

EBV and Lymphomagenesis
Abstract
:Simple Summary
Abstract
1. Introduction
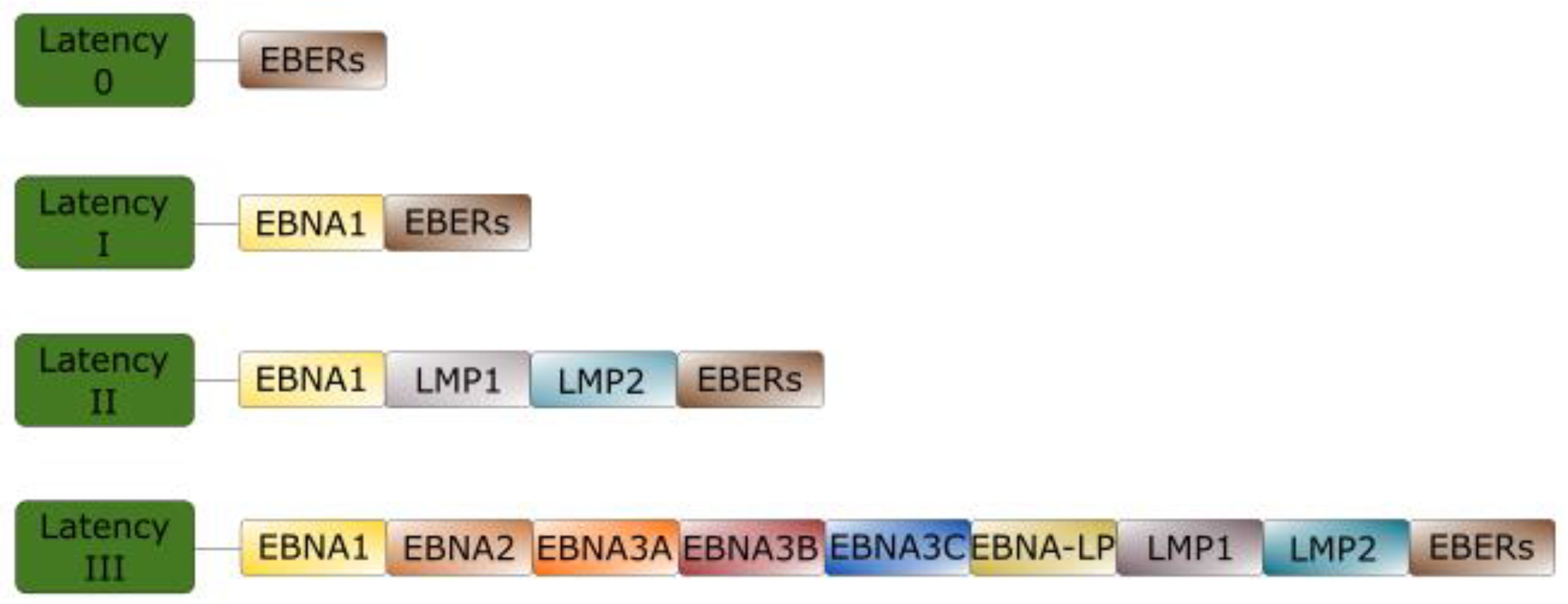
2. EBV Latency
Brief Overview of EBV Latent Proteins
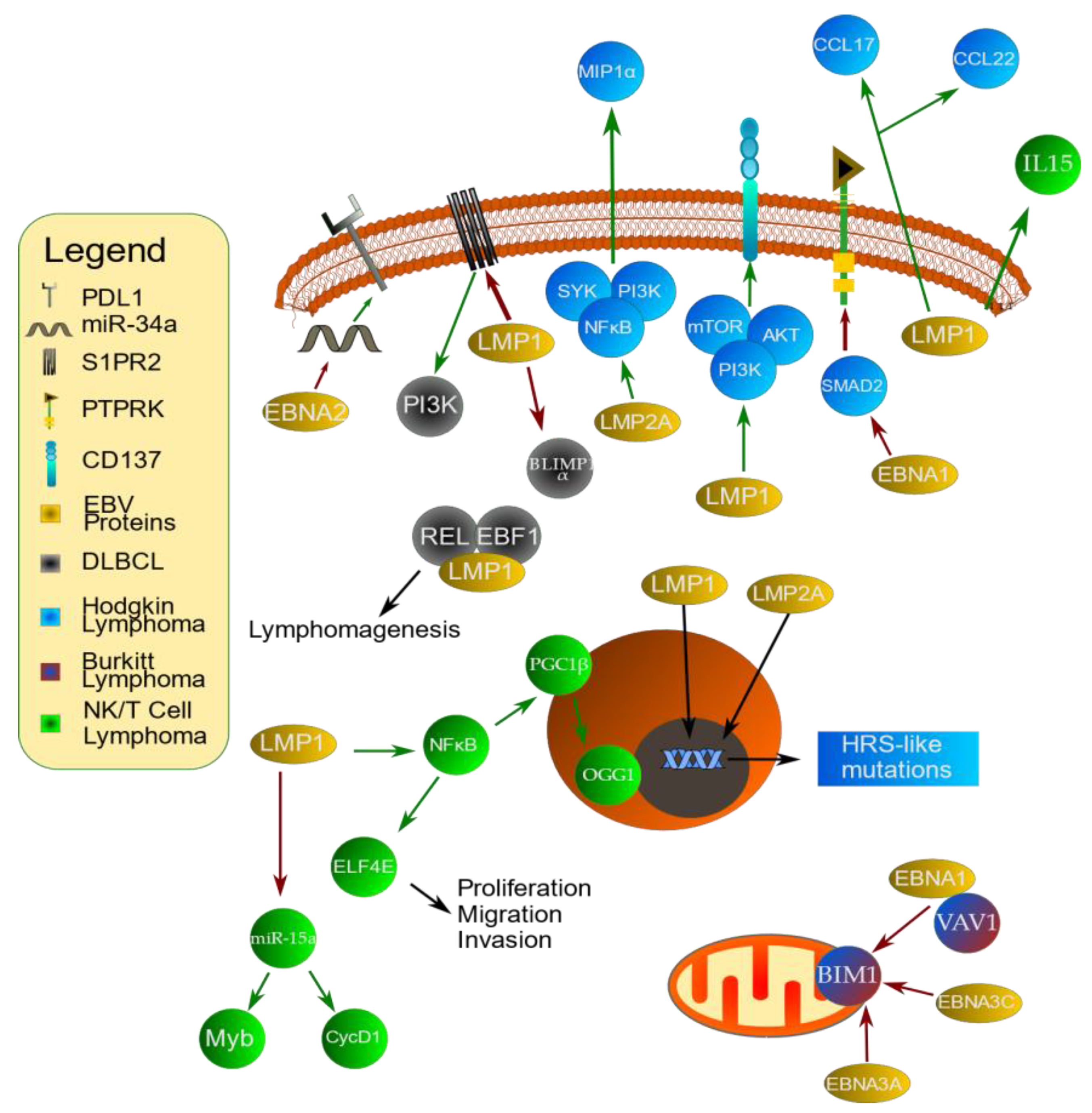
3. EBV and Oncogenesis
3.1. Diffuse Large B-Cell Lymphoma
Management
3.2. Classic Hodgkin Lymphoma
Management
3.3. Burkitt Lymphoma
Management
3.4. NK/T-Cell Lymphoma
Management
3.5. Primary CNS Lymphoma
Management
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dunmire, S.K.; Verghese, P.S.; Balfour, H.H., Jr. Primary Epstein-Barr virus infection. J. Clin. Virol. 2018, 102, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Mohseni, M.; Boniface, M.P.; Graham, C. Mononucleosis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Naughton, P.; Healy, M.; Enright, F.; Lucey, B. Infectious Mononucleosis: Diagnosis and clinical interpretation. Br. J. Biomed Sci. 2021, 78, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Abbott, R.J.; Pachnio, A.; Pedroza-Pacheco, I.; Leese, A.M.; Begum, J.; Long, H.M.; Croom-Carter, D.; Stacey, A.; Moss, P.A.H.; Hislop, A.D.; et al. Asymptomatic Primary Infection with Epstein-Barr Virus: Observations on Young Adult Cases. J. Virol. 2017, 91, 21. [Google Scholar] [CrossRef] [Green Version]
- Guo, R.; Gewurz, B.E. Epigenetic control of the Epstein-Barr lifecycle. Curr. Opin. Virol. 2022, 52, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.J. Epstein-Barr Virus and Cancer. Annu. Rev. Pathol. 2019, 14, 29–53. [Google Scholar] [CrossRef] [PubMed]
- Malpica, L.; Marques-Piubelli, M.L.; Beltran, B.E.; Chavez, J.C.; Miranda, R.N.; Castillo, J.J. EBV-positive diffuse large B-cell lymphoma, not otherwise specified: 2022 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2022, 97, 951–965. [Google Scholar] [CrossRef]
- Jiang, P.; Nolte, I.M.; Hepkema, B.G.; Stulp, M.; van den Berg, A.; Diepstra, A. Killer Cell Immunoglobulin-Like Receptor Haplotype B Modulates Susceptibility to EBV-Associated Classic Hodgkin Lymphoma. Front. Immunol. 2022, 13, 829943. [Google Scholar] [CrossRef]
- Shafiee, A.; Shamsi, S.; Kohandel Gargari, O.; Beiky, M.; Allahkarami, M.M.; Miyanaji, A.B.; Aghajanian, S.; Mozhgani, S.H. EBV associated T- and NK-cell lymphoproliferative diseases: A comprehensive overview of clinical manifestations and novel therapeutic insights. Rev. Med. Virol. 2022, 32, e2328. [Google Scholar] [CrossRef]
- Dharnidharka, V.R.; Webster, A.C.; Martinez, O.M.; Preiksaitis, J.K.; Leblond, V.; Choquet, S. Post-transplant lymphoproliferative disorders. Nat. Rev. Dis. Primers 2016, 2, 15088. [Google Scholar] [CrossRef]
- Patel, P.D.; Alghareeb, R.; Hussain, A.; Maheshwari, M.V.; Khalid, N. The Association of Epstein-Barr Virus With Cancer. Cureus 2022, 14, e26314. [Google Scholar] [CrossRef]
- Gandhi, M.K.; Hoang, T.; Law, S.C.; Brosda, S.; O’Rourke, K.; Tobin, J.W.D.; Vari, F.; Murigneux, V.; Fink, L.; Gunawardana, J.; et al. EBV-associated primary CNS lymphoma occurring after immunosuppression is a distinct immunobiological entity. Blood 2021, 137, 1468–1477. [Google Scholar] [CrossRef] [PubMed]
- Shechter, O.; Sausen, D.G.; Gallo, E.S.; Dahari, H.; Borenstein, R. Epstein-Barr Virus (EBV) Epithelial Associated Malignancies: Exploring Pathologies and Current Treatments. Int. J. Mol. Sci. 2022, 23, 14389. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Kono, K. Landscape of EBV-positive gastric cancer. Gastric Cancer 2021, 24, 983–989. [Google Scholar] [CrossRef]
- Chang, E.T.; Ye, W.; Zeng, Y.X.; Adami, H.O. The Evolving Epidemiology of Nasopharyngeal Carcinoma. Cancer Epidemiol. Biomark. Prev. 2021, 30, 1035–1047. [Google Scholar] [CrossRef]
- Wong, Y.; Meehan, M.T.; Burrows, S.R.; Doolan, D.L.; Miles, J.J. Estimating the global burden of Epstein-Barr virus-related cancers. J. Cancer Res. Clin. Oncol. 2022, 148, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Khan, G.; Hashim, M.J. Global burden of deaths from Epstein-Barr virus attributable malignancies 1990–2010. Infect. Agent Cancer 2014, 9, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, G.; Fitzmaurice, C.; Naghavi, M.; Ahmed, L.A. Global and regional incidence, mortality and disability-adjusted life-years for Epstein-Barr virus-attributable malignancies, 1990–2017. BMJ Open 2020, 10, e037505. [Google Scholar] [CrossRef]
- Munz, C. Latency and lytic replication in Epstein-Barr virus-associated oncogenesis. Nat. Rev. Microbiol. 2019, 17, 691–700. [Google Scholar] [CrossRef] [Green Version]
- Khanal, D.; Singh, T.; Rabinstein, A. Epstein Barr Virus Encephalitis in Adults: A Case Series (P1.293). Neurology 2016, 86, P1.293. [Google Scholar]
- Almazyad, A.; Alabdulaaly, L.; Noonan, V.; Woo, S.B. Oral hairy leukoplakia: A series of 45 cases in immunocompetent patients. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2021, 132, 210–216. [Google Scholar] [CrossRef]
- Al-Mansour, Z.; Nelson, B.P.; Evens, A.M. Post-transplant lymphoproliferative disease (PTLD): Risk factors, diagnosis, and current treatment strategies. Curr. Hematol. Malig. Rep. 2013, 8, 173–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.Y.; Yang, Y.X.; Kuo, K.; Li, H.Q.; Shen, X.N.; Chen, S.D.; Cui, M.; Tan, L.; Dong, Q.; Yu, J.T. Herpesvirus infections and Alzheimer’s disease: A Mendelian randomization study. Alzheimers Res. Ther. 2021, 13, 158. [Google Scholar] [CrossRef] [PubMed]
- Pyzik, A.; Grywalska, E.; Matyjaszek-Matuszek, B.; Ludian, J.; Kiszczak-Bochynska, E.; Smolen, A.; Rolinski, J.; Pyzik, D. Does the Epstein-Barr Virus Play a Role in the Pathogenesis of Graves’ Disease? Int. J. Mol. Sci. 2019, 20, 3145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houen, G.; Trier, N.H. Epstein-Barr Virus and Systemic Autoimmune Diseases. Front. Immunol. 2020, 11, 587380. [Google Scholar] [CrossRef] [PubMed]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Pellett, P.E.; Roizman, B. Herpesviridae. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1802–1822. [Google Scholar]
- Longnecker, R.M.; Kieff, E.; Cohen, J.I. Epstein-Barr virus. In Field’s Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1898–1959. [Google Scholar]
- Hutt-Fletcher, L.M. EBV glycoproteins: Where are we now? Future Virol. 2015, 10, 1155–1162. [Google Scholar] [CrossRef] [Green Version]
- Connolly, S.A.; Jardetzky, T.S.; Longnecker, R. The structural basis of herpesvirus entry. Nat. Rev. Microbiol. 2021, 19, 110–121. [Google Scholar] [CrossRef]
- Lake, C.M.; Hutt-Fletcher, L.M. Epstein-Barr virus that lacks glycoprotein gN is impaired in assembly and infection. J. Virol. 2000, 74, 11162–11172. [Google Scholar] [CrossRef] [Green Version]
- Gram, A.M.; Oosenbrug, T.; Lindenbergh, M.F.; Bull, C.; Comvalius, A.; Dickson, K.J.; Wiegant, J.; Vrolijk, H.; Lebbink, R.J.; Wolterbeek, R.; et al. The Epstein-Barr Virus Glycoprotein gp150 Forms an Immune-Evasive Glycan Shield at the Surface of Infected Cells. PLoS Pathog. 2016, 12, e1005550. [Google Scholar] [CrossRef]
