

SRSF5 Regulates the Expression of BQ323636.1 to Modulate Tamoxifen Resistance in ER-Positive Breast Cancer
 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mammalian Cell Culture, Transfection and Stable Cell Line Establishment
2.2. Chemicals, siRNA, shRNA and Plasmids
2.3. Real-Time qPCR and PCR-Based Splicing Assay
2.4. Cell Viability Assay
2.5. Western Blot and Co-Immunoprecipiation
2.6. RNA–Protein Interaction
2.7. Xenograft
2.8. Tissue Microarray and Immunohistochemistry
2.9. Statistical Analysis
3. Results
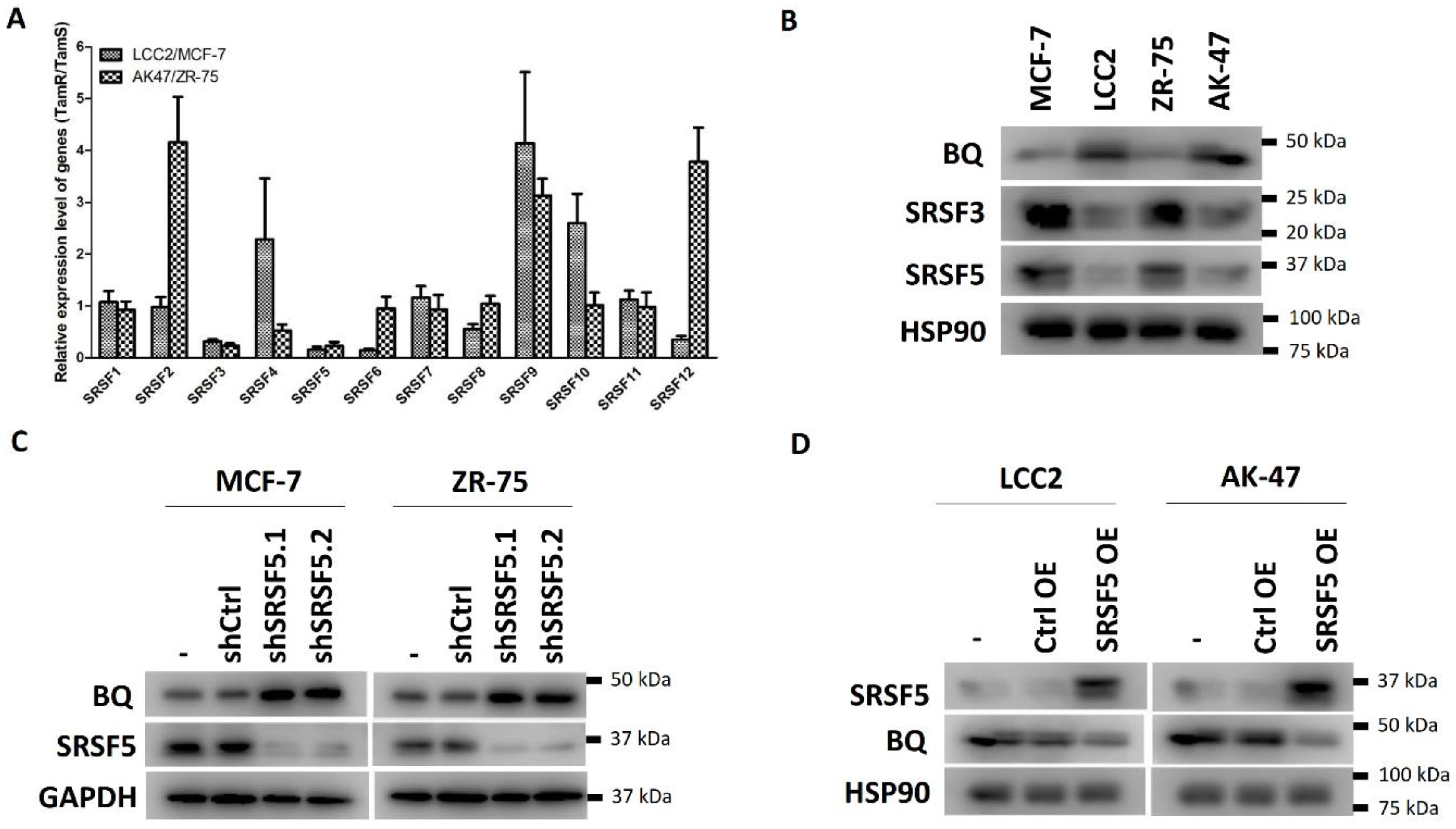
3.1. SRSF5 Modulated the Expression of BQ
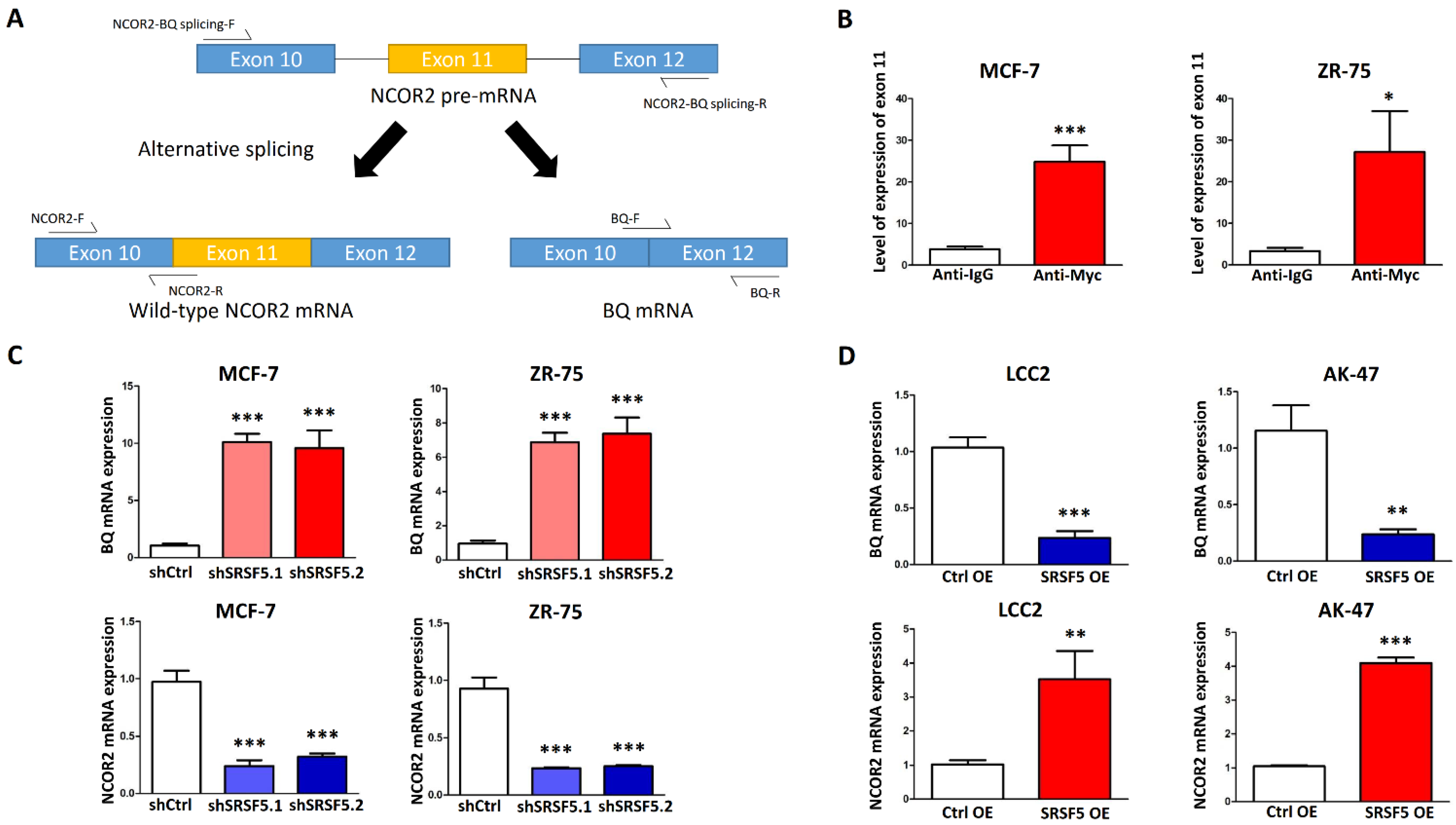
3.2. SRSF5 Bound to Exon 11 of NCOR2 to Favour the Synthesis of Wild-Type NCOR2 mRNA
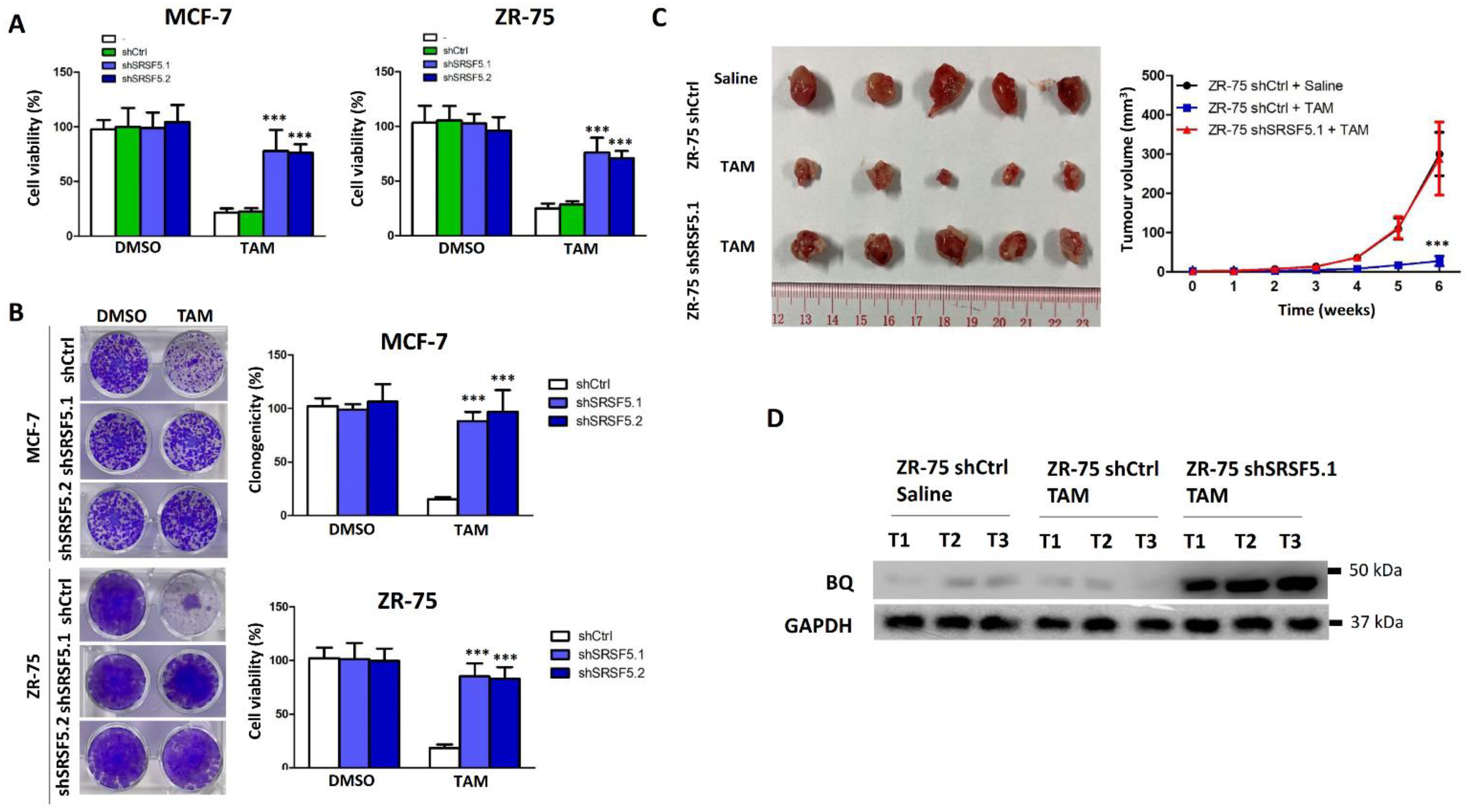
3.3. SRSF5 Could Modulate TAM Response
3.4. Clinical Significance of SRSF5 in ER +ve Breast Cancer
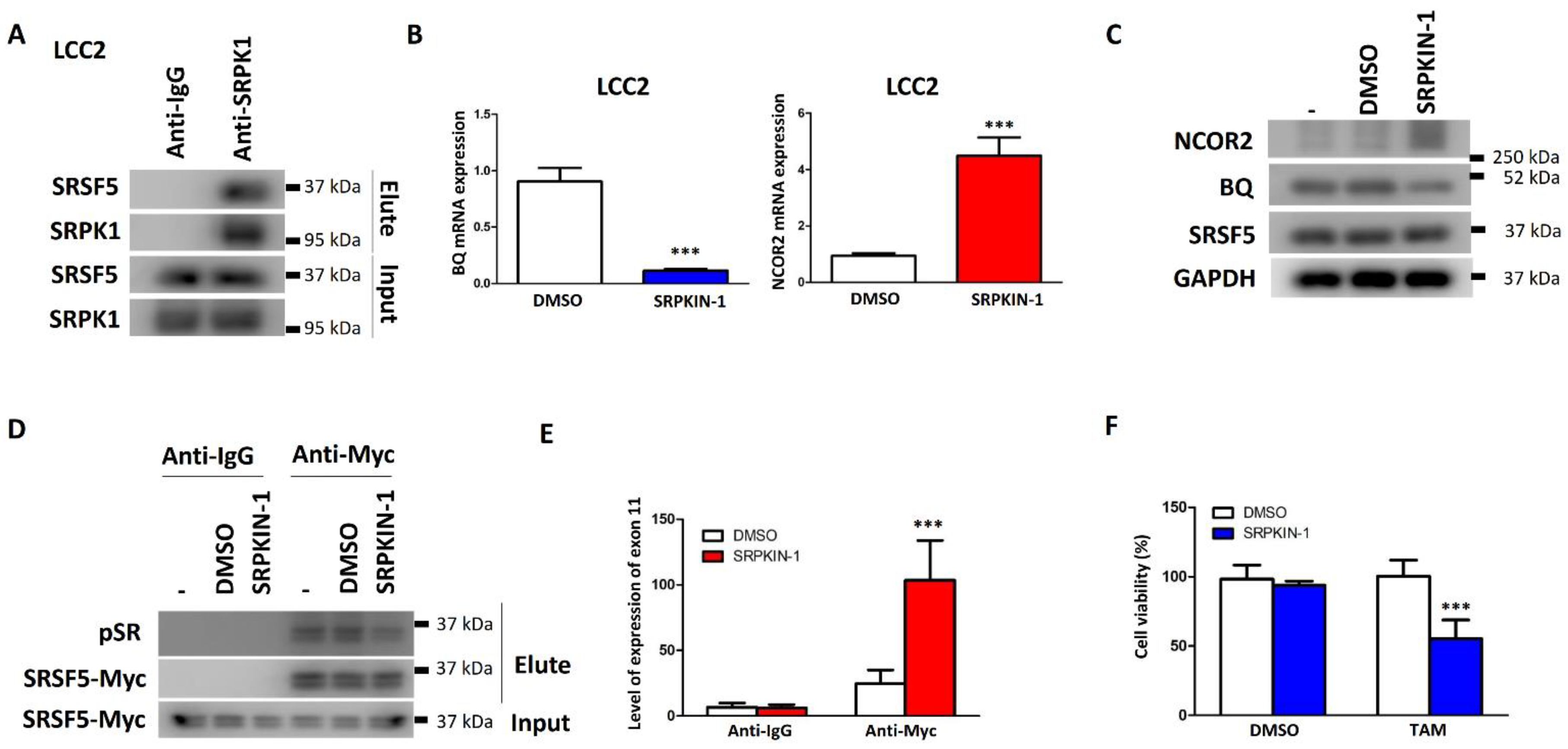
3.5. Inhibition of SRPK1 Could Reduce BQ Expression and TAM Resistance by Altering SRSF5 Activity
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yussof, I.; Mohd Tahir, N.A.; Hatah, E.; Mohamed Shah, N. Factors influencing five-year adherence to adjuvant endocrine therapy in breast cancer patients: A systematic review. Breast 2022, 62, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Davies, C.; Godwin, J.; Gray, R.; Clarke, M.; Darby, S.; McGale, P.; Wang, Y.C.; Peto, R.; Pan, H.C.; Cutter, D.; et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: Patient-level meta-analysis of randomised trials. Lancet 2011, 378, 771–784. [Google Scholar] [CrossRef] [Green Version]
