
Extracellular Vesicles Act as Carriers for Cargo Delivery and Regulate Wnt Signaling in the Hepatocellular Carcinoma Tumor Microenvironment
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Hepatocellular Carcinoma (HCC)
1.2. Extracellular Vesicles (EVs) and Tumor Microenvironment (TME)
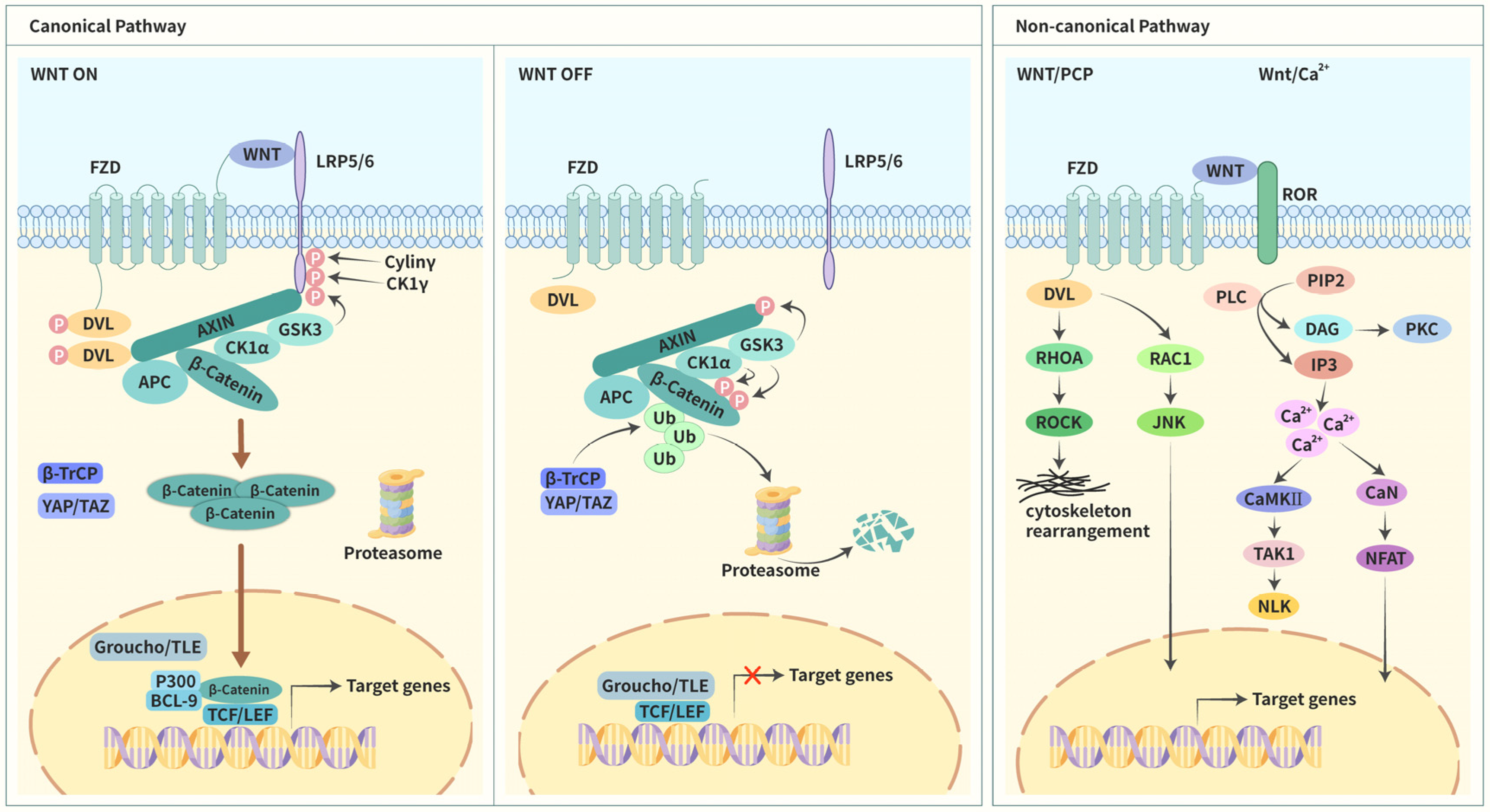
1.3. Wnt Signaling
1.3.1. Canonical Pathway
1.3.2. Noncanonical Pathways
2. EVs Regulate Wnt Signaling in HCC
2.1. Evs Regulate Wnt Signaling via Transport between Different Types of HCC Cells

{kind=link}
{kind=link}
| Molecules | Origin | Recipient Cells | Effects on Cell Behaviors | Mechanisms | References |
|---|---|---|---|---|---|
| MiR-1273f | Hypoxic HCC cells | Normoxic HCC cells | Promote proliferation, migration, invasion, and EMT | Upregulate LHX6/β-catenin | [70] |
| MiR-25 | High-metastatic HCC cells | Low-metastatic HCC cells | Reduce apoptosis and promote proliferation, migration, and invasion | Upregulate SIK1/β-catenin | [78] |
| DDX55 | HCC cells | HCC cells | Promote cell cycle progression and EMT. | Upregulate PI3K/Akt/GSK-3β/β-catenin | [79] |
| Endothelial cells | Promote angiogenesis | Upregulate PI3K/Akt/GSK-3β/β-catenin | [79] | ||
| PIgR | Advanced HCC cells | Early HCC cells | Acquire stem cell properties and promote migration and invasion | Upregulate PDK1/Akt/GSK-3β/β-catenin | [80] |
| Not mentioned | High-metastatic HCC cells | Low-metastatic HCC cells | Promote proliferation and migration | Upregulate Akt/GSK-3β/β-catenin and JNK1/2 signaling | [82] |
2.2. EVs Modulate Wnt Signaling through Transport between HCC and Stromal Cells and between Different Types of Stromal Cells
| Molecules | Origin | Recipient Cells | Effects on Cell Behaviors | Mechanisms | References |
|---|---|---|---|---|---|
| Gremlin 1 | CAFs | HCC cells | Promote migration, invasion, and EMT and reduce sensitivity to sorafenib | Upregulate Wnt/β-catenin | [83] |
| miR-320a | CAFs | HCC cells | Inhibit proliferation | Downregulate Wnt/β-catenin | [84,85] |
| MiR-92a-2-5p | M2 macrophages | HCC cells | Promote invasion | Upregulate PHLPP/p-AKT/β-catenin | [88] |
| MiR-21-5p | M2 macrophages | CD8+ T cells | Induce exhaustion and reduce intratumoral immune infiltration | Upregulate YAP/β-catenin | [92] |
2.3. EVs Modulate β-Catenin Signaling in HCC by Altering the Cellular Localization of β-Catenin
3. EVs-Based Biological Nanoparticles Modulate Wnt Signaling in HCC Cells
4. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wong, C.-M.; Tsang, F.H.-C.; Ng, I.O.-L. Non-coding RNAs in hepatocellular carcinoma: Molecular functions and pathological implications. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Affo, S.; Yu, L.-X.; Schwabe, R.F. The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 153–186. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, E.; Sarkar, D. Emerging Therapies for Hepatocellular Carcinoma (HCC). Cancers 2022, 14, 2798. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Ward, E.M.; Johnson, C.J.; Cronin, K.A.; Ma, J.; Ryerson, A.B.; Mariotto, A.; Lake, A.J.; Wilson, R.; Sherman, R.L.; et al. Annual Report to the Nation on the Status of Cancer, 1975–2014, Featuring Survival. Gynecol. Oncol. 2017, 109, djx030. [Google Scholar] [CrossRef] [Green Version]