- Bu, G.L.; Xie, C.; Kang, Y.F.; Zeng, M.S.; Sun, C. How EBV Infects: The Tropism and Underlying Molecular Mechanism for Viral Infection. Viruses 2022, 14, 2372. [Google Scholar] [CrossRef]
- Murata, T.; Sugimoto, A.; Inagaki, T.; Yanagi, Y.; Watanabe, T.; Sato, Y.; Kimura, H. Molecular Basis of Epstein-Barr Virus Latency Establishment and Lytic Reactivation. Viruses 2021, 13, 2344. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Sato, Y.; Kimura, H. Modes of infection and oncogenesis by the Epstein-Barr virus. Rev. Med. Virol. 2014, 24, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Sausen, D.G.; Bhutta, M.S.; Gallo, E.S.; Dahari, H.; Borenstein, R. Stress-Induced Epstein-Barr Virus Reactivation. Biomolecules 2021, 11, 1380. [Google Scholar] [CrossRef] [PubMed]
- Marques-Piubelli, M.L.; Salas, Y.I.; Pachas, C.; Becker-Hecker, R.; Vega, F.; Miranda, R.N. Epstein-Barr virus-associated B-cell lymphoproliferative disorders and lymphomas: A review. Pathology 2020, 52, 40–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massini, G.; Siemer, D.; Hohaus, S. EBV in Hodgkin Lymphoma. Mediterr. J. Hematol. Infect. Dis. 2009, 1, e2009013. [Google Scholar] [CrossRef]
- Kanda, T. EBV-Encoded Latent Genes. Adv. Exp. Med. Biol. 2018, 1045, 377–394. [Google Scholar] [CrossRef]
- Frappier, L. Ebna1. Curr. Top Microbiol. Immunol. 2015, 391, 3–34. [Google Scholar] [CrossRef]
- Dheekollu, J.; Wiedmer, A.; Ayyanathan, K.; Deakyne, J.S.; Messick, T.E.; Lieberman, P.M. Cell-cycle-dependent EBNA1-DNA crosslinking promotes replication termination at oriP and viral episome maintenance. Cell 2021, 184, 643–654.e613. [Google Scholar] [CrossRef]
- Wang, Y.; Du, S.; Zhu, C.; Wang, C.; Yu, N.; Lin, Z.; Gan, J.; Guo, Y.; Huang, X.; He, Y.; et al. STUB1 is targeted by the SUMO-interacting motif of EBNA1 to maintain Epstein-Barr Virus latency. PLoS Pathog. 2020, 16, e1008447. [Google Scholar] [CrossRef] [Green Version]
- Frappier, L. Contributions of Epstein-Barr nuclear antigen 1 (EBNA1) to cell immortalization and survival. Viruses 2012, 4, 1537–1547. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Nagy, N.; Masucci, M.G. The Epstein-Barr virus nuclear antigen-1 upregulates the cellular antioxidant defense to enable B-cell growth transformation and immortalization. Oncogene 2020, 39, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Alfieri, C.; Birkenbach, M.; Kieff, E. Early events in Epstein-Barr virus infection of human B lymphocytes. Virology 1991, 181, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Pich, D.; Mrozek-Gorska, P.; Bouvet, M.; Sugimoto, A.; Akidil, E.; Grundhoff, A.; Hamperl, S.; Ling, P.D.; Hammerschmidt, W. First Days in the Life of Naive Human B Lymphocytes Infected with Epstein-Barr Virus. mBio 2019, 10, e01723-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J.I.; Wang, F.; Mannick, J.; Kieff, E. Epstein-Barr virus nuclear protein 2 is a key determinant of lymphocyte transformation. Proc. Natl. Acad. Sci. USA 1989, 86, 9558–9562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, H.; Nagata, K.; Kato, K.; Kanai, K.; Yamamoto, K.; Okuno, K.; Kuwamoto, S.; Higaki-Mori, H.; Sugihara, H.; Kato, M.; et al. EBNA-2 -deleted Epstein-Barr virus from P3HR-1 can infect rabbits with lower efficiency than prototype Epstein-Barr virus from B95-8. Intervirology 2013, 56, 114–121. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Romero-Masters, J.C.; Huebner, S.; Ohashi, M.; Hayes, M.; Bristol, J.A.; Nelson, S.E.; Eichelberg, M.R.; Van Sciver, N.; Ranheim, E.A.; et al. EBNA2-deleted Epstein-Barr virus (EBV) isolate, P3HR1, causes Hodgkin-like lymphomas and diffuse large B cell lymphomas with type II and Wp-restricted latency types in humanized mice. PLoS Pathog. 2020, 16, e1008590. [Google Scholar] [CrossRef]
- Shannon-Lowe, C.; Baldwin, G.; Feederle, R.; Bell, A.; Rickinson, A.; Delecluse, H.J. Epstein-Barr virus-induced B-cell transformation: Quantitating events from virus binding to cell outgrowth. J. Gen. Virol. 2005, 86, 3009–3019. [Google Scholar] [CrossRef]
- Cordier, M.; Calender, A.; Billaud, M.; Zimber, U.; Rousselet, G.; Pavlish, O.; Banchereau, J.; Tursz, T.; Bornkamm, G.; Lenoir, G.M. Stable transfection of Epstein-Barr virus (EBV) nuclear antigen 2 in lymphoma cells containing the EBV P3HR1 genome induces expression of B-cell activation molecules CD21 and CD23. J. Virol. 1990, 64, 1002–1013. [Google Scholar] [CrossRef] [Green Version]
- Yanagi, Y.; Okuno, Y.; Narita, Y.; Masud, H.; Watanabe, T.; Sato, Y.; Kanda, T.; Kimura, H.; Murata, T. RNAseq analysis identifies involvement of EBNA2 in PD-L1 induction during Epstein-Barr virus infection of primary B cells. Virology 2021, 557, 44–54. [Google Scholar] [CrossRef]
- Anastasiadou, E.; Stroopinsky, D.; Alimperti, S.; Jiao, A.L.; Pyzer, A.R.; Cippitelli, C.; Pepe, G.; Severa, M.; Rosenblatt, J.; Etna, M.P.; et al. Epstein-Barr virus-encoded EBNA2 alters immune checkpoint PD-L1 expression by downregulating miR-34a in B-cell lymphomas. Leukemia 2019, 33, 132–147. [Google Scholar] [CrossRef] [Green Version]
- Su, C.; Lu, F.; Soldan, S.S.; Lamontagne, R.J.; Tang, H.Y.; Napoletani, G.; Farrell, P.J.; Tempera, I.; Kossenkov, A.V.; Lieberman, P.M. EBNA2 driven enhancer switching at the CIITA-DEXI locus suppresses HLA class II gene expression during EBV infection of B-lymphocytes. PLoS Pathog. 2021, 17, e1009834. [Google Scholar] [CrossRef] [PubMed]
- Kempkes, B.; Ling, P.D. EBNA2 and its coactivator EBNA-LP. In Epstein Barr Virus Volume 2: One Herpes Virus: Many Diseases; Münz, C., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 35–59. [Google Scholar]
- Kang, M.S.; Kieff, E. Epstein-Barr virus latent genes. Exp. Mol. Med. 2015, 47, e131. [Google Scholar] [CrossRef] [Green Version]
- Lu, F.; Chen, H.S.; Kossenkov, A.V.; DeWispeleare, K.; Won, K.J.; Lieberman, P.M. EBNA2 Drives Formation of New Chromosome Binding Sites and Target Genes for B-Cell Master Regulatory Transcription Factors RBP-jkappa and EBF1. PLoS Pathog. 2016, 12, e1005339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manet, E.; Polveche, H.; Mure, F.; Mrozek-Gorska, P.; Roisne-Hamelin, F.; Hammerschmidt, W.; Auboeuf, D.; Gruffat, H. Modulation of alternative splicing during early infection of human primary B lymphocytes with Epstein-Barr virus (EBV): A novel function for the viral EBNA-LP protein. Nucleic Acids Res. 2021, 49, 10657–10676. [Google Scholar] [CrossRef] [PubMed]