- Ring, A.; Dowsett, M. Mechanisms of tamoxifen resistance. Endocr. Relat. Cancer 2004, 11, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, S.; Farahmand, L.; Teymourzadeh, A.; Majidzadeh-A, K. Clinical Evidence on the Magnitude of Change in Growth Pathway Activity in Relation to Tamoxifen Resistance is Required. Curr. Cancer Drug Targets 2018, 18, 668–676. [Google Scholar] [CrossRef]
- Jeselsohn, R.; Cornwell, M.; Pun, M.; Buchwalter, G.; Nguyen, M.; Bango, C.; Huang, Y.; Kuang, Y.N.; Paweletz, C.; Fu, X.Y.; et al. Embryonic transcription factor SOX9 drives breast cancer endocrine resistance. Proc. Natl. Acad. Sci. USA 2017, 114, E4482–E4491. [Google Scholar] [CrossRef] [Green Version]
- Piva, M.; Domenici, G.; Iriondo, O.; Rabano, M.; Simoes, B.M.; Comaills, V.; Barredo, I.; Lopez-Ruiz, J.A.; Zabalza, I.; Kypta, R.; et al. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Mol. Med. 2014, 6, 66–79. [Google Scholar] [CrossRef]
- Yu, P.; Li, A.X.; Chen, X.S.; Tian, M.; Wang, H.Y.; Wang, X.L.; Zhang, Y.; Wang, K.S.; Cheng, Y. PKM2-c-Myc-Survivin Cascade Regulates the Cell Proliferation, Migration, and Tamoxifen Resistance in Breast Cancer. Front. Pharmacol. 2020, 11, 550469. [Google Scholar] [CrossRef] [PubMed]
- Lui, A.; New, J.; Ogony, J.; Thomas, S.; Lewis-Wambi, J. Everolimus downregulates estrogen receptor and induces autophagy in aromatase inhibitor-resistant breast cancer cells. BMC Cancer 2016, 16, 487. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.H.; Liu, Y.J.; Zhang, C.; Chu, J.J.; Wu, Y.Q.; Li, Y.D.; Liu, J.Q.; Li, Q.; Li, S.Y.; Shi, Q.F.; et al. Tamoxifen-resistant breast cancer cells are resistant to DNA-damaging chemotherapy because of upregulated BARD1 and BRCA1. Nat. Commun. 