
Computational Methods Summarizing Mutational Patterns in Cancer: Promise and Limitations for Clinical Applications

Abstract
Simple Summary
Abstract
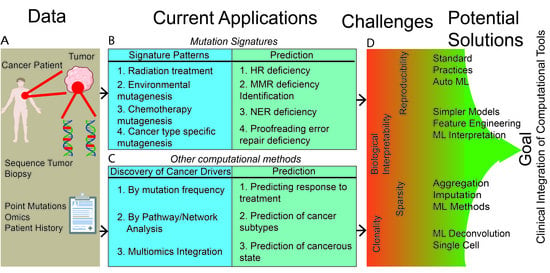
1. Introduction
| Targeted therapies | Therapies targeting a specific protein associated with a disease |
| Synthetic lethality | A type of interaction wherein a single event is tolerable but co-occurrence of two or more events is lethal |
| Driver mutation | A mutation that provides a selective advantage to a cell and transforms a cell into a cancerous state |
| Passenger mutation | A mutation that is a result, but not a direct cause, of a cell becoming cancerous |
| Mutagenic process | Anything that causes damage to DNA or induces mutations in DNA, such as UV light, radiation, or alkylating agents |
| Non-negative matrix factorization (in progress) | Unsupervised mathematical method wherein a single large nonnegative matrix is decomposed into two or more smaller matrices |
| COSMIC | Catalogue of Somatic Mutations in Cancer: https://cancer.sanger.ac.uk/cosmic, accessed on 12 March 2023 |
| Signature 1 | Mutation signature associated with age |
| Signature 2 | Mutation signature associated with the mutagenic effects of APOBEC activity |
| Signature 4 | Mutation signature associated with tobacco smoke |
| Signature 7 | Mutation signature associated with UV exposure |
| Signature 10 | Mutation signature associated with POLE proofreading errors |
| Signature 16 | Mutation signature associated with alcohol consumption |
| Signature 18 | Mutation signature associated with the mutagenic effects of the MUTYH gene |
| DDR | DNA damage repair, a network of processes that repairs damaged DNA |
| MMR | Mismatch repair, a DDR pathway involved in detecting and repairing DNA mismatches |
| BER | Base-excision repair, a pathway that repairs typically small-scale mutations by first removing only the base and leaving an abasic site, which is later removed and replaced with other nucleotides |
| NER | Nucleotide-excision repair, a pathway that repairs mutations by entirely removing mutated sections of DNA |
| HR | Homologous recombination, a pathway repairing double-strand DNA damage that uses another strand of DNA as a template for repair |
| NHEJ | Non-homologous end joining, a pathway repairing double-strand DNA damage that involves attaching two strands of broken DNA together. |
| Logistic regression | A regression model for supervised classification |
| LASSO logistic regression | A regression model that uses L1 regularization |
| Random rorest | An ensemble machine-learning model that combines decision trees produced by bagging |
| ICI | Immune-checkpoint inhibitors, a class of cancer drugs that suppresses pro-tumor immune-system regulatory effects |
| Supervised learning | Machine-learning strategies wherein the classes of outcomes are known |
| Unsupervised learning | Machine-learning strategies wherein the task of the model is to cluster the data into previously unidentified classes or discover the underlying classes |
| Neural network | A machine-learning model that connects the input data to a desired output classification, where nodes connected by edges apply non-linear transformations to the data passed through the network |
| Deep learning | Machine-learning models that are composed of multiple layers of neural networks stacked over one another (giving rise to the term “deep”) |
| Overfitting | Fitting a particular data point too well and therefore failing to predict on other data |
| Underfitting | Not fitting the data well enough and inferring simplified decision rules that may not be optimized for any dataset |
| Graph convolutional networks | Neural-network architectures that represent graph data for learning tasks |
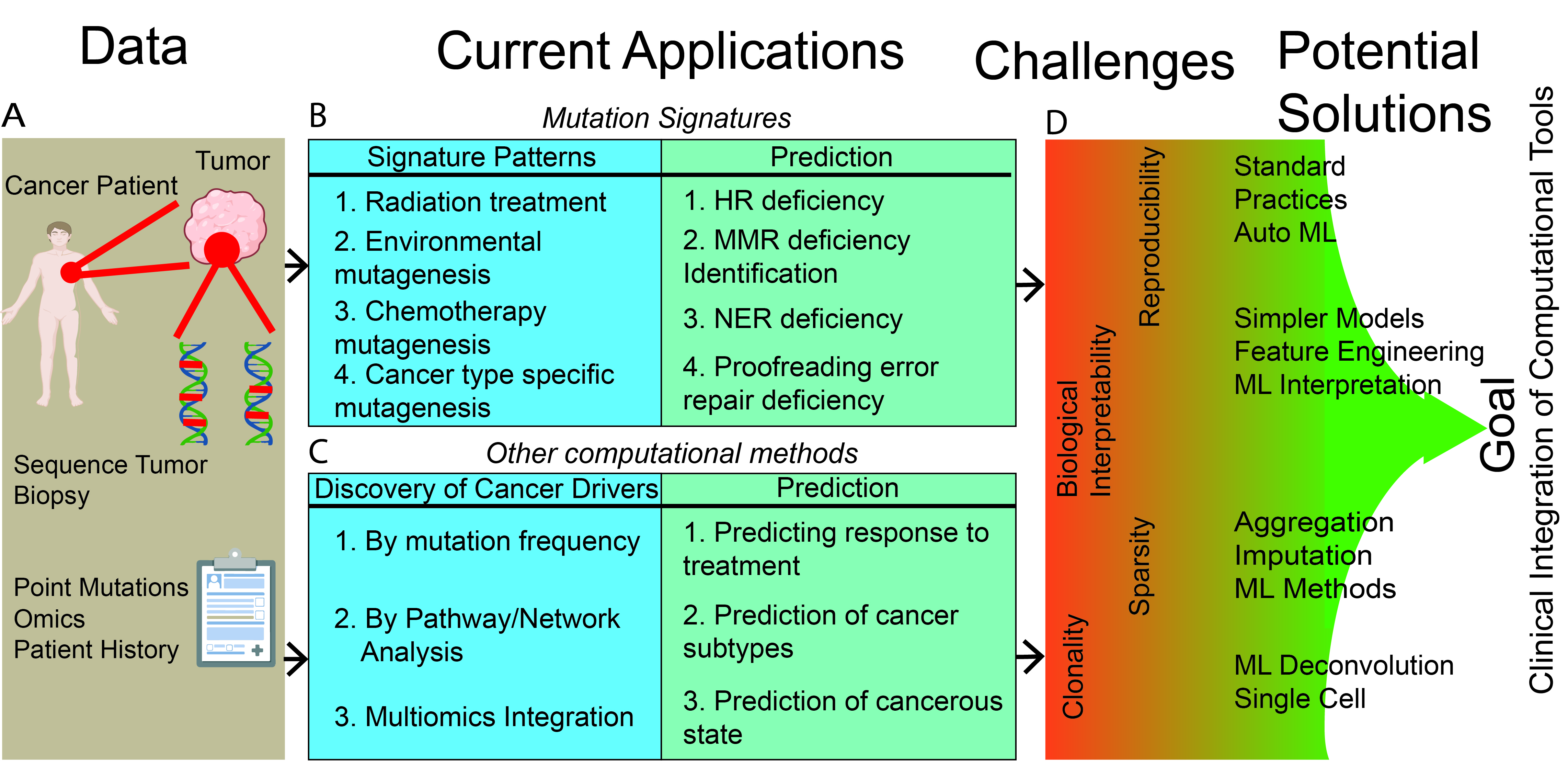
2. Mutation-Signatures Background
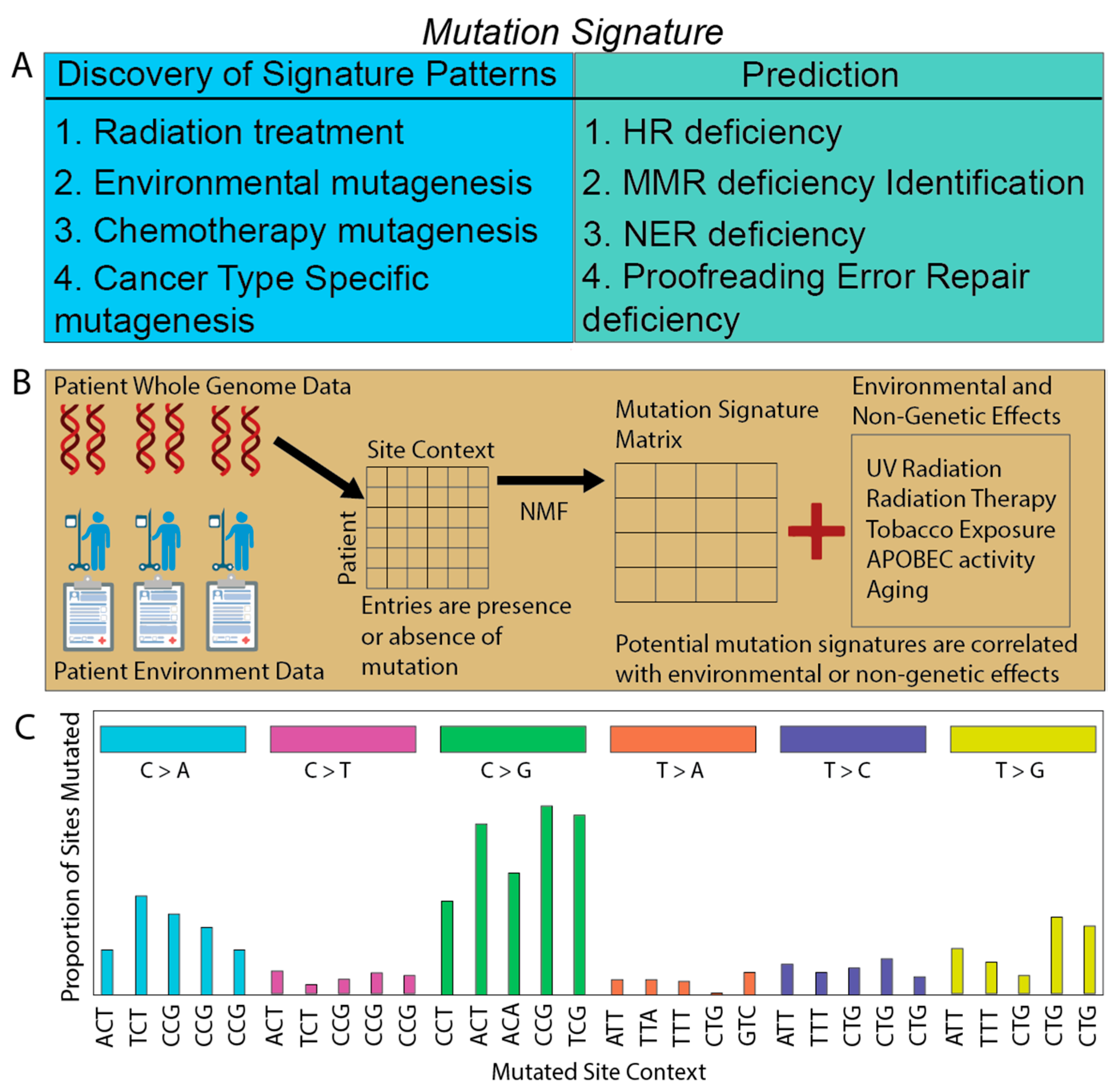
2.1. Deriving Signatures of Mutations
2.2. Associating Mutation Signatures with Carcinogenic Processes
3. Clinical Applications of Mutation Signatures: Promises and Challenges
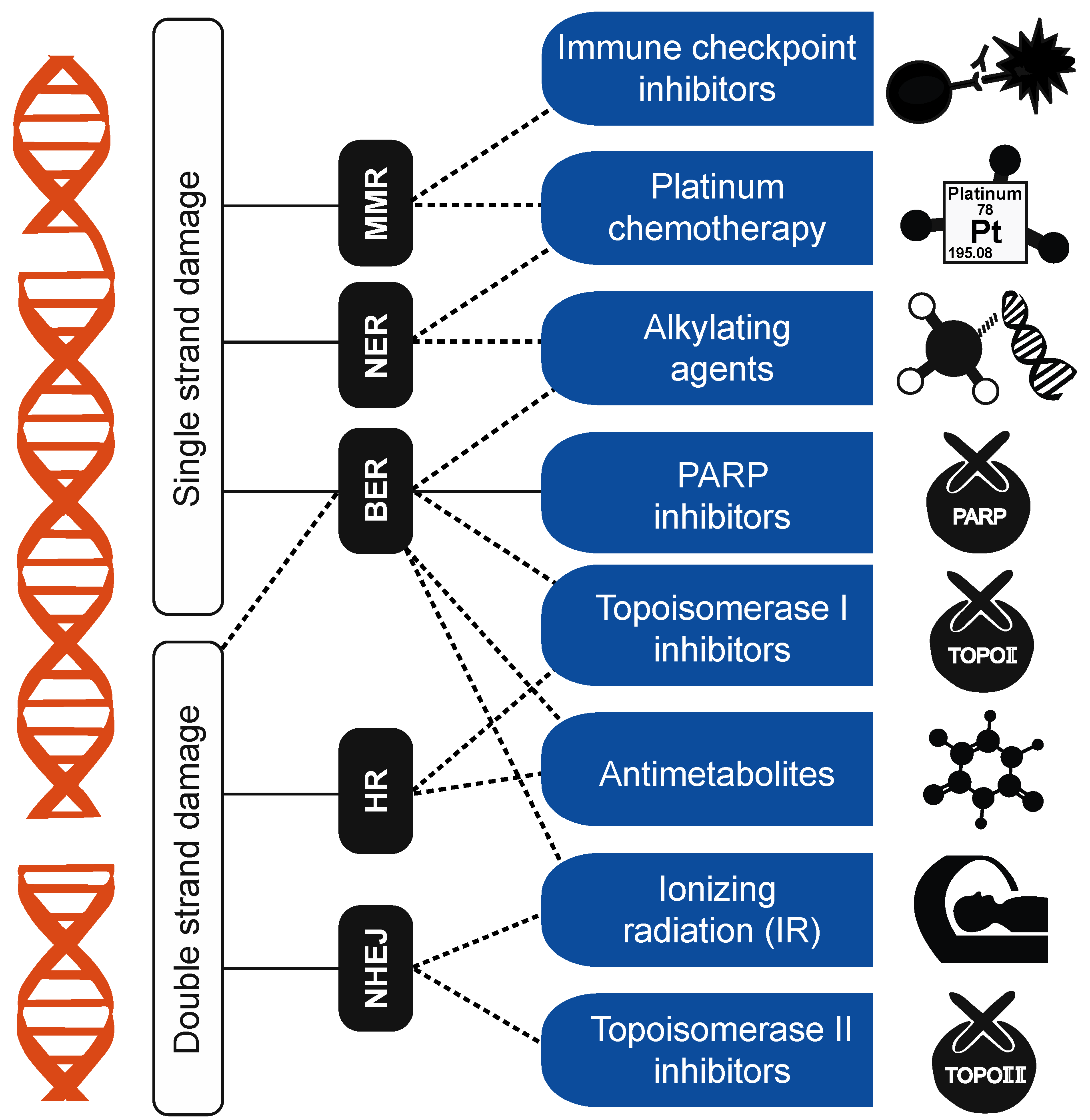
3.1. DNA-Damage-Repair Footprints and Clinical Applications of Mutation Signatures
3.2. Mutation Signatures as Clinical-Discovery Tools
4. Beyond Mutation Signatures: Computational Approaches to Infer Clinically Relevant Patterns of Mutations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Task Category | Sub-Category | Clinical Relevance | Example Methods |
|---|---|---|---|
| Identifying cancer drivers | Cancer drivers by mutation frequency | Cancer-driver discovery; Obtaining cancer drivers for prognosis, cancer identification, and treatment | Methods based on mutation frequencies: MutSigCV [104], Invex [105], Music [106] |
| Amino-acid and functional-impact changes: Chasm [107,108,109], polyphen2 [110], SIFT [112,113] | |||
| Protein structure: MSEA [114], iPACT [115], GraphPac [116] | |||
| Phosphorylation-site mutation: ActiveDriver [117] | |||
| Cancer drivers by pathway | Heat diffusion: HotNet2 [118] | ||
| Mutated neighbors: MUFFIN [119] | |||
| Curated pathways and gene-factor modeling: Paradigm [120] | |||
| Network-based modules [121] | |||
| Network-based coding and non-coding modules [122] | |||
| Deep-learning cancer-driver analysis [123] | |||
| Computational-saturation mutagenesis [124] | |||
| Exploring mutated pathways | Predicting outcomes using pathways | Patient-prognosis prediction | Identification of genes associated with DNA-damage response and clinical outcomes [135] |
| Patient response to immunotherapy | Machine learning on clinical mutation data to predict patient response to ICI in melanoma and other cancers [132] | ||
| Deep learning on pathway information, mutations, and copy-number variation to predict melanoma outcomes [136] | |||
| Detecting drug targets through pathways | Drug discovery and tailored treatments [133] | ||
| Pathways of cancer subtypes | Cancer-subtype identification | Cancer-subtype identification and prognosis [134] | |
| Prediction of patient response, tumor type, and histology | Gene-network-based stratification using mutation data for prediction [131] | ||
| Identifying complex patterns of multiple mutations | Inferring interactions between mutations | Interactions conferring sensitivity | Mutual-exclusivity analysis of genes [139,140] |
| Epistatic effects of genes [140,141] | |||
| Clustering samples | Cancer-type identification | Unsupervised NMF and supervised ML to identify cancer subtypes [143] | |
| Applying deep-learning neural network to passenger mutations to classify metastatic cancers of unknown origin [144] | |||
| Identification of tumors vs. healthy tissues | Gene-combination analysis [138] | ||
| Inferring order of mutations | Inferring timing of mutations | Mutation patterns to infer order of mutation events [145,146,147,148,149] | |
| Determining timing for predicting clinical outcomes | Mutation timing to predict clinical outcome [150] | ||
| Clonality analysis for outcome prediction [151,152] | |||
| Machine learning to predict outcome through mutational time series [153] | |||
| Multiomics approach: integrating mutations with other data types | Multiomics outcome prediction | Chemotherapy response or resistance | Using SVM on mutations, copy number, and expression for chemotherapy prediction [155] |
| ICI response or resistance | Genomic and transcriptomic information for response or resistance to ICI [156] | ||
| Prediction of patient outcomes | Mutation, interaction, and pathway information to identify ovarian-cancer outcomes [158] | ||
| Mutation-burden prediction for ICI therapy | Lung-cancer mutation-burden prediction using a multiomics approach [160] | ||
| Cancer classification | Identification of cancerous vs. non-cancerous cells | Identification of HCC cells from normal cells through mutation and expression information [157] | |
| Identification of drug targets | Drug repositioning | Mutations, expression, epigenetics, drug targets, and deep learning for drug repositioning [159] |
5. Major Challenges for Clinical Utility of Complex and Data-Driven Mutational Patterns
6. Summary
| Method Name | Method Description | Code/Tool | Reference | Review Section |
|---|---|---|---|---|
| IntOGen | A method to access the database of mutational-cancer drivers | https://www.intogen.org/search | [2] | 1 |
| SigProfiler | Framework for deciphering mutation signatures from mutational catalogues of cancer genomes | https://www.mathworks.com/matlabcentral/fileexchange/38724-sigprofiler | [24,25,26,27] | 2.1 |
| MutSpec | Somatic-mutation analysis in human and mouse | https://toolshed.g2.bx.psu.edu/ | [31] | 2.1 |