- Szymula, A.; Palermo, R.D.; Bayoumy, A.; Groves, I.J.; Ba Abdullah, M.; Holder, B.; White, R.E. Epstein-Barr virus nuclear antigen EBNA-LP is essential for transforming naive B cells, and facilitates recruitment of transcription factors to the viral genome. PLoS Pathog. 2018, 14, e1006890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharjee, S.; Ghosh Roy, S.; Bose, P.; Saha, A. Role of EBNA-3 Family Proteins in EBV Associated B-cell Lymphomagenesis. Front. Microbiol. 2016, 7, 457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomkinson, B.; Robertson, E.; Kieff, E. Epstein-Barr virus nuclear proteins EBNA-3A and EBNA-3C are essential for B-lymphocyte growth transformation. J. Virol. 1993, 67, 2014–2025. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.; Divisconte, M.; Jiang, X.; Quink, C.; Wang, F. Epstein-Barr virus with the latent infection nuclear antigen 3B completely deleted is still competent for B-cell growth transformation in vitro. J. Virol. 2005, 79, 4506–4509. [Google Scholar] [CrossRef] [Green Version]
- Saha, A.; Robertson, E.S. Mechanisms of B-Cell Oncogenesis Induced by Epstein-Barr Virus. J. Virol. 2019, 93, e00238-19. [Google Scholar] [CrossRef] [Green Version]
- Tursiella, M.L.; Bowman, E.R.; Wanzeck, K.C.; Throm, R.E.; Liao, J.; Zhu, J.; Sample, C.E. Epstein-Barr virus nuclear antigen 3A promotes cellular proliferation by repression of the cyclin-dependent kinase inhibitor p21WAF1/CIP1. PLoS Pathog. 2014, 10, e1004415. [Google Scholar] [CrossRef] [Green Version]
- Allday, M.J.; Bazot, Q.; White, R.E. The EBNA3 Family: Two Oncoproteins and a Tumour Suppressor that Are Central to the Biology of EBV in B Cells. Curr. Top Microbiol. Immunol. 2015, 391, 61–117. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Singh, R.K.; Shukla, S.K.; Lang, F.; Zhang, S.; Robertson, E.S. Epstein-Barr Virus Nuclear Antigen 3C Facilitates Cell Proliferation by Regulating Cyclin D2. J. Virol. 2018, 92, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, R.E.; Ramer, P.C.; Naresh, K.N.; Meixlsperger, S.; Pinaud, L.; Rooney, C.; Savoldo, B.; Coutinho, R.; Bodor, C.; Gribben, J.; et al. EBNA3B-deficient EBV promotes B cell lymphomagenesis in humanized mice and is found in human tumors. J. Clin. Investig. 2012, 122, 1487–1502. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Liebowitz, D.; Kieff, E. An EBV membrane protein expressed in immortalized lymphocytes transforms established rodent cells. Cell 1985, 43, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Kieser, A.; Sterz, K.R. The Latent Membrane Protein 1 (LMP1). Curr. Top Microbiol. Immunol. 2015, 391, 119–149. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kamranvar, S.A.; Masucci, M.G. Oxidative stress enables Epstein-Barr virus-induced B-cell transformation by posttranscriptional regulation of viral and cellular growth-promoting factors. Oncogene 2016, 35, 3807–3816. [Google Scholar] [CrossRef] [PubMed]
- Kume, A.; Shinozaki-Ushiku, A.; Kunita, A.; Kondo, A.; Ushiku, T. Enhanced PD-L1 Expression in LMP1-positive Cells of Epstein-Barr Virus-associated Malignant Lymphomas and Lymphoproliferative Disorders: A Single-cell Resolution Analysis With Multiplex Fluorescence Immunohistochemistry and In Situ Hybridization. Am. J. Surg. Pathol. 2022, 46, 1386–1396. [Google Scholar] [CrossRef]
- Xu-Monette, Z.Y.; Zhou, J.; Young, K.H. PD-1 expression and clinical PD-1 blockade in B-cell lymphomas. Blood 2018, 131, 68–83. [Google Scholar] [CrossRef] [Green Version]
- Xie, W.; Medeiros, L.J.; Li, S.; Yin, C.C.; Khoury, J.D.; Xu, J. PD-1/PD-L1 Pathway and Its Blockade in Patients with Classic Hodgkin Lymphoma and Non-Hodgkin Large-Cell Lymphomas. Curr. Hematol. Malig. Rep. 2020, 15, 372–381. [Google Scholar] [CrossRef]
- Soni, V.; Cahir-McFarland, E.; Kieff, E. LMP1 TRAFficking activates growth and survival pathways. Adv. Exp. Med. Biol. 2007, 597, 173–187. [Google Scholar] [CrossRef]
- Vaysberg, M.; Lambert, S.L.; Krams, S.M.; Martinez, O.M. Activation of the JAK/STAT pathway in Epstein Barr virus+-associated posttransplant lymphoproliferative disease: Role of interferon-gamma. Am. J. Transplant. 2009, 9, 2292–2302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kung, C.P.; Meckes, D.G., Jr.; Raab-Traub, N. Epstein-Barr virus LMP1 activates EGFR, STAT3, and ERK through effects on PKCdelta. J. Virol. 2011, 85, 4399–4408. [Google Scholar] [CrossRef] [Green Version]
- Vrzalikova, K.; Ibrahim, M.; Nagy, E.; Vockerodt, M.; Perry, T.; Wei, W.; Woodman, C.; Murray, P. Co-Expression of the Epstein-Barr Virus-Encoded Latent Membrane Proteins and the Pathogenesis of Classic Hodgkin Lymphoma. Cancers 2018, 10, 285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fruehling, S.; Longnecker, R. The immunoreceptor tyrosine-based activation motif of Epstein-Barr virus LMP2A is essential for blocking BCR-mediated signal transduction. Virology 1997, 235, 241–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fish, K.; Comoglio, F.; Shaffer, A.L., 3rd; Ji, Y.; Pan, K.T.; Scheich, S.; Oellerich, A.; Doebele, C.; Ikeda, M.; Schaller, S.J.; et al. Rewiring of B cell receptor signaling by Epstein-Barr virus LMP2A. Proc. Natl. Acad. Sci. USA 2020, 117, 26318–26327. [Google Scholar] [CrossRef] [PubMed]
- Rechsteiner, M.P.; Berger, C.; Zauner, L.; Sigrist, J.A.; Weber, M.; Longnecker, R.; Bernasconi, M.; Nadal, D. Latent membrane protein 2B regulates susceptibility to induction of lytic Epstein-Barr virus infection. J. Virol. 2008, 82, 1739–1747. [Google Scholar] [CrossRef] [Green Version]
- Cen, O.; Longnecker, R. Latent Membrane Protein 2 (LMP2). Curr. Top Microbiol. Immunol. 2015, 391, 151–180. [Google Scholar] [CrossRef]
- Shannon-Lowe, C.; Rickinson, A.B.; Bell, A.I. Epstein-Barr virus-associated lymphomas. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160271. [Google Scholar] [CrossRef] [Green Version]
- Vockerodt, M.; Yap, L.F.; Shannon-Lowe, C.; Curley, H.; Wei, W.; Vrzalikova, K.; Murray, P.G. The Epstein-Barr virus and the pathogenesis of lymphoma. J. Pathol. 2015, 235, 312–322. [Google Scholar] [CrossRef]
- Beltran, B.E.; Castro, D.; Paredes, S.; Miranda, R.N.; Castillo, J.J. EBV-positive diffuse large B-cell lymphoma, not otherwise specified: 2020 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2020, 95, 435–445. [Google Scholar] [CrossRef]
- Oyama, T.; Ichimura, K.; Suzuki, R.; Suzumiya, J.; Ohshima, K.; Yatabe, Y.; Yokoi, T.; Kojima, M.; Kamiya, Y.; Taji, H.; et al. Senile EBV+ B-cell lymphoproliferative disorders: A clinicopathologic study of 22 patients. Am. J. Surg. Pathol. 2003, 27, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, A.; Kohno, K.; Ishikawa, E.; Suzuki, Y.; Shimada, S.; Eladl, A.E.; Elsayed, A.A.; Daroontum, T.; Satou, A.; Takahara, T.; et al. Age-related EBV-associated B-cell lymphoproliferative disorders and other EBV+ lymphoproliferative diseases: New insights into immune escape and immunodeficiency through staining with anti-PD-L1 antibody clone SP142. Pathol. Int. 2020, 70, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Sadighi Akha, A.A. Aging and the immune system: An overview. J. Immunol. Methods 2018, 463, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Nan, F.F.; Zhang, L.; Li, L.; Li, X.; Sun, Z.C.; Zhang, X.D.; Li, Z.M.; Li, S.C.; Jia, S.S.; Xiao, S.; et al. Clinical features and survival impact of EBV-positive diffuse large B-Cell lymphoma with different age cutoffs. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 8947–8956. [Google Scholar] [CrossRef] [PubMed]