- Shiani, A.; Narayanan, S.; Pena, L.; Friedman, M. The Role of Diagnosis and Treatment of Underlying Liver Disease for the Prognosis of Primary Liver Cancer. Cancer Control. 2017, 24, 073274817729240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, P. The Nature and Significance of Platelet Products in Human Plasma. Br. J. Haematol. 1967, 13, 269–288. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Yu, D.; Li, Y.; Wang, M.; Gu, J.; Xu, W.; Cai, H.; Fang, X.; Zhang, X. Exosomes as a new frontier of cancer liquid biopsy. Mol. Cancer 2022, 21, 56. [Google Scholar] [CrossRef]
- Tricarico, C.; Clancy, J.; D’Souza-Schorey, C. Biology and biogenesis of shed microvesicles. Small GTPases 2016, 8, 220–232. [Google Scholar] [CrossRef] [Green Version]
- Harding, C.; Heuser, J.; Stahl, P. Endocytosis and intracellular processing of transferrin and colloidal gold-transferrin in rat reticulocytes: Demonstration of a pathway for receptor shedding. Eur. J. Cell Biol. 1984, 35, 256–263. [Google Scholar]
- Pan, B.T.; Teng, K.; Wu, C.; Adam, M.; Johnstone, R.M. Electron microscopic evidence for externalization of the transferrin receptor in vesicular form in sheep reticulocytes. J. Cell Biol. 1985, 101, 942–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yáñez-Mó, M.; Siljander, P.R.-M.; Andreu, Z.; Bedina Zavec, A.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabhan, J.F.; Hu, R.; Oh, R.S.; Cohen, S.N.; Lu, Q. Formation and release of arrestin domain-containing protein 1-mediated microvesicles (ARMMs) at plasma membrane by recruitment of TSG101 protein. Proc. Natl. Acad. Sci. USA 2012, 109, 4146–4151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnstone, R.M.; Adam, M.; Hammond, J.R.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar] [CrossRef]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Lo Cicero, A.; Stahl, P.D.; Raposo, G. Extracellular vesicles shuffling intercellular messages: For good or for bad. Curr. Opin. Cell Biol. 2015, 35, 69–77. [Google Scholar] [CrossRef]
- Słomka, A.; Mocan, T.; Wang, B.; Nenu, I.; Urban, S.K.; Gonzalez-Carmona, M.A.; Schmidt-Wolf, I.G.H.; Lukacs-Kornek, V.; Strassburg, C.P.; Spârchez, Z.; et al. EVs as Potential New Therapeutic Tool/Target in Gastrointestinal Cancer and HCC. Cancers 2020, 12, 3019. [Google Scholar] [CrossRef]
- Lee, Y.-T.; Tran, B.; Wang, J.; Liang, I.; You, S.; Zhu, Y.; Agopian, V.; Tseng, H.-R.; Yang, J. The Role of Extracellular Vesicles in Disease Progression and Detection of Hepatocellular Carcinoma. Cancers 2021, 13, 3076. [Google Scholar] [CrossRef]
- Von Felden, J.; Garcia-Lezana, T.; Dogra, N.; Gonzalez-Kozlova, E.; Ahsen, M.E.; Craig, A.; Gifford, S.; Wunsch, B.; Smith, J.T.; Kim, S.; et al. Unannotated small RNA clusters associated with circulating extracellular vesicles detect early stage liver cancer. Gut 2021, 71, 2069–2080. [Google Scholar] [CrossRef]