| MutSignatures | Cancer-mutation-signatures analysis | https://github.com/dami82/mutSignatures | [32] | 2.1 |
| SigneR | Bayesian approach to discover mutation signatures | http://bioconductor.org/packages/release/bioc/html/signeR.html | [35] | 2.1 |
| pmsignature | Probabilistic model to infer and visualize cancer-mutation signatures | https://github.com/friend1ws/pmsignature https://friend1ws.shinyapps.io/pmsignature_shiny/ | [36] | 2.1 |
| SomaticSignatures | Inferring characteristics of mutation signatures | https://www.bioconductor.org/packages/release/bioc/html/SomaticSignatures.html | [38] | 2.1 |
| Helmsman | Mutation-signature analysis | https://github.com/carjed/helmsman | [39] | 2.1 |
| deconstructSigs | Mutation signature by machine learning | https://github.com/raerose01/deconstructSigs | [40] | 2.1 |
| SignatureEstimation | Discovering the existence of mutation signatures in cancer | https://www.ncbi.nlm.nih.gov/CBBresearch/Przytycka/index.cgi#signatureestimation | [41] | 2.1 |
| Signal | Mutation-signature analysis | https://github.com/Nik-Zainal-Group/signature.tools.lib | [42] | 2.1 |
| MutationalPatterns | Comprehensive analysis of mutation processes across the genome | http://bioconductor.org/packages/release/bioc/html/MutationalPatterns.html | [43] | 2.1 |
| Identification of mutation signatures | https://github.com/team113sanger/mouse-mutatation-signatures | [55] | 2.2 | |
| CHORD | Classifier identifying homologous recombination deficiency across cancers | https://github.com/UMCUGenetics/CHORD | [68] | 3.1 |
| SigMA | Identification of mutation signatures | https://github.com/parklab/SigMA | [70] | 3.1 |
| mutfootprints | Identification of mutation footprint of and for cancer treatment | https://bitbucket.org/bbglab/mutfootprints/src/master/ | [88] | 3.2 |
| Identification of mutation signatures | https://github.com/UMCUGenetics/5FU | [89] | 3.2 | |
| CUPLR | Classification of primary-tumor identity of metastatic tumors | https://github.com/UMCUGenetics/CUPLR | [98] | 3.2 |
| MutSigCV | Identification of mutated genes in cancer | https://software.broadinstitute.org/cancer/cga/mutsig | [104] | 4 |
| inVex | Identification of positive selection for non-silent mutations | https://software.broadinstitute.org/cancer/cga/invex | [105] | 4 |
| MuSiC | Identification of mutational relevance in cancer genome | http://gmt.genome.wustl.edu/ | [106] | 4 |
| CHASM | Identification of important biological single-nucleotide mutations in cancer | http://wiki.chasmsoftware.org/index.php/Main_Page | [107,108,109] | 4 |
| PolyPhen-2 | Classification of missense-mutation damaging effects on protein | http://genetics.bwh.harvard.edu/pph2/ | [110] | 4 |
| e-Driver | Identification of protein functional regions driving cancer | https://github.com/eduardporta/e-Driver | [111] | |
| SIFT | Classification of amino-acid-substitution impact on proteins | https://sift.bii.a-star.edu.sg/ | [112,113] | 4 |
| MSEA | Classification of cancer genes based on patterns of mutation hotspots | https://github.com/bsml320/MSEA | [114] | 4 |
| iPAC | Identification of non-random somatic mutations in proteins | http://www.bioconductor.org/packages/2.12/bioc/html/iPAC.html | [115] | 4 |
| GraphPAC | Identification of non-random somatic mutations in proteins | http://bioconductor.org/packages/release/bioc/html/GraphPAC.html | [116] | 4 |
| ActiveDriver | Effect of mutation on post-translational signaling | http://www.baderlab.org/Software/ActiveDriver | [117] | 4 |
| HotNet2 | Identification of rare somatic-mutation combinations in pathways and protein complexes | http://compbio-research.cs.brown.edu/pancancer/hotnet2/#!/ http://compbio.cs.brown.edu/software/ | [118] | 4 |
| MUFFINN | Cancer-gene detection through network analysis of somatic mutations | http://www.inetbio.org/muffinn/ | [119] | 4 |
| boostDM | Identification of driver mutations in cancer genes from observed mutations in human tumors | https://zenodo.org/record/4813082#.Y9L38dLMKV4 | [124] | 4 |
| DEOGEN2/MutaFrame | Classification of single-amino-acid variant loss in human proteins | http://babylone.3bio.ulb.ac.be/MutaFrame/ | [126] | 4 |
| PrimateAI | Classification of clinical impact of human mutations | https://basespace.illumina.com/s/cPgCSmecvhb4 | [128] | 4 |
| Classification of immune-checkpoint-inhibitor therapy response | https://github.com/AuslanderLab/Mutated_pathway_ICI_prediction | [132] | 4 | |
| Identification of associations between driver mutations and chromosomal aberrations | https://github.com/noamaus/INTERPLAY-TUMOR-CODES | [135] | 4 | |
| KP-NET | Classification of immunotherapy response | https://github.com/0219zhang/KP-NET | [136] | 4 |
| Causal identifications of individual instances of cancer | https://bitbucket.org/sajal000/multihit-combinations/src/master/ | [138] | 4 | |
| CLICnet | Identification of somatic-mutation combinations that predict cancer survival | https://github.com/gussow/clicnet | [142] | 4 |
| Classification of primary and metastatic tumors | https://github.com/ICGC-TCGA-PanCancer/TumorType-WGS | [144] | 4 | |
| SMASH | Identification of somatic-mutation associations | https://github.com/Sun-lab/SMASH | [152] | 4 |
| Learning evolution of a tumor through mutational time series | https://github.com/noamaus/LSTM-Mutational-series | [153] | 4 | |
| Classification outcomes of checkpoint inhibition by tumor and immune-signal combination | https://zenodo.org/record/5528497#.Y9Ps1dLMKV4 | [156] | 4 | |
| DeepDRK | Drug response prediction | https://github.com/wangyc82/DeepDRK | [159] | 4 |
| MetAML | Prediction of metagenomics-based tasks | https://github.com/segatalab/metaml | [176] | 5 |
| Generalization in machine learning for dataset characteristics | https://github.com/pietrobarbiero/dataset-characteristics | [177] | 5 | |
| Auptimizer | Hyperparameter optimization | https://github.com/LGE-ARC-AdvancedAI/auptimizer | [178] | 5 |
| TPOT | Automated ML–tree-based optimization pipeline | https://github.com/EpistasisLab/tpot | [181,182] | 5 |
| Hyperband | Hyperparameter optimization | https://github.com/automl/pylearningcurvepredictor | [183] | 5 |
| DanQ | Classification of the function of DNA de novo mutations from sequences | http://github.com/uci-cbcl/DanQ | [188] | 5 |
| An explainable machine learning tool of severity-level predictions of COVID-19 patients | https://github.com/freddygabbay/covid19explainableML | [196] | 5 | |
| DeepLIFT | An explainable machine-learning tool | https://github.com/kundajelab/deeplift | [197] | 5 |
| SpliceRover | Classification of donor and acceptor splice site | http://bioit2.irc.ugent.be/rover/splicerover/ | [199] | 5 |
| RIDDLE | Imputation technique using deep learning | https://github.com/jisungk/RIDDLE | [200] | 5 |
| P-NET | Classification of prostate cancer | https://github.com/marakeby/pnet_prostate_paper | [203] | 5 |
| SHAP | An explainable machine learning tool | https://github.com/slundberg/shap | [204] | 5 |
| devCellPy | Classification of cell types across complex annotation hierarchies | https://github.com/devCellPy-Team/devCellPy | [205] | 5 |
| BCrystal | An interpretable sequence-based protein-crystallization predictor | https://github.com/raghvendra5688/BCrystal | [206] | 5 |
| MetaNet | Metastatic-risk assessment of a primary tumor | https://github.com/WangLabHKUST/METANET-analysis | [207] | 5 |
| Ocelot | Prediction of relationships across histone modifications | https://github.com/GuanLab/Ocelot | [208] | 5 |
| DeepHF | Optimization of CRISPR guide RNA design using deep learning for two high-fidelity Cas9 variants | https://github.com/izhangcd/DeepHF http://www.deephf.com/#/home | [209] | 5 |
| MTGCN | Identification of cancer-driver genes | https://github.com/weiba/MTGCN | [213] | 5 |
| GNNExplainer | An explainable graph neural-network tool | https://github.com/RexYing/gnn-model-explainer | [215] | 5 |
| SBMClone | Identification of tumor clones in sparse single-cell-mutation data | https://github.com/raphael-group/SBMClone | [221] | 5 |
| Mix-MMM | Identification of mutation signatures from sparse mutation data | https://github.com/itaysason/Mix-MMM | [222] | 5 |
| JDINAC | Identification of differential interaction patterns of network activation using high-dimensional sparse omics data | https://github.com/jijiadong/JDINAC | [223] | 5 |
| MoGP | Identification of patterns in amyotrophic lateral-sclerosis progression from sparse longitudinal data | https://github.com/fraenkel-lab/mogp | [225] | 5 |
| Multi-cancer analysis of clonality in paired primary tumors and metastases | https://github.com/cancersysbio/pan-metastasis | [251] | 5 | |
