
α-Mangostin Promotes In Vitro and In Vivo Degradation of Androgen Receptor and AR-V7 Splice Variant in Prostate Cancer Cells
 , , , ,
, , , , 
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. MTT Assays
2.3. ELISA Assays
2.4. Western Blotting
2.5. Quantitative PCR (qPCR)
2.6. Immunoprecipitations
2.7. Plasmid Transfections
2.8. Site Directed Mutagenesis
2.9. Protein Purification
2.10. Surface Plasmon Resonance (SPR)
2.11. In Vivo 22Rv1 Xenograft
2.12. Immunohistochemistry
2.13. Calculations and Statistical Analysis
3. Results
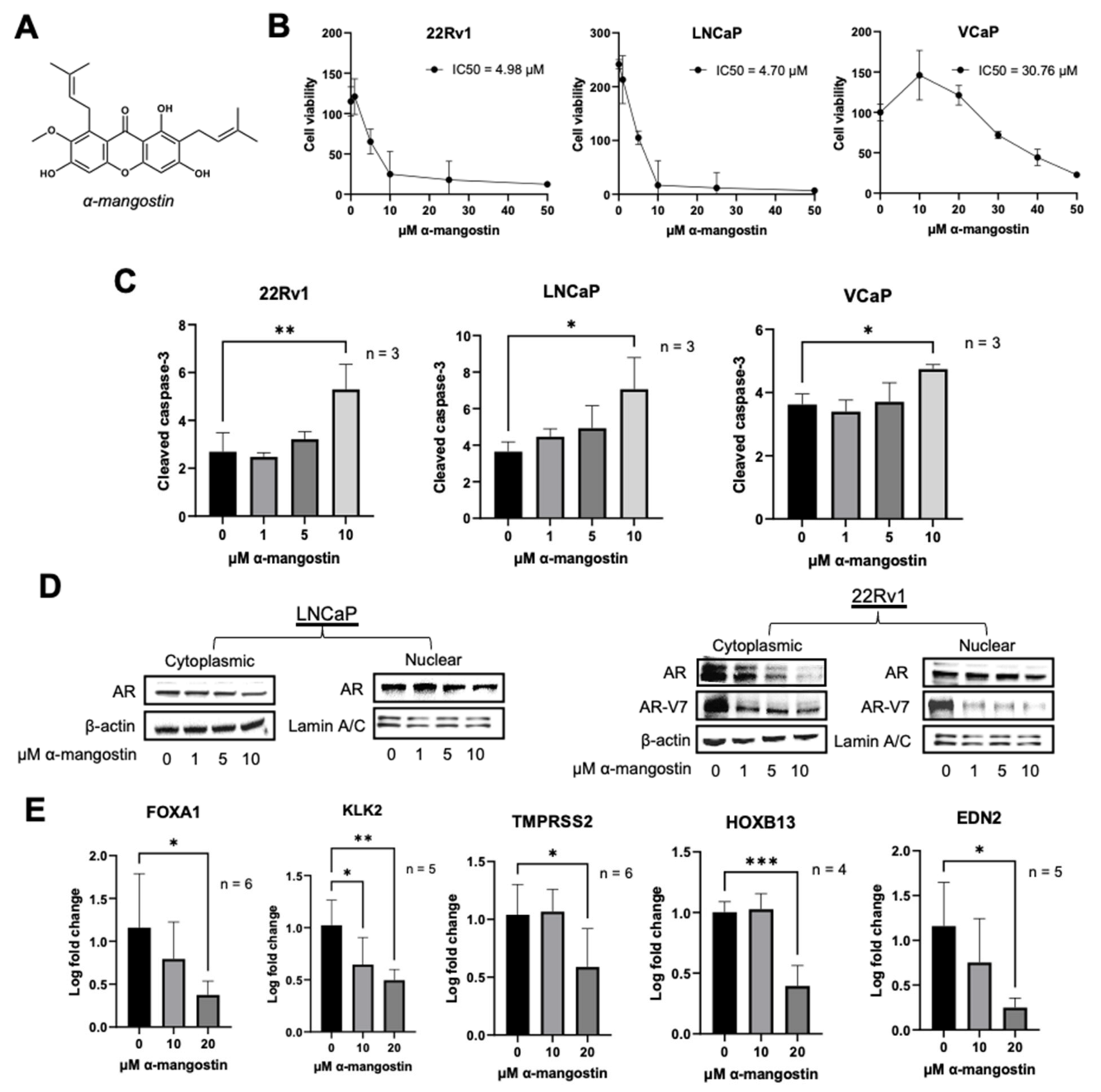
3.1. α-Mangostin Promotes Apoptosis and Inhibits Nuclear Translocation
3.2. α-Mangostin Promotes Ubiquitination and Degradation of AR and AR-V7 via the Proteasome
3.3. α-Mangostin Decreases Mutant AR Expression and Promotes the Expression of BiP
3.4. α-Mangostin Increased BiP Expression, Promoted a Protein-Protein Interaction between AR and BiP, and Binds to BiP
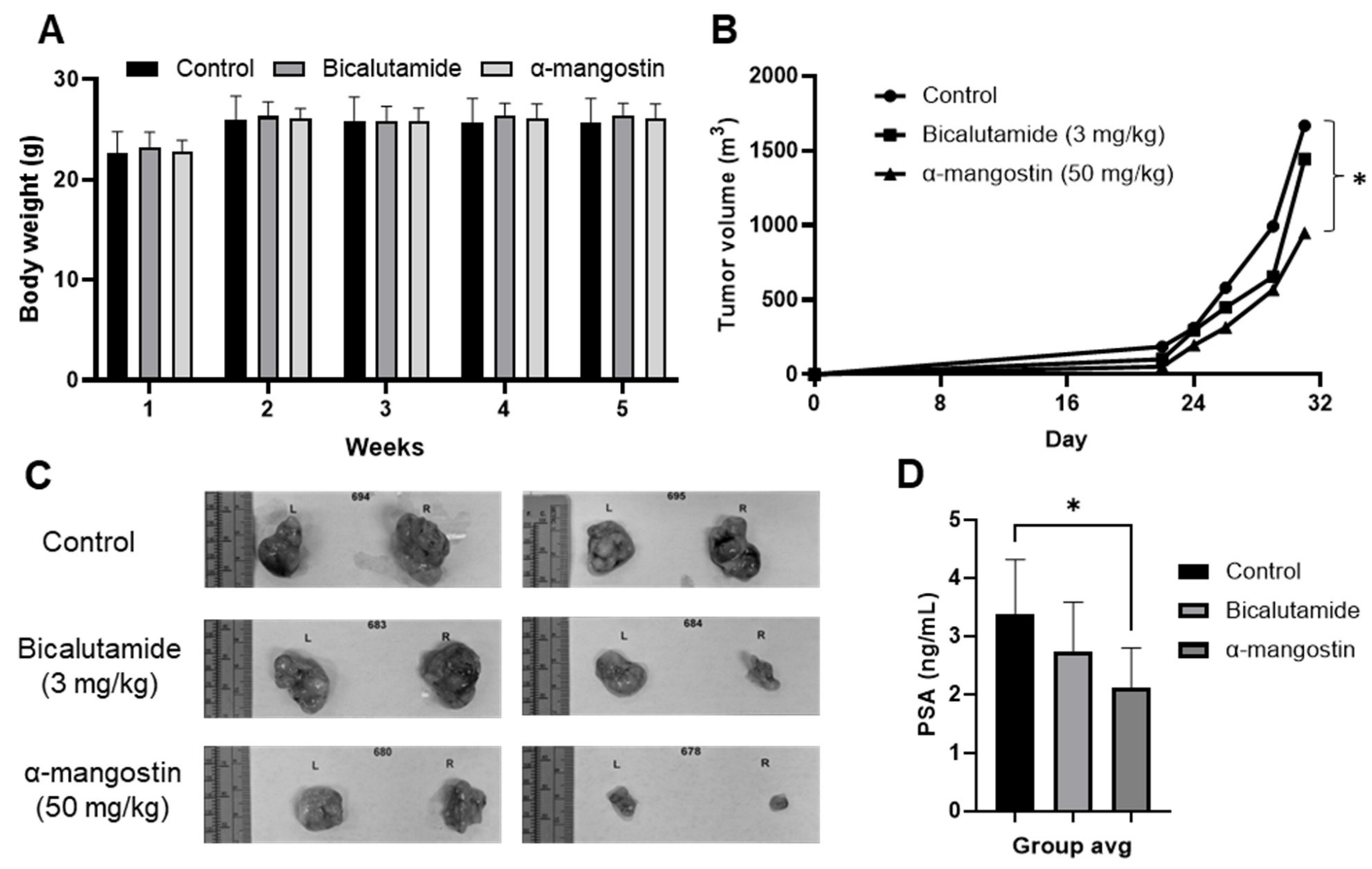
3.5. α-Mangostin Is More Effective than Bicalutamide at Slowing Tumor Growth and Cancer Growth in a 22Rν1 Xenograft Study
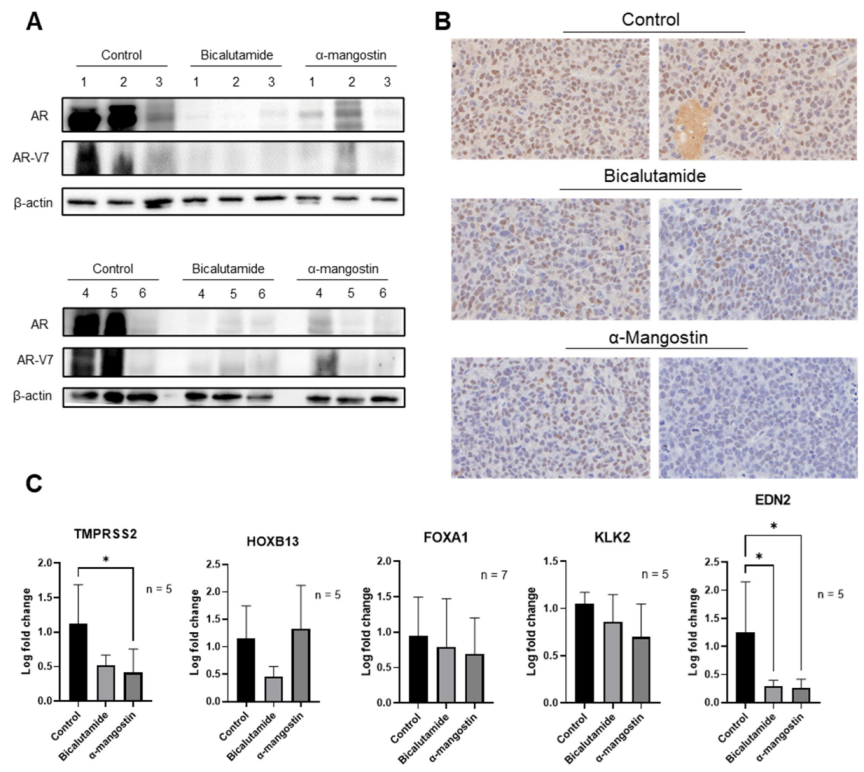
3.6. α-Mangostin Decreased AR and AR-V7 Protein Expression and Related Genes In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandhu, G.S.; Nepple, K.G.; Tanagho, Y.S.; Andriole, G.L. Prostate cancer chemoprevention. Semin. Oncol. 2013, 40, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, G.; Singh, C.K.; Ndiaye, M.A.; Fedorowicz, S.; Molot, A.; Ahmad, N. Prostate cancer chemoprevention by natural agents: Clinical evidence and potential implications. Cancer Lett. 2018, 422, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.J.; Kim, J.; Yu, J. Androgen receptor genomic regulation. Transl. Androl. Urol. 2013, 2, 157–177. [Google Scholar] [CrossRef] [PubMed]
- Grosse, A.; Bartsch, S.; Baniahmad, A. Androgen receptor-mediated gene repression. Mol. Cell. Endocrinol. 2012, 352, 46–56. [Google Scholar] [CrossRef]
- Fujita, K.; Nonomura, N. Role of Androgen Receptor in Prostate Cancer: A Review. World J. Mens. Health 2019, 37, 288–295. [Google Scholar] [CrossRef]
- Eisermann, K.; Wang, D.; Jing, Y.; Pascal, L.E.; Wang, Z. Androgen receptor gene mutation, rearrangement, polymorphism. Transl. Androl. Urol. 2013, 2, 137–147. [Google Scholar] [CrossRef]
- Evans, A.J. Treatment effects in prostate cancer. Mod. Pathol. 2018, 31, 110–121. [Google Scholar] [CrossRef] [Green Version]