- Urban, S.K.; Sanger, H.; Krawczyk, M.; Julich-Haertel, H.; Willms, A.; Ligocka, J.; Azkargorta, M.; Mocan, T.; Kahlert, C.; Kruk, B.; et al. Synergistic effects of extracellular vesicle phenotyping and AFP in hepatobiliary cancer differentiation. Liver Int. 2020, 40, 3103–3116. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Cheah, Y.K. The Interplay between MicroRNAs and Cellular Components of Tumour Microenvironment (TME) on Non-Small-Cell Lung Cancer (NSCLC) Progression. J. Immunol. Res. 2019, 2019, 3046379. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Minciacchi, V.R.; Freeman, M.R.; Di Vizio, D. Extracellular Vesicles in Cancer: Exosomes, Microvesicles and the Emerging Role of Large Oncosomes. Semin. Cell Dev. Biol. 2015, 40, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Jahan, S.; Mukherjee, S.; Ali, S.; Bhardwaj, U.; Choudhary, R.K.; Balakrishnan, S.; Naseem, A.; Mir, S.A.; Banawas, S.; Alaidarous, M.; et al. Pioneer Role of Extracellular Vesicles as Modulators of Cancer Initiation in Progression, Drug Therapy, and Vaccine Prospects. Cells 2022, 11, 490. [Google Scholar] [CrossRef]
- Yang, J.; Chen, J.; Liang, H.; Yu, Y. Nasopharyngeal cancer cell-derived exosomal PD-L1 inhibits CD8 + T-cell activity and promotes immune escape. Cancer Sci. 2022, 113, 3044–3054. [Google Scholar] [CrossRef]
- Moon, B.; Chang, S. Exosome as a Delivery Vehicle for Cancer Therapy. Cells 2022, 11, 316. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R. Wingless, a new mutant in D. melanogaster. Drosoph. Inf. Serv. 1973, 50, 134. [Google Scholar]
- Nüsslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef]
- Sharma, R.; Chopra, V. Effect of the wingless (wg1) mutation on wing and haltere development in Drosophila melanogaster. Dev. Biol. 1976, 48, 461–465. [Google Scholar] [CrossRef]
- Nusse, R.; Varmus, H.E. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell 1982, 31, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; van Ooyen, A.; Cox, D.; Fung, Y.K.T.; Varmus, H. Mode of proviral activation of a putative mammary oncogene (int-1) on mouse chromosome 15. Nature 1984, 307, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, A.S.; Grosschedl, R.; Guzman, R.C.; Parslow, T.; Varmus, H.E. Expression of the int-1 gene in transgenic mice is associated with mammary gland hyperplasia and adenocarcinomas in male and female mice. Cell 1988, 55, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Rijsewijk, F.; Schuermann, M.; Wagenaar, E.; Parren, P.; Weigel, D.; Nusse, R. The Drosophila homology of the mouse mammary oncogene int-1 is identical to the segment polarity gene wingless. Cell 1987, 50, 649–657. [Google Scholar] [CrossRef]
- McMahon, A.P.; Moon, R.T. Ectopic expression of the proto-oncogene int-1 in Xenopus embryos leads to duplication of the embryonic axis. Cell 1989, 58, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.W.; Nilbert, M.C.; Su, L.-K.; Vogelstein, B.; Bryan, T.M.; Levy, D.B.; Smith, K.J.; Preisinger, A.C.; Hedge, P.; McKechnie, D.; et al. Identification of FAP Locus Genes from Chromosome 5q21. Science 1991, 253, 661–665. [Google Scholar] [CrossRef]
- Nishisho, I.; Nakamura, Y.; Miyoshi, Y.; Miki, Y.; Ando, H.; Horii, A.; Koyama, K.; Utsunomiya, J.; Baba, S.; Hedge, P.; et al. Mutations of Chromosome 5q21 Genes in FAP and Colorectal Cancer Patients. Science 1991, 253, 665–669. [Google Scholar] [CrossRef]
- Rubinfeld, B.; Souza, B.; Albert, I.; Müller, O.; Chamberlain, S.H.; Masiarz, F.R.; Munemitsu, S.; Polakis, P. Association of the APC Gene Product with β-Catenin. Science 1993, 262, 1731–1734. [Google Scholar] [CrossRef]
- Su, L.-K.; Vogelstein, B.; Kinzler, K.W. Association of the APC Tumor Suppressor Protein with Catenins. Science 1993, 262, 1734–1737. [Google Scholar] [CrossRef]