| CHESS | Spatial stochastic tumor-growth model to simulate multi-region sequencing data derived from spatial sampling of neoplasm | https://github.com/kchkhaidze/CHESS.cpp | [256] | 5 |
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Hoeck, A.; Tjoonk, N.H.; van Boxtel, R.; Cuppen, E. Portrait of a Cancer: Mutational Signature Analyses for Cancer Diagnostics. BMC Cancer 2019, 19, 457. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Jiménez, F.; Muiños, F.; Sentís, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A Compendium of Mutational Cancer Driver Genes. Nat. Rev. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef] [PubMed]
- Kamel, H.F.M.; Al-Amodi, H.S.A.B. Exploitation of Gene Expression and Cancer Biomarkers in Paving the Path to Era of Personalized Medicine. Genom. Proteom. Bioinform. 2017, 15, 220–235. [Google Scholar] [CrossRef]
- Gayther, S.A.; Warren, W.; Mazoyer, S.; Russell, P.A.; Harrington, P.A.; Chiano, M.; Seal, S.; Hamoudi, R.; van Rensburg, E.J.; Dunning, A.M.; et al. Germline Mutations of the BRCA1 Gene in Breast and Ovarian Cancer Families Provide Evidence for a Genotype–Phenotype Correlation. Nat. Genet. 1995, 11, 428–433. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different Roles in a Common Pathway of Genome Protection. Nat. Rev. Cancer 2012, 12, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Turk, A.; Wisinski, K.B. PARP Inhibition in BRCA-Mutant Breast Cancer. Cancer 2018, 124, 2498–2506. [Google Scholar] [CrossRef] [PubMed]
- Proietti, I.; Skroza, N.; Michelini, S.; Mambrin, A.; Balduzzi, V.; Bernardini, N.; Marchesiello, A.; Tolino, E.; Volpe, S.; Maddalena, P.; et al. BRAF Inhibitors: Molecular Targeting and Immunomodulatory Actions. Cancers 2020, 12, 1823. [Google Scholar] [CrossRef]
- Hertzman Johansson, C.; Egyhazi Brage, S. BRAF Inhibitors in Cancer Therapy. Pharmacol. Ther. 2014, 142, 176–182. [Google Scholar] [CrossRef]
- Liu, J.; Kang, R.; Tang, D. The KRAS-G12C Inhibitor: Activity and Resistance. Cancer Gene 2022, 29, 875–878. [Google Scholar] [CrossRef]
- Rosell, R.; Aguilar, A.; Pedraz, C.; Chaib, I. KRAS Inhibitors, Approved. Nat. Cancer 2021, 2, 1254–1256. [Google Scholar] [CrossRef]
- Cerchione, C.; Romano, A.; Daver, N.; DiNardo, C.; Jabbour, E.J.; Konopleva, M.; Ravandi-Kashani, F.; Kadia, T.; Martelli, M.P.; Isidori, A.; et al. IDH1/IDH2 Inhibition in Acute Myeloid Leukemia. Front. Oncol. 2021, 11, 639387. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Turcan, S. From Laboratory Studies to Clinical Trials: Temozolomide Use in IDH-Mutant Gliomas. Cells 2021, 10, 1225. [Google Scholar] [CrossRef] [PubMed]
- Kwak, E.L.; Bang, Y.-J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.-H.I.; Dezube, B.J.; Jänne, P.A.; Costa, D.B.; et al. Anaplastic Lymphoma Kinase Inhibition in Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, B.; Palmer, R.H. The Role of the ALK Receptor in Cancer Biology. Ann. Oncol. 2016, 27, iii4–iii15. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Kim, D.-W.; Mehra, R.; Tan, D.S.W.; Felip, E.; Chow, L.Q.M.; Camidge, D.R.; Vansteenkiste, J.; Sharma, S.; De Pas, T.; et al. Ceritinib in ALK-Rearranged Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2014, 370, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of Chemotherapy plus a Monoclonal Antibody against HER2 for Metastatic Breast Cancer That Overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Swain, S.M.; Kim, S.-B.; Cortés, J.; Ro, J.; Semiglazov, V.; Campone, M.; Ciruelos, E.; Ferrero, J.-M.; Schneeweiss, A.; Knott, A.; et al. Pertuzumab, Trastuzumab, and Docetaxel for.r HER2-Positive Metastatic Breast Cancer (CLEOPATRA Study): Overall Survival Results from a Randomised, Double-Blind, Placebo-Controlled, Phase 3 Study. Lancet Oncol. 2013, 14, 461–471. [Google Scholar] [CrossRef]
- Cameron, D.; Casey, M.; Press, M.; Lindquist, D.; Pienkowski, T.; Romieu, C.G.; Chan, S.; Jagiello-Gruszfeld, A.; Kaufman, B.; Crown, J.; et al. A Phase III Randomized Comparison of Lapatinib plus Capecitabine versus Capecitabine Alone in Women with Advanced Breast Cancer That Has Progressed on Trastuzumab: Updated Efficacy and Biomarker Analyses. Breast Cancer Res. Treat. 2008, 112, 533–543. [Google Scholar] [CrossRef]
- Martin, M.; Bonneterre, J.; Geyer, C.E.; Ito, Y.; Ro, J.; Lang, I.; Kim, S.-B.; Germa, C.; Vermette, J.; Wang, K.; et al. A Phase Two Randomised Trial of Neratinib Monotherapy versus Lapatinib plus Capecitabine Combination Therapy in Patients with HER2+ Advanced Breast Cancer. Eur. J. Cancer 2013, 49, 3763–3772. [Google Scholar] [CrossRef]
- Johnston, S.; Pippen, J.; Pivot, X.; Lichinitser, M.; Sadeghi, S.; Dieras, V.; Gomez, H.L.; Romieu, G.; Manikhas, A.; Kennedy, M.J.; et al. Lapatinib Combined With Letrozole Versus Letrozole and Placebo As First-Line Therapy for Postmenopausal Hormone Receptor–Positive Metastatic Breast Cancer. JCO 2009, 27, 5538–5546. [Google Scholar] [CrossRef]
- Villanueva, C.; Romieu, G.; Salvat, J.; Chaigneau, L.; Merrouche, Y.; N’Guyen, T.; Vuillemin, A.T.; Demarchi, M.; Dobi, E.; Pivot, X. Phase II Study Assessing Lapatinib Added to Letrozole in Patients with Progressive Disease under Aromatase Inhibitor in Metastatic Breast Cancer—Study BES 06. Target. Oncol. 2013, 8, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.K. DNA Damage, Mutagenesis and Cancer. Int. J. Mol. Sci. 2018, 19, 970. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational Processes Molding the Genomes of 21 Breast Cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The Repertoire of Mutational Signatures in Human Cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Baez-Ortega, A.; Gori, K. Computational Approaches for Discovery of Mutational Signatures in Cancer. Brief. Bioinform. 2019, 20, 77–88. [Google Scholar] [CrossRef]
- Omichessan, H.; Severi, G.; Perduca, V. Computational Tools to Detect Signatures of Mutational Processes in DNA from Tumours: A Review and Empirical Comparison of Performance. PLoS ONE 2019, 14, e0221235. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Jones, P.H.; Wedge, D.C.; Sale, J.E.; Campbell, P.J.; Nik-Zainal, S.; Stratton, M.R. Clock-like Mutational Processes in Human Somatic Cells. Nat. Genet. 2015, 47, 1402–1407. [Google Scholar] [CrossRef]
- Lee, D.D.; Seung, H.S. Learning the Parts of Objects by Non-Negative Matrix Factorization. Nature 1999, 401, 788–791. [Google Scholar] [CrossRef]
- Paatero, P.; Tapper, U. Positive Matrix Factorization: A Non-Negative Factor Model with Optimal Utilization of Error Estimates of Data Values. Environmetrics 1994, 5, 111–126. [Google Scholar] [CrossRef]
- Ardin, M.; Cahais, V.; Castells, X.; Bouaoun, L.; Byrnes, G.; Herceg, Z.; Zavadil, J.; Olivier, M. MutSpec: A Galaxy Toolbox for Streamlined Analyses of Somatic Mutation Spectra in Human and Mouse Cancer Genomes. BMC Bioinform. 2016, 17, 170. [Google Scholar] [CrossRef] [PubMed]
- Fantini, D.; Vidimar, V.; Yu, Y.; Condello, S.; Meeks, J.J. MutSignatures: An R Package for Extraction and Analysis of Cancer Mutational Signatures. Sci. Rep. 2020, 10, 18217. [Google Scholar] [CrossRef] [PubMed]
- Kasar, S.; Kim, J.; Improgo, R.; Tiao, G.; Polak, P.; Haradhvala, N.; Lawrence, M.S.; Kiezun, A.; Fernandes, S.M.; Bahl, S.; et al. Whole-Genome Sequencing Reveals Activation-Induced Cytidine Deaminase Signatures during Indolent Chronic Lymphocytic Leukaemia Evolution. Nat. Commun. 2015, 6, 8866. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Mouw, K.W.; Polak, P.; Braunstein, L.Z.; Kamburov, A.; Kwiatkowski, D.J.; Rosenberg, J.E.; Van Allen, E.M.; D’Andrea, A.; Getz, G. Somatic ERCC2 Mutations Are Associated with a Distinct Genomic Signature in Urothelial Tumors. Nat. Genet. 2016, 48, 600–606. [Google Scholar] [CrossRef]
- Rosales, R.A.; Drummond, R.D.; Valieris, R.; Dias-Neto, E.; da Silva, I.T. SigneR: An Empirical Bayesian Approach to Mutational Signature Discovery. Bioinformatics 2017, 33, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, Y.; Tremmel, G.; Miyano, S.; Stephens, M. A Simple Model-Based Approach to Inferring and Visualizing Cancer Mutation Signatures. PLoS Genet. 2015, 11, e1005657. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Illingworth, C.J.; Campbell, P.J.; Mustonen, V. EMu: Probabilistic Inference of Mutational Processes and Their Localization in the Cancer Genome. Genome Biol. 2013, 14, R39. [Google Scholar] [CrossRef]
- Gehring, J.S.; Fischer, B.; Lawrence, M.; Huber, W. SomaticSignatures: Inferring Mutational Signatures from Single-Nucleotide Variants. Bioinformatics 2015, 31, 3673–3675. [Google Scholar] [CrossRef]
- Carlson, J.; Li, J.Z.; Zöllner, S. Helmsman: Fast and Efficient Mutation Signature Analysis for Massive Sequencing Datasets. BMC Genom. 2018, 19, 845. [Google Scholar] [CrossRef]