- Estébanez-Perpiñá, E.; Bevan, C.L.; McEwan, I.J. Eighty Years of Targeting Androgen Receptor Activity in Prostate Cancer: The Fight Goes on. Cancers 2021, 13, 509. [Google Scholar] [CrossRef]
- Chism, D.D.; De Silva, D.; Whang, Y.E. Mechanisms of acquired resistance to androgen receptor targeting drugs in castration-resistant prostate cancer. Expert Rev. Anticancer. Ther. 2014, 14, 1369–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, K.T.; Huitema, A.D.R.; Chau, C.H.; Figg, W.D. Resistance to second-generation androgen receptor antagonists in prostate cancer. Nat. Rev. Urol. 2021, 18, 209–226. [Google Scholar] [CrossRef]
- Hörnberg, E.; Ylitalo, E.B.; Crnalic, S.; Antti, H.; Stattin, P.; Widmark, A.; Bergh, A.; Wikström, P. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS ONE 2011, 6, e19059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Mol, E.; Fenwick, R.B.; Phang, C.T.; Buzón, V.; Szulc, E.; de la Fuente, A.; Escobedo, A.; García, J.; Bertoncini, C.W.; Estébanez-Perpiñá, E.; et al. EPI-001, A Compound Active against Castration-Resistant Prostate Cancer, Targets Transactivation Unit 5 of the Androgen Receptor. ACS Chem. Biol. 2016, 11, 2499–2505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.C.; Banuelos, C.A.; Mawji, N.R.; Wang, J.; Kato, M.; Haile, S.; McEwan, I.J.; Plymate, S.; Sadar, M.D. Targeting Androgen Receptor Activation Function-1 with EPI to Overcome Resistance Mechanisms in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2016, 22, 4466–4477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, R.J.; Mawji, N.R.; Wang, J.; Wang, G.; Haile, S.; Myung, J.K.; Watt, K.; Tam, T.; Yang, Y.C.; Bañuelos, C.A.; et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell 2010, 17, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Ovalle-Magallanes, B.; Eugenio-Perez, D.; Pedraza-Chaverri, J. Medicinal properties of mangosteen (Garcinia mangostana L.): A comprehensive update. Food Chem. Toxicol. 2017, 109, 102–122. [Google Scholar] [CrossRef]
- Chin, Y.W.; Kinghorn, A.D. Structural Characterization, Biological Effects, and Synthetic Studies on Xanthones from Mangosteen (Garcinia mangostana), a Popular Botanical Dietary Supplement. Mini. Rev. Org. Chem. 2008, 5, 355–364. [Google Scholar] [CrossRef] [Green Version]
- Shan, T.; Ma, Q.; Guo, K.; Liu, J.; Li, W.; Wang, F.; Wu, E. Xanthones from mangosteen extracts as natural chemopreventive agents: Potential anticancer drugs. Curr. Mol. Med. 2011, 11, 666–677. [Google Scholar] [CrossRef]
