrinol. 2019, 15, 261–273. [Google Scholar] [CrossRef]
- Breton, J.; Galmiche, M.; Déchelotte, P. Dysbiotic Gut Bacteria in Obesity: An Overview of the Metabolic Mechanisms and Therapeutic Perspectives of Next-Generation Probiotics. Microorganisms 2022, 10, 452. [Google Scholar] [CrossRef]
- Liu, B.N.; Liu, X.T.; Liang, Z.H.; Wang, J.H. Gut microbiota in obesity. World J. Gastroenterol. 2021, 27, 3837–3850. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Ni, Q.; Sun, W.; Li, L.; Feng, X. The links between gut microbiota and obesity and obesity related diseases. Biomed. Pharmacother. 2022, 147, 112678. [Google Scholar] [CrossRef] [PubMed]
- Al-Assal, K.; Martinez, A.C.; Torrinhas, R.S.; Cardinelli, C.; Waitzberg, D. Gut microbiota and obesity. Clin. Nutr. Exp. 2018, 20, 60–64. [Google Scholar] [CrossRef]
- de La Serre, C.B.; Ellis, C.L.; Lee, J.; Hartman, A.L.; Rutledge, J.C.; Raybould, H.E. Propensity to high-fat diet-induced obesity in rats is associated with changes in the gut microbiota and gut inflammation. Am. J. Physiol. -Gastrointest. Liver Physiol. 2010, 299, G440–G448. [Google Scholar] [CrossRef]
- Chen, D.; Yang, Z.; Chen, X.; Huang, Y.; Yin, B.; Guo, F.; Zhao, H.; Huang, J.; Wu, Y.; Gu, R.; et al. Effect of Lactobacillus rhamnosus hsryfm 1301 on the gut microbiota and lipid metabolism in rats fed a high-fat diet. J. Microbiol. Biotechnol. 2015, 25, 687–695. [Google Scholar] [CrossRef]
- Kim, K.-A.; Gu, W.; Lee, I.-A.; Joh, E.-H.; Kim, D.-H. High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS ONE 2012, 7, e47713. [Google Scholar] [CrossRef]
- Hamilton, M.K.; Boudry, G.; Lemay, D.G.; Raybould, H.E. Changes in intestinal barrier function and gut microbiota in high-fat diet-fed rats are dynamic and region dependent. Am. J. Physiol. -Gastrointest. Liver Physiol. 2015, 308, G840–G851. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Furet, J.-P.; Kong, L.-C.; Tap, J.; Poitou, C.; Basdevant, A.; Bouillot, J.-L.; Mariat, D.; Corthier, G.; Doré, J.; Henegar, C.; et al. Differential adaptation of human gut microbiota to bariatric surgery–induced weight loss: Links with metabolic and low-grade inflammation markers. Diabetes 2010, 59, 3049–3057. [Google Scholar] [CrossRef]
- Kong, L.-C.; Tap, J.; Aron-Wisnewsky, J.; Pelloux, V.; Basdevant, A.; Bouillot, J.-L.; Zucker, J.-D.; Dore, J.; Clement, K. Gut microbiota after gastric bypass in human obesity: Increased richness and associations of bacterial genera with adipose tissue genes. Am. J. Clin. Nutr. 2013, 98, 16–24. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Manchester, J.K.; Semenkovich, C.F.; Gordon, J.I. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. USA 2007, 104, 979–984. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Tremaroli, V.; Karlsson, F.; Werling, M.; Ståhlman, M.; Kovatcheva-Datchary, P.; Olbers, T.; Fändriks, L.; le Roux, C.W.; Nielsen, J.; Bäckhed, F. Roux-en-Y gastric bypass and vertical banded gastroplasty induce long-term changes on the human gut microbiome contributing to fat mass regulation. Cell Metab. 2015, 22, 228–238. [Google Scholar] [CrossRef]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojärvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916.e917. [Google Scholar] [CrossRef]
- Yan, H.; Qin, Q.; Chen, J.; Yan, S.; Li, T.; Gao, X.; Yang, Y.; Li, A.; Ding, S. Gut Microbiome Alterations in Patients With Visceral Obesity Based on Quantitative Computed Tomography. Front. Microbiol. 2022, 11, 1451. [Google Scholar] [CrossRef]
- Sánchez-Alcoholado, L.; Ordóñez, R.; Otero, A.; Plaza-Andrade, I.; Laborda-Illanes, A.; Medina, J.A.; Ramos-Molina, B.; Gómez-Millán, J.; Queipo-Ortuño, M.I. Gut Microbiota-Mediated Inflammation and Gut Permeability in Patients with Obesity and Colorectal Cancer. Int. J. Mol. Sci. 2020, 21, 6782. [Google Scholar] [CrossRef]
- Shoji, M.; Sasaki, Y.; Abe, Y.; Nishise, S.; Yaoita, T.; Yagi, M.; Mizumoto, N.; Kon, T.; Onozato, Y.; Sakai, T.; et al. Characteristics of the gut microbiome profile in obese patients with colorectal cancer. JGH Open 2021, 5, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wei, H.; Zhou, Y.; Szeto, C.H.; Li, C.; Lin, Y.; Coker, O.O.; Lau, H.C.H.; Chan, A.W.H.; Sung, J.J.Y.; et al. High-Fat Diet Promotes Colorectal Tumorigenesis Through Modulating Gut Microbiota and Metabolites. Gastroenterology 2022, 162, 135–149.e132. [Google Scholar] [CrossRef] [PubMed]
- Balamurugan, R.; Rajendiran, E.; George, S.; Samuel, G.V.; Ramakrishna, B.S. Real-time polymerase chain reaction quantification of specific butyrate-producing bacteria, Desulfovibrio and Enterococcus faecalis in the feces of patients with colorectal cancer. J. Gastroenterol. Hepatol. 2008, 23, 1298–1303. [Google Scholar] [CrossRef]
- Wu, N.; Yang, X.; Zhang, R.; Li, J.; Xiao, X.; Hu, Y.; Chen, Y.; Yang, F.; Lu, N.; Wang, Z.; et al. Dysbiosis signature of fecal microbiota in colorectal cancer patients. Microb. Ecol. 2013, 66, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Cai, G.; Qiu, Y.; Fei, N.; Zhang, M.; Pang, X.; Jia, W.; Cai, S.; Zhao, L. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 2012, 6, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Coker, O.O.; Liu, C.; Wu, W.K.K.; Wong, S.H.; Jia, W.; Sung, J.J.Y.; Yu, J. Altered gut metabolites and microbiota interactions are implicated in colorectal carcinogenesis and can be non-invasive diagnostic biomarkers. Microbiome 2022, 10, 35. [Google Scholar] [CrossRef]