- Korinek, V.; Barker, N.; Morin, P.J.; van Wichen, D.; de Weger, R.; Kinzler, K.W.; Vogelstein, B.; Clevers, H. Constitutive Transcriptional Activation by a β-Catenin-Tcf Complex in APC −/− Colon Carcinoma. Science 1997, 275, 1784–1787. [Google Scholar] [CrossRef] [Green Version]
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [Green Version]
- Steinhart, Z.; Angers, S. Wnt signaling in development and tissue homeostasis. Development 2018, 145, dev146589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pai, S.G.; Carneiro, B.A.; Mota, J.M.; Costa, R.; Leite, C.A.; Barroso-Sousa, R.; Kaplan, J.B.; Chae, Y.K.; Giles, F.J. Wnt/beta-catenin pathway: Modulating anticancer immune response. J. Hematol. Oncol. 2017, 10, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajos-Michniewicz, A.; Czyz, M. WNT Signaling in Melanoma. Int. J. Mol. Sci. 2020, 21, 4852. [Google Scholar] [CrossRef]
- Masuda, T.; Ishitani, T. Context-dependent regulation of the β-catenin transcriptional complex supports diverse functions of Wnt/β-catenin signaling. J. Biochem. 2016, 161, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Taciak, B.; Pruszynska, I.; Kiraga, L.; Bialasek, M.; Krol, M. Wnt signaling pathway in development and cancer. J. Physiol. Pharmacol. 2018, 69, 185–196. [Google Scholar] [CrossRef]
- Polakis, P. Wnt Signaling in Cancer. Cold Spring Harb. Perspect. Biol. 2012, 4, a008052. [Google Scholar] [CrossRef] [Green Version]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V.; et al. YAP/TAZ Incorporation in the β-Catenin Destruction Complex Orchestrates the Wnt Response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef] [Green Version]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uzdensky, A.; Demyanenko, S.; Bibov, M. Signal Transduction in Human Cutaneous Melanoma and Target Drugs. Curr. Cancer Drug Targets 2013, 13, 843–866. [Google Scholar] [CrossRef] [PubMed]
- Lien, W.-H.; Fuchs, E. Wnt some lose some: Transcriptional governance of stem cells by Wnt/β-catenin signaling. Genes Dev. 2014, 28, 1517–1532. [Google Scholar] [CrossRef] [Green Version]
- Kretzschmar, K.; Clevers, H. Wnt/β-catenin signaling in adult mammalian epithelial stem cells. Dev. Biol. 2017, 428, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Virshup, D.M. Wnt Signaling and Drug Resistance in Cancer. Mol. Pharmacol. 2020, 97, 72–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-E.; Huang, H.; Zhao, M.; Zhang, X.; Zhang, A.; Semonov, M.V.; MacDonald, B.T.; Zhang, X.; Abreu, J.G.; Peng, L.; et al. Wnt Stabilization of β-Catenin Reveals Principles for Morphogen Receptor-Scaffold Assemblies. Science 2013, 340, 867–870. [Google Scholar] [CrossRef] [Green Version]
- Willert, K.; Shibamoto, S.; Nusse, R. Wnt-induced dephosphorylation of Axin releases beta -catenin from the Axin complex. Genes Dev. 1999, 13, 1768–1773. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, S.; Kishida, S.; Yamamoto, H.; Murai, H.; Koyama, S.; Kikuchi, A. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J. 1998, 17, 1371–1384. [Google Scholar] [CrossRef]
- Goswami, V.G.; Patel, B.D. Recent updates on Wnt signaling modulators: A patent review (2014–2020). Expert Opin. Ther. Patents 2021, 31, 1009–1043. [Google Scholar] [CrossRef]