- Monabati, A.; Vahedi, A.; Safaei, A.; Noori, S.; Mokhtari, M.; Vahedi, L.; Zamani, M. Epstein-Barr Virus-Positive Diffuse Large B-Cell Lymphoma: Is It Different between Over and Under 50 Years of Age? Asian Pac. J. Cancer Prev. 2016, 17, 2285–2289. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, J.; Dong, K.; Lin, F.; Long, M.; Ouyang, Y.; Wei, J.; Chen, X.; Weng, Y.; He, T.; et al. Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell Signal. 2015, 27, 443–452. [Google Scholar] [CrossRef]
- Green, J.A.; Cyster, J.G. S1PR2 links germinal center confinement and growth regulation. Immunol. Rev. 2012, 247, 36–51. [Google Scholar] [CrossRef] [Green Version]
- Stelling, A.; Hashwah, H.; Bertram, K.; Manz, M.G.; Tzankov, A.; Muller, A. The tumor suppressive TGF-beta/SMAD1/S1PR2 signaling axis is recurrently inactivated in diffuse large B-cell lymphoma. Blood 2018, 131, 2235–2246. [Google Scholar] [CrossRef] [Green Version]
- Vockerodt, M.; Vrzalikova, K.; Ibrahim, M.; Nagy, E.; Margielewska, S.; Hollows, R.; Lupino, L.; Tooze, R.; Care, M.; Simmons, W.; et al. Regulation of S1PR2 by the EBV oncogene LMP1 in aggressive ABC-subtype diffuse large B-cell lymphoma. J. Pathol. 2019, 248, 142–154. [Google Scholar] [CrossRef] [Green Version]
- Sommermann, T.; Yasuda, T.; Ronen, J.; Wirtz, T.; Weber, T.; Sack, U.; Caeser, R.; Zhang, J.; Li, X.; Chu, V.T.; et al. Functional interplay of Epstein-Barr virus oncoproteins in a mouse model of B cell lymphomagenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 14421–14432. [Google Scholar] [CrossRef]
- Vrzalikova, K.; Vockerodt, M.; Leonard, S.; Bell, A.; Wei, W.; Schrader, A.; Wright, K.L.; Kube, D.; Rowe, M.; Woodman, C.B.; et al. Down-regulation of BLIMP1alpha by the EBV oncogene, LMP-1, disrupts the plasma cell differentiation program and prevents viral replication in B cells: Implications for the pathogenesis of EBV-associated B-cell lymphomas. Blood 2011, 117, 5907–5917. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.D.; Tsai, M.H.; Romero-Masters, J.C.; Ranheim, E.A.; Huebner, S.M.; Bristol, J.A.; Delecluse, H.J.; Kenney, S.C. Latent Membrane Protein 1 (LMP1) and LMP2A Collaborate To Promote Epstein-Barr Virus-Induced B Cell Lymphomas in a Cord Blood-Humanized Mouse Model but Are Not Essential. J. Virol. 2017, 91, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagel, S.; MacLeod, R.A.F.; Meyer, C.; Kaufmann, M.; Drexler, H.G. NKL homeobox gene activities in B-cell development and lymphomas. PLoS ONE 2018, 13, e0205537. [Google Scholar] [CrossRef] [PubMed]
- Nagel, S.; Uphoff, C.C.; Dirks, W.G.; Pommerenke, C.; Meyer, C.; Drexler, H.G. Epstein-Barr virus (EBV) activates NKL homeobox gene HLX in DLBCL. PLoS ONE 2019, 14, e0216898. [Google Scholar] [CrossRef]
- Montes-Moreno, S.; Odqvist, L.; Diaz-Perez, J.A.; Lopez, A.B.; de Villambrosia, S.G.; Mazorra, F.; Castillo, M.E.; Lopez, M.; Pajares, R.; Garcia, J.F.; et al. EBV-positive diffuse large B-cell lymphoma of the elderly is an aggressive post-germinal center B-cell neoplasm characterized by prominent nuclear factor-kB activation. Mod. Pathol. 2012, 25, 968–982. [Google Scholar] [CrossRef] [Green Version]
- Chabay, P. Advances in the Pathogenesis of EBV-Associated Diffuse Large B Cell Lymphoma. Cancers 2021, 13, 2717. [Google Scholar] [CrossRef]
- Murthy, S.L.; Hitchcock, M.A.; Endicott-Yazdani, T.R.; Watson, J.T.; Krause, J.R. Epstein-Barr virus-positive diffuse large B-cell lymphoma. Bayl. Univ. Med. Cent. Proc. 2017, 30, 443–444. [Google Scholar] [CrossRef] [Green Version]
- Pei, Y.; Wong, J.H.Y.; Robertson, E.S. Targeted Therapies for Epstein-Barr Virus-Associated Lymphomas. Cancers 2020, 12, 2565. [Google Scholar] [CrossRef]
- Yoon, S.E.; Kim, S.J.; Yoon, D.H.; Koh, Y.; Mun, Y.C.; Do, Y.R.; Choi, Y.S.; Yang, D.H.; Kim, M.K.; Lee, G.W.; et al. A phase II study of ibrutinib in combination with rituximab-cyclophosphamide-doxorubicin hydrochloride-vincristine sulfate-prednisone therapy in Epstein-Barr virus-positive, diffuse large B cell lymphoma (54179060LYM2003: IVORY study): Results of the final analysis. Ann. Hematol. 2020, 99, 1283–1291. [Google Scholar] [CrossRef] [Green Version]
- Tilly, H.; Morschhauser, F.; Sehn, L.H.; Friedberg, J.W.; Trneny, M.; Sharman, J.P.; Herbaux, C.; Burke, J.M.; Matasar, M.; Rai, S.; et al. Polatuzumab Vedotin in Previously Untreated Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2022, 386, 351–363. [Google Scholar] [CrossRef]
- Zou, P.; Kawada, J.; Pesnicak, L.; Cohen, J.I. Bortezomib induces apoptosis of Epstein-Barr virus (EBV)-transformed B cells and prolongs survival of mice inoculated with EBV-transformed B cells. J. Virol. 2007, 81, 10029–10036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.H.; Kosek, J.; Wang, M.; Heise, C.; Schafer, P.H.; Chopra, R. Lenalidomide efficacy in activated B-cell-like subtype diffuse large B-cell lymphoma is dependent upon IRF4 and cereblon expression. Br. J. Haematol. 2013, 160, 487–502. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, E.; Lakhotia, R.; Pittaluga, S.; Muppidi, J.; Phelan, J.; Evans, S.; Pradhan, A.; Hillsman, A.; Steinberg, S.; Jaffe, E.S.; et al. Phase 2 Study of Nivolumab in Epstein-Barr Virus (EBV)-Positive Lymphoproliferative Disorders and EBV-Positive Non-Hodgkin Lymphomas. Blood 2021, 138, 4504. [Google Scholar] [CrossRef]
- Leen, A.M.; Rooney, C.M.; Foster, A.E. Improving T cell therapy for cancer. Annu. Rev. Immunol. 2007, 25, 243–265. [Google Scholar] [CrossRef] [PubMed]
- Sattva, N.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.; Miklos, D.; Jacobson, C.; Braunshweig, I.; Oluwole, O.; Siddiqui, T.; Lin, Y.; et al. Kte-C19 (anti-CD19 CAR T Cells) Induces Complete Remissions in Patients with Refractory Diffuse Large B-Cell Lymphoma (DLBCL): Results from the Pivotal Phase 2 Zuma-1. Blood 2016, 128, LBA-6. [Google Scholar]
- Wudhikarn, K.; Alarcon Tomas, A.; Flynn, J.R.; Devlin, S.M.; Brower, J.; Bachanova, V.; Nastoupil, L.J.; McGuirk, J.P.; Maziarz, R.T.; Oluwole, O.O.; et al. Low toxicity and excellent outcomes in patients with DLBCL without residual lymphoma at the time of CD19 CAR T-cell therapy. Blood Adv. 2022. [Google Scholar] [CrossRef] [PubMed]
- Viracta Therapeutics, Inc. An Open-Label, Phase 2 Trial of Nanatinostat in Combination with Valganciclovir in Patients With Epstein-Barr Virus-Positive (EBV+) Relapsed/Refractory Lymphomas. 2023. Available online: https://clinicaltrials.gov/ct2/show/NCT05011058 (accessed on 8 February 2023).