2018, 9, 1595. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Deng, K.; Huang, J.; Zeng, R.; Zuo, J. Progress in the Understanding of the Mechanism of Tamoxifen Resistance in Breast Cancer. Front. Pharmacol. 2020, 11, 592912. [Google Scholar] [CrossRef]
- Sette, C.; Ladomery, M.; Ghigna, C. Alternative splicing: Role in cancer development and progression. Int. J. Cell Biol. 2013, 2013, 421606. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.D.; Gong, C.; Lau, S.L.Y.; Yang, N.; Wong, O.G.W.; Cheung, A.N.Y.; Tsang, J.W.H.; Chan, K.Y.K.; Khoo, U.S. SpliceArray Profiling of Breast Cancer Reveals a Novel Variant of NCOR2/SMRT That Is Associated with Tamoxifen Resistance and Control of ER alpha Transcriptional Activity. Cancer Res. 2013, 73, 246–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, C.; Man, E.P.S.; Tsoi, H.; Lee, T.K.W.; Lee, P.; Ma, S.T.; Wong, L.S.; Luk, M.Y.; Rakha, E.A.; Green, A.R.; et al. BQ323636.1, a Novel Splice Variant to NCOR2, as a Predictor for Tamoxifen-Resistant Breast Cancer. Clin. Cancer Res. 2018, 24, 3681–3691. [Google Scholar] [CrossRef] [Green Version]
- Tsoi, H.; Man, E.P.; Leung, M.H.; Mok, K.C.; Chau, K.M.; Wong, L.S.; Chan, W.L.; Chan, S.Y.; Luk, M.Y.; Cheng, C.N.; et al. KPNA1 regulates nuclear import of NCOR2 splice variant BQ323636.1 to confer tamoxifen resistance in breast cancer. Clin. Transl. Med. 2021, 11, e554. [Google Scholar] [CrossRef]