- Ghosh, N.; Hossain, U.; Mandal, A.; Sil, P.C. The Wnt signaling pathway: A potential therapeutic target against cancer. Ann. N. Y. Acad. Sci. 2019, 1443, 54–74. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2016, 36, 1461–1473. [Google Scholar] [CrossRef]
- James, R.G.; Conrad, W.H.; Moon, R.T. β-Catenin-Independent Wnt Pathways: Signals, Core Proteins, and Effectors. Methods Mol. Biol. 2008, 468, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Marinou, K.; Christodoulides, C.; Antoniades, C.; Koutsilieris, M. Wnt signaling in cardiovascular physiology. Trends Endocrinol. Metab. 2012, 23, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Farb, M.G.; Karki, S.; Park, S.-Y.; Saggese, S.M.; Carmine, B.; Hess, D.T.; Apovian, C.; Fetterman, J.L.; Bretón-Romero, R.; Hamburg, N.; et al. WNT5A-JNK regulation of vascular insulin resistance in human obesity. Vasc. Med. 2016, 21, 489–496. [Google Scholar] [CrossRef] [Green Version]
- Ackers, I.; Malgor, R. Interrelationship of canonical and non-canonical Wnt signalling pathways in chronic metabolic diseases. Diabetes Vasc. Dis. Res. 2017, 15, 3–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, C.M.R.; Chan, C.K.; Veltri, A.; Lien, W.-H. Wnt Signaling Pathways in Keratinocyte Carcinomas. Cancers 2019, 11, 1216. [Google Scholar] [CrossRef] [Green Version]
- Luga, V.; Zhang, L.; Viloria-Petit, A.M.; Ogunjimi, A.A.; Inanlou, M.R.; Chiu, E.; Buchanan, M.; Hosein, A.N.; Basik, M.; Wrana, J.L. Exosomes Mediate Stromal Mobilization of Autocrine Wnt-PCP Signaling in Breast Cancer Cell Migration. Cell 2012, 151, 1542–1556. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.-J.; Sun, S.-G.; Tang, X.-Y.; Lin, Y.-Y.; Hua, K.-Q. Extracellular vesicular Wnt7b mediates HPV E6-induced cervical cancer angiogenesis by activating the β-catenin signaling pathway. J. Exp. Clin. Cancer Res. 2020, 39, 1–17. [Google Scholar] [CrossRef]
- Azad, M.B.; Chen, Y.; Henson, E.S.; Cizeau, J.; McMillan-Ward, E.; Israels, S.J.; Gibson, S.B. Hypoxia induces autophagic cell death in apoptosis-competent cells through a mechanism involving BNIP3. Autophagy 2008, 4, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Henze, A.-T.; Mazzone, M. The impact of hypoxia on tumor-associated macrophages. J. Clin. Investig. 2016, 126, 3672–3679. [Google Scholar] [CrossRef]
- Yu, Y.; Min, Z.; Zhihang, Z.; Linhong, M.; Tao, R.; Yan, L.; Song, H. Hypoxia-induced exosomes promote hepatocellular carcinoma proliferation and metastasis via miR-1273f transfer. Exp. Cell Res. 2019, 385, 111649. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Xie, L. LHX6 inhibits breast cancer cell proliferation and invasion via repression of the Wnt/β-catenin signaling pathway. Mol. Med. Rep. 2015, 12, 4634–4639. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Cai, K.; Sun, K.; Gui, J.; Liang, J. MiR-1290 promotes proliferation, migration, and invasion of glioma cells by targeting LHX6. J. Cell. Physiol. 2017, 233, 6621–6629. [Google Scholar] [CrossRef]
- Yang, J.; Han, F.; Liu, W.; Zhang, M.; Huang, Y.; Hao, X.; Jiang, X.; Yin, L.; Chen, H.; Cao, J.; et al. LHX6, An Independent Prognostic Factor, Inhibits Lung Adenocarcinoma Progression through Transcriptional Silencing of β-catenin. J. Cancer 2017, 8, 2561–2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Feng, X.; Liu, H.; Tong, R.; Wu, J.; Li, C.; Yu, H.; Chen, Y.; Cheng, Q.; Chen, J.; et al. High-metastatic cancer cells derived exosomal miR92a-3p promotes epithelial-mesenchymal transition and metastasis of low-metastatic cancer cells by regulating PTEN/Akt pathway in hepatocellular carcinoma. Oncogene 2020, 39, 6529–6543. [Google Scholar] [CrossRef]