- Rosenthal, R.; McGranahan, N.; Herrero, J.; Taylor, B.S.; Swanton, C. DeconstructSigs: Delineating Mutational Processes in Single Tumors Distinguishes DNA Repair Deficiencies and Patterns of Carcinoma Evolution. Genome Biol. 2016, 17, 31. [Google Scholar] [CrossRef]
- Huang, X.; Wojtowicz, D.; Przytycka, T.M. Detecting Presence of Mutational Signatures in Cancer with Confidence. Bioinformatics 2018, 34, 330–337. [Google Scholar] [CrossRef]
- Degasperi, A.; Amarante, T.D.; Czarnecki, J.; Shooter, S.; Zou, X.; Glodzik, D.; Morganella, S.; Nanda, A.S.; Badja, C.; Koh, G.; et al. A Practical Framework and Online Tool for Mutational Signature Analyses Show Intertissue Variation and Driver Dependencies. Nat. Cancer 2020, 1, 249–263. [Google Scholar] [CrossRef]
- Blokzijl, F.; Janssen, R.; van Boxtel, R.; Cuppen, E. MutationalPatterns: Comprehensive Genome-Wide Analysis of Mutational Processes. Genome Med. 2018, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Di Noia, J.M.; Neuberger, M.S. Molecular Mechanisms of Antibody Somatic Hypermutation. Annu. Rev. Biochem. 2007, 76, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.; Roberts, S.A.; Klimczak, L.J.; Sterling, J.F.; Saini, N.; Malc, E.P.; Kim, J.; Kwiatkowski, D.J.; Fargo, D.C.; Mieczkowski, P.A.; et al. An APOBEC3A Hypermutation Signature Is Distinguishable from the Signature of Background Mutagenesis by APOBEC3B in Human Cancers. Nat. Genet. 2015, 47, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Petljak, M.; Alexandrov, L.B.; Brammeld, J.S.; Price, S.; Wedge, D.C.; Grossmann, S.; Dawson, K.J.; Ju, Y.S.; Iorio, F.; Tubio, J.M.C.; et al. Characterizing Mutational Signatures in Human Cancer Cell Lines Reveals Episodic APOBEC Mutagenesis. Cell 2019, 176, 1282–1294.e20. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Ruiz, P.; Ito, T.; Sellers, W.R. Targeting Pan-Essential Genes in Cancer: Challenges and Opportunities. Cancer Cell 2021, 39, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Buisson, R.; Lawrence, M.S.; Benes, C.H.; Zou, L. APOBEC3A and 3B Activities Render Cancer Cells Susceptible to ATR Inhibition. Cancer Res. 2017, 77, 4567–4578. [Google Scholar] [CrossRef] [PubMed]
- Moody, S.; Senkin, S.; Islam, S.M.A.; Wang, J.; Nasrollahzadeh, D.; Cortez Cardoso Penha, R.; Fitzgerald, S.; Bergstrom, E.N.; Atkins, J.; He, Y.; et al. Mutational Signatures in Esophageal Squamous Cell Carcinoma from Eight Countries with Varying Incidence. Nat. Genet. 2021, 53, 1553–1563. [Google Scholar] [CrossRef]
- Pilati, C.; Shinde, J.; Alexandrov, L.B.; Assié, G.; André, T.; Hélias-Rodzewicz, Z.; Ducoudray, R.; Le Corre, D.; Zucman-Rossi, J.; Emile, J.-F.; et al. Mutational Signature Analysis Identifies MUTYH Deficiency in Colorectal Cancers and Adrenocortical Carcinomas. J. Pathol. 2017, 242, 10–15. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Ju, Y.S.; Haase, K.; Van Loo, P.; Martincorena, I.; Nik-Zainal, S.; Totoki, Y.; Fujimoto, A.; Nakagawa, H.; Shibata, T.; et al. Mutational Signatures Associated with Tobacco Smoking in Human Cancer. Science 2016, 354, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Haradhvala, N.J.; Kim, J.; Maruvka, Y.E.; Polak, P.; Rosebrock, D.; Livitz, D.; Hess, J.M.; Leshchiner, I.; Kamburov, A.; Mouw, K.W.; et al. Distinct Mutational Signatures Characterize Concurrent Loss of Polymerase Proofreading and Mismatch Repair. Nat. Commun. 2018, 9, 1746. [Google Scholar] [CrossRef] [PubMed]
- Behjati, S.; Gundem, G.; Wedge, D.C.; Roberts, N.D.; Tarpey, P.S.; Cooke, S.L.; Van Loo, P.; Alexandrov, L.B.; Ramakrishna, M.; Davies, H.; et al. Mutational Signatures of Ionizing Radiation in Second Malignancies. Nat. Commun. 2016, 7, 12605. [Google Scholar] [CrossRef] [PubMed]
- Rose Li, Y.; Halliwill, K.D.; Adams, C.J.; Iyer, V.; Riva, L.; Mamunur, R.; Jen, K.-Y.; del Rosario, R.; Fredlund, E.; Hirst, G.; et al. Mutational Signatures in Tumours Induced by High and Low Energy Radiation in Trp53 Deficient Mice. Nat. Commun. 2020, 11, 394. [Google Scholar] [CrossRef]
- Riva, L.; Pandiri, A.R.; Li, Y.R.; Droop, A.; Hewinson, J.; Quail, M.A.; Iyer, V.; Shepherd, R.; Herbert, R.A.; Campbell, P.J.; et al. The Mutational Signature Profile of Known and Suspected Human Carcinogens in Mice. Nat. Genet. 2020, 52, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Zhou, P.-K. DNA Damage Repair: Historical Perspectives, Mechanistic Pathways and Clinical Translation for Targeted Cancer Therapy. Signal Transduct. Target. 2021, 6, 254. [Google Scholar] [CrossRef] [PubMed]
- Alhmoud, J.F.; Woolley, J.F.; Al Moustafa, A.-E.; Malki, M.I. DNA Damage/Repair Management in Cancers. Cancers 2020, 12, 1050. [Google Scholar] [CrossRef]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic Instability—An Evolving Hallmark of Cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic Instabilities in Human Cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef]
- Turgeon, M.-O.; Perry, N.J.S.; Poulogiannis, G. DNA Damage, Repair, and Cancer Metabolism. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef]
- Li, L.; Guan, Y.; Chen, X.; Yang, J.; Cheng, Y. DNA Repair Pathways in Cancer Therapy and Resistance. Front. Pharmacol. 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, V.; Dantzer, F.; Ame, J.-C.; de Murcia, G. Poly(ADP-Ribose): Novel Functions for an Old Molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect Is a Predictor of BRCA1 and BRCA2 Deficiency Based on Mutational Signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef]
- Staaf, J.; Glodzik, D.; Bosch, A.; Vallon-Christersson, J.; Reuterswärd, C.; Häkkinen, J.; Degasperi, A.; Amarante, T.D.; Saal, L.H.; Hegardt, C.; et al. Whole-Genome Sequencing of Triple-Negative Breast Cancers in a Population-Based Clinical Study. Nat. Med. 2019, 25, 1526–1533. [Google Scholar] [CrossRef]
- Nones, K.; Johnson, J.; Newell, F.; Patch, A.M.; Thorne, H.; Kazakoff, S.H.; de Luca, X.M.; Parsons, M.T.; Ferguson, K.; Reid, L.E.; et al. Whole-Genome Sequencing Reveals Clinically Relevant Insights into the Aetiology of Familial Breast Cancers. Ann. Oncol. 2019, 30, 1071–1079. [Google Scholar] [CrossRef]
- Zhao, E.Y.; Shen, Y.; Pleasance, E.; Kasaian, K.; Leelakumari, S.; Jones, M.; Bose, P.; Ch’ng, C.; Reisle, C.; Eirew, P.; et al. Homologous Recombination Deficiency and Platinum-Based Therapy Outcomes in Advanced Breast Cancer. Clin. Cancer Res. 2017, 23, 7521–7530. [Google Scholar] [CrossRef]
- Nguyen, L.W.M.; Martens, J.; Van Hoeck, A.; Cuppen, E. Pan-Cancer Landscape of Homologous Recombination Deficiency. Nat. Commun. 2020, 11, 5584. [Google Scholar] [CrossRef]
- Chopra, N.; Tovey, H.; Pearson, A.; Cutts, R.; Toms, C.; Proszek, P.; Hubank, M.; Dowsett, M.; Dodson, A.; Daley, F.; et al. Homologous Recombination DNA Repair Deficiency and PARP Inhibition Activity in Primary Triple Negative Breast Cancer. Nat. Commun. 2020, 11, 2662. [Google Scholar] [CrossRef]
- Gulhan, D.C.; Lee, J.J.-K.; Melloni, G.E.M.; Cortés-Ciriano, I.; Park, P.J. Detecting the Mutational Signature of Homologous Recombination Deficiency in Clinical Samples. Nat. Genet. 2019, 51, 912–919. [Google Scholar] [CrossRef]
- Toh, M.; Ngeow, J. Homologous Recombination Deficiency: Cancer Predispositions and Treatment Implications. Oncol. 2021, 26, e1526–e1537. [Google Scholar] [CrossRef] [PubMed]
- Buisson, R.; Joshi, N.; Rodrigue, A.; Ho, C.K.; Kreuzer, J.; Foo, T.K.; Hardy, E.J.-L.; Dellaire, G.; Haas, W.; Xia, B.; et al. Coupling of Homologous Recombination and the Checkpoint by ATR. Mol. Cell 2017, 65, 336–346. [Google Scholar] [CrossRef]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.-M.; Nguyen, H.D.; Ho, C.K.; Todorova Kwan, T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR Inhibition Disrupts Rewired Homologous Recombination and Fork Protection Pathways in PARP Inhibitor-Resistant BRCA-Deficient Cancer Cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR Inhibition Overcomes PARP Inhibitor and Platinum Resistance in Ovarian Cancer Models. Nat. Commun. 2020, 11, 3726. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Stewart, J.; Porta, N.; Toms, C.; Leary, A.; Lheureux, S.; Khalique, S.; Tai, J.; Attygalle, A.; Vroobel, K.; et al. ATARI Trial: ATR Inhibitor in Combination with Olaparib in Gynecological Cancers with ARID1A Loss or No Loss (ENGOT/GYN1/NCRI). Int. J. Gynecol. Cancer 2021, 31, 1471–1475. [Google Scholar] [CrossRef]
- FDA. FDA Approves First-Line Immunotherapy for Patients with MSI-H/DMMR Metastatic Colorectal Cancer. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-line-immunotherapy-patients-msi-hdmmr-metastatic-colorectal-cancer (accessed on 22 August 2022).