- Tsoi, H.; Shi, L.; Leung, M.-H.; Man, E.P.S.; So, Z.-Q.; Chan, W.-L.; Khoo, U.-S. Overexpression of BQ323636.1 Modulated AR/IL-8/CXCR1 Axis to Confer Tamoxifen Resistance in ER-Positive Breast Cancer. Life 2022, 12, 93. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, H.; Man, E.P.S.; Chau, K.M.; Khoo, U.S. Targeting the IL-6/STAT3 Signalling Cascade to Reverse Tamoxifen Resistance in Estrogen Receptor Positive Breast Cancer. Cancers 2021, 13, 1511. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [Green Version]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Dvinge, H. Regulation of alternative mRNA splicing: Old players and new perspectives. FEBS Lett. 2018, 592, 2987–3006. [Google Scholar] [CrossRef] [Green Version]
- Chao, Y.; Jiang, Y.; Zhong, M.; Wei, K.; Hu, C.; Qin, Y.; Zuo, Y.; Yang, L.; Shen, Z.; Zou, C. Regulatory roles and mechanisms of alternative RNA splicing in adipogenesis and human metabolic health. Cell Biosci. 2021, 11, 66. [Google Scholar] [CrossRef]
- Shepard, P.J.; Hertel, K.J. The SR protein family. Genome Biol. 2009, 10, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerasuolo, A.; Buonaguro, L.; Buonaguro, F.M.; Tornesello, M.L. The Role of RNA Splicing Factors in Cancer: Regulation of Viral and Human Gene Expression in Human Papillomavirus-Related Cervical Cancer. Front. Cell Dev. Biol. 2020, 8, 474. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qian, J.; Gu, C.; Yang, Y. Alternative splicing and cancer: A systematic review. Signal Transduct. Target. Ther. 2021, 6, 78. [Google Scholar] [CrossRef] [PubMed]
- Karni, R.; de Stanchina, E.; Lowe, S.W.; Sinha, R.; Mu, D.; Krainer, A.R. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat. Struct. Mol. Biol. 2007, 14, 185–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, C.L.; Cheng, Y.M.; Liu, Y.G.; Chen, L.L.; Liu, L.N.; Wei, N.; Xie, Z.Q.; Wu, W.W.; Feng, Y. SRSF2 Regulates Alternative Splicing to Drive Hepatocellular Carcinoma Development. Cancer Res. 2017, 77, 1168–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, R.; Ajiro, M.; Yu, L.L.; McCoy, P.; Zheng, Z.M. Oncogenic splicing factor SRSF3 regulates ILF3 alternative splicing to promote cancer cell proliferation and transformation. RNA 2019, 25, 630–644. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.S.; Jia, R.; Bian, Z. SRSF5 functions as a novel oncogenic splicing factor and is upregulated by oncogene SRSF3 in oral squamous cell carcinoma. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Aubol, B.E.; Wu, G.W.; Keshwani, M.M.; Movassat, M.; Fattet, L.; Hertel, K.J.; Fu, X.D.; Adams, J.A. Release of SR Proteins from CLK1 by SRPK1: A Symbiotic Kinase System for Phosphorylation Control of Pre-mRNA Splicing. Mol. Cell 2016, 63, 218–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Roosmalen, W.; Le Devedec, S.E.; Golani, O.; Smid, M.; Pulyakhina, I.; Timmermans, A.M.; Look, M.P.; Zi, D.; Pont, C.; de Graauw, M.; et al. Tumor cell migration screen identifies SRPK1 as breast cancer metastasis determinant. J. Clin. Investig. 2015, 125, 1648–1664. [Google Scholar] [CrossRef] [Green Version]
- Nikas, I.P.; Themistocleous, S.C.; Paschou, S.A.; Tsamis, K.I.; Ryu, H.S. Serine-Arginine Protein Kinase 1 (SRPK1) as a Prognostic Factor and Potential Therapeutic Target in Cancer: Current Evidence and Future Perspectives. Cells 2020, 9, 19. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Zhou, Z.H.; Subhramanyam, C.S.; Cao, Q.; Heng, Z.S.L.; Liu, W.; Fu, X.D.; Hu, Q.D. SRPK1 acetylation modulates alternative splicing to regulate cisplatin resistance in breast cancer cells. Commun. Biol. 2020, 3, 268. [Google Scholar] [CrossRef] [PubMed]