- Li, R.; Wang, Y.; Zhang, X.; Feng, M.; Ma, J.; Li, J.; Yang, X.; Fang, F.; Xia, Q.; Zhang, Z.; et al. Exosome-mediated secretion of LOXL4 promotes hepatocellular carcinoma cell invasion and metastasis. Mol. Cancer 2019, 18, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Chen, G.; Lin, X.; Xing, X.; Cai, Z.; Liu, X.; Liu, J. Role of exosomes in hepatocellular carcinoma cell mobility alteration. Oncol. Lett. 2017, 14, 8122–8131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.; Tang, Y.; Wu, W.; Ouyang, Y.; Tan, D.; Huang, Y. Exosomal microRNA-25 released from cancer cells targets SIK1 to promote hepatocellular carcinoma tumorigenesis. Dig. Liver Dis. 2021, 54, 954–963. [Google Scholar] [CrossRef]
- Yu, B.; Zhou, S.; Long, D.; Ning, Y.; Yao, H.; Zhou, E.; Wang, Y. DDX55 promotes hepatocellular carcinoma progression by interacting with BRD4 and participating in exosome-mediated cell-cell communication. Cancer Sci. 2022, 113, 3002–3017. [Google Scholar] [CrossRef]
- Tey, S.K.; Wong, S.W.K.; Chan, J.Y.T.; Mao, X.; Ng, T.H.; Yeung, C.L.S.; Leung, Z.; Fung, H.L.; Tang, A.H.N.; Wong, D.K.H.; et al. Patient pIgR-enriched extracellular vesicles drive cancer stemness, tumorigenesis and metastasis in hepatocellular carcinoma. J. Hepatol. 2021, 76, 883–895. [Google Scholar] [CrossRef]
- Dall’Olio, F.; Chiricolo, M. Sialyltransferases in cancer. Glycoconj. J. 2001, 18, 841–850. [Google Scholar] [CrossRef]
- Wang, L.; Chen, X.; Wang, L.; Wang, S.; Li, W.; Liu, Y.; Zhang, J. Knockdown of ST6Gal-I expression in human hepatocellular carcinoma cells inhibits their exosome-mediated proliferation- and migration-promoting effects. IUBMB Life 2021, 73, 1378–1391. [Google Scholar] [CrossRef]
- Qin, W.; Wang, L.; Tian, H.; Wu, X.; Xiao, C.; Pan, Y.; Fan, M.; Tai, Y.; Liu, W.; Zhang, Q.; et al. CAF-derived exosomes transmitted Gremlin-1 promotes cancer progression and decreases the sensitivity of hepatoma cells to sorafenib. Mol. Carcinog. 2022, 61, 764–775. [Google Scholar] [CrossRef]
- Lu, C.; Liao, Z.; Cai, M.; Zhang, G. MicroRNA-320a downregulation mediates human liver cancer cell proliferation through the Wnt/β-catenin signaling pathway. Oncol. Lett. 2016, 13, 573–578. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Li, X.; Sun, W.; Yue, S.; Yang, J.; Li, J.; Ma, B.; Wang, J.; Yang, X.; Pu, M.; et al. Loss of exosomal miR-320a from cancer-associated fibroblasts contributes to HCC proliferation and metastasis. Cancer Lett. 2017, 397, 33–42. [Google Scholar] [CrossRef]
- Gordon, S.; Martinez, F.O. Alternative Activation of Macrophages: Mechanism and Functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [Green Version]
- Talmadge, J.E.; Donkor, M.; Scholar, E. Inflammatory cell infiltration of tumors: Jekyll or Hyde. Cancer Metastasis Rev. 2007, 26, 373–400. [Google Scholar] [CrossRef]
- Liu, G.; Ouyang, X.; Sun, Y.; Xiao, Y.; You, B.; Gao, Y.; Yeh, S.; Li, Y.; Chang, C. The miR-92a-2-5p in exosomes from macrophages increases liver cancer cells invasion via altering the AR/PHLPP/p-AKT/β-catenin signaling. Cell Death Differ. 2020, 27, 3258–3272. [Google Scholar] [CrossRef]