- Song, X.; An, Y.; Chen, D.; Zhang, W.; Wu, X.; Li, C.; Wang, S.; Dong, W.; Wang, B.; Liu, T.; et al. Microbial metabolite deoxycholic acid promotes vasculogenic mimicry formation in intestinal carcinogenesis. Cancer Sci. 2022, 113, 459–477. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Xu, Q.; Sun, L.; Ye, Y.; Ji, G. Short-chain fatty acids administration is protective in colitis-associated colorectal cancer development. J. Nutr. Biochem. 2018, 57, 103–109. [Google Scholar] [CrossRef]
- Zagato, E.; Pozzi, C.; Bertocchi, A.; Schioppa, T.; Saccheri, F.; Guglietta, S.; Fosso, B.; Melocchi, L.; Nizzoli, G.; Troisi, J.; et al. Endogenous murine microbiota member Faecalibaculum rodentium and its human homologue protect from intestinal tumour growth. Nat. Microbiol. 2020, 5, 511–524. [Google Scholar] [CrossRef]
- Zeng, H.; Hamlin, S.K.; Safratowich, B.D.; Cheng, W.H.; Johnson, L.K. Superior inhibitory efficacy of butyrate over propionate and acetate against human colon cancer cell proliferation via cell cycle arrest and apoptosis: Linking dietary fiber to cancer prevention. Nutr. Res. 2020, 83, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Jin, D.; Huang, S.; Wu, J.; Xu, M.; Liu, T.; Dong, W.; Liu, X.; Wang, S.; Zhong, W.; et al. Clostridium butyricum, a butyrate-producing probiotic, inhibits intestinal tumor development through modulating Wnt signaling and gut microbiota. Cancer Lett. 2020, 469, 456–467. [Google Scholar] [CrossRef]
- Fu, T.; Coulter, S.; Yoshihara, E.; Oh, T.G.; Fang, S.; Cayabyab, F.; Zhu, Q.; Zhang, T.; Leblanc, M.; Liu, S.; et al. FXR Regulates Intestinal Cancer Stem Cell Proliferation. Cell 2019, 176, 1098–1112.e1018. [Google Scholar] [CrossRef]
- Yin, Y.; Wang, M.; Gu, W.; Chen, L. Intestine-specific FXR agonists as potential therapeutic agents for colorectal cancer. Biochem. Pharmacol. 2021, 186, 114430. [Google Scholar] [CrossRef]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef]
- Million, M.; Maraninchi, M.; Henry, M.; Armougom, F.; Richet, H.; Carrieri, P.; Valero, R.; Raccah, D.; Vialettes, B.; Raoult, D. Obesity-associated gut microbiota is enriched in Lactobacillus reuteri and depleted in Bifidobacterium animalis and Methanobrevibacter smithii. Int. J. Obes. (2005) 2012, 36, 817–825. [Google Scholar] [CrossRef]
- Yu, C.; Liu, S.; Chen, L.; Shen, J.; Niu, Y.; Wang, T.; Zhang, W.; Fu, L. Effect of exercise and butyrate supplementation on microbiota composition and lipid metabolism. J. Endocrinol. 2019, 243, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Tazi, A.; Araujo, J.R.; Mulet, C.; Arena, E.T.; Nigro, G.; Pédron, T.; Sansonetti, P.J. Disentangling Host-Microbiota Regulation of Lipid Secretion by Enterocytes: Insights from Commensals Lactobacillus paracasei and Escherichia coli. mBio 2018, 9, e01493-18. [Google Scholar] [CrossRef] [PubMed]
- Aronsson, L.; Huang, Y.; Parini, P.; Korach-André, M.; Håkansson, J.; Gustafsson, J.; Pettersson, S.; Arulampalam, V.; Rafter, J. Decreased fat storage by Lactobacillus paracasei is associated with increased levels of angiopoietin-like 4 protein (ANGPTL4). PLoS ONE 2010, 5, e13087. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Wang, B.; Kaliannan, K.; Wang, X.; Lang, H.; Hui, S.; Huang, L.; Zhang, Y.; Zhou, M.; Chen, M.; et al. Gut Microbiota Mediates the Protective Effects of Dietary Capsaicin against Chronic Low-Grade Inflammation and Associated Obesity Induced by High-Fat Diet. mBio 2017, 8, e00470-17. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Hong, J.; Li, H.; Hu, Y.; Jia, L.; Cai, D.; Zhao, R. Butyrate stimulates adipose lipolysis and mitochondrial oxidative phosphorylation through histone hyperacetylation-associated β(3) -adrenergic receptor activation in high-fat diet-induced obese mice. Exp. Physiol. 2017, 102, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Frost, G.; Sleeth, M.L.; Sahuri-Arisoylu, M.; Lizarbe, B.; Cerdan, S.; Brody, L.; Anastasovska, J.; Ghourab, S.; Hankir, M.; Zhang, S.; et al. The short-chain fatty acid acetate reduces appetite via a central homeostatic mechanism. Nat. Commun. 2014, 5, 3611. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Bäckhed, F.; Fulton, L.; Gordon, J.I. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 2008, 3, 213–223. [Google Scholar] [CrossRef]
- Aoun, A.; Darwish, F.; Hamod, N. The Influence of the Gut Microbiome on Obesity in Adults and the Role of Probiotics, Prebiotics, and Synbiotics for Weight Loss. Prev. Nutr. Food Sci. 2020, 25, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Hong, J.; Xu, X.; Feng, Q.; Zhang, D.; Gu, Y.; Shi, J.; Zhao, S.; Liu, W.; Wang, X. Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention. Nat. Med. 2017, 23, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Proctor, L.M. The Human Microbiome Project in 2011 and beyond. Cell Host Microbe 2011, 10, 287–291. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.J.; Rawls, J.F.; Randall, T.A.; Burcall, L.; Mpande, C.; Jenkins, N.; Jovov, B.; Abdo, Z.; Sandler, R.S.; Keku, T.O. Molecular characterization of mucosal adherent bacteria and associations with colorectal adenomas. Gut Microbes 2010, 1, 138–147. [Google Scholar] [CrossRef]
- McCoy, A.N.; Araújo-Pérez, F.; Azcárate-Peril, A.; Yeh, J.J.; Sandler, R.S.; Keku, T.O. Fusobacterium is associated with colorectal adenomas. PLoS ONE 2013, 8, e53653. [Google Scholar] [CrossRef]
- Schulz, M.D.; Atay, Ç.; Heringer, J.; Romrig, F.K.; Schwitalla, S.; Aydin, B.; Ziegler, P.K.; Varga, J.; Reindl, W.; Pommerenke, C. High-fat-diet-mediated dysbiosis promotes intestinal carcinogenesis independently of obesity. Nature 2014, 514, 508–512. [Google Scholar] [CrossRef]