- Gutierrez-Orozco, F.; Failla, M.L. Biological activities and bioavailability of mangosteen xanthones: A critical review of the current evidence. Nutrients 2013, 5, 3163–3183. [Google Scholar] [CrossRef] [Green Version]
- Nauman, M.C.; Johnson, J.J. The purple mangosteen (Garcinia mangostana): Defining the anticancer potential of selected xanthones. Pharmacol. Res. 2022, 175, 106032. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Petiwala, S.M.; Pierce, D.R.; Nonn, L.; Johnson, J.J. Selective modulation of endoplasmic reticulum stress markers in prostate cancer cells by a standardized mangosteen fruit extract. PLoS ONE 2013, 8, e81572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Petiwala, S.M.; Nonn, L.; Johnson, J.J. Inhibition of CHOP accentuates the apoptotic effect of α-mangostin from the mangosteen fruit (Garcinia mangostana) in 22Rv1 prostate cancer cells. Biochem. Biophys. Res. Commun. 2014, 453, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Ramaiya, A.; Li, G.; Petiwala, S.M.; Johnson, J.J. Single dose oral pharmacokinetic profile of α-mangostin in mice. Curr. Drug. Targets 2012, 13, 1698–1704. [Google Scholar] [CrossRef]
- Petiwala, S.M.; Li, G.; Ramaiya, A.; Kumar, A.; Gill, R.K.; Saksena, S.; Johnson, J.J. Pharmacokinetic characterization of mangosteen (Garcinia mangostana) fruit extract standardized to α-mangostin in C57BL/6 mice. Nutr. Res. 2014, 34, 336–345. [Google Scholar] [CrossRef]
- Johnson, J.J.; Petiwala, S.M.; Syed, D.N.; Rasmussen, J.T.; Adhami, V.M.; Siddiqui, I.A.; Kohl, A.M.; Mukhtar, H. α-Mangostin, a xanthone from mangosteen fruit, promotes cell cycle arrest in prostate cancer and decreases xenograft tumor growth. Carcinogenesis 2012, 33, 413–419. [Google Scholar] [CrossRef] [Green Version]
- Nauman, M.C.; Tocmo, R.; Vemu, B.; Veenstra, J.P.; Johnson, J.J. Inhibition of CDK2/CyclinE1 by xanthones from the mangosteen (Garcinia mangostana): A structure-activity relationship study. Nat. Prod. Res. 2020, 35, 5429–5433. [Google Scholar] [CrossRef]
- Vemu, B.; Nauman, M.C.; Veenstra, J.P.; Johnson, J.J. Structure activity relationship of xanthones for inhibition of Cyclin Dependent Kinase 4 from mangosteen (Garcinia mangostana L.). Int. J. Nutr. 2019, 4, 38–45. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Petiwala, S.M.; Yan, M.; Won, J.H.; Petukhov, P.A.; Johnson, J.J. Gartanin, an isoprenylated xanthone from the mangosteen fruit (Garcinia mangostana), is an androgen receptor degradation enhancer. Mol. Nutr. Food Res. 2016, 60, 1458–1469. [Google Scholar] [CrossRef]