- Zou, X.; Koh, G.C.C.; Nanda, A.S.; Degasperi, A.; Urgo, K.; Roumeliotis, T.I.; Agu, C.A.; Badja, C.; Momen, S.; Young, J.; et al. A Systematic CRISPR Screen Defines Mutational Mechanisms Underpinning Signatures Caused by Replication Errors and Endogenous DNA Damage. Nat. Cancer 2021, 2, 643–657. [Google Scholar] [CrossRef]
- Brady, S.W.; Gout, A.M.; Zhang, J. Therapeutic and Prognostic Insights from the Analysis of Cancer Mutational Signatures. Trends Genet. 2022, 38, 194–208. [Google Scholar] [CrossRef]
- Nowak, J.A.; Yurgelun, M.B.; Bruce, J.L.; Rojas-Rudilla, V.; Hall, D.L.; Shivdasani, P.; Garcia, E.P.; Agoston, A.T.; Srivastava, A.; Ogino, S.; et al. Detection of Mismatch Repair Deficiency and Microsatellite Instability in Colorectal Adenocarcinoma by Targeted Next-Generation Sequencing. J. Mol. Diagn. 2017, 19, 84–91. [Google Scholar] [CrossRef]
- Li, B.; Brady, S.W.; Ma, X.; Shen, S.; Zhang, Y.; Li, Y.; Szlachta, K.; Dong, L.; Liu, Y.; Yang, F.; et al. Therapy-Induced Mutations Drive the Genomic Landscape of Relapsed Acute Lymphoblastic Leukemia. Blood 2020, 135, 41–55. [Google Scholar] [CrossRef]
- Esteller, M.; Levine, R.; Baylin, S.B.; Ellenson, L.H.; Herman, J.G. MLH1 Promoter Hypermethylation Is Associated with the Microsatellite Instability Phenotype in Sporadic Endometrial Carcinomas. Oncogene 1998, 17, 2413–2417. [Google Scholar] [CrossRef]
- Picco, G.; Cattaneo, C.M.; van Vliet, E.J.; Crisafulli, G.; Rospo, G.; Consonni, S.; Vieira, S.F.; Rodríguez, I.S.; Cancelliere, C.; Banerjee, R.; et al. Werner Helicase Is a Synthetic-Lethal Vulnerability in Mismatch Repair–Deficient Colorectal Cancer Refractory to Targeted Therapies, Chemotherapy, and Immunotherapy. Cancer Discov. 2021, 11, 1923–1937. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.M.; Shibue, T.; McFarland, J.M.; Gaeta, B.; Ghandi, M.; Dumont, N.; Gonzalez, A.; McPartlan, J.S.; Li, T.; Zhang, Y.; et al. WRN Helicase Is a Synthetic Lethal Target in Microsatellite Unstable Cancers. Nature 2019, 568, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Connor, A.A.; Denroche, R.E.; Jang, G.H.; Timms, L.; Kalimuthu, S.N.; Selander, I.; McPherson, T.; Wilson, G.W.; Chan-Seng-Yue, M.A.; Borozan, I.; et al. Association of Distinct Mutational Signatures With Correlates of Increased Immune Activity in Pancreatic Ductal Adenocarcinoma. JAMA Oncol. 2017, 3, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Jager, M.; Blokzijl, F.; Kuijk, E.; Bertl, J.; Vougioukalaki, M.; Janssen, R.; Besselink, N.; Boymans, S.; de Ligt, J.; Pedersen, J.S.; et al. Deficiency of Nucleotide Excision Repair Is Associated with Mutational Signature Observed in Cancer. Genome Res. 2019, 29, 1067–1077. [Google Scholar] [CrossRef]
- Mehnert, J.M.; Panda, A.; Zhong, H.; Hirshfield, K.; Damare, S.; Lane, K.; Sokol, L.; Stein, M.N.; Rodriguez-Rodriquez, L.; Kaufman, H.L.; et al. Immune Activation and Response to Pembrolizumab in POLE-Mutant Endometrial Cancer. J. Clin. Investig. 2016, 126, 2334–2340. [Google Scholar] [CrossRef]
- Howitt, B.E.; Shukla, S.A.; Sholl, L.M.; Ritterhouse, L.L.; Watkins, J.C.; Rodig, S.; Stover, E.; Strickland, K.C.; D’Andrea, A.D.; Wu, C.J.; et al. Association of Polymerase e–Mutated and Microsatellite-Instable Endometrial Cancers With Neoantigen Load, Number of Tumor-Infiltrating Lymphocytes, and Expression of PD-1 and PD-L1. JAMA Oncol. 2015, 1, 1319–1323. [Google Scholar] [CrossRef]
- Pich, O.; Muiños, F.; Lolkema, M.P.; Steeghs, N.; Gonzalez-Perez, A.; Lopez-Bigas, N. The Mutational Footprints of Cancer Therapies. Nat. Genet. 2019, 51, 1732–1740. [Google Scholar] [CrossRef]
- Christensen, S.; Van der Roest, B.; Besselink, N.; Janssen, R.; Boymans, S.; Martens, J.W.M.; Yaspo, M.-L.; Priestley, P.; Kuijk, E.; Cuppen, E.; et al. 5-Fluorouracil Treatment Induces Characteristic T>G Mutations in Human Cancer. Nat. Commun. 2019, 10, 4571. [Google Scholar] [CrossRef]
- Hoang, M.L.; Chen, C.-H.; Sidorenko, V.S.; He, J.; Dickman, K.G.; Yun, B.H.; Moriya, M.; Niknafs, N.; Douville, C.; Karchin, R.; et al. Mutational Signature of Aristolochic Acid Exposure as Revealed by Whole-Exome Sequencing. Sci. Transl. Med. 2013, 5, 197ra102. [Google Scholar] [CrossRef]
- Poon, S.L.; Huang, M.N.; Choo, Y.; McPherson, J.R.; Yu, W.; Heng, H.L.; Gan, A.; Myint, S.S.; Siew, E.Y.; Ler, L.D.; et al. Mutation Signatures Implicate Aristolochic Acid in Bladder Cancer Development. Genome Med. 2015, 7, 38. [Google Scholar] [CrossRef]
- Poon, S.L.; Pang, S.-T.; McPherson, J.R.; Yu, W.; Huang, K.K.; Guan, P.; Weng, W.-H.; Siew, E.Y.; Liu, Y.; Heng, H.L.; et al. Genome-Wide Mutational Signatures of Aristolochic Acid and Its Application as a Screening Tool. Sci. Transl. Med. 2013, 5, 197ra101. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Tan, W.; Ling, Z.; Xi, R.; Shao, M.; Chen, M.; Luo, Y.; Zhao, Y.; Liu, Y.; Huang, X.; et al. Genomic Analysis of Oesophageal Squamous-Cell Carcinoma Identifies Alcohol Drinking-Related Mutation Signature and Genomic Alterations. Nat. Commun. 2017, 8, 15290. [Google Scholar] [CrossRef] [PubMed]
- Li, X.C.; Wang, M.Y.; Yang, M.; Dai, H.J.; Zhang, B.F.; Wang, W.; Chu, X.L.; Wang, X.; Zheng, H.; Niu, R.F.; et al. A Mutational Signature Associated with Alcohol Consumption and Prognostically Significantly Mutated Driver Genes in Esophageal Squamous Cell Carcinoma. Ann. Oncol. 2018, 29, 938–944. [Google Scholar] [CrossRef]
- Letouzé, E.; Shinde, J.; Renault, V.; Couchy, G.; Blanc, J.-F.; Tubacher, E.; Bayard, Q.; Bacq, D.; Meyer, V.; Semhoun, J.; et al. Mutational Signatures Reveal the Dynamic Interplay of Risk Factors and Cellular Processes during Liver Tumorigenesis. Nat. Commun. 2017, 8, 1315. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Li, P.; He, F.; Wei, G.; Zhou, Z.; Su, Z.; Ni, T. Comprehensive Analysis Reveals Distinct Mutational Signature and Its Mechanistic Insights of Alcohol Consumption in Human Cancers. Brief. Bioinform. 2021, 22, bbaa066. [Google Scholar] [CrossRef] [PubMed]
- Secrier, M.; Li, X.; de Silva, N.; Eldridge, M.D.; Contino, G.; Bornschein, J.; MacRae, S.; Grehan, N.; O’Donovan, M.; Miremadi, A.; et al. Mutational Signatures in Esophageal Adenocarcinoma Define Etiologically Distinct Subgroups with Therapeutic Relevance. Nat. Genet. 2016, 48, 1131–1141. [Google Scholar] [CrossRef]
- Nguyen, L.; Van Hoeck, A.; Cuppen, E. Machine Learning-Based Tissue of Origin Classification for Cancer of Unknown Primary Diagnostics Using Genome-Wide Mutation Features. Nat. Commun. 2022, 13, 4013. [Google Scholar] [CrossRef]
- Wang, K.; Tepper, J.E. Radiation Therapy-Associated Toxicity: Etiology, Management, and Prevention. CA A Cancer J. Clin. 2021, 71, 437–454. [Google Scholar] [CrossRef]
- Majeed, H.; Gupta, V. Adverse Effects Of Radiation Therapy. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Cheng, F.; Zhao, J.; Zhao, Z. Advances in Computational Approaches for Prioritizing Driver Mutations and Significantly Mutated Genes in Cancer Genomes. Brief. Bioinform. 2016, 17, 642–656. [Google Scholar] [CrossRef]
- Tsimberidou, A.M.; Fountzilas, E.; Nikanjam, M.; Kurzrock, R. Review of Precision Cancer Medicine: Evolution of the Treatment Paradigm. Cancer Treat. Rev. 2020, 86, 102019. [Google Scholar] [CrossRef]
- Moscow, J.A.; Fojo, T.; Schilsky, R.L. The Evidence Framework for Precision Cancer Medicine. Nat. Rev. Clin. Oncol. 2018, 15, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational Heterogeneity in Cancer and the Search for New Cancer-Associated Genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.-P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A Landscape of Driver Mutations in Melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Dees, N.D.; Zhang, Q.; Kandoth, C.; Wendl, M.C.; Schierding, W.; Koboldt, D.C.; Mooney, T.B.; Callaway, M.B.; Dooling, D.; Mardis, E.R.; et al. MuSiC: Identifying Mutational Significance in Cancer Genomes. Genome Res. 2012, 22, 1589–1598. [Google Scholar] [CrossRef]
- Carter, H.; Chen, S.; Isik, L.; Tyekucheva, S.; Velculescu, V.E.; Kinzler, K.W.; Vogelstein, B.; Karchin, R. Cancer-Specific High-Throughput Annotation of Somatic Mutations: Computational Prediction of Driver Missense Mutations. Cancer Res. 2009, 69, 6660–6667. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.C.; Kim, D.; Carter, H.; Diekhans, M.; Ryan, M.C.; Karchin, R. CHASM and SNVBox: Toolkit for Detecting Biologically Important Single Nucleotide Mutations in Cancer. Bioinformatics 2011, 27, 2147–2148. [Google Scholar] [CrossRef]
- Carter, H.; Samayoa, J.; Hruban, R.H.; Karchin, R. Prioritization of Driver Mutations in Pancreatic Cancer Using Cancer-Specific High-Throughput Annotation of Somatic Mutations (CHASM). Cancer Biol. Ther. 2010, 10, 582–587. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Porta-Pardo, E.; Godzik, A. E-Driver: A Novel Method to Identify Protein Regions Driving Cancer. Bioinformatics 2014, 30, 3109–3114. [Google Scholar] [CrossRef]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT Web Server: Predicting Effects of Amino Acid Substitutions on Proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the Effects of Coding Non-Synonymous Variants on Protein Function Using the SIFT Algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Jia, P.; Wang, Q.; Chen, Q.; Hutchinson, K.E.; Pao, W.; Zhao, Z. MSEA: Detection and Quantification of Mutation Hotspots through Mutation Set Enrichment Analysis. Genome Biol. 2014, 15, 489. [Google Scholar] [CrossRef] [PubMed]
- Ryslik, G.A.; Cheng, Y.; Cheung, K.-H.; Modis, Y.; Zhao, H. Utilizing Protein Structure to Identify Non-Random Somatic Mutations. BMC Bioinform. 2013, 14, 190. [Google Scholar] [CrossRef] [PubMed]
- Ryslik, G.A.; Cheng, Y.; Cheung, K.-H.; Modis, Y.; Zhao, H. A Graph Theoretic Approach to Utilizing Protein Structure to Identify Non-Random Somatic Mutations. BMC Bioinform. 2014, 15, 86. [Google Scholar] [CrossRef]
- Reimand, J.; Bader, G.D. Systematic Analysis of Somatic Mutations in Phosphorylation Signaling Predicts Novel Cancer Drivers. Mol. Syst. Biol. 2013, 9, 637. [Google Scholar] [CrossRef]
- Leiserson, M.D.M.; Vandin, F.; Wu, H.-T.; Dobson, J.R.; Eldridge, J.V.; Thomas, J.L.; Papoutsaki, A.; Kim, Y.; Niu, B.; McLellan, M.; et al. Pan-Cancer Network Analysis Identifies Combinations of Rare Somatic Mutations across Pathways and Protein Complexes. Nat. Genet. 2015, 47, 106–114. [Google Scholar] [CrossRef]
- Cho, A.; Shim, J.E.; Kim, E.; Supek, F.; Lehner, B.; Lee, I. MUFFINN: Cancer Gene Discovery via Network Analysis of Somatic Mutation Data. Genome Biol. 2016, 17, 129. [Google Scholar] [CrossRef]
- Vaske, C.J.; Benz, S.C.; Sanborn, J.Z.; Earl, D.; Szeto, C.; Zhu, J.; Haussler, D.; Stuart, J.M. Inference of Patient-Specific Pathway Activities from Multi-Dimensional Cancer Genomics Data Using PARADIGM. Bioinformatics 2010, 26, i237–i245. [Google Scholar] [CrossRef]