- Zheng, C.; Zheng, L.; Yoo, J.-K.; Guo, H.; Zhang, Y.; Guo, X.; Kang, B.; Hu, R.; Huang, J.Y.; Zhang, Q.; et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017, 169, 1342–1356. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Zheng, B.; Goswami, S.; Meng, L.; Zhang, D.; Cao, C.; Li, T.; Zhu, F.; Ma, L.; Zhang, Z.; et al. PD1Hi CD8+ T cells correlate with exhausted signature and poor clinical outcome in hepatocellular carcinoma. J. Immunother. Cancer 2019, 7, 1–15. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Pu, J.; Xu, Z.; Nian, J.; Fang, Q.; Yang, M.; Huang, Y.; Li, W.; Bin Ge, B.; Wang, J.; Wei, H. M2 macrophage-derived extracellular vesicles facilitate CD8+T cell exhaustion in hepatocellular carcinoma via the miR-21-5p/YOD1/YAP/β-catenin pathway. Cell Death Discov. 2021, 7, 182. [Google Scholar] [CrossRef]
- Sehrawat, S.; Koenig, P.-A.; Kirak, O.; Schlieker, C.; Fankhauser, M.; Ploegh, H.L. A catalytically inactive mutant of the deubiquitylase YOD-1 enhances antigen cross-presentation. Blood 2013, 121, 1145–1156. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Lu, X.; Dey, P.; Deng, P.; Wu, C.C.; Jiang, S.; Fang, Z.; Zhao, K.; Konaparthi, R.; Hua, S.; et al. Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov. 2016, 6, 80–95. [Google Scholar] [CrossRef] [Green Version]
- Murakami, S.; Shahbazian, D.; Surana, R.; Zhang, W.; Chen, H.; Graham, G.T.; White, S.M.; Weiner, L.M.; Yi, C. Yes-associated protein mediates immune reprogramming in pancreatic ductal adenocarcinoma. Oncogene 2017, 36, 1232–1244. [Google Scholar] [CrossRef] [Green Version]
- Heuberger, J.; Birchmeier, W. Interplay of Cadherin-Mediated Cell Adhesion and Canonical Wnt Signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002915. [Google Scholar] [CrossRef]
- McCrea, P.D.; Maher, M.T.; Gottardi, C.J. Nuclear Signaling from Cadherin Adhesion Complexes. Curr. Top. Dev. Biol. 2015, 112, 129–196. [Google Scholar] [CrossRef] [Green Version]
- Han, Q.; Lv, L.; Wei, J.; Lei, X.; Lin, H.; Li, G.; Cao, J.; Xie, J.; Yang, W.; Wu, S.; et al. Vps4A mediates the localization and exosome release of β-catenin to inhibit epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2019, 457, 47–59. [Google Scholar] [CrossRef]
- Yang, W.; Wei, J.; Lv, L.; Xie, J.; Li, A.; Zheng, Z.; Zhuo, W.; Liu, P.; Min, J. p62 Promotes Malignancy of Hepatocellular Carcinoma by Regulating the Secretion of Exosomes and the Localization of β-Catenin. Front. Biosci. 2022, 27, 89. [Google Scholar] [CrossRef]
- Bahmani, L.; Ullah, M. Different Sourced Extracellular Vesicles and Their Potential Applications in Clinical Treatments. Cells 2022, 11, 1989. [Google Scholar] [CrossRef]
- Greish, K. Enhanced permeability and retention of macromolecular drugs in solid tumors: A royal gate for targeted anticancer nanomedicines. J. Drug Target. 2007, 15, 457–464. [Google Scholar] [CrossRef]
- Matsuda, A.; Ishiguro, K.; Yan, I.K.; Patel, T. Extracellular Vesicle-Based Therapeutic Targeting of β-Catenin to Modulate Anticancer Immune Responses in Hepatocellular Cancer. Hepatol. Commun. 2019, 3, 525–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, T.; Budhu, A.; Forgues, M.; Wang, X.W. Activation of Hepatic Stem Cell Marker EpCAM by Wnt–β-Catenin Signaling in Hepatocellular Carcinoma. Cancer Res. 2007, 67, 10831–10839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Farmer, R.W.; Yang, Y.; Martin, R.C.G. Epithelial cell adhesion molecule in human hepatocellular carcinoma cell lines: A target of chemoresistence. BMC Cancer 2016, 16, 228. [Google Scholar] [CrossRef] [Green Version]