- Huang, Y.; Tocmo, R.; Nauman, M.C.; Haughan, M.A.; Johnson, J.J. Defining the Cholesterol Lowering Mechanism of Bergamot (Citrus bergamia) Extract in HepG2 and Caco-2 Cells. Nutrients 2021, 13, 3156. [Google Scholar] [CrossRef]
- Leestemaker, Y.; de Jong, A.; Witting, K.F.; Penning, R.; Schuurman, K.; Rodenko, B.; Zaal, E.A.; van de Kooij, B.; Laufer, S.; Heck, A.J.R.; et al. Proteasome Activation by Small Molecules. Cell Chem. Biol. 2017, 24, 725–736.e727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petiwala, S.M.; Berhe, S.; Li, G.; Puthenveetil, A.G.; Rahman, O.; Nonn, L.; Johnson, J.J. Rosemary (Rosmarinus officinalis) extract modulates CHOP/GADD153 to promote androgen receptor degradation and decreases xenograft tumor growth. PLoS ONE 2014, 9, e89772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiota, M.; Eto, M. Current status of primary pharmacotherapy and future perspectives toward upfront therapy for metastatic hormone-sensitive prostate cancer. Int. J. Urol. 2016, 23, 360–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jernberg, E.; Bergh, A.; Wikström, P. Clinical relevance of androgen receptor alterations in prostate cancer. Endocr. Connect. 2017, 6, R146–R161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, K.; McManus, J.M.; Sharifi, N. Hormonal Therapy for Prostate Cancer. Endocr. Rev. 2021, 42, 354–373. [Google Scholar] [CrossRef]
- Ehsani, M.; David, F.O.; Baniahmad, A. Androgen Receptor-Dependent Mechanisms Mediating Drug Resistance in Prostate Cancer. Cancers 2021, 13, 1534. [Google Scholar] [CrossRef]
- Einstein, D.J.; Arai, S.; Balk, S.P. Targeting the androgen receptor and overcoming resistance in prostate cancer. Curr. Opin. Oncol. 2019, 31, 175–182. [Google Scholar] [CrossRef]
- Salehi, B.; Fokou, P.V.T.; Yamthe, L.R.T.; Tali, B.T.; Adetunji, C.O.; Rahavian, A.; Mudau, F.N.; Martorell, M.; Setzer, W.N.; Rodrigues, C.F.; et al. Phytochemicals in Prostate Cancer: From Bioactive Molecules to Upcoming Therapeutic Agents. Nutrients 2019, 11, 1483. [Google Scholar] [CrossRef] [Green Version]
- Asadi-Samani, M.; Rafieian-Kopaei, M.; Lorigooini, Z.; Shirzad, H. A screening of growth inhibitory activity of Iranian medicinal plants on prostate cancer cell lines. Biomedicine 2018, 8, 8. [Google Scholar] [CrossRef] [Green Version]