- Iranzo, J.; Martincorena, I.; Koonin, E.V. Cancer-Mutation Network and the Number and Specificity of Driver Mutations. Proc. Natl. Acad. Sci. USA 2018, 115, E6010–E6019. [Google Scholar] [CrossRef]
- Reyna, M.A.; Haan, D.; Paczkowska, M.; Verbeke, L.P.C.; Vazquez, M.; Kahraman, A.; Pulido-Tamayo, S.; Barenboim, J.; Wadi, L.; Dhingra, P.; et al. Pathway and Network Analysis of More than 2500 Whole Cancer Genomes. Nat. Commun. 2020, 11, 729. [Google Scholar] [CrossRef]
- Sherman, M.A.; Yaari, A.U.; Priebe, O.; Dietlein, F.; Loh, P.-R.; Berger, B. Genome-Wide Mapping of Somatic Mutation Rates Uncovers Drivers of Cancer. Nat. Biotechnol. 2022, 40, 1634–1643. [Google Scholar] [CrossRef] [PubMed]
- Muiños, F.; Martínez-Jiménez, F.; Pich, O.; Gonzalez-Perez, A.; Lopez-Bigas, N. In Silico Saturation Mutagenesis of Cancer Genes. Nature 2021, 596, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, J.; Wang, Y.; Ng, P.K.-S.; Tsang, Y.H.; Shaw, K.R.; Mills, G.B.; Liang, H. Comprehensive Assessment of Computational Algorithms in Predicting Cancer Driver Mutations. Genome Biol. 2020, 21, 43. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, D.; Tanyalcin, I.; Ferté, J.; Gazzo, A.; Orlando, G.; Lenaerts, T.; Rooman, M.; Vranken, W. DEOGEN2: Prediction and Interactive Visualization of Single Amino Acid Variant Deleteriousness in Human Proteins. Nucleic Acids Res. 2017, 45, W201–W206. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.e18. [Google Scholar] [CrossRef]
- Sundaram, L.; Gao, H.; Padigepati, S.R.; McRae, J.F.; Li, Y.; Kosmicki, J.A.; Fritzilas, N.; Hakenberg, J.; Dutta, A.; Shon, J.; et al. Predicting the Clinical Impact of Human Mutation with Deep Neural Networks. Nat. Genet. 2018, 50, 1161–1170. [Google Scholar] [CrossRef]
- Mallik, S.; Zhao, Z. Graph- and Rule-Based Learning Algorithms: A Comprehensive Review of Their Applications for Cancer Type Classification and Prognosis Using Genomic Data. Brief. Bioinform. 2020, 21, 368–394. [Google Scholar] [CrossRef]
- Zhang, W.; Chien, J.; Yong, J.; Kuang, R. Network-Based Machine Learning and Graph Theory Algorithms for Precision Oncology. npj Precis. Oncol. 2017, 1, 25. [Google Scholar] [CrossRef]
- Hofree, M.; Shen, J.P.; Carter, H.; Gross, A.; Ideker, T. Network-Based Stratification of Tumor Mutations. Nat. Methods 2013, 10, 1108–1115. [Google Scholar] [CrossRef]
- Patterson, A.; Auslander, N. Mutated Processes Predict Immune Checkpoint Inhibitor Therapy Benefit in Metastatic Melanoma. Nat. Commun. 2022, 13, 5151. [Google Scholar] [CrossRef]
- Zolotovskaia, M.A.; Sorokin, M.I.; Emelianova, A.A.; Borisov, N.M.; Kuzmin, D.V.; Borger, P.; Garazha, A.V.; Buzdin, A.A. Pathway Based Analysis of Mutation Data Is Efficient for Scoring Target Cancer Drugs. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Kuijjer, M.L.; Paulson, J.N.; Salzman, P.; Ding, W.; Quackenbush, J. Cancer Subtype Identification Using Somatic Mutation Data. Br. J. Cancer 2018, 118, 1492–1501. [Google Scholar] [CrossRef] [PubMed]
- Auslander, N.; Wolf, Y.I.; Koonin, E.V. Interplay between DNA Damage Repair and Apoptosis Shapes Cancer Evolution through Aneuploidy and Microsatellite Instability. Nat. Commun. 2020, 11, 1234. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Cao, L.; Li, S.; Wang, L.; Song, Y.; Huang, Y.; Xu, Z.; He, J.; Wang, M.; Li, K. Biologically Interpretable Deep Learning to Predict Response to Immunotherapy in Advanced Melanoma Using Mutations and Copy Number Variations. Res. Sq. 2022. preprint. [Google Scholar]
- Zou, J.; Huss, M.; Abid, A.; Mohammadi, P.; Torkamaani, A.; Telenti, A. A Primer on Deep Learning in Genomics. Nat. Genet. 2019, 51, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Kinney, N.A.; Varghese, R.T.; Garner, H.R.; Feng, W.; Anandakrishnan, R. Differentiating between Cancer and Normal Tissue Samples Using Multi-Hit Combinations of Genetic Mutations. Sci. Rep. 2019, 9, 1005. [Google Scholar] [CrossRef] [PubMed]
- Leiserson, M.D.; Wu, H.-T.; Vandin, F.; Raphael, B.J. CoMEt: A Statistical Approach to Identify Combinations of Mutually Exclusive Alterations in Cancer. Genome Biol. 2015, 16, 160. [Google Scholar] [CrossRef]
- Ciriello, G.; Cerami, E.; Sander, C.; Schultz, N. Mutual Exclusivity Analysis Identifies Oncogenic Network Modules. Genome Res. 2012, 22, 398–406. [Google Scholar] [CrossRef]
- van de Haar, J.; Canisius, S.; Yu, M.K.; Voest, E.E.; Wessels, L.F.A.; Ideker, T. Identifying Epistasis in Cancer Genomes: A Delicate Affair. Cell 2019, 177, 1375–1383. [Google Scholar] [CrossRef]
- Gussow, A.B.; Koonin, E.V.; Auslander, N. Identification of Combinations of Somatic Mutations That Predict Cancer Survival and Immunotherapy Benefit. NAR Cancer 2021, 3, zcab017. [Google Scholar] [CrossRef]
- Vural, S.; Wang, X.; Guda, C. Classification of Breast Cancer Patients Using Somatic Mutation Profiles and Machine Learning Approaches. BMC Syst. Biol. 2016, 10, 62. [Google Scholar] [CrossRef] [PubMed]
- Jiao, W.; Atwal, G.; Polak, P.; Karlic, R.; Cuppen, E.; Danyi, A.; de Ridder, J.; van Herpen, C.; Lolkema, M.P.; Steeghs, N.; et al. A Deep Learning System Accurately Classifies Primary and Metastatic Cancers Using Passenger Mutation Patterns. Nat. Commun. 2020, 11, 728. [Google Scholar] [CrossRef] [PubMed]
- Gerstung, M.; Jolly, C.; Leshchiner, I.; Dentro, S.C.; Gonzalez, S.; Rosebrock, D.; Mitchell, T.J.; Rubanova, Y.; Anur, P.; Yu, K.; et al. The Evolutionary History of 2,658 Cancers. Nature 2020, 578, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Jolly, C.; Van Loo, P. Timing Somatic Events in the Evolution of Cancer. Genome Biol. 2018, 19, 95. [Google Scholar] [CrossRef] [PubMed]
- Attolini, C.S.-O.; Cheng, Y.-K.; Beroukhim, R.; Getz, G.; Abdel-Wahab, O.; Levine, R.L.; Mellinghoff, I.K.; Michor, F. A Mathematical Framework to Determine the Temporal Sequence of Somatic Genetic Events in Cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 17604–17609. [Google Scholar] [CrossRef]
- Cheng, Y.-K.; Beroukhim, R.; Levine, R.L.; Mellinghoff, I.K.; Holland, E.C.; Michor, F. A Mathematical Methodology for Determining the Temporal Order of Pathway Alterations Arising during Gliomagenesis. PLoS Comput. Biol. 2012, 8, e1002337. [Google Scholar] [CrossRef]
- Desper, R.; Jiang, F.; Kallioniemi, O.P.; Moch, H.; Papadimitriou, C.H.; Schäffer, A.A. Distance-Based Reconstruction of Tree Models for Oncogenesis. J. Comput. Biol. 2000, 7, 789–803. [Google Scholar] [CrossRef]
- Bozic, I.; Nowak, M.A. Timing and Heterogeneity of Mutations Associated with Drug Resistance in Metastatic Cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 15964–15968. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, J.; Jia, P.; Li, X.; Pei, G.; Wang, C.; Fang, X.; Zhao, Z.; Cai, Z.; Yi, X.; et al. Clonal Architectures Predict Clinical Outcome in Clear Cell Renal Cell Carcinoma. Nat. Commun. 2019, 10, 1245. [Google Scholar] [CrossRef]
- Little, P.; Lin, D.-Y.; Sun, W. Associating Somatic Mutations to Clinical Outcomes: A Pan-Cancer Study of Survival Time. Genome Med. 2019, 11, 37. [Google Scholar] [CrossRef]
- Auslander, N.; Wolf, Y.I.; Koonin, E.V. In Silico Learning of Tumor Evolution through Mutational Time Series. Proc. Natl. Acad. Sci. USA 2019, 116, 9501–9510. [Google Scholar] [CrossRef] [PubMed]
- Yoo, B.C.; Kim, K.-H.; Woo, S.M.; Myung, J.K. Clinical Multi-Omics Strategies for the Effective Cancer Management. J. Proteom. 2018, 188, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Dorman, S.N.; Baranova, K.; Knoll, J.H.M.; Urquhart, B.L.; Mariani, G.; Carcangiu, M.L.; Rogan, P.K. Genomic Signatures for Paclitaxel and Gemcitabine Resistance in Breast Cancer Derived by Machine Learning. Mol. Oncol. 2016, 10, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Freeman, S.S.; Sade-Feldman, M.; Kim, J.; Stewart, C.; Gonye, A.L.K.; Ravi, A.; Arniella, M.B.; Gushterova, I.; LaSalle, T.J.; Blaum, E.M.; et al. Combined Tumor and Immune Signals from Genomes or Transcriptomes Predict Outcomes of Checkpoint Inhibition in Melanoma. Cell Rep. Med. 2022, 3, 100500. [Google Scholar] [CrossRef]
- Cheng, B.; Zhou, P.; Chen, Y. Machine-Learning Algorithms Based on Personalized Pathways for a Novel Predictive Model for the Diagnosis of Hepatocellular Carcinoma. BMC Bioinform. 2022, 23, 248. [Google Scholar] [CrossRef]
- Kim, D.; Li, R.; Lucas, A.; Verma, S.S.; Dudek, S.M.; Ritchie, M.D. Using Knowledge-Driven Genomic Interactions for Multi-Omics Data Analysis: Metadimensional Models for Predicting Clinical Outcomes in Ovarian Carcinoma. J. Am. Med. Inform. Assoc. 2017, 24, 577–587. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Y.; Chen, S.; Wang, J. DeepDRK: A Deep Learning Framework for Drug Repurposing through Kernel-Based Multi-Omics Integration. Brief. Bioinform. 2021, 22, bbab048. [Google Scholar] [CrossRef]
- Chaudhary, K.; Poirion, O.B.; Lu, L.; Garmire, L.X. Deep Learning–Based Multi-Omics Integration Robustly Predicts Survival in Liver Cancer. Clin. Cancer Res. 2018, 24, 1248–1259. [Google Scholar] [CrossRef]
- Wang, J.; Chen, P.; Su, M.; Zhong, G.; Zhang, S.; Gou, D. Integrative Modeling of Multiomics Data for Predicting Tumor Mutation Burden in Patients with Lung Cancer. BioMed Res. Int. 2022, 2022, e2698190. [Google Scholar] [CrossRef]
- Olivier, M.; Asmis, R.; Hawkins, G.A.; Howard, T.D.; Cox, L.A. The Need for Multi-Omics Biomarker Signatures in Precision Medicine. Int. J. Mol. Sci. 2019, 20, 4781. [Google Scholar] [CrossRef]
- Lewis, J.; Breeze, C.E.; Charlesworth, J.; Maclaren, O.J.; Cooper, J. Where next for the Reproducibility Agenda in Computational Biology? BMC Syst. Biol. 2016, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Garijo, D.; Kinnings, S.; Xie, L.; Xie, L.; Zhang, Y.; Bourne, P.E.; Gil, Y. Quantifying Reproducibility in Computational Biology: The Case of the Tuberculosis Drugome. PLoS ONE 2013, 8, e80278. [Google Scholar] [CrossRef] [PubMed]
- Niven, D.J.; McCormick, T.J.; Straus, S.E.; Hemmelgarn, B.R.; Jeffs, L.; Barnes, T.R.M.; Stelfox, H.T. Reproducibility of Clinical Research in Critical Care: A Scoping Review. BMC Med. 2018, 16, 26. [Google Scholar] [CrossRef]
- Greener, J.G.; Kandathil, S.M.; Moffat, L.; Jones, D.T. A Guide to Machine Learning for Biologists. Nat. Rev. Mol. Cell Biol 2022, 23, 40–55. [Google Scholar] [CrossRef]
- Cook, J.A.; Ranstam, J. Overfitting. Br. J. Surg. 2016, 103, 1814. [Google Scholar] [CrossRef]
- Chicco, D. Ten Quick Tips for Machine Learning in Computational Biology. BioData Min. 2017, 10, 35. [Google Scholar] [CrossRef] [PubMed]
- Papin, J.A.; Gabhann, F.M.; Sauro, H.M.; Nickerson, D.; Rampadarath, A. Improving Reproducibility in Computational Biology Research. PLoS Comput. Biol. 2020, 16, e1007881. [Google Scholar] [CrossRef]
- Sandve, G.K.; Nekrutenko, A.; Taylor, J.; Hovig, E. Ten Simple Rules for Reproducible Computational Research. PLoS Comput. Biol. 2013, 9, e1003285. [Google Scholar] [CrossRef]
- Piccolo, S.R.; Frampton, M.B. Tools and Techniques for Computational Reproducibility. GigaScience 2016, 5, 30. [Google Scholar] [CrossRef]