- Haverkos, B.M.; Alpdogan, O.; Baiocchi, R.; Brammer, J.E.; Feldman, T.; Capra, M.; Brem, E.A.; Nair, S.M.; Scheinberg, P.; Pereira, J.; et al. Nanatinostat (Nstat) and Valganciclovir (VGCV) in Relapsed/Refractory (R/R) Epstein-Barr Virus-Positive (EBV+) Lymphomas: Final Results from the Phase 1b/2 VT3996-201 Study. Blood 2021, 138, 623. [Google Scholar] [CrossRef]
- The First Affiliated Hospital with Nanjing Medical University. Sintilimab in Combination With R-CHOP in Patients With Treatment-Naive EBV-Positive DLBCL, NOS. 2023. Available online: https://clinicaltrials.gov/ct2/show/NCT04181489 (accessed on 9 February 2023).
- Aggarwal, P.; Limaiem, F. Reed sternberg cells. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Murray, P.G.; Young, L.S. An etiological role for the Epstein-Barr virus in the pathogenesis of classical Hodgkin lymphoma. Blood 2019, 134, 591–596. [Google Scholar] [CrossRef]
- Stein, H.; Herbst, H.; Anagnostopoulos, I.; Niedobitek, G.; Dallenbach, F.; Kratzsch, H.C. The nature of Hodgkin and Reed-Sternberg cells, their association with EBV, and their relationship to anaplastic large-cell lymphoma. Ann. Oncol. 1991, 2 (Suppl. S2), 33–38. [Google Scholar] [CrossRef]
- Weniger, M.A.; Kuppers, R. Molecular biology of Hodgkin lymphoma. Leukemia 2021, 35, 968–981. [Google Scholar] [CrossRef]
- Mundo, L.; Del Porro, L.; Granai, M.; Siciliano, M.C.; Mancini, V.; Santi, R.; Marcar, L.; Vrzalikova, K.; Vergoni, F.; Di Stefano, G.; et al. Correction: Frequent traces of EBV infection in Hodgkin and non-Hodgkin lymphomas classified as EBV-negative by routine methods: Expanding the landscape of EBV-related lymphomas. Mod. Pathol. 2020, 33, 2637. [Google Scholar] [CrossRef] [PubMed]
- Huppmann, A.R.; Nicolae, A.; Slack, G.W.; Pittaluga, S.; Davies-Hill, T.; Ferry, J.A.; Harris, N.L.; Jaffe, E.S.; Hasserjian, R.P. EBV may be expressed in the LP cells of nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL) in both children and adults. Am. J. Surg. Pathol. 2014, 38, 316–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vockerodt, M.; Morgan, S.L.; Kuo, M.; Wei, W.; Chukwuma, M.B.; Arrand, J.R.; Kube, D.; Gordon, J.; Young, L.S.; Woodman, C.B.; et al. The Epstein-Barr virus oncoprotein, latent membrane protein-1, reprograms germinal centre B cells towards a Hodgkin’s Reed-Sternberg-like phenotype. J. Pathol. 2008, 216, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Hertel, C.B.; Zhou, X.G.; Hamilton-Dutoit, S.J.; Junker, S. Loss of B cell identity correlates with loss of B cell-specific transcription factors in Hodgkin/Reed-Sternberg cells of classical Hodgkin lymphoma. Oncogene 2002, 21, 4908–4920. [Google Scholar] [CrossRef] [Green Version]
- Aravinth, S.P.; Rajendran, S.; Li, Y.; Wu, M.; Yi Wong, A.H.; Schwarz, H. Epstein-Barr virus-encoded LMP1 induces ectopic CD137 expression on Hodgkin and Reed-Sternberg cells via the PI3K-AKT-mTOR pathway. Leuk. Lymphoma 2019, 60, 2697–2704. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.T.; Pang, W.L.; Chong, S.M.; Castella, A.; Al-Salam, S.; Tan, T.E.; Moh, M.C.; Koh, L.K.; Gan, S.U.; Cheng, C.K.; et al. Expression of CD137 on Hodgkin and Reed-Sternberg cells inhibits T-cell activation by eliminating CD137 ligand expression. Cancer Res. 2013, 73, 652–661. [Google Scholar] [CrossRef] [Green Version]
- Jorapur, A.; Marshall, L.A.; Jacobson, S.; Xu, M.; Marubayashi, S.; Zibinsky, M.; Hu, D.X.; Robles, O.; Jackson, J.J.; Baloche, V.; et al. EBV+ tumors exploit tumor cell-intrinsic and -extrinsic mechanisms to produce regulatory T cell-recruiting chemokines CCL17 and CCL22. PLoS Pathog. 2022, 18, e1010200. [Google Scholar] [CrossRef]
- Nakayama, T.; Hieshima, K.; Nagakubo, D.; Sato, E.; Nakayama, M.; Kawa, K.; Yoshie, O. Selective induction of Th2-attracting chemokines CCL17 and CCL22 in human B cells by latent membrane protein 1 of Epstein-Barr virus. J. Virol. 2004, 78, 1665–1674. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, T.; Matsuo, K.; Hashida, Y.; Kitahata, K.; Ujihara, T.; Taniguchi, A.; Yoshie, O.; Nakayama, T.; Daibata, M. Epstein-Barr virus-positive pyothorax-associated lymphoma expresses CCL17 and CCL22 chemokines that attract CCR4-expressing regulatory T cells. Cancer Lett. 2019, 453, 184–192. [Google Scholar] [CrossRef]
- Lin, H.C.; Chang, Y.; Chen, R.Y.; Hung, L.Y.; Chen, P.C.; Chen, Y.P.; Medeiros, L.J.; Chiang, P.M.; Chang, K.C. Epstein-Barr virus latent membrane protein-1 upregulates autophagy and promotes viability in Hodgkin lymphoma: Implications for targeted therapy. Cancer Sci. 2021, 112, 1589–1602. [Google Scholar] [CrossRef]
- Wu, J.; Liu, H.; Hu, T.; Wang, S. Gene expression trend changes in breast cancer populations over two decades: Insights from The Cancer Genome Atlas database. Hereditas 2022, 159, 18. [Google Scholar] [CrossRef] [PubMed]
- Nagai, L.A.E.; Park, S.J.; Nakai, K. Analyzing the 3D chromatin organization coordinating with gene expression regulation in B-cell lymphoma. BMC Med. Genom. 2019, 11, 127. [Google Scholar] [CrossRef]
- Spina, V.; Bruscaggin, A.; Cuccaro, A.; Martini, M.; Di Trani, M.; Forestieri, G.; Manzoni, M.; Condoluci, A.; Arribas, A.; Terzi-Di-Bergamo, L.; et al. Circulating tumor DNA reveals genetics, clonal evolution, and residual disease in classical Hodgkin lymphoma. Blood 2018, 131, 2413–2425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portis, T.; Dyck, P.; Longnecker, R. Epstein-Barr Virus (EBV) LMP2A induces alterations in gene transcription similar to those observed in Reed-Sternberg cells of Hodgkin lymphoma. Blood 2003, 102, 4166–4178. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.J.; Longnecker, R. Epstein-Barr virus latent membrane protein 2A exploits Notch1 to alter B-cell identity in vivo. Blood 2009, 113, 108–116. [Google Scholar] [CrossRef] [Green Version]
- Incrocci, R.; McAloon, J.; Montesano, M.; Bardahl, J.; Vagvala, S.; Stone, A.; Swanson-Mungerson, M. Epstein-Barr virus LMP2A utilizes Syk and PI3K to activate NF-kappaB in B-cell lymphomas to increase MIP-1alpha production. J. Med. Virol. 2019, 91, 845–855. [Google Scholar] [CrossRef]
- Vrzalikova, K.; Pugh, M.; Mundo, L.; Murray, P. The contributions of ebv to the pathogenesis of classical hodgkin lymphoma. Ann. Lymphoma 2021, 5, 30. [Google Scholar] [CrossRef]
- Flavell, J.R.; Baumforth, K.R.; Wood, V.H.; Davies, G.L.; Wei, W.; Reynolds, G.M.; Morgan, S.; Boyce, A.; Kelly, G.L.; Young, L.S.; et al. Down-regulation of the TGF-beta target gene, PTPRK, by the Epstein-Barr virus encoded EBNA1 contributes to the growth and survival of Hodgkin lymphoma cells. Blood 2008, 111, 292–301. [Google Scholar] [CrossRef]
- Baumforth, K.R.; Birgersdotter, A.; Reynolds, G.M.; Wei, W.; Kapatai, G.; Flavell, J.R.; Kalk, E.; Piper, K.; Lee, S.; Machado, L.; et al. Expression of the Epstein-Barr virus-encoded Epstein-Barr virus nuclear antigen 1 in Hodgkin’s lymphoma cells mediates Up-regulation of CCL20 and the migration of regulatory T cells. Am. J. Pathol. 2008, 173, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Ansell, S.M. Hodgkin Lymphoma: Diagnosis and Treatment. Mayo Clin. Proc. 2015, 90, 1574–1583. [Google Scholar] [CrossRef] [Green Version]
- Engert, A. Treatment of elderly Hodgkin lymphoma patients. Hematol. Oncol. 2019, 37 (Suppl. S1), 92–94. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, R.F.; Stark, G.L.; White, J.; Angus, B.; Alexander, F.E.; Krajewski, A.S.; Freeland, J.; Taylor, G.M.; Taylor, P.R.; for the Scotland and Newcastle Epidemiology of Hodgkin Disease Study Group; et al. Impact of tumor Epstein-Barr virus status on presenting features and outcome in age-defined subgroups of patients with classic Hodgkin lymphoma: A population-based study. Blood 2005, 106, 2444–2451. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Rodig, S.; Juszczynski, P.; Ouyang, J.; Sinha, P.; O’Donnell, E.; Neuberg, D.; Shipp, M.A. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: Implications for targeted therapy. Clin. Cancer Res. 2012, 18, 1611–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prince, H.M.; Hutchings, M.; Domingo-Domenech, E.; Eichenauer, D.A.; Advani, R. Anti-CD30 antibody-drug conjugate therapy in lymphoma: Current knowledge, remaining controversies, and future perspectives. Ann. Hematol. 2023, 102, 13–29. [Google Scholar] [CrossRef]
- Advani, R.H.; Moskowitz, A.J.; Bartlett, N.L.; Vose, J.M.; Ramchandren, R.; Feldman, T.A.; LaCasce, A.S.; Christian, B.A.; Ansell, S.M.; Moskowitz, C.H.; et al. Brentuximab vedotin in combination with nivolumab in relapsed or refractory Hodgkin lymphoma: 3-year study results. Blood 2021, 138, 427–438. [Google Scholar] [CrossRef]
- Kersten, M.J.; Driessen, J.; Zijlstra, J.M.; Plattel, W.J.; Morschhauser, F.; Lugtenburg, P.J.; Brice, P.; Hutchings, M.; Gastinne, T.; Liu, R.; et al. Combining brentuximab vedotin with dexamethasone, high-dose cytarabine and cisplatin as salvage treatment in relapsed or refractory Hodgkin lymphoma: The phase II HOVON/LLPC Transplant BRaVE study. Haematologica 2021, 106, 1129–1137. [Google Scholar] [CrossRef] [Green Version]
- Straus, D.J.; Dlugosz-Danecka, M.; Alekseev, S.; Illes, A.; Picardi, M.; Lech-Maranda, E.; Feldman, T.; Smolewski, P.; Savage, K.J.; Bartlett, N.L.; et al. Brentuximab vedotin with chemotherapy for stage III/IV classical Hodgkin lymphoma: 3-year update of the ECHELON-1 study. Blood 2020, 135, 735–742. [Google Scholar] [CrossRef]
- Meier, J.A.; Savoldo, B.; Grover, N.S. The Emerging Role of CAR T Cell Therapy in Relapsed/Refractory Hodgkin Lymphoma. J. Pers. Med. 2022, 12, 197. [Google Scholar] [CrossRef]
- Ramos, C.A.; Grover, N.S.; Beaven, A.W.; Lulla, P.D.; Wu, M.F.; Ivanova, A.; Wang, T.; Shea, T.C.; Rooney, C.M.; Dittus, C.; et al. Anti-CD30 CAR-T Cell Therapy in Relapsed and Refractory Hodgkin Lymphoma. J. Clin. Oncol. 2020, 38, 3794–3804. [Google Scholar] [CrossRef]
- Zhang, P.; Yang, X.; Cao, Y.; Wang, J.; Zhou, M.; Chen, L.; Wei, J.; Mao, Z.; Wang, D.; Xiao, Y.; et al. Autologous stem cell transplantation in tandem with Anti-CD30 CAR T-cell infusion in relapsed/refractory CD30(+) lymphoma. Exp. Hematol. Oncol. 2022, 11, 72. [Google Scholar] [CrossRef]
- Taylor, G.S.; Long, H.M.; Brooks, J.M.; Rickinson, A.B.; Hislop, A.D. The immunology of Epstein-Barr virus-induced disease. Annu. Rev. Immunol. 2015, 33, 787–821. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Snapper, C.M. Epstein Barr Virus: Development of Vaccines and Immune Cell Therapy for EBV-Associated Diseases. Front. Immunol. 2021, 12, 734471. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Xie, C.; Lung, H.L.; Lo, K.W.; Law, G.L.; Mak, N.K.; Wong, K.L. EBNA1-targeted inhibitors: Novel approaches for the treatment of Epstein-Barr virus-associated cancers. Theranostics 2018, 8, 5307–5319. [Google Scholar] [CrossRef] [PubMed]
- Roschewski, M.; Staudt, L.M.; Wilson, W.H. Burkitt’s Lymphoma. N. Engl. J. Med. 2022, 387, 1111–1122. [Google Scholar] [CrossRef]
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus Particles in Cultured Lymphoblasts from Burkitt’s Lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef]
- Shannon-Lowe, C.; Rickinson, A. The Global Landscape of EBV-Associated Tumors. Front. Oncol. 2019, 9, 713. [Google Scholar] [CrossRef] [Green Version]
- Summerauer, A.M.; Jaggi, V.; Ogwang, R.; Traxel, S.; Colombo, L.; Amundsen, E.; Eyer, T.; Subramanian, B.; Fehr, J.; Mantel, P.Y.; et al. Epstein-Barr virus and malaria upregulate AID and APOBEC3 enzymes, but only AID seems to play a major mutagenic role in Burkitt lymphoma. Eur. J. Immunol. 2022, 52, 1273–1284. [Google Scholar] [CrossRef] [PubMed]
- Rochford, R. Reframing Burkitt lymphoma: Virology not epidemiology defines clinical variants. Ann. Lymphoma 2021, 5, 22. [Google Scholar] [CrossRef]
- Noh, K.W.; Park, J.; Kang, M.S. Targeted disruption of EBNA1 in EBV-infected cells attenuated cell growth. BMB Rep. 2016, 49, 226–231. [Google Scholar] [CrossRef]
- Wang, R.; Wang, J.; Zhang, N.; Wan, Y.; Liu, Y.; Zhang, L.; Pan, S.; Zhang, C.; Zhang, H.; Cao, Y. The interaction between Vav1 and EBNA1 promotes survival of Burkitt’s lymphoma cells by down-regulating the expression of Bim. Biochem. Biophys. Res. Commun. 2019, 511, 787–793. [Google Scholar] [CrossRef]
- Prieto-Sanchez, R.M.; Hernandez, J.A.; Garcia, J.L.; Gutierrez, N.C.; San Miguel, J.; Bustelo, X.R.; Hernandez, J.M. Overexpression of the VAV proto-oncogene product is associated with B-cell chronic lymphocytic leukaemia displaying loss on 13q. Br. J. Haematol. 2006, 133, 642–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderton, E.; Yee, J.; Smith, P.; Crook, T.; White, R.E.; Allday, M.J. Two Epstein-Barr virus (EBV) oncoproteins cooperate to repress expression of the proapoptotic tumour-suppressor Bim: Clues to the pathogenesis of Burkitt’s lymphoma. Oncogene 2008, 27, 421–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccaluga, P.P.; Navari, M.; De Falco, G.; Ambrosio, M.R.; Lazzi, S.; Fuligni, F.; Bellan, C.; Rossi, M.; Sapienza, M.R.; Laginestra, M.A.; et al. Virus-encoded microRNA contributes to the molecular profile of EBV-positive Burkitt lymphomas. Oncotarget 2016, 7, 224–240. [Google Scholar] [CrossRef] [PubMed]
- Vereide, D.T.; Seto, E.; Chiu, Y.F.; Hayes, M.; Tagawa, T.; Grundhoff, A.; Hammerschmidt, W.; Sugden, B. Epstein-Barr virus maintains lymphomas via its miRNAs. Oncogene 2014, 33, 1258–1264. [Google Scholar] [CrossRef] [Green Version]
- Asadi, M.; Taghizadeh, S.; Kaviani, E.; Vakili, O.; Taheri-Anganeh, M.; Tahamtan, M.; Savardashtaki, A. Caspase-3: Structure, function, and biotechnological aspects. Biotechnol. Appl. Biochem. 2022, 69, 1633–1645. [Google Scholar] [CrossRef]
- Harold, C.; Cox, D.; Riley, K.J. Epstein-Barr viral microRNAs target caspase 3. Virol. J. 2016, 13, 145. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.M.; Yu, Y.; Zhao, H.P. EBV-BART-6-3p and cellular microRNA-197 compromise the immune defense of host cells in EBV-positive Burkitt lymphoma. Mol. Med. Rep. 2017, 15, 1877–1883. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Bu, Y.; Liang, Y.; Zhang, F.; Zhang, H.; Li, S. Epstein-Barr Virus (EBV)-BamHI-A Rightward Transcript (BART)-6 and Cellular MicroRNA-142 Synergistically Compromise Immune Defense of Host Cells in EBV-Positive Burkitt Lymphoma. Med. Sci. Monit. 2016, 22, 4114–4120. [Google Scholar] [CrossRef] [Green Version]
- Yehia, L.; Keel, E.; Eng, C. The Clinical Spectrum of PTEN Mutations. Annu. Rev. Med. 2020, 71, 103–116. [Google Scholar] [CrossRef] [Green Version]
- Crombie, J.; LaCasce, A. The treatment of Burkitt lymphoma in adults. Blood 2021, 137, 743–750. [Google Scholar] [CrossRef]
- Evens, A.M.; Danilov, A.; Jagadeesh, D.; Sperling, A.; Kim, S.H.; Vaca, R.; Wei, C.; Rector, D.; Sundaram, S.; Reddy, N.; et al. Burkitt lymphoma in the modern era: Real-world outcomes and prognostication across 30 US cancer centers. Blood 2021, 137, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Lv, K.; Yin, T.; Yu, M.; Chen, Z.; Zhou, Y.; Li, F. Treatment Advances in EBV Related Lymphoproliferative Diseases. Front. Oncol. 2022, 12, 838817. [Google Scholar] [CrossRef]
- Maramattom, L.V.; Hari, P.; Burns, L.J.; Carreras, J.; Arcese, W.; Cairo, M.S.; Costa, L.; Fenske, T.; Lill, M.; Freytes, C.; et al. Autologous and allogeneic transplantation for burkitt lymphoma outcomes and changes in utilization: A report from the center for international blood and marrow transplant research. Biol. Blood Marrow Transplant. 2013, 19, 173–179. [Google Scholar] [CrossRef] [Green Version]
- Short, N.J.; Kantarjian, H.M.; Ko, H.; Khoury, J.D.; Ravandi, F.; Thomas, D.A.; Garcia-Manero, G.; Khouri, M.; Cortes, J.E.; Wierda, W.G.; et al. Outcomes of adults with relapsed or refractory Burkitt and high-grade B-cell leukemia/lymphoma. Am. J. Hematol. 2017, 92, E114–E117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avigdor, A.; Shouval, R.; Jacoby, E.; Davidson, T.; Shimoni, A.; Besser, M.; Nagler, A. CAR T cells induce a complete response in refractory Burkitt Lymphoma. Bone Marrow Transpl. 2018, 53, 1583–1585. [Google Scholar] [CrossRef]
- Dalton, T.; Doubrovina, E.; Pankov, D.; Reynolds, R.; Scholze, H.; Selvakumar, A.; Vizconde, T.; Savalia, B.; Dyomin, V.; Weigel, C.; et al. Epigenetic reprogramming sensitizes immunologically silent EBV+ lymphomas to virus-directed immunotherapy. Blood 2020, 135, 1870–1881. [Google Scholar] [CrossRef]
- Wang, H.; Fu, B.B.; Gale, R.P.; Liang, Y. NK-/T-cell lymphomas. Leukemia 2021, 35, 2460–2468. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [Green Version]
- Montes-Mojarro, I.A.; Fend, F.; Quintanilla-Martinez, L. EBV and the Pathogenesis of NK/T Cell Lymphoma. Cancers 2021, 13, 1414. [Google Scholar] [CrossRef]
- Feng, J.; Chen, Q.; Zhang, P.; Huang, X.; Xie, W.; Zhang, H.; Yao, P. Latent Membrane Protein 1 Promotes Tumorigenesis Through Upregulation of PGC1beta Signaling Pathway. Stem Cell Rev. Rep. 2021, 17, 1486–1499. [Google Scholar] [CrossRef]
- Deblois, G.; St-Pierre, J.; Giguere, V. The PGC-1/ERR signaling axis in cancer. Oncogene 2013, 32, 3483–3490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Li, L.; Chen, Q.; Li, M.; Feng, J.; Sun, Y.; Zhao, R.; Zhu, Y.; Lv, Y.; Zhu, Z.; et al. PGC1beta regulates multiple myeloma tumor growth through LDHA-mediated glycolytic metabolism. Mol. Oncol. 2018, 12, 1579–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Feng, J.; Wu, J.; Yu, Z.; Zhang, W.; Chen, Y.; Yao, P.; Zhang, H. HKDC1 C-terminal based peptides inhibit extranodal natural killer/T-cell lymphoma by modulation of mitochondrial function and EBV suppression. Leukemia 2020, 34, 2736–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Zhao, Y.; Shi, H.; Ma, C.; Wei, L. LMP1 promotes nasal NK/T-cell lymphoma cell function by eIF4E via NF-kappaB pathway. Oncol. Rep. 2015, 34, 3264–3271. [Google Scholar] [CrossRef] [Green Version]
- Dong, Q.; Dong, L.; Liu, S.; Kong, Y.; Zhang, M.; Wang, X. Tumor-Derived Exosomal eIF4E as a Biomarker for Survival Prediction in Patients with Non-Small Cell Lung Cancer. Med. Sci. Monit. 2020, 26, e923210. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Sun, H.; Li, Y.; Bai, X.; Dong, X.; Zhao, N.; Meng, J.; Sun, B.; Zhang, D. High expression of eIF4E is associated with tumor macrophage infiltration and leads to poor prognosis in breast cancer. BMC Cancer 2021, 21, 1305. [Google Scholar] [CrossRef] [PubMed]
- Komabayashi, Y.; Kishibe, K.; Nagato, T.; Ueda, S.; Takahara, M.; Harabuchi, Y. Downregulation of miR-15a due to LMP1 promotes cell proliferation and predicts poor prognosis in nasal NK/T-cell lymphoma. Am. J. Hematol. 2014, 89, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Ishii, H.; Takahara, M.; Nagato, T.; Kis, L.L.; Nagy, N.; Kishibe, K.; Harabuchi, Y.; Klein, E. Monocytes enhance cell proliferation and LMP1 expression of nasal natural killer/T-cell lymphoma cells by cell contact-dependent interaction through membrane-bound IL-15. Int. J. Cancer 2012, 130, 48–58. [Google Scholar] [CrossRef]
- Tse, E.; Zhao, W.L.; Xiong, J.; Kwong, Y.L. How we treat NK/T-cell lymphomas. J. Hematol. Oncol. 2022, 15, 74. [Google Scholar] [CrossRef]
- Ando, M.; Sugimoto, K.; Kitoh, T.; Sasaki, M.; Mukai, K.; Ando, J.; Egashira, M.; Schuster, S.M.; Oshimi, K. Selective apoptosis of natural killer-cell tumours by l-asparaginase. Br. J. Haematol. 2005, 130, 860–868. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. T-Cell Lymphomas (Version 1.2023). Available online: https://www.nccn.org/professionals/physician_gls/pdf/t-cell.pdf (accessed on 25 February 2023).
- Kim, W.S.; Oki, Y.; Kim, S.J.; Yoon, S.E.; Ardeshna, K.M.; Lin, Y.; Ruan, J.; Porcu, P.; Brammer, J.E.; Jacobsen, E.D.; et al. Autologous EBV-specific T cell treatment results in sustained responses in patients with advanced extranodal NK/T lymphoma: Results of a multicenter study. Ann. Hematol. 2021, 100, 2529–2539. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, L.P.; Rouce, R.; Gottschalk, S.; Torrano, V.; Carrum, G.; Wu, M.F.; Hoq, F.; Grilley, B.; Marcogliese, A.M.; Hanley, P.J.; et al. EBV/LMP-specific T cells maintain remissions of T- and B-cell EBV lymphomas after allogeneic bone marrow transplantation. Blood 2018, 132, 2351–2361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanate, A.S.; DiGilio, A.; Ahn, K.W.; Al Malki, M.; Jacobsen, E.; Steinberg, A.; Hamerschlak, N.; Kharfan-Dabaja, M.; Salit, R.; Ball, E.; et al. Allogeneic haematopoietic cell transplantation for extranodal natural killer/T-cell lymphoma, nasal type: A CIBMTR analysis. Br. J. Haematol. 2018, 182, 916–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.G.; Kim, N.; Sohn, H.J.; Lee, S.K.; Oh, S.T.; Lee, H.J.; Cho, H.I.; Yim, H.W.; Jung, S.E.; Park, G.; et al. Long-term Outcome of Extranodal NK/T Cell Lymphoma Patients Treated With Postremission Therapy Using EBV LMP1 and LMP2a-specific CTLs. Mol. Ther. 2015, 23, 1401–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grommes, C.; DeAngelis, L.M. Primary CNS Lymphoma. J. Clin. Oncol. 2017, 35, 2410–2418. [Google Scholar] [CrossRef] [PubMed]
- Henkenberens, C.; Franzke, A.; Raab, P.; Oschlies, I.; Klapper, W.; Christiansen, H. Primary EBV-positive Hodgkin’s lymphoma of the CNS under azathioprine treatment: Case report and review of the literature. Strahlenther. Onkol. 2014, 190, 847–852. [Google Scholar] [CrossRef]
- Radke, J.; Ishaque, N.; Koll, R.; Gu, Z.; Schumann, E.; Sieverling, L.; Uhrig, S.; Hubschmann, D.; Toprak, U.H.; Lopez, C.; et al. The genomic and transcriptional landscape of primary central nervous system lymphoma. Nat. Commun. 2022, 13, 2558. [Google Scholar] [CrossRef]
- Prado, I.P.; Barbiero, F.; Baehring, J.; Becker, K.; Corbin, Z. The impact of Epstein-Barr virus status on primary CNS lymphoma survival. J. Clin. Oncol. 2019, 37, e13528. [Google Scholar] [CrossRef]
- Ferreri, A.J.M.; Calimeri, T.; Conte, G.M.; Cattaneo, D.; Fallanca, F.; Ponzoni, M.; Scarano, E.; Curnis, F.; Nonis, A.; Lopedote, P.; et al. R-CHOP preceded by blood-brain barrier permeabilization with engineered tumor necrosis factor-alpha in primary CNS lymphoma. Blood 2019, 134, 252–262. [Google Scholar] [CrossRef]
- Grommes, C.; Pastore, A.; Palaskas, N.; Tang, S.S.; Campos, C.; Schartz, D.; Codega, P.; Nichol, D.; Clark, O.; Hsieh, W.Y.; et al. Ibrutinib Unmasks Critical Role of Bruton Tyrosine Kinase in Primary CNS Lymphoma. Cancer Discov. 2017, 7, 1018–1029. [Google Scholar] [CrossRef] [Green Version]
- Nayak, L.; Abrey, L.E.; Drappatz, J.; Gilbert, M.R.; Reardon, D.A.; Wen, P.Y.; Prados, M.; Deangelis, L.M.; Omuro, A.; for the North American Brain Tumor Consortium. Multicenter phase II study of rituximab and temozolomide in recurrent primary central nervous system lymphoma. Leuk Lymphoma 2013, 54, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Raizer, J.J.; Rademaker, A.; Evens, A.M.; Rice, L.; Schwartz, M.; Chandler, J.P.; Getch, C.C.; Tellez, C.; Grimm, S.A. Pemetrexed in the treatment of relapsed/refractory primary central nervous system lymphoma. Cancer 2012, 118, 3743–3748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, L.; Thiel, E.; Klasen, H.A.; Birkmann, J.; Jahnke, K.; Martus, P.; Korfel, A. Prospective trial on topotecan salvage therapy in primary CNS lymphoma. Ann. Oncol. 2006, 17, 1141–1145. [Google Scholar] [CrossRef]
- Reni, M.; Zaja, F.; Mason, W.; Perry, J.; Mazza, E.; Spina, M.; Bordonaro, R.; Ilariucci, F.; Faedi, M.; Corazzelli, G.; et al. Temozolomide as salvage treatment in primary brain lymphomas. Br. J. Cancer 2007, 96, 864–867. [Google Scholar] [CrossRef] [Green Version]
- Batchelor, T.T.; Grossman, S.A.; Mikkelsen, T.; Ye, X.; Desideri, S.; Lesser, G.J. Rituximab monotherapy for patients with recurrent primary CNS lymphoma. Neurology 2011, 76, 929–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haque, T.; Wilkie, G.M.; Jones, M.M.; Higgins, C.D.; Urquhart, G.; Wingate, P.; Burns, D.; McAulay, K.; Turner, M.; Bellamy, C.; et al. Allogeneic cytotoxic T-cell therapy for EBV-positive posttransplantation lymphoproliferative disease: Results of a phase 2 multicenter clinical trial. Blood 2007, 110, 1123–1131. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.E.; Moss, D.J. Treatment options for post-transplant lymphoproliferative disorder and other Epstein-Barr virus-associated malignancies. Tissue Antigens 2004, 63, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Slobod, K.S.; Taylor, G.H.; Sandlund, J.T.; Furth, P.; Helton, K.J.; Sixbey, J.W. Epstein-Barr virus-targeted therapy for AIDS-related primary lymphoma of the central nervous system. Lancet 2000, 356, 1493–1494. [Google Scholar] [CrossRef] [PubMed]
- Rozman, M.; Korac, P.; Jambrosic, K.; Zidovec Lepej, S. Progress in Prophylactic and Therapeutic EBV Vaccine Development Based on Molecular Characteristics of EBV Target Antigens. Pathogens 2022, 11, 864. [Google Scholar] [CrossRef]
- Kerr, J.R. Epstein-Barr virus (EBV) reactivation and therapeutic inhibitors. J. Clin. Pathol. 2019, 72, 651–658. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Cancer | Therapy | Notes | Citation |
|---|---|---|---|
| DLBCL | Rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone (R-CHOP) | Rituximab: anti-CD20 monoclonal antibody Standard of care | [7] |
| R-CHOP + ibrutinib | Ibrutinib: BTK inhibitor included because LMP1 mimics BTK-dependent B-cell receptor | [103] | |
| Rituximab, cyclophosphamide, doxorubicin, polatuzumab vedotin, prednisone (pola-R-CHP) | Polatuzumab vedotin: anti-79b monoclonal antibody | [104] | |
| Bortezomib | Apoptosis of EBV-transformed B cells in mice | [105] | |
| Lenalidomide | Inhibits IRF4 and BCR-NF-κB | [106] | |
| Nivolumab | IgG4 monoclonal antibody targeting PD-1 | [107] | |
| Antigen-specific T cells | [108,109,110] | ||
| Nanatinostat + valganciclovir | Nanatinostat: selective for isoforms of class I HDACs | [111,112] | |
| R-CHOP + Sintilimab | Sintilimab: anti-PD-1 antibody | [113] | |
| Classic Hodgkin Lymphoma | Doxorubicin, bleomycin, vinblastine, dacarbazine (ABVD) | Standard of care, can be used in combination with radiation | [137] |
| Bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, prednisone (BEACOPP) | Standard of care, can be used in combination with radiation | [137] | |
| Nitrogen mustard, vincristine, procarbazine, prednisone (MOPP) | Standard of care, can be used in combination with radiation | [137] | |
| Anti-PD-1 pathway immune checkpoint inhibitors | [140] | ||
| Brentuximab vedotin | Anti-CD30 antibody | [141,142,143,144] | |
| T-cell therapy | [145,146,147,148] | ||
| EBV vaccination | [148] | ||
| Therapies targeting EBV latent proteins/EBNA1 inhibitors | [149,150] | ||
| Burkitt Lymphoma | Rituximab, etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin (R-EPOCH) | Standard of care Low dose adjusted form for elderly/HIV+ patients | [167,168,169] |
| Cyclophosphomide, doxorubicin, vincristine, methotrexate, ifosfamide cytarabine, etoposide (CODOX-M/IVAC) | Standard of care | [167,168,169] | |
| Hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone alternating with high-dose methotrexate and cytarabine (hyperCVAD) | Standard of care | [167,168,169] | |
| Autologous/allogenic hematopoietic stem cell transplant | Salvage therapy | [170] | |
| T-cell therapy | Case report of salvage therapy | [172] | |
| Decitabine | Epigenetic induction of immunogenic EBV proteins | [173] | |
| NK/T-cell lymphoma | Asparaginase | Key component of therapy; causes NK/T cell apoptosis | [186,187] |
| Steroid (dexamethasone), methotrexate, ifosfamide, pegaspargase, etoposide (SMILE) | Standard of care | [188] | |
| Gemcitabine, pegaspargase, oxaliplatin (P-GEMOX) | Standard of care | [188] | |
| Dexamethasone, cisplatin, gemcitabine, pegaspargase (DDGP) | Standard of care | [188] | |
| Balataleucel-T | Autologous EBV-specific CTLs; studied in salvage/adjuvant setting | [189] | |
| T-cell therapy targeting LMP | [190,192] | ||
| Allogeneic hematopoietic cell transplantation | [191] | ||
| Primary CNS lymphoma | Rituximab + high-dose methotrexate | Included in induction therapy | [193] |
| Rituximab, HD MTX, vincristine, procarbazine (R-MVP) | Induction chemotherapy | [193] | |
| Rituximab, HD MTX, temozolomide (R-MT) | Induction chemotherapy | [193] | |
| Thiotepa, rituximab, HD MTX, high-dose cytarabine (MATRix) | Induction chemotherapy | [193] | |
| Rituximab, temozolomide | Salvage chemotherapy | [199] | |
| Single agent salvage therapy: temozolomide, rituximab, pemetrexed, topotecan | [200,201,202,203] | ||
| Ibrutinib | BTK inhibitor | [198] | |
| EBV-specific CTLs | Examined in PTLD, but may be applicable in CNS lymphoma | [12,204,205] | |
| Hydroxyurea | Case report | [206] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sausen, D.G.; Basith, A.; Muqeemuddin, S. EBV and Lymphomagenesis. Cancers 2023, 15, 2133. https://doi.org/10.3390/cancers15072133
Sausen DG, Basith A, Muqeemuddin S. EBV and Lymphomagenesis. Cancers. 2023; 15(7):2133. https://doi.org/10.3390/cancers15072133
Chicago/Turabian StyleSausen, Daniel G., Ayeman Basith, and Syed Muqeemuddin. 2023. "EBV and Lymphomagenesis" Cancers 15, no. 7: 2133. https://doi.org/10.3390/cancers15072133