- Zackular, J.P.; Baxter, N.T.; Iverson, K.D.; Sadler, W.D.; Petrosino, J.F.; Chen, G.Y.; Schloss, P.D. The gut microbiome modulates colon tumorigenesis. MBio 2013, 4, e00692-13. [Google Scholar] [CrossRef]
- Zackular, J.P.; Baxter, N.T.; Chen, G.Y.; Schloss, P.D. Manipulation of the gut microbiota reveals role in colon tumorigenesis. MSphere 2016, 1, e00001-15. [Google Scholar] [CrossRef]
- Bullman, S.; Pedamallu, C.S.; Sicinska, E.; Clancy, T.E.; Zhang, X.; Cai, D.; Neuberg, D.; Huang, K.; Guevara, F.; Nelson, T. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 2017, 358, 1443–1448. [Google Scholar] [CrossRef]
- Gagnière, J.; Raisch, J.; Veziant, J.; Barnich, N.; Bonnet, R.; Buc, E.; Bringer, M.-A.; Pezet, D.; Bonnet, M. Gut microbiota imbalance and colorectal cancer. World J. Gastroenterol. 2016, 22, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Tjalsma, H.; Boleij, A.; Marchesi, J.R.; Dutilh, B.E. A bacterial driver-passenger model for colorectal cancer: Beyond the usual suspects. Nat. Rev. Microbiol. 2012, 10, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.; Yang, L.; Pei, Z. Gut Microbiota, Fusobacteria, and Colorectal Cancer. Diseases 2018, 6, 109. [Google Scholar] [CrossRef] [PubMed]
- Campisciano, G.; de Manzini, N.; Delbue, S.; Cason, C.; Cosola, D.; Basile, G.; Ferrante, P.; Comar, M.; Palmisano, S. The Obesity-Related Gut Bacterial and Viral Dysbiosis Can Impact the Risk of Colon Cancer Development. Microorganisms 2020, 8, 431. [Google Scholar] [CrossRef]
- Ranjbar, M.; Salehi, R.; Haghjooy Javanmard, S.; Rafiee, L.; Faraji, H.; Jafarpor, S.; Ferns, G.A.; Ghayour-Mobarhan, M.; Manian, M.; Nedaeinia, R. The dysbiosis signature of Fusobacterium nucleatum in colorectal cancer-cause or consequences? A systematic review. Cancer Cell Int. 2021, 21, 194. [Google Scholar] [CrossRef]
- Wong, S.H.; Yu, J. Gut microbiota in colorectal cancer: Mechanisms of action and clinical applications. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 690–704. [Google Scholar] [CrossRef] [PubMed]
- Ternes, D.; Karta, J.; Tsenkova, M.; Wilmes, P.; Haan, S.; Letellier, E. Microbiome in Colorectal Cancer: How to Get from Meta-omics to Mechanism? Trends Microbiol. 2020, 28, 401–423. [Google Scholar] [CrossRef]
- Pleguezuelos-Manzano, C.; Puschhof, J.; Rosendahl Huber, A.; van Hoeck, A.; Wood, H.M.; Nomburg, J.; Gurjao, C.; Manders, F.; Dalmasso, G.; Stege, P.B. Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature 2020, 580, 269–273. [Google Scholar] [CrossRef]
- Martin, O.C.; Bergonzini, A.; d’Amico, F.; Chen, P.; Shay, J.W.; Dupuy, J.; Svensson, M.; Masucci, M.G.; Frisan, T. Infection with genotoxin-producing Salmonella enterica synergises with loss of the tumour suppressor APC in promoting genomic instability via the PI3K pathway in colonic epithelial cells. Cell. Microbiol. 2019, 21, e13099. [Google Scholar] [CrossRef]
- Chew, S.S.; Tan, L.T.; Law, J.W.; Pusparajah, P.; Goh, B.H.; Ab Mutalib, N.S.; Lee, L.H. Targeting Gut Microbial Biofilms-A Key to Hinder Colon Carcinogenesis? Cancers 2020, 12, 2272. [Google Scholar] [CrossRef]
- Li, S.; Konstantinov, S.R.; Smits, R.; Peppelenbosch, M.P. Bacterial Biofilms in Colorectal Cancer Initiation and Progression. Trends Mol. Med. 2017, 23, 18–30. [Google Scholar] [CrossRef]
- Wei, W.; Riley, N.M.; Yang, A.C.; Kim, J.T.; Terrell, S.M.; Li, V.L.; Garcia-Contreras, M.; Bertozzi, C.R.; Long, J.Z. Cell type-selective secretome profiling in vivo. Nat. Chem. Biol. 2021, 17, 326–334. [Google Scholar] [CrossRef]
- Ranganathan, S.; Garg, G. Secretome: Clues into pathogen infection and clinical applications. Genome Med. 2009, 1, 113. [Google Scholar] [CrossRef]
- Kamoun, S. A catalogue of the effector secretome of plant pathogenic oomycetes. Annu. Rev. Phytopathol. 2006, 44, 41–60. [Google Scholar] [CrossRef] [PubMed]
- Sphyris, N.; Hodder, M.C.; Sansom, O.J. Subversion of Niche-Signalling Pathways in Colorectal Cancer: What Makes and Breaks the Intestinal Stem Cell. Cancers 2021, 13, 1000. [Google Scholar] [CrossRef] [PubMed]
- Marzano, M.; Fosso, B.; Piancone, E.; Defazio, G.; Pesole, G.; De Robertis, M. Stem Cell Impairment at the Host-Microbiota Interface in Colorectal Cancer. Cancers 2021, 13, 996. [Google Scholar] [CrossRef]
- Mokhles, S.; Macbeth, F.; Farewell, V.; Fiorentino, F.; Williams, N.; Younes, R.; Takkenberg, J.; Treasure, T. Meta-analysis of colorectal cancer follow-up after potentially curative resection. J. Br. Surg. 2016, 103, 1259–1268. [Google Scholar] [CrossRef]
- Scarpa, M.; Ruffolo, C.; Erroi, F.; Fiorot, A.; Basato, S.; Pozza, A.; Canal, F.; Massani, M.; Cavallin, F.; Antoniutti, M.; et al. Obesity is a Risk Factor for Multifocal Disease and Recurrence after Colorectal Cancer Surgery: A Case-Control Study. Anticancer. Res. 2014, 34, 5735–5741. [Google Scholar]
- Lin, K.; Jaspan, V.; Wilder, E.; Chang, S.; Popov, V. 299 Obesity Is Risk Factor for Colorectal Cancer Recurrence: A Systematic Review and Meta-Analysis. Off. J. Am. Coll. Gastroenterol. 2019, 114, S174–S176. [Google Scholar] [CrossRef]
- Koliarakis, I.; Athanasakis, E.; Sgantzos, M.; Mariolis-Sapsakos, T.; Xynos, E.; Chrysos, E.; Souglakos, J.; Tsiaoussis, J. Intestinal Microbiota in Colorectal Cancer Surgery. Cancers 2020, 12, 3011. [Google Scholar] [CrossRef] [PubMed]
- Facciorusso, A.; Di Maso, M.; Serviddio, G.; Vendemiale, G.; Spada, C.; Costamagna, G.; Muscatiello, N. Factors Associated With Recurrence of Advanced Colorectal Adenoma After Endoscopic Resection. Clin. Gastroenterol. Hepatol. 2016, 14, 1148–1154.e1144. [Google Scholar] [CrossRef]
- Said, A.H.; Raufman, J.-P.; Xie, G. The role of matrix metalloproteinases in colorectal cancer. Cancers 2014, 6, 366–375. [Google Scholar] [CrossRef]