- Katoh, M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: Cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review). Int. J. Oncol. 2017, 51, 1357–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishiguro, K.; Yan, I.K.; Lewis-Tuffin, L.; Patel, T. Targeting Liver Cancer Stem Cells Using Engineered Biological Nanoparticles for the Treatment of Hepatocellular Cancer. Hepatol. Commun. 2020, 4, 298–313. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Ye, M.; Zhang, Y.; Li, W.; Li, M.; Wang, H.; Qiu, X.; Xu, Y.; Yin, J.; Hu, Q.; et al. MicroRNA-375-3p enhances chemosensitivity to 5-fluorouracil by targeting thymidylate synthase in colorectal cancer. Cancer Sci. 2020, 111, 1528–1541. [Google Scholar] [CrossRef]
- Yang, D.; Yan, R.; Zhang, X.; Zhu, Z.; Wang, C.; Liang, C.; Zhang, X. Deregulation of MicroRNA-375 inhibits cancer proliferation migration and chemosensitivity in pancreatic cancer through the association of HOXB3. Am. J. Transl. Res. 2016, 8, 1551–1559. [Google Scholar] [PubMed]
- Fu, H.; Fu, L.; Xie, C.; Zuo, W.-S.; Liu, Y.-S.; Zheng, M.-Z.; Yu, J.-M. miR-375 inhibits cancer stem cell phenotype and tamoxifen resistance by degrading HOXB3 in human ER-positive breast cancer. Oncol. Rep. 2017, 37, 1093–1099. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Liu, J.; Fan, Q.; Yu, J.; Ren, X.; Wang, X. Extracellular Vesicle-Encapsulated MicroRNA-375 from Bone Marrow-Derived Mesenchymal Stem Cells Inhibits Hepatocellular Carcinoma Progression through Regulating HOXB3-Mediated Wnt/β-Catenin Pathway. Anal. Cell. Pathol. 2022, 2022, 9302496. [Google Scholar] [CrossRef]
- Golpanian, S.; Wolf, A.; Hatzistergos, K.E.; Hare, J.M. Rebuilding the Damaged Heart: Mesenchymal Stem Cells, Cell-Based Therapy, and Engineered Heart Tissue. Physiol. Rev. 2016, 96, 1127–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamizo, A.; Marini, F.; Amano, T.; Khan, A.; Studeny, M.; Gumin, J.; Chen, J.; Hentschel, S.; Vecil, G.; Dembinski, J.; et al. Human Bone Marrow–Derived Mesenchymal Stem Cells in the Treatment of Gliomas. Cancer Res. 2005, 65, 3307–3318. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, R.; Xu, Y.; Yu, L.; Meng, N.; Wang, H.; Cui, Y.; Yam, J.W.P. Extracellular Vesicles Act as Carriers for Cargo Delivery and Regulate Wnt Signaling in the Hepatocellular Carcinoma Tumor Microenvironment. Cancers 2023, 15, 2088. https://doi.org/10.3390/cancers15072088
He R, Xu Y, Yu L, Meng N, Wang H, Cui Y, Yam JWP. Extracellular Vesicles Act as Carriers for Cargo Delivery and Regulate Wnt Signaling in the Hepatocellular Carcinoma Tumor Microenvironment. Cancers. 2023; 15(7):2088. https://doi.org/10.3390/cancers15072088
Chicago/Turabian StyleHe, Risheng, Yi Xu, Liang Yu, Nanfeng Meng, Hang Wang, Yunfu Cui, and Judy Wai Ping Yam. 2023. "Extracellular Vesicles Act as Carriers for Cargo Delivery and Regulate Wnt Signaling in the Hepatocellular Carcinoma Tumor Microenvironment" Cancers 15, no. 7: 2088. https://doi.org/10.3390/cancers15072088