- Shang, Z.; Niu, Y.; Cai, Q.; Chen, J.; Tian, J.; Yeh, S.; Lai, K.P.; Chang, C. Human kallikrein 2 (KLK2) promotes prostate cancer cell growth via function as a modulator to promote the ARA70-enhanced androgen receptor transactivation. Tumour. Biol. 2014, 35, 1881–1890. [Google Scholar] [CrossRef]
- Bonk, S.; Kluth, M.; Jansen, K.; Hube-Magg, C.; Makrypidi-Fraune, G.; Höflmayer, D.; Weidemann, S.; Möller, K.; Uhlig, R.; Büscheck, F.; et al. Reduced KLK2 expression is a strong and independent predictor of poor prognosis in ERG-negative prostate cancer. Prostate 2020, 80, 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, Y.; Zhang, J.; Hu, Q.; Zhi, F.; Zhang, S.; Mao, D.; Zhang, Y.; Liang, H. Significance of the TMPRSS2:ERG gene fusion in prostate cancer. Mol. Med. Rep. 2017, 16, 5450–5458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosquera, J.M.; Mehra, R.; Regan, M.M.; Perner, S.; Genega, E.M.; Bueti, G.; Shah, R.B.; Gaston, S.; Tomlins, S.A.; Wei, J.T.; et al. Prevalence of TMPRSS2-ERG fusion prostate cancer among men undergoing prostate biopsy in the United States. Clin. Cancer Res. 2009, 15, 4706–4711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Au, C.C.; Jamalruddin, M.A.B.; Abou-Ghali, N.E.; Mukhtar, E.; Portella, L.; Berger, A.; Worroll, D.; Vatsa, P.; Rickman, D.S.; et al. AR-V7 exhibits non-canonical mechanisms of nuclear import and chromatin engagement in castrate-resistant prostate cancer. Elife 2022, 11. [Google Scholar] [CrossRef]
- Chen, Z.; Wu, D.; Thomas-Ahner, J.M.; Lu, C.; Zhao, P.; Zhang, Q.; Geraghty, C.; Yan, P.S.; Hankey, W.; Sunkel, B.; et al. Diverse AR-V7 cistromes in castration-resistant prostate cancer are governed by HoxB13. Proc. Natl. Acad. Sci. USA 2018, 115, 6810–6815. [Google Scholar] [CrossRef] [Green Version]
- Navarro, H.I.; Goldstein, A.S. HoxB13 mediates AR-V7 activity in prostate cancer. Proc. Natl. Acad. Sci. USA 2018, 115, 6528–6529. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Löhr, C.V.; Fischer, K.; Dashwood, W.M.; Greenwood, J.A.; Ho, E.; Williams, D.E.; Ashktorab, H.; Dashwood, M.R.; Dashwood, R.H. Epigenetic inactivation of endothelin-2 and endothelin-3 in colon cancer. Int. J. Cancer 2013, 132, 1004–1012. [Google Scholar] [CrossRef] [Green Version]
- Basil, P.; Robertson, M.J.; Bingman, W.E., 3rd; Dash, A.K.; Krause, W.C.; Shafi, A.A.; Piyarathna, B.; Coarfa, C.; Weigel, N.L. Cistrome and transcriptome analysis identifies unique androgen receptor (AR) and AR-V7 splice variant chromatin binding and transcriptional activities. Sci. Rep. 2022, 12, 5351. [Google Scholar] [CrossRef]