- Yu, T.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell 2017, 170, 548–563.e516. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Rhee, K.-J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.-R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef]
- Lucas, C.; Barnich, N.; Nguyen, H.T.T. Microbiota, inflammation and colorectal cancer. Int. J. Mol. Sci. 2017, 18, 1310. [Google Scholar] [CrossRef]
- Soh, J.S.; Jo, S.I.; Lee, H.; Do, E.-j.; Hwang, S.W.; Park, S.H.; Ye, B.D.; Byeon, J.-S.; Yang, S.-K.; Kim, J.H.; et al. Immunoprofiling of Colitis-associated and Sporadic Colorectal Cancer and its Clinical Significance. Sci. Rep. 2019, 9, 6833. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Brooks, R.S.; Ciappio, E.D.; Kim, S.J.; Crott, J.W.; Bennett, G.; Greenberg, A.S.; Mason, J.B. Diet-induced obesity elevates colonic TNF-α in mice and is accompanied by an activation of Wnt signaling: A mechanism for obesity-associated colorectal cancer. J. Nutr. Biochem. 2012, 23, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Alcoholado, L.; Ramos-Molina, B.; Otero, A.; Laborda-Illanes, A.; Ordóñez, R.; Medina, J.A.; Gómez-Millán, J.; Queipo-Ortuño, M.I. The role of the gut microbiome in colorectal cancer development and therapy response. Cancers 2020, 12, 1406. [Google Scholar] [CrossRef]
- Sun, J.; Kato, I. Gut microbiota, inflammation and colorectal cancer. Genes Dis. 2016, 3, 130–143. [Google Scholar] [CrossRef]
- Siddiqui, R.; Boghossian, A.; Alharbi, A.M.; Alfahemi, H.; Khan, N.A. The pivotal role of the gut microbiome in colorectal cancer. Biology 2022, 11, 1642. [Google Scholar] [CrossRef]
- Ding, S.; Chi, M.M.; Scull, B.P.; Rigby, R.; Schwerbrock, N.M.; Magness, S.; Jobin, C.; Lund, P.K. High-fat diet: Bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS ONE 2010, 5, e12191. [Google Scholar] [CrossRef] [PubMed]
- Devkota, S.; Wang, Y.; Musch, M.W.; Leone, V.; Fehlner-Peach, H.; Nadimpalli, A.; Antonopoulos, D.A.; Jabri, B.; Chang, E.B. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature 2012, 487, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Conlon, M.A.; Bird, A.R. The impact of diet and lifestyle on gut microbiota and human health. Nutrients 2014, 7, 17–44. [Google Scholar] [CrossRef] [PubMed]
- Serino, M.; Luche, E.; Gres, S.; Baylac, A.; Bergé, M.; Cenac, C.; Waget, A.; Klopp, P.; Iacovoni, J.; Klopp, C.; et al. Metabolic adaptation to a high-fat diet is associated with a change in the gut microbiota. Gut 2012, 61, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Ghanim, H.; Abuaysheh, S.; Sia, C.L.; Korzeniewski, K.; Chaudhuri, A.; Fernandez-Real, J.M.; Dandona, P. Increase in plasma endotoxin concentrations and the expression of Toll-like receptors and suppressor of cytokine signaling-3 in mononuclear cells after a high-fat, high-carbohydrate meal: Implications for insulin resistance. Diabetes Care 2009, 32, 2281–2287. [Google Scholar] [CrossRef]
- Pendyala, S.; Neff, L.M.; Suarez-Farinas, M.; Holt, P.R. Diet-induced weight loss reduces colorectal inflammation: Implications for colorectal carcinogenesis. Am. J. Clin. Nutr. 2011, 93, 234–242. [Google Scholar] [CrossRef]
- Caesar, R.; Reigstad, C.S.; Bäckhed, H.K.; Reinhardt, C.; Ketonen, M.; Lundén, G.Ö.; Cani, P.D.; Bäckhed, F. Gut-derived lipopolysaccharide augments adipose macrophage accumulation but is not essential for impaired glucose or insulin tolerance in mice. Gut 2012, 61, 1701–1707. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Wang, K.; Mucida, D.; Stewart, C.A.; Schnabl, B.; Jauch, D.; Taniguchi, K.; Yu, G.Y.; Osterreicher, C.H.; Hung, K.E.; et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012, 491, 254–258. [Google Scholar] [CrossRef]
- Hersoug, L.G.; Møller, P.; Loft, S. Gut microbiota-derived lipopolysaccharide uptake and trafficking to adipose tissue: Implications for inflammation and obesity. Obes. Rev. 2016, 17, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Puppa, M.J.; White, J.P.; Sato, S.; Cairns, M.; Baynes, J.W.; Carson, J.A. Gut barrier dysfunction in the Apc(Min/+) mouse model of colon cancer cachexia. Biochim. Et Biophys. Acta 2011, 1812, 1601–1606. [Google Scholar] [CrossRef] [PubMed]
- Malesza, I.J.; Malesza, M.; Walkowiak, J.; Mussin, N.; Walkowiak, D.; Aringazina, R.; Bartkowiak-Wieczorek, J.; Mądry, E. High-Fat, Western-Style Diet, Systemic Inflammation, and Gut Microbiota: A Narrative Review. Cells 2021, 10, 3164. [Google Scholar] [CrossRef] [PubMed]
- Sanmiguel, C.; Gupta, A.; Mayer, E.A. Gut Microbiome and Obesity: A Plausible Explanation for Obesity. Curr. Obes. Rep. 2015, 4, 250–261. [Google Scholar] [CrossRef]
- Tsukumo, D.M.; Carvalho-Filho, M.A.; Carvalheira, J.B.; Prada, P.O.; Hirabara, S.M.; Schenka, A.A.; Araújo, E.P.; Vassallo, J.; Curi, R.; Velloso, L.A. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes 2007, 56, 1986–1998. [Google Scholar] [CrossRef]
- Kolb, R.; Sutterwala, F.S.; Zhang, W. Obesity and cancer: Inflammation bridges the two. Curr. Opin. Pharmacol. 2016, 29, 77–89. [Google Scholar] [CrossRef]
- Fan, X.; Jin, Y.; Chen, G.; Ma, X.; Zhang, L. Gut Microbiota Dysbiosis Drives the Development of Colorectal Cancer. Digestion 2021, 102, 508–515. [Google Scholar] [CrossRef]
- Cheng, Y.; Ling, Z.; Li, L. The Intestinal Microbiota and Colorectal Cancer. Front. Immunol. 2020, 11, 615056. [Google Scholar] [CrossRef]
- Fung, K.Y.; Cosgrove, L.; Lockett, T.; Head, R.; Topping, D.L. A review of the potential mechanisms for the lowering of colorectal oncogenesis by butyrate. Br. J. Nutr. 2012, 108, 820–831. [Google Scholar] [CrossRef] [PubMed]
- Fung, K.Y.; Ooi, C.C.; Lewanowitsch, T.; Tan, S.; Tan, H.T.; Lim, T.K.; Lin, Q.; Williams, D.B.; Lockett, T.J.; Cosgrove, L.J. Identification of potential pathways involved in induction of apoptosis by butyrate and 4-benzoylbutyrate in HT29 colorectal cancer cells. J. Proteome Res. 2012, 11, 6019–6029. [Google Scholar] [CrossRef]
- Ohara, T.; Mori, T. Antiproliferative Effects of Short-chain Fatty Acids on Human Colorectal Cancer Cells <em>via</em> Gene Expression Inhibition. J. Anticancer. Res. 2019, 39, 4659–4666. [Google Scholar] [CrossRef]
- Fernández, J.; Ledesma, E.; Monte, J.; Millán, E.; Costa, P.; de la Fuente, V.G.; García, M.T.F.; Martínez-Camblor, P.; Villar, C.J.; Lombó, F. Traditional processed meat products re-designed towards inulin-rich functional foods reduce polyps in two colorectal cancer animal models. Sci. Rep. 2019, 9, 14783. [Google Scholar] [CrossRef]
- Yuille, S.; Reichardt, N.; Panda, S.; Dunbar, H.; Mulder, I.E. Human gut bacteria as potent class I histone deacetylase inhibitors in vitro through production of butyric acid and valeric acid. PLoS ONE 2018, 13, e0201073. [Google Scholar] [CrossRef]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Parada Venegas, D.; De la Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef]
- Kumari, R.; Ahuja, V.; Paul, J. Fluctuations in butyrate-producing bacteria in ulcerative colitis patients of North India. World J. Gastroenterol. 2013, 19, 3404. [Google Scholar] [CrossRef]
- Takahashi, K.; Nishida, A.; Fujimoto, T.; Fujii, M.; Shioya, M.; Imaeda, H.; Inatomi, O.; Bamba, S.; Andoh, A.; Sugimoto, M. Reduced abundance of butyrate-producing bacteria species in the fecal microbial community in Crohn’s disease. Digestion 2016, 93, 59–65. [Google Scholar] [CrossRef]
- Wang, W.; Chen, L.; Zhou, R.; Wang, X.; Song, L.; Huang, S.; Wang, G.; Xia, B. Increased proportions of Bifidobacterium and the Lactobacillus group and loss of butyrate-producing bacteria in inflammatory bowel disease. J. Clin. Microbiol. 2014, 52, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-M.; Yu, Y.-N.; Wang, J.-L.; Lin, Y.-W.; Kong, X.; Yang, C.-Q.; Yang, L.; Liu, Z.-J.; Yuan, Y.-Z.; Liu, F. Decreased dietary fiber intake and structural alteration of gut mirobiota in patients with advanced colorectal adenoma. Am. Clin. Nutr. 2013, 97, 1044–1052. [Google Scholar] [CrossRef]
- Saffarian, A.; Mulet, C.; Regnault, B.; Amiot, A.; Tran-Van-Nhieu, J.; Ravel, J.; Sobhani, I.; Sansonetti, P.J.; Pedron, T. Crypt-and mucosa-associated core microbiotas in humans and their alteration in colon cancer patients. MBio 2019, 10, e01315–e01319. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.G.; Rao, S.; Weir, T.L.; O’Malia, J.; Bazan, M.; Brown, R.J.; Ryan, E.P. Metabolomics and metabolic pathway networks from human colorectal cancers, adjacent mucosa, and stool. Cancer Metab. 2016, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Salcedo, R.; Worschech, A.; Cardone, M.; Jones, Y.; Gyulai, Z.; Dai, R.-M.; Wang, E.; Ma, W.; Haines, D.; O’hUigin, C.J. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: Role of interleukin 18. J. Exp. Med. 2010, 207, 1625–1636. [Google Scholar] [CrossRef] [PubMed]
- Vaishnava, S.; Behrendt, C.L.; Ismail, A.S.; Eckmann, L.; Hooper, L.V. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc. Natl. Acad. Sci. USA 2008, 105, 20858–20863. [Google Scholar] [CrossRef] [PubMed]
- Dupaul-Chicoine, J.; Yeretssian, G.; Doiron, K.; Bergstrom, K.S.; McIntire, C.R.; LeBlanc, P.M.; Meunier, C.; Turbide, C.; Gros, P.; Beauchemin, N. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity 2010, 32, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Allen, I.C.; TeKippe, E.M.; Woodford, R.-M.T.; Uronis, J.M.; Holl, E.K.; Rogers, A.B.; Herfarth, H.H.; Jobin, C.; Ting, J.P.-Y. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 2010, 207, 1045–1056. [Google Scholar] [CrossRef]
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011, 145, 745–757. [Google Scholar] [CrossRef]
- Mirzaei, R.; Afaghi, A.; Babakhani, S.; Sohrabi, M.R.; Hosseini-Fard, S.R.; Babolhavaeji, K.; Khani Ali Akbari, S.; Yousefimashouf, R.; Karampoor, S. Role of microbiota-derived short-chain fatty acids in cancer development and prevention. Biomed. Pharmacother. 2021, 139, 111619. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Yang, Y.; Wang, H.; Wang, H.; Yu, X.; Lu, Y.; Shen, S.; Teng, L. Gut microbiota shifts in patients with gastric cancer in perioperative period. Medicine 2019, 98, e16626. [Google Scholar] [CrossRef]
- O’keefe, S.J. Diet, microorganisms and their metabolites, and colon cancer. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 691–706. [Google Scholar] [CrossRef]
- Dalal, N.; Jalandra, R.; Bayal, N.; Yadav, A.K.; Harshulika; Sharma, M.; Makharia, G.K.; Kumar, P.; Singh, R.; Solanki, P.R.; et al. Gut microbiota-derived metabolites in CRC progression and causation. J. Cancer Res. Clin. Oncol 2021, 147, 3141–3155. [Google Scholar] [CrossRef]
- Zhang, W.; An, Y.; Qin, X.; Wu, X.; Wang, X.; Hou, H.; Song, X.; Liu, T.; Wang, B.; Huang, X.; et al. Gut Microbiota-Derived Metabolites in Colorectal Cancer: The Bad and the Challenges. Front. Oncol. 2021, 11, 739648. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, S.; Zhou, W.; Hu, D.; Xu, H.; Ji, G. Secondary Bile Acids and Tumorigenesis in Colorectal Cancer. Front. Oncol. 2022, 12, 813745. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Bruce, W.R. Increase by deoxycholic acid of the colonic nuclear damage induced by known carcinogens in C57BL/6J mice. J. Natl. Cancer Inst. 1986, 76, 1129–1132. [Google Scholar]
- Payne, C.M.; Bernstein, C.; Dvorak, K.; Bernstein, H. Hydrophobic bile acids, genomic instability, Darwinian selection, and colon carcinogenesis. Clin. Exp. Gastroenterol. 2008, 1, 19–47. [Google Scholar] [CrossRef] [PubMed]
- Barrasa, J.I.; Olmo, N.; Pérez-Ramos, P.; Santiago-Gómez, A.; Lecona, E.; Turnay, J.; Antonia Lizarbe, M. Deoxycholic and chenodeoxycholic bile acids induce apoptosis via oxidative stress in human colon adenocarcinoma cells. Apoptosis 2011, 16, 1054–1067. [Google Scholar] [CrossRef]
- Qiao, D.; Gaitonde, S.V.; Qi, W.; Martinez, J.D. Deoxycholic acid suppresses p53 by stimulating proteasome-mediated p53 protein degradation. Carcinogenesis 2001, 22, 957–964. [Google Scholar] [CrossRef]
- Zheng, W.; Lee, S.-A. Well-done meat intake, heterocyclic amine exposure, and cancer risk. Nutr. Cancer 2009, 61, 437–446. [Google Scholar] [CrossRef]
- Cross, A.J.; Sinha, R. Meat-related mutagens/carcinogens in the etiology of colorectal cancer. Environ. Mol. Mutagen. 2004, 44, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Ollberding, N.J.; Wilkens, L.R.; Henderson, B.E.; Kolonel, L.N.; Le Marchand, L. Meat consumption, heterocyclic amines and colorectal cancer risk: The Multiethnic Cohort Study. Int. J. Cancer 2012, 131, E1125–E1133. [Google Scholar] [CrossRef]
- Butler, L.M.; Sinha, R.; Millikan, R.C.; Martin, C.F.; Newman, B.; Gammon, M.D.; Ammerman, A.S.; Sandler, R.S. Heterocyclic Amines, Meat Intake, and Association with Colon Cancer in a Population-based Study. Am. J. Epidemiol. 2003, 157, 434–445. [Google Scholar] [CrossRef] [PubMed]
- Marchand, L. The role of heterocyclic aromatic amines in colorectal cancer: The evidence from epidemiologic studies. Genes Environ. 2021, 43, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Murata, M.; Kobayashi, M.; Kawanishi, S. Mechanism of oxidative DNA damage induced by a heterocyclic amine, 2-amino-3,8-dimethylimidazo[4,5f]quinoxaline. Jpn. J. Cancer Res. Gann 1999, 90, 268–275. [Google Scholar] [CrossRef]
- Hasegawa, R.; Sano, M.; Tamano, S.; Imaida, K.; Shirai, T.; Nagao, M.; Sugimura, T.; Ito, N. Dose-dependence of 2-amino-1-methy1–6-phenylimidazo [4, 5-b]-pyridine (PhIP) carcinogenicity in rats. Carcinogenesis 1993, 14, 2553–2557. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Hold, G.L.; Flint, H.J.J.N.r.m. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Fang, Y.J.; Abulimiti, A.; Yang, X.; Li, L.; Liu, K.Y.; Zhang, X.; Feng, X.L.; Chen, Y.M.; Zhang, C.X. Dietary Polyamines Intake and Risk of Colorectal Cancer: A Case-Control Study. Nutrients 2020, 12, 3575. [Google Scholar] [CrossRef]
- Thomas, A.M.; Manghi, P.; Asnicar, F.; Pasolli, E.; Armanini, F.; Zolfo, M.; Beghini, F.; Manara, S.; Karcher, N.; Pozzi, C. Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat. Med. 2019, 25, 667–678. [Google Scholar] [CrossRef]
- Bell, H.N.; Huber, A.K.; Singhal, R.; Korimerla, N.; Rebernick, R.J.; Kumar, R.; El-derany, M.O.; Sajjakulnukit, P.; Das, N.K.; Kerk, S.A.; et al. Microenvironmental ammonia enhances T cell exhaustion in colorectal cancer. Cell Metab. 2023, 35, 134–149.e136. [Google Scholar] [CrossRef]
- Zhu, D.; Jiang, Y.; Cao, H.; Yang, J.; Shu, Y.; Feng, H.; Yang, X.; Sun, X.; Shao, M. Lactate: A regulator of immune microenvironment and a clinical prognosis indicator in colorectal cancer. Front. Immunol. 2022, 13, 876195. [Google Scholar] [CrossRef]
- Gao, X.; Zhou, S.; Qin, Z.; Li, D.; Zhu, Y.; Ma, D. Upregulation of HMGB1 in tumor-associated macrophages induced by tumor cell-derived lactate further promotes colorectal cancer progression. J. Transl. Med. 2023, 21, 53. [Google Scholar] [CrossRef]
- Whisner, C.M.; Athena Aktipis, C. The Role of the Microbiome in Cancer Initiation and Progression: How Microbes and Cancer Cells Utilize Excess Energy and Promote One Another’s Growth. Curr. Nutr. Rep. 2019, 8, 42–51. [Google Scholar] [CrossRef]
- Ott, B.; Skurk, T.; Hastreiter, L.; Lagkouvardos, I.; Fischer, S.; Büttner, J.; Kellerer, T.; Clavel, T.; Rychlik, M.; Haller, D. Effect of caloric restriction on gut permeability, inflammation markers, and fecal microbiota in obese women. Sci. Rep. 2017, 7, 11955. [Google Scholar] [CrossRef]
- Fraumene, C.; Manghina, V.; Cadoni, E.; Marongiu, F.; Abbondio, M.; Serra, M.; Palomba, A.; Tanca, A.; Laconi, E.; Uzzau, S. Caloric restriction promotes rapid expansion and long-lasting increase of Lactobacillus in the rat fecal microbiota. Gut Microbes 2018, 9, 104–114. [Google Scholar] [CrossRef]
- O’Flanagan, C.H.; Smith, L.A.; McDonell, S.B.; Hursting, S.D. When less may be more: Calorie restriction and response to cancer therapy. BMC Med. 2017, 15, 1–9. [Google Scholar] [CrossRef]
- Rogers, C.J.; Prabhu, K.S.; Vijay-Kumar, M. The microbiome and obesity-an established risk for certain types of cancer. Cancer J. 2014, 20, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Vigneri, P.G.; Tirrò, E.; Pennisi, M.S.; Massimino, M.; Stella, S.; Romano, C.; Manzella, L. The Insulin/IGF System in Colorectal Cancer Development and Resistance to Therapy. Front. Oncol. 2015, 5, 230. [Google Scholar] [CrossRef] [PubMed]
- Castellarin, M.; Warren, R.L.; Freeman, J.D.; Dreolini, L.; Krzywinski, M.; Strauss, J.; Barnes, R.; Watson, P.; Allen-Vercoe, E.; Moore, R.A. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012, 22, 299–306. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, X.; Cao, Y.; Fang, J.Y.; Hong, J.; Chen, H. Fecal Fusobacterium nucleatum for the diagnosis of colorectal tumor: A systematic review and meta-analysis. Cancer Med. 2019, 8, 480–491. [Google Scholar] [CrossRef]
- Mima, K.; Nishihara, R.; Qian, Z.R.; Cao, Y.; Sukawa, Y.; Nowak, J.; Yang, J.; Dou, R.; Masugi, Y.; Song, M.; et al. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut 2015, 65, 1973–1980. [Google Scholar] [CrossRef]
- Yusuf, F.; Adewiah, S.; Fatchiyah, F. The Level Short Chain Fatty Acids and HSP 70 in Colorectal Cancer and Non-Colorectal Cancer. Acta Inform. Med. 2018, 26, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Niccolai, E.; Baldi, S.; Ricci, F.; Russo, E.; Nannini, G.; Menicatti, M.; Poli, G.; Taddei, A.; Bartolucci, G.; Calabrò, A.S.; et al. Evaluation and comparison of short chain fatty acids composition in gut diseases. World J. Gastroenterol. 2019, 25, 5543–5558. [Google Scholar] [CrossRef]
- Olovo, C.V.; Huang, X.; Zheng, X.; Xu, M. Faecal microbial biomarkers in early diagnosis of colorectal cancer. J. Cell. Mol. Med. 2021, 25, 10783–10797. [Google Scholar] [CrossRef]
- Bhalla, P.; Rengaswamy, R.; Karunagaran, D.; Suraishkumar, G.; Sahoo, S. Metabolic modeling of host–microbe interactions for therapeutics in colorectal cancer. NPJ Syst. Biol. Appl. 2022, 8, 1–11. [Google Scholar] [CrossRef]
- Sivamaruthi, B.S.; Kesika, P.; Chaiyasut, C. The Role of Probiotics in Colorectal Cancer Management. Evid. -Based Complement. Altern. Med. Ecam 2020, 2020, 3535982. [Google Scholar] [CrossRef]
- Quévrain, E.; Maubert, M.; Michon, C.; Chain, F.; Marquant, R.; Tailhades, J.; Miquel, S.; Carlier, L.; Bermúdez-Humarán, L.; Pigneur, B. Identification of an anti-inflammatory protein from Faecalibacterium prausnitzii, a commensal bacterium deficient in Crohn’s disease. Gut 2016, 65, 415–425. [Google Scholar] [CrossRef]
- Liu, Z.; Qin, H.; Yang, Z.; Xia, Y.; Liu, W.; Yang, J.; Jiang, Y.; Zhang, H.; Yang, Z.; Wang, Y.; et al. Randomised clinical trial: The effects of perioperative probiotic treatment on barrier function and post-operative infectious complications in colorectal cancer surgery—A double-blind study. Aliment. Pharmacol. Ther. 2011, 33, 50–63. [Google Scholar] [CrossRef]
- Ishikawa, H.; Akedo, I.; Otani, T.; Suzuki, T.; Nakamura, T.; Takeyama, I.; Ishiguro, S.; Miyaoka, E.; Sobue, T.; Kakizoe, T. Randomized trial of dietary fiber and Lactobacillus casei administration for prevention of colorectal tumors. Int. J. Cancer 2005, 116, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Kahouli, I.; Malhotra, M.; Westfall, S.; Alaoui-Jamali, M.A.; Prakash, S.J.A.M. Design and validation of an orally administrated active L. fermentum-L. acidophilus probiotic formulation using colorectal cancer Apc Min/+ mouse model. Appl. Microbiol. Biotechnol. 2017, 101, 1999–2019. [Google Scholar] [CrossRef]
- Verma, A.; Shukla, G. Probiotics Lactobacillus rhamnosus GG, Lactobacillus acidophilus suppresses DMH-induced procarcinogenic fecal enzymes and preneoplastic aberrant crypt foci in early colon carcinogenesis in Sprague Dawley rats. Nutr. Cancer 2013, 65, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wang, C.; Ye, L.; Yang, W.; Huang, H.; Meng, F.; Shi, S.; Ding, Z.J. Anti-tumour immune effect of oral administration of Lactobacillus plantarum to CT26 tumour-bearing mice. J. Biosci. 2015, 40, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Lenoir, M.; Del Carmen, S.; Cortes-Perez, N.G.; Lozano-Ojalvo, D.; Muñoz-Provencio, D.; Chain, F.; Langella, P.; De Moreno De LeBlanc, A.; LeBlanc, J.G.; Bermúdez-Humarán, L.G. RETRACTED ARTICLE: Lactobacillus casei BL23 regulates Treg and Th17 T-cell populations and reduces DMH-associated colorectal cancer. J. Gastroenterol. 2016, 51, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Do, E.j.; Hwang, S.W.; Kim, S.Y.; Ryu, Y.M.; Cho, E.A.; Chung, E.J.; Park, S.; Lee, H.J.; Byeon, J.S.; Ye, B.D.; et al. Suppression of colitis-associated carcinogenesis through modulation of IL-6/STAT3 pathway by balsalazide and VSL# 3. J. Gastroenterol. Hepatol. 2016, 31, 1453–1461. [Google Scholar]
- Hibberd, A.A.; Lyra, A.; Ouwehand, A.C.; Rolny, P.; Lindegren, H.; Cedgård, L.; Wettergren, Y. Intestinal microbiota is altered in patients with colon cancer and modified by probiotic intervention. BMJ Open Gastroenterol. 2017, 4, e000145. [Google Scholar] [CrossRef]
- Theodoropoulos, G.E.; Memos, N.A.; Peitsidou, K.; Karantanos, T.; Spyropoulos, B.G.; Zografos, G. Synbiotics and gastrointestinal function-related quality of life after elective colorectal cancer resection. Ann. Gastroenterol. Q. Publ. Hell. Soc. Gastroenterol. 2016, 29, 56. [Google Scholar]
- Kotzampassi, K.; Stavrou, G.; Damoraki, G.; Georgitsi, M.; Basdanis, G.; Tsaousi, G.; Giamarellos-Bourboulis, E.J. A four-probiotics regimen reduces postoperative complications after colorectal surgery: A randomized, double-blind, placebo-controlled study. World J. Surg. 2015, 39, 2776–2783. [Google Scholar] [CrossRef]
- Golkhalkhali, B.; Rajandram, R.; Paliany, A.S.; Ho, G.F.; Wan Ishak, W.Z.; Johari, C.S.; Chin, K.F. Strain-specific probiotic (microbial cell preparation) and omega-3 fatty acid in modulating quality of life and inflammatory markers in colorectal cancer patients: A randomized controlled trial. Asia-Pac. J. Clin. Oncol. 2018, 14, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.K.; Said, S.; Rajandram, R.; Wang, Z.; Roslani, A.C.; Chin, K.F. Pre-surgical administration of microbial cell preparation in colorectal cancer patients: A randomized controlled trial. World J. Surg. 2016, 40, 1985–1992. [Google Scholar] [CrossRef]
- Yang, Y.; Xia, Y.; Chen, H.; Hong, L.; Feng, J.; Yang, J.; Yang, Z.; Shi, C.; Wu, W.; Gao, R. The effect of perioperative probiotics treatment for colorectal cancer: Short-term outcomes of a randomized controlled trial. Oncotarget 2016, 7, 8432. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-H.; Huang, M.-J.; Zhang, X.-W.; Wang, L.; Huang, N.-Q.; Peng, H.; Lan, P.; Peng, J.-S.; Yang, Z.; Xia, Y. The effects of perioperative probiotic treatment on serum zonulin concentration and subsequent postoperative infectious complications after colorectal cancer surgery: A double-center and double-blind randomized clinical trial. Am. J. Clin. Nutr. 2013, 97, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Harjai, K.; Shukla, G. Effect of bacteriocin and exopolysaccharides isolated from probiotic on P. aeruginosa PAO1 biofilm. Folia Microbiol. 2018, 63, 181–190. [Google Scholar] [CrossRef]
- Mahdhi, A.; Leban, N.; Chakroun, I.; Bayar, S.; Mahdouani, K.; Majdoub, H.; Kouidhi, B. Use of extracellular polysaccharides, secreted by Lactobacillus plantarum and Bacillus spp., as reducing indole production agents to control biofilm formation and efflux pumps inhibitor in Escherichia coli. Microb. Pathog. 2018, 125, 448–453. [Google Scholar] [CrossRef]
- Azcarate-Peril, M.A.; Ritter, A.J.; Savaiano, D.; Monteagudo-Mera, A.; Anderson, C.; Magness, S.T.; Klaenhammer, T.R. Impact of short-chain galactooligosaccharides on the gut microbiome of lactose-intolerant individuals. Proc. Natl. Acad. Sci. USA 2017, 114, E367–E375. [Google Scholar] [CrossRef]
- Maier, T.V.; Lucio, M.; Lee, L.H.; VerBerkmoes, N.C.; Brislawn, C.J.; Bernhardt, J.; Lamendella, R.; McDermott, J.E.; Bergeron, N.; Heinzmann, S.S. Impact of dietary resistant starch on the human gut microbiome, metaproteome, and metabolome. MBio 2017, 8, e01343-17. [Google Scholar] [CrossRef] [PubMed]
- Everard, A.; Lazarevic, V.; Derrien, M.; Girard, M.; Muccioli, G.G.; Neyrinck, A.M.; Possemiers, S.; Van Holle, A.; François, P.; de Vos, W.M. Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes 2011, 60, 2775–2786. [Google Scholar] [CrossRef]
- Jansma, J.; Brinkman, F.; van Hemert, S.; El Aidy, S.J.P.i.N.-P.; Psychiatry, B. Targeting the endocannabinoid system with microbial interventions to improve gut integrity. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2021, 106, 110169. [Google Scholar] [CrossRef]
- Mahdavi, M.; Laforest-Lapointe, I.; Massé, E. Preventing Colorectal Cancer through Prebiotics. Microorganisms 2021, 9, 1325. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.W.; Li, R.Q.; An, J.X.; Xie, T.Q.; Han, Z.Y.; Xu, R.; Fang, Y.; Zhang, X.Z. Prebiotics-encapsulated probiotic spores regulate gut microbiota and suppress colon cancer. Adv. Mater. 2020, 32, 2004529. [Google Scholar] [CrossRef] [PubMed]
- Qamar, T.R.; Syed, F.; Nasir, M.; Rehman, H.; Zahid, M.N.; Liu, R.H.; Iqbal, S. Novel Combination of Prebiotics Galacto-Oligosaccharides and Inulin-Inhibited Aberrant Crypt Foci Formation and Biomarkers of Colon Cancer in Wistar Rats. Nutrients 2016, 8, 4658. [Google Scholar] [CrossRef]
- Abbasi, A.; Rad, A.H.; Maleki, L.A.; Kafil, H.S.; Baghbanzadeh, A. Antigenotoxicity and Cytotoxic Potentials of Cell-Free Supernatants Derived from Saccharomyces cerevisiae var. boulardii on HT-29 Human Colon Cancer Cell Lines. Probiotics Antimicrob. Proteins 2023, 1–13. [Google Scholar] [CrossRef]
- Elham, N.; Naheed, M.; Elahe, M.; Hossein, M.M.; Majid, T. Selective Cytotoxic effect of Probiotic, Paraprobiotic and Postbiotics of L. casei strains against Colorectal Cancer Cells: Invitro studies. Braz. J. Pharm. Sci. 2022, 58, e19400. [Google Scholar] [CrossRef]
- Surawicz, C.M.; Brandt, L.J.; Binion, D.G.; Ananthakrishnan, A.N.; Curry, S.R.; Gilligan, P.H.; McFarland, L.V.; Mellow, M.; Zuckerbraun, B.S. Guidelines for diagnosis, treatment, and prevention ofclostridium difficileInfections. Off. J. Am. Coll. Gastroenterol. 2013, 108, 478–498. [Google Scholar] [CrossRef] [PubMed]
- Smits, L.P.; Bouter, K.E.; De Vos, W.M.; Borody, T.J.; Nieuwdorp, M. Therapeutic potential of fecal microbiota transplantation. Gastroenterology 2013, 145, 946–953. [Google Scholar] [CrossRef] [PubMed]
- Rosshart, S.P.; Vassallo, B.G.; Angeletti, D.; Hutchinson, D.S.; Morgan, A.P.; Takeda, K.; Hickman, H.D.; McCulloch, J.A.; Badger, J.H.; Ajami, N.J. Wild mouse gut microbiota promotes host fitness and improves disease resistance. Cell 2017, 171, 1015–1028.e1013. [Google Scholar] [CrossRef]
- Wong, S.H.; Zhao, L.; Zhang, X.; Nakatsu, G.; Han, J.; Xu, W.; Xiao, X.; Kwong, T.N.; Tsoi, H.; Wu, W.K. Gavage of fecal samples from patients with colorectal cancer promotes intestinal carcinogenesis in germ-free and conventional mice. Gastroenterology 2017, 153, 1621–1633.e1626. [Google Scholar] [CrossRef]
- Huang, J.; Zheng, X.; Kang, W.; Hao, H.; Mao, Y.; Zhang, H.; Chen, Y.; Tan, Y.; He, Y.; Zhao, W.; et al. Metagenomic and metabolomic analyses reveal synergistic effects of fecal microbiota transplantation and anti-PD-1 therapy on treating colorectal cancer. Front. Immunol. 2022, 13, 874922. [Google Scholar] [CrossRef] [PubMed]
- Kabwe, M.; Meehan-Andrews, T.; Ku, H.; Petrovski, S.; Batinovic, S.; Chan, H.T.; Tucci, J. Lytic Bacteriophage EFA1 Modulates HCT116 Colon Cancer Cell Growth and Upregulates ROS Production in an Enterococcus faecalis Co-culture System. Front. Microbiol. 2021, 12, 650849. [Google Scholar] [CrossRef]
- Dong, X.; Pan, P.; Zheng, D.W.; Bao, P.; Zeng, X.; Zhang, X.Z. Bioinorganic hybrid bacteriophage for modulation of intestinal microbiota to remodel tumor-immune microenvironment against colorectal cancer. Sci. Adv. 2020, 6, eaba1590. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).