- Heil, B.J.; Hoffman, M.M.; Markowetz, F.; Lee, S.-I.; Greene, C.S.; Hicks, S.C. Reproducibility Standards for Machine Learning in the Life Sciences. Nat. Methods 2021, 18, 1132–1135. [Google Scholar] [CrossRef]
- Beam, A.L.; Manrai, A.K.; Ghassemi, M. Challenges to the Reproducibility of Machine Learning Models in Health Care. JAMA 2020, 323, 305–306. [Google Scholar] [CrossRef] [PubMed]
- McDermott, M.B.A.; Wang, S.; Marinsek, N.; Ranganath, R.; Foschini, L.; Ghassemi, M. Reproducibility in Machine Learning for Health Research: Still a Ways to Go. Sci. Transl. Med. 2021, 13, eabb1655. [Google Scholar] [CrossRef] [PubMed]
- Doshi-Velez, F.; Kim, B. Considerations for Evaluation and Generalization in Interpretable Machine Learning. In Explainable and Interpretable Models in Computer Vision and Machine Learning; Escalante, H.J., Escalera, S., Guyon, I., Baró, X., Güçlütürk, Y., Güçlü, U., van Gerven, M., Eds.; The Springer Series on Challenges in Machine Learning; Springer International Publishing: Cham, Switzerland, 2018; pp. 3–17. ISBN 978-3-319-98131-4. [Google Scholar]
- Pasolli, E.; Truong, D.T.; Malik, F.; Waldron, L.; Segata, N. Machine Learning Meta-Analysis of Large Metagenomic Datasets: Tools and Biological Insights. PLoS Comput. Biol. 2016, 12, e1004977. [Google Scholar] [CrossRef] [PubMed]
- Barbiero, P.; Squillero, G.; Tonda, A. Modeling Generalization in Machine Learning: A Methodological and Computational Study. arXiv 2020, arXiv:2006.15680. [Google Scholar]
- Liu, J.; Tripathi, S.; Kurup, U.; Shah, M. Auptimizer—An Extensible, Open-Source Framework for Hyperparameter Tuning. In Proceedings of the IEEE International Conference on Big Data (Big Data), Los Angeles, CA, USA, 9–12 December 2019. [Google Scholar]
- Liang, J.; Meyerson, E.; Hodjat, B.; Fink, D.; Mutch, K.; Miikkulainen, R. Evolutionary Neural AutoML for Deep Learning. In Proceedings of the Genetic and Evolutionary Computation Conference, Prague, Czech Republic, 13–17 July 2019. [Google Scholar]
- Chen, B.; Wu, H.; Mo, W.; Chattopadhyay, I.; Lipson, H. Autostacker: A Compositional Evolutionary Learning System. In Proceedings of the Genetic and Evolutionary Computation Conference, Kyoto, Japan, 15–19 July 2018. [Google Scholar]
- Olson, R.S.; Urbanowicz, R.J.; Andrews, P.C.; Lavender, N.A.; Kidd, L.C.; Moore, J.H. Automating Biomedical Data Science through Tree-Based Pipeline Optimization. In Applications of Evolutionary Computation: 19th European Conference, EvoApplications 2016, Porto, Portugal, 30 March–1 April 2016; Springer International Publishing: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Olson, R.S.; Bartley, N.; Urbanowicz, R.J.; Moore, J.H. Evaluation of a Tree-Based Pipeline Optimization Tool for Automating Data Science. In Proceedings of the Genetic and Evolutionary Computation Conference, Denver, CO, USA, 20–24 July 2016. [Google Scholar]
- Li, L.; Jamieson, K.; DeSalvo, G.; Rostamizadeh, A.; Talwalkar, A. Hyperband: A Novel Bandit-Based Approach to Hyperparameter Optimization. J. Mach. Learn. Res. 2018, 18, 6765–6816. [Google Scholar]
- Xanthopoulos, I.; Tsamardinos, I.; Christophides, V.; Simon, E.; Salinger, A. Putting the Human Back in the AutoML Loop. In Proceedings of the Workshops of the EDBT/ICDT 2020 Joint Conference, Copenhagen, Denmark, 30 March 2020. [Google Scholar]
- Baker, B.; Gupta, O.; Raskar, R.; Naik, N. Accelerating Neural Architecture Search Using Performance Prediction. arXiv 2017, arXiv:1705.10823. [Google Scholar]
- Errington, T.M.; Iorns, E.; Gunn, W.; Tan, F.E.; Lomax, J.; Nosek, B.A. An Open Investigation of the Reproducibility of Cancer Biology Research. eLife 2014, 3, e04333. [Google Scholar] [CrossRef]
- Nosek, B.A.; Errington, T.M. Making Sense of Replications. eLife 2017, 6, e23383. [Google Scholar] [CrossRef]
- Quang, D.; Xie, X. DanQ: A Hybrid Convolutional and Recurrent Deep Neural Network for Quantifying the Function of DNA Sequences. Nucleic Acids Res. 2016, 44, e107. [Google Scholar] [CrossRef]
- Azodi, C.B.; Tang, J.; Shiu, S.-H. Opening the Black Box: Interpretable Machine Learning for Geneticists. Trends Genet. 2020, 36, 442–455. [Google Scholar] [CrossRef]
- Molnar, C.; Casalicchio, G.; Bischl, B. Interpretable Machine Learning—A Brief History, State-of-the-Art and Challenges. In Proceedings of the ECML PKDD 2020 Workshops, Ghent, Belgium, 14–18 September 2020; Koprinska, I., Kamp, M., Appice, A., Loglisci, C., Antonie, L., Zimmermann, A., Guidotti, R., Özgöbek, Ö., Ribeiro, R.P., Gavaldà, R., et al., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 417–431. [Google Scholar]
- Ahmad, M.A.; Eckert, C.; Teredesai, A. Interpretable Machine Learning in Healthcare. In Proceedings of the 2018 ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics, Washington, DC, USA, 29 August–1 September 2018; Association for Computing Machinery: New York, NY, USA, 2018; pp. 559–560. [Google Scholar]
- Wang, F.; Kaushal, R.; Khullar, D. Should Health Care Demand Interpretable Artificial Intelligence or Accept “Black Box” Medicine? Ann. Intern. Med. 2020, 172, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Watson, D.S.; Krutzinna, J.; Bruce, I.N.; Griffiths, C.E.; McInnes, I.B.; Barnes, M.R.; Floridi, L. Clinical Applications of Machine Learning Algorithms: Beyond the Black Box. BMJ 2019, 364, l886. [Google Scholar] [CrossRef] [PubMed]
- Linardatos, P.; Papastefanopoulos, V.; Kotsiantis, S. Explainable AI: A Review of Machine Learning Interpretability Methods. Entropy 2021, 23, 18. [Google Scholar] [CrossRef] [PubMed]
- Palatnik de Sousa, I.; Maria Bernardes Rebuzzi Vellasco, M.; Costa da Silva, E. Local Interpretable Model-Agnostic Explanations for Classification of Lymph Node Metastases. Sensors 2019, 19, 2969. [Google Scholar] [CrossRef] [PubMed]
- Gabbay, F.; Bar-Lev, S.; Montano, O.; Hadad, N. A LIME-Based Explainable Machine Learning Model for Predicting the Severity Level of COVID-19 Diagnosed Patients. Appl. Sci. 2021, 11, 10417. [Google Scholar] [CrossRef]
- Shrikumar, A.; Greenside, P.; Kundaje, A. Learning Important Features Through Propagating Activation Differences. In Proceedings of the 34th International Conference on Machine Learning, Sydney, Australia, 6–11 August 2017. [Google Scholar]
- Washburn, J.D.; Mejia-Guerra, M.K.; Ramstein, G.; Kremling, K.A.; Valluru, R.; Buckler, E.S.; Wang, H. Evolutionarily Informed Deep Learning Methods for Predicting Relative Transcript Abundance from DNA Sequence. Proc. Natl. Acad. Sci. USA 2019, 116, 5542–5549. [Google Scholar] [CrossRef]
- Zuallaert, J.; Godin, F.; Kim, M.; Soete, A.; Saeys, Y.; De Neve, W. SpliceRover: Interpretable Convolutional Neural Networks for Improved Splice Site Prediction. Bioinformatics 2018, 34, 4180–4188. [Google Scholar] [CrossRef]
- Kim, J.-S.; Gao, X.; Rzhetsky, A. RIDDLE: Race and Ethnicity Imputation from Disease History with Deep LEarning. PLoS Comput. Biol. 2018, 14, e1006106. [Google Scholar] [CrossRef]
- Kong, L.; Chen, Y.; Xu, F.; Xu, M.; Li, Z.; Fang, J.; Zhang, L.; Pian, C. Mining Influential Genes Based on Deep Learning. BMC Bioinform. 2021, 22, 27. [Google Scholar] [CrossRef]
- Chen, L.; Capra, J.A. Learning and Interpreting the Gene Regulatory Grammar in a Deep Learning Framework. PLoS Comput. Biol. 2020, 16, e1008334. [Google Scholar] [CrossRef]
- Elmarakeby, H.A.; Hwang, J.; Arafeh, R.; Crowdis, J.; Gang, S.; Liu, D.; AlDubayan, S.H.; Salari, K.; Kregel, S.; Richter, C.; et al. Biologically Informed Deep Neural Network for Prostate Cancer Discovery. Nature 2021, 598, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, S.M.; Lee, S.-I. A Unified Approach to Interpreting Model Predictions. In Proceedings of the Advances in Neural Information Processing Systems, Long Beach, CA, USA, 4–9 December 2017; Curran Associates, Inc.: Red Hook, NY, USA, 2017; Volume 30. [Google Scholar]
- Galdos, F.X.; Xu, S.; Goodyer, W.R.; Duan, L.; Huang, Y.V.; Lee, S.; Zhu, H.; Lee, C.; Wei, N.; Lee, D.; et al. DevCellPy Is a Machine Learning-Enabled Pipeline for Automated Annotation of Complex Multilayered Single-Cell Transcriptomic Data. Nat. Commun. 2022, 13, 5271. [Google Scholar] [CrossRef] [PubMed]
- Elbasir, A.; Mall, R.; Kunji, K.; Rawi, R.; Islam, Z.; Chuang, G.-Y.; Kolatkar, P.R.; Bensmail, H. BCrystal: An Interpretable Sequence-Based Protein Crystallization Predictor. Bioinformatics 2020, 36, 1429–1438. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Mu, Q.; Qiu, F.; Li, X.; Xu, W.; Yu, J.; Fu, W.; Cao, Y.; Wang, J. Machine Learning of Genomic Features in Organotropic Metastases Stratifies Progression Risk of Primary Tumors. Nat. Commun. 2021, 12, 6692. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Guan, Y. Asymmetric Predictive Relationships across Histone Modifications. Nat. Mach. Intell. 2022, 4, 288–299. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, C.; Wang, B.; Li, B.; Wang, Q.; Liu, D.; Wang, H.; Zhou, Y.; Shi, L.; Lan, F.; et al. Optimized CRISPR Guide RNA Design for Two High-Fidelity Cas9 Variants by Deep Learning. Nat. Commun. 2019, 10, 4284. [Google Scholar] [CrossRef]
- Camacho, D.M.; Collins, K.M.; Powers, R.K.; Costello, J.C.; Collins, J.J. Next-Generation Machine Learning for Biological Networks. Cell 2018, 173, 1581–1592. [Google Scholar] [CrossRef]
- Auslander, N.; Gussow, A.B.; Koonin, E.V. Incorporating Machine Learning into Established Bioinformatics Frameworks. Int. J. Mol. Sci. 2021, 22, 2903. [Google Scholar] [CrossRef]
- Yang, X.; Wang, W.; Ma, J.-L.; Qiu, Y.-L.; Lu, K.; Cao, D.-S.; Wu, C.-K. BioNet: A Large-Scale and Heterogeneous Biological Network Model for Interaction Prediction with Graph Convolution. Brief. Bioinform. 2022, 23, bbab491. [Google Scholar] [CrossRef]
- Peng, W.; Tang, Q.; Dai, W.; Chen, T. Improving Cancer Driver Gene Identification Using Multi-Task Learning on Graph Convolutional Network. Brief. Bioinform. 2022, 23, bbab432. [Google Scholar] [CrossRef]
- Chu, Y.; Wang, X.; Dai, Q.; Wang, Y.; Wang, Q.; Peng, S.; Wei, X.; Qiu, J.; Salahub, D.R.; Xiong, Y.; et al. MDA-GCNFTG: Identifying MiRNA-Disease Associations Based on Graph Convolutional Networks via Graph Sampling through the Feature and Topology Graph. Brief. Bioinform. 2021, 22, bbab165. [Google Scholar] [CrossRef] [PubMed]
- Ying, R.; Bourgeois, D.; You, J.; Zitnik, M.; Leskovec, J. GNNExplainer: Generating Explanations for Graph Neural Networks. arXiv 2019, arXiv:1903.03894. [Google Scholar]
- Carvalho, D.V.; Pereira, E.M.; Cardoso, J.S. Machine Learning Interpretability: A Survey on Methods and Metrics. Electronics 2019, 8, 832. [Google Scholar] [CrossRef]
- Wyatt, K.D.; Branda, M.E.; Anderson, R.T.; Pencille, L.J.; Montori, V.M.; Hess, E.P.; Ting, H.H.; LeBlanc, A. Peering into the Black Box: A Meta-Analysis of How Clinicians Use Decision Aids during Clinical Encounters. Implement. Sci. 2014, 9, 26. [Google Scholar] [CrossRef]
- Doshi-Velez, F.; Kim, B. Towards A Rigorous Science of Interpretable Machine Learning. arXiv 2017, arXiv:1702.08608. [Google Scholar]
- Vidyasagar, M. Machine Learning Methods in the Computational Biology of Cancer. Proc. R. Soc. A Math. Phys. Eng. Sci. 2014, 470, 20140081. [Google Scholar] [CrossRef] [PubMed]
- Danyi, A.; Jager, M.; de Ridder, J. Cancer Type Classification in Liquid Biopsies Based on Sparse Mutational Profiles Enabled through Data Augmentation and Integration. Life 2022, 12, 1. [Google Scholar] [CrossRef]
- Myers, M.A.; Zaccaria, S.; Raphael, B.J. Identifying Tumor Clones in Sparse Single-Cell Mutation Data. Bioinformatics 2020, 36, i186–i193. [Google Scholar] [CrossRef]
- Sason, I.; Chen, Y.; Leiserson, M.D.M.; Sharan, R. A Mixture Model for Signature Discovery from Sparse Mutation Data. Genome Med. 2021, 13, 173. [Google Scholar] [CrossRef]
- Ji, J.; He, D.; Feng, Y.; He, Y.; Xue, F.; Xie, L. JDINAC: Joint Density-Based Non-Parametric Differential Interaction Network Analysis and Classification Using High-Dimensional Sparse Omics Data. Bioinformatics 2017, 33, 3080–3087. [Google Scholar] [CrossRef]
- Xu, B.; Li, X.; Gao, X.; Jia, Y.; Liu, J.; Li, F.; Zhang, Z. DeNOPA: Decoding Nucleosome Positions Sensitively with Sparse ATAC-Seq Data. Brief. Bioinform. 2022, 23, bbab469. [Google Scholar] [CrossRef]
- Ramamoorthy, D.; Severson, K.; Ghosh, S.; Sachs, K.; Glass, J.D.; Fournier, C.N.; Herrington, T.M.; Berry, J.D.; Ng, K.; Fraenkel, E. Identifying Patterns in Amyotrophic Lateral Sclerosis Progression from Sparse Longitudinal Data. Nat. Comput. Sci. 2022, 2, 605–616. [Google Scholar] [CrossRef]
- Suresh, S.; Saraswathi, S.; Sundararajan, N. Performance Enhancement of Extreme Learning Machine for Multi-Category Sparse Data Classification Problems. Eng. Appl. Artif. Intell. 2010, 23, 1149–1157. [Google Scholar] [CrossRef]
- Ransohoff, D.F. Rules of Evidence for Cancer Molecular-Marker Discovery and Validation. Nat. Rev. Cancer 2004, 4, 309–314. [Google Scholar] [CrossRef]
- Fang, J. A Critical Review of Five Machine Learning-Based Algorithms for Predicting Protein Stability Changes upon Mutation. Brief. Bioinform. 2020, 21, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Giudice, G.; Petsalaki, E. Proteomics and Phosphoproteomics in Precision Medicine: Applications and Challenges. Brief. Bioinform. 2019, 20, 767–777. [Google Scholar] [CrossRef]
- Li, J.; Liu, L.; Le, T.D.; Liu, J. Accurate Data-Driven Prediction Does Not Mean High Reproducibility. Nat. Mach. Intell. 2020, 2, 13–15. [Google Scholar] [CrossRef]
- Kim, E.; Ilic, N.; Shrestha, Y.; Zou, L.; Kamburov, A.; Zhu, C.; Yang, X.; Lubonja, R.; Tran, N.; Nguyen, C.; et al. Systematic Functional Interrogation of Rare Cancer Variants Identifies Oncogenic Alleles. Cancer Discov. 2016, 6, 714–726. [Google Scholar] [CrossRef]
- Dogruluk, T.; Tsang, Y.H.; Espitia, M.; Chen, F.; Chen, T.; Chong, Z.; Appadurai, V.; Dogruluk, A.; Eterovic, A.K.; Bonnen, P.E.; et al. Identification of Variant-Specific Functions of PIK3CA by Rapid Phenotyping of Rare Mutations. Cancer Res. 2015, 75, 5341–5354. [Google Scholar] [CrossRef]
- Kumar, P.; Gangal, A.; Kumari, S. Prognosis of Breast Cancer by Implementing Machine Learning Algorithms Using Modified Bootstrap Aggregating. In Innovations in Computational Intelligence and Computer Vision; Sharma, M.K., Dhaka, V.S., Perumal, T., Dey, N., Tavares, J.M.R.S., Eds.; Springer: Singapore, 2021; pp. 561–569. [Google Scholar]
- Roth, H.R.; Lu, L.; Liu, J.; Yao, J.; Seff, A.; Cherry, K.; Kim, L.; Summers, R.M. Improving Computer-Aided Detection Using Convolutional Neural Networks and Random View Aggregation. IEEE Trans. Med. Imaging 2016, 35, 1170–1181. [Google Scholar] [CrossRef]
- Suthar, B.; Patel, H.; Goswami, A. A Survey: Classification of Imputation Methods in Data Mining. Int. J. Emerg. Technol. Adv. Eng. 2012, 2, 309–312. [Google Scholar]
- Houari, R.; Bounceur, A.; Tari, A.K.; Kecha, M.T. Handling Missing Data Problems with Sampling Methods. In Proceedings of the 2014 International Conference on Advanced Networking Distributed Systems and Applications, Bejaia, Algeria, 17–19 June 2014; pp. 99–104. [Google Scholar]
- Ayilara, O.F.; Zhang, L.; Sajobi, T.T.; Sawatzky, R.; Bohm, E.; Lix, L.M. Impact of Missing Data on Bias and Precision When Estimating Change in Patient-Reported Outcomes from a Clinical Registry. Health Qual. Life Outcomes 2019, 17, 106. [Google Scholar] [CrossRef] [PubMed]
- Ludbrook, J. Outlying Observations and Missing Values: How Should They Be Handled? Clin. Exp. Pharmacol. Physiol. 2008, 35, 670–678. [Google Scholar] [CrossRef]
- Langkamp, D.L.; Lehman, A.; Lemeshow, S. Techniques for Handling Missing Data in Secondary Analyses of Large Surveys. Acad. Pediatr. 2010, 10, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Donders, A.R.T.; van der Heijden, G.J.M.G.; Stijnen, T.; Moons, K.G.M. Review: A Gentle Introduction to Imputation of Missing Values. J. Clin. Epidemiol. 2006, 59, 1087–1091. [Google Scholar] [CrossRef] [PubMed]
- Baraldi, A.N.; Enders, C.K. An Introduction to Modern Missing Data Analyses. J. Sch. Psychol. 2010, 48, 5–37. [Google Scholar] [CrossRef]
- Graham, J.W. Missing Data Analysis: Making It Work in the Real World. Annu. Rev. Psychol. 2009, 60, 549–576. [Google Scholar] [CrossRef]
- Lin, J.; Li, N.; Alam, M.A.; Ma, Y. Data-Driven Missing Data Imputation in Cluster Monitoring System Based on Deep Neural Network. Appl. Intell. 2020, 50, 860–877. [Google Scholar] [CrossRef]
- Choudhury, A.; Kosorok, M.R. Missing Data Imputation for Classification Problems. arXiv 2020, arXiv:2002.10709. [Google Scholar]
- Khan, S.I.; Hoque, A.S.M.L. SICE: An Improved Missing Data Imputation Technique. J. Big Data 2020, 7, 37. [Google Scholar] [CrossRef]
- Al-Helali, B.; Chen, Q.; Xue, B.; Zhang, M. A New Imputation Method Based on Genetic Programming and Weighted KNN for Symbolic Regression with Incomplete Data. Soft. Comput. 2021, 25, 5993–6012. [Google Scholar] [CrossRef]
- Peng, D.; Zou, M.; Liu, C.; Lu, J. RESI: A Region-Splitting Imputation Method for Different Types of Missing Data. Expert Syst. Appl. 2021, 168, 114425. [Google Scholar] [CrossRef]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Zhao, L.; Shi, Y.; Zhang, R.; Long, Q.; Bai, S.; Luo, Q.; Lin, Y.; Zou, X.; Ghazanfar, S.; et al. Clonal Evolution in Liver Cancer at Single-Cell and Single-Variant Resolution. J. Hematol. Oncol. 2021, 14, 22. [Google Scholar] [CrossRef] [PubMed]
- Biermann, J.; Parris, T.Z.; Nemes, S.; Danielsson, A.; Engqvist, H.; Werner Rönnerman, E.; Forssell-Aronsson, E.; Kovács, A.; Karlsson, P.; Helou, K. Clonal Relatedness in Tumour Pairs of Breast Cancer Patients. Breast Cancer Res. 2018, 20, 96. [Google Scholar] [CrossRef]
- Hu, Z.; Li, Z.; Ma, Z.; Curtis, C. Multi-Cancer Analysis of Clonality and the Timing of Systemic Spread in Paired Primary Tumors and Metastases. Nat. Genet. 2020, 52, 701–708. [Google Scholar] [CrossRef]
- Wang, E.; Zou, J.; Zaman, N.; Beitel, L.K.; Trifiro, M.; Paliouras, M. Cancer Systems Biology in the Genome Sequencing Era: Part 2, Evolutionary Dynamics of Tumor Clonal Networks and Drug Resistance. Semin. Cancer Biol. 2013, 23, 286–292. [Google Scholar] [CrossRef]
- Zare, H.; Wang, J.; Hu, A.; Weber, K.; Smith, J.; Nickerson, D.; Song, C.; Witten, D.; Blau, C.A.; Noble, W.S. Inferring Clonal Composition from Multiple Sections of a Breast Cancer. PLoS Comput. Biol. 2014, 10, e1003703. [Google Scholar] [CrossRef]
- Ha, G.; Roth, A.; Khattra, J.; Ho, J.; Yap, D.; Prentice, L.M.; Melnyk, N.; McPherson, A.; Bashashati, A.; Laks, E.; et al. TITAN: Inference of Copy Number Architectures in Clonal Cell Populations from Tumor Whole-Genome Sequence Data. Genome Res. 2014, 24, 1881–1893. [Google Scholar] [CrossRef]
- Roth, A.; Khattra, J.; Yap, D.; Wan, A.; Laks, E.; Biele, J.; Ha, G.; Aparicio, S.; Bouchard-Côté, A.; Shah, S.P. PyClone: Statistical Inference of Clonal Population Structure in Cancer. Nat. Methods 2014, 11, 396–398. [Google Scholar] [CrossRef]
- Chkhaidze, K.; Heide, T.; Werner, B.; Williams, M.J.; Huang, W.; Caravagna, G.; Graham, T.A.; Sottoriva, A. Spatially Constrained Tumour Growth Affects the Patterns of Clonal Selection and Neutral Drift in Cancer Genomic Data. PLoS Comput. Biol. 2019, 15, e1007243. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.K.; De, S. An Assessment of Computational Methods for Estimating Purity and Clonality Using Genomic Data Derived from Heterogeneous Tumor Tissue Samples. Brief. Bioinform. 2015, 16, 232–241. [Google Scholar] [CrossRef] [PubMed]




| Category | Descriptive Mutational Process | Clinical Use |
|---|---|---|
| Clinically relevant DDR pathways | Homologous recombination (HR) | Biomarker for PARP-inhibitor sensitivity [64,65,66] |
| Biomarker for platinum-treatment sensitivity [67] | ||
| Biomarker for ATRi-inhibitor sensitivity [71,73,74,75] | ||
| Mismatch repair (MMR) | Immune-checkpoint-inhibitor biomarker [77] | |
| Identification of Werner-helicase-sensitive patients [78,82,83] | ||
| Potential biomarker for antitumor immune activation [84] | ||
| Nucleotide excision repair (NER) | Biomarker for platinum-treatment sensitivity [34,85] | |
| Biomarker of ERCC2 deficiency [34,85] | ||
| Proofreading errors | Biomarker of POLE deficiency [86,87] | |
| Characterization of clinically relevant phenomena | Radiation treatment | Identification of radiation-driver tumors [53] |
| Identification of genes with potential contra-indications of radiation therapy [54,88] | ||
| Chemotherapy | Tumorigenic effects of 5-FU [88,89] | |
| Tumorigenic effects of platinum and capecitabine treatments | ||
| Environmental | Screening for aristolochic-acid damage [90,91,92] | |
| Alcohol-consumption signatures across cancers [93,94,95,96] | ||
| Cancer-type specific mutagenesis | Identification of different subtypes of esophageal cancer [97] | |
| Identification of secondary tumors of unknown origin [98] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patterson, A.; Elbasir, A.; Tian, B.; Auslander, N. Computational Methods Summarizing Mutational Patterns in Cancer: Promise and Limitations for Clinical Applications. Cancers 2023, 15, 1958. https://doi.org/10.3390/cancers15071958
Patterson A, Elbasir A, Tian B, Auslander N. Computational Methods Summarizing Mutational Patterns in Cancer: Promise and Limitations for Clinical Applications. Cancers. 2023; 15(7):1958. https://doi.org/10.3390/cancers15071958
Chicago/Turabian StylePatterson, Andrew, Abdurrahman Elbasir, Bin Tian, and Noam Auslander. 2023. "Computational Methods Summarizing Mutational Patterns in Cancer: Promise and Limitations for Clinical Applications" Cancers 15, no. 7: 1958. https://doi.org/10.3390/cancers15071958
APA StylePatterson, A., Elbasir, A., Tian, B., & Auslander, N. (2023). Computational Methods Summarizing Mutational Patterns in Cancer: Promise and Limitations for Clinical Applications. Cancers, 15(7), 1958. https://doi.org/10.3390/cancers15071958