- Krause, W.C.; Shafi, A.A.; Nakka, M.; Weigel, N.L. Androgen receptor and its splice variant, AR-V7, differentially regulate FOXA1 sensitive genes in LNCaP prostate cancer cells. Int. J. Biochem. Cell Biol. 2014, 54, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Stelloo, S.; Nevedomskaya, E.; van der Poel, H.G.; de Jong, J.; van Leenders, G.J.; Jenster, G.; Wessels, L.F.; Bergman, A.M.; Zwart, W. Androgen receptor profiling predicts prostate cancer outcome. EMBO Mol. Med. 2015, 7, 1450–1464. [Google Scholar] [CrossRef]
- De Laere, B.; Rajan, P.; Grönberg, H.; Dirix, L.; Lindberg, J. Androgen Receptor Burden and Poor Response to Abiraterone or Enzalutamide in TP53 Wild-Type Metastatic Castration-Resistant Prostate Cancer. JAMA Oncol. 2019, 5, 1060–1062. [Google Scholar] [CrossRef] [PubMed]
- Descotes, J.L. Diagnosis of prostate cancer. Asian J. Urol. 2019, 6, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.; You, Z. In vitro and in vivo model systems used in prostate cancer research. J. Biol. Methods 2015, 2, e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, A.; Coleman, I.; Yuan, W.; Sprenger, C.; Dolling, D.; Rodrigues, D.N.; Russo, J.W.; Figueiredo, I.; Bertan, C.; Seed, G.; et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J. Clin. Investig. 2019, 129, 192–208. [Google Scholar] [CrossRef] [Green Version]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Wang, H.; Chen, Y.; Nakazawa, M.; Nadal, R.; Paller, C.J.; Denmeade, S.R.; Carducci, M.A.; et al. Androgen Receptor Splice Variant 7 and Efficacy of Taxane Chemotherapy in Patients With Metastatic Castration-Resistant Prostate Cancer. JAMA Oncol. 2015, 1, 582–591. [Google Scholar] [CrossRef] [Green Version]
- Rathkopf, D.E.; Smith, M.R.; Ryan, C.J.; Berry, W.R.; Shore, N.D.; Liu, G.; Higano, C.S.; Alumkal, J.J.; Hauke, R.; Tutrone, R.F.; et al. Androgen receptor mutations in patients with castration-resistant prostate cancer treated with apalutamide. Ann. Oncol. 2017, 28, 2264–2271. [Google Scholar] [CrossRef]
- Lallous, N.; Volik, S.V.; Awrey, S.; Leblanc, E.; Tse, R.; Murillo, J.; Singh, K.; Azad, A.A.; Wyatt, A.W.; LeBihan, S.; et al. Functional analysis of androgen receptor mutations that confer anti-androgen resistance identified in circulating cell-free DNA from prostate cancer patients. Genome Biol. 2016, 17, 10. [Google Scholar] [CrossRef] [Green Version]
- Kregel, S.; Wang, C.; Han, X.; Xiao, L.; Fernandez-Salas, E.; Bawa, P.; McCollum, B.L.; Wilder-Romans, K.; Apel, I.J.; Cao, X.; et al. Androgen receptor degraders overcome common resistance mechanisms developed during prostate cancer treatment. Neoplasia 2020, 22, 111–119. [Google Scholar] [CrossRef]
- Kim, G.Y.; Song, C.W.; Yang, Y.S.; Lee, N.R.; Yoo, H.S.; Son, S.H.; Lee, S.J.; Park, J.S.; Lee, J.K.; Inn, K.S.; et al. Chemical Degradation of Androgen Receptor (AR) Using Bicalutamide Analog-Thalidomide PROTACs. Molecules 2021, 26, 2525. [Google Scholar] [CrossRef]
- Han, X.; Zhao, L.; Xiang, W.; Qin, C.; Miao, B.; McEachern, D.; Wang, Y.; Metwally, H.; Wang, L.; Matvekas, A.; et al. Strategies toward Discovery of Potent and Orally Bioavailable Proteolysis Targeting Chimera Degraders of Androgen Receptor for the Treatment of Prostate Cancer. J. Med. Chem. 2021, 64, 12831–12854. [Google Scholar] [CrossRef]
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 2015, 17, 829–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilda, M.; Alfaro-Valdés, F.B.-B.; Casanova-Morales, N.; Quiroga-Roger, D.; Wilson, C.A.M. Endoplasmic Reticulum; IntechOpen: London, UK, 2018. [Google Scholar]
- McCarty, D.J.; Huang, W.; Kane, M.A.; Purushottamachar, P.; Gediya, L.K.; Njar, V.C.O. Novel galeterone analogs act independently of AR and AR-V7 for the activation of the unfolded protein response and induction of apoptosis in the CWR22Rv1 prostate cancer cell model. Oncotarget 2017, 8, 88501–88516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurusinghe, K.; Mishra, A.; Mishra, S. Glucose-regulated protein 78 substrate-binding domain alters its conformation upon EGCG inhibitor binding to nucleotide-binding domain: Molecular dynamics studies. Sci. Rep. 2018, 8, 5487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pobre, K.F.R.; Poet, G.J.; Hendershot, L.M. The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: Getting by with a little help from ERdj friends. J. Biol. Chem. 2019, 294, 2098–2108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, M.; Reinstein, J.; Buchner, J. Modulation of the ATPase cycle of BiP by peptides and proteins. J. Mol. Biol. 2003, 330, 137–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, T.; Giblin, D.; Gross, M.L. Structural determinant of chemical reactivity and potential health effects of quinones from natural products. Chem. Res. Toxicol. 2011, 24, 1527–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolton, J.L.; Dunlap, T.L.; Dietz, B.M. Formation and biological targets of botanical o-quinones. Food Chem. Toxicol. 2018, 120, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Dunsmore, L.; Navo, C.D.; Becher, J.; de Montes, E.G.; Guerreiro, A.; Hoyt, E.; Brown, L.; Zelenay, V.; Mikutis, S.; Cooper, J.; et al. Controlled masking and targeted release of redox-cycling ortho-quinones via a C-C bond-cleaving 1,6-elimination. Nat. Chem. 2022, 14, 754–765. [Google Scholar] [CrossRef]
- Ghods, A.; Gilbert, J.; Baker, J.R.; Russell, C.C.; Sakoff, J.A.; McCluskey, A. A focused library synthesis and cytotoxicity of quinones derived from the natural product bolinaquinone. R Soc. Open Sci. 2018, 5, 171189. [Google Scholar] [CrossRef] [Green Version]
- Brito, J.A.; Sousa, F.L.; Stelter, M.; Bandeiras, T.M.; Vonrhein, C.; Teixeira, M.; Pereira, M.M.; Archer, M. Structural and functional insights into sulfide:quinone oxidoreductase. Biochemistry 2009, 48, 5613–5622. [Google Scholar] [CrossRef]
- Ilic, D.; Djulbegovic, M.; Jung, J.H.; Hwang, E.C.; Zhou, Q.; Cleves, A.; Agoritsas, T.; Dahm, P. Prostate cancer screening with prostate-specific antigen (PSA) test: A systematic review and meta-analysis. BMJ 2018, 362, k3519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David MK, L.S. Prostate Specific Antigen; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nauman, M.C.; Won, J.H.; Petiwala, S.M.; Vemu, B.; Lee, H.; Sverdlov, M.; Johnson, J.J. α-Mangostin Promotes In Vitro and In Vivo Degradation of Androgen Receptor and AR-V7 Splice Variant in Prostate Cancer Cells. Cancers 2023, 15, 2118. https://doi.org/10.3390/cancers15072118
Nauman MC, Won JH, Petiwala SM, Vemu B, Lee H, Sverdlov M, Johnson JJ. α-Mangostin Promotes In Vitro and In Vivo Degradation of Androgen Receptor and AR-V7 Splice Variant in Prostate Cancer Cells. Cancers. 2023; 15(7):2118. https://doi.org/10.3390/cancers15072118
Chicago/Turabian StyleNauman, Mirielle C., Jong Hoon Won, Sakina M. Petiwala, Bhaskar Vemu, Hyun Lee, Maria Sverdlov, and Jeremy J. Johnson. 2023. "α-Mangostin Promotes In Vitro and In Vivo Degradation of Androgen Receptor and AR-V7 Splice Variant in Prostate Cancer Cells" Cancers 15, no. 7: 2118. https://doi.org/10.3390/cancers15072118
APA StyleNauman, M. C., Won, J. H., Petiwala, S. M., Vemu, B., Lee, H., Sverdlov, M., & Johnson, J. J. (2023). α-Mangostin Promotes In Vitro and In Vivo Degradation of Androgen Receptor and AR-V7 Splice Variant in Prostate Cancer Cells. Cancers, 15(7), 2118. https://doi.org/10.3390/cancers15072118