- Malvi, P.; Janostiak, R.; Chava, S.; Manrai, P.; Yoon, E.; Singh, K.; Harigopal, M.; Gupta, R.; Wajapeyee, N. LIMK2 promotes the metastatic progression of triple-negative breast cancer by activating SRPK1. Oncogenesis 2020, 9, 77. [Google Scholar] [CrossRef] [PubMed]
- Gehring, N.H.; Roignant, J.Y. Anything but Ordinary—Emerging Splicing Mechanisms in Eukaryotic Gene Regulation. Trends Genet. 2021, 37, 355–372. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yang, C.; Wang, Z.W.; Hu, J.F.; Pan, J.J.; Liao, C.Y.; Zhang, J.Q.; Chen, J.Z.; Huang, Y.; Huang, L.; et al. CLK1/SRSF5 pathway induces aberrant exon skipping of METTL14 and Cyclin L2 and promotes growth and metastasis of pancreatic cancer. J. Hematol. Oncol. 2021, 14, 60. [Google Scholar] [CrossRef]
- Muraki, M.; Ohkawara, B.; Hosoya, T.; Onogi, H.; Koizumi, J.; Koizumi, T.; Sumi, K.; Yomoda, J.; Murray, M.V.; Kimura, H.; et al. Manipulation of alternative splicing by a newly developed inhibitor of Clks. J. Biol. Chem. 2004, 279, 24246–24254. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Hatcher, J.M.; Wu, G.W.; Zeng, C.Y.; Zhu, J.; Meng, F.; Patel, S.; Wang, W.Q.; Ficarro, S.B.; Leggett, A.L.; Powell, C.E.; et al. SRPKIN-1: A Covalent SRPK1/2 Inhibitor that Potently Converts VEGF from Pro-angiogenic to Anti-angiogenic Isoform. Cell Chem. Biol. 2018, 25, 460–470.e6. [Google Scholar] [CrossRef] [Green Version]
- Williams, K.E.; Anderton, D.L.; Lee, M.P.; Pentecost, B.T.; Arcaro, K.F. High-density array analysis of DNA methylation in Tamoxifen-resistant breast cancer cell lines. Epigenetics 2014, 9, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.M.; Zhang, Z.; Cenciarini, M.E.; Proietti, C.J.; Amasino, M.; Hong, T.; Yang, M.; Liao, Y.J.; Chiang, H.C.; Kaklamani, V.G.; et al. Tamoxifen Resistance in Breast Cancer Is Regulated by the EZH2-ER alpha-GREB1 Transcriptional Axis. Cancer Res. 2018, 78, 671–684. [Google Scholar] [CrossRef] [Green Version]
- Barazetti, J.F.; Jucoski, T.S.; Carvalho, T.M.; Veiga, R.N.; Kohler, A.F.; Baig, J.; Al Bizri, H.; Gradia, D.F.; Mader, S.; de Oliveira, J.C. From Micro to Long: Non-Coding RNAs in Tamoxifen Resistance of Breast Cancer Cells. Cancers 2021, 13, 3688. [Google Scholar] [CrossRef]
- Dong, C.; Wu, J.; Chen, Y.; Nie, J.N.; Chen, C.S. Activation of PI3K/AKT/mTOR Pathway Causes Drug Resistance in Breast Cancer. Front. Pharmacol. 2021, 12, 628690. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heckler, M.M.; Thakor, H.; Schafer, C.C.; Riggins, R.B. ERK/MAPK regulates ERR gamma expression, transcriptional activity and receptor-mediated tamoxifen resistance in ER plus breast cancer. FEBS J. 2014, 281, 2431–2442. [Google Scholar] [CrossRef] [Green Version]
- Peng, W.X.; Huang, J.G.; Yang, L.; Gong, A.H.; Mo, Y.Y. Linc-RoR promotes MAPK/ERK signaling and confers estrogen-independent growth of breast cancer. Mol. Cancer 2017, 16, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.H.; Wester, L.; He, J.C.; Geiger, T.; Moerkens, M.; Siddappa, R.; Helmijr, J.A.; Timmermans, M.M.; Look, M.P.; van Deurzen, C.H.M.; et al. IGF1R signaling drives antiestrogen resistance through PAK2/PIX activation in luminal breast cancer. Oncogene 2018, 37, 1869–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahem, S.M.; Epsi, N.J.; Coffman, F.D.; Mitrofanova, A. Genome-wide analysis of therapeutic response uncovers molecular pathways governing tamoxifen resistance in ER+ breast cancer. Ebiomedicine 2020, 61, 103047. [Google Scholar] [CrossRef] [PubMed]
- Lago, S.; Nadai, M.; Cernilogar, F.M.; Kazerani, M.; Moreno, H.D.; Schotta, G.; Richter, S.N. Promoter G-quadruplexes and transcription factors cooperate to shape the cell type-specific transcriptome. Nat. Commun. 2021, 12, 3885. [Google Scholar] [CrossRef]
- Vaquero-Garcia, J.; Barrera, A.; Gazzara, M.R.; Gonzalez-Vallinas, J.; Lahens, N.F.; Hogenesch, J.B.; Lynch, K.W.; Barash, Y. A new view of transcriptome complexity and regulation through the lens of local splicing variations. Elife 2016, 5, e11752. [Google Scholar] [CrossRef]
- Nowakowski, T.J.; Rani, N.; Golkaram, M.; Zhou, H.J.R.; Alvarado, B.; Huch, K.; West, J.A.; Leyrat, A.; Pollen, A.A.; Kriegstein, A.R.; et al. Regulation of cell-type-specific transcriptomes by microRNA networks during human brain development. Nat. Neurosci. 2018, 21, 1784–1792. [Google Scholar] [CrossRef]
- Moroney, J.B.; Vasudev, A.; Pertsemlidis, A.; Zan, H.; Casali, P. Integrative transcriptome and chromatin landscape analysis reveals distinct epigenetic regulations in human memory B cells. Nat. Commun. 2020, 11, 5435. [Google Scholar] [CrossRef]
- Duan, H.M.; Ge, W.H.; Zhang, A.F.; Xi, Y.; Chen, Z.H.; Luo, D.D.; Cheng, Y.; Fan, K.S.; Horvath, S.; Sofroniew, M.V.; et al. Transcriptome analyses reveal molecular mechanisms underlying functional recovery after spinal cord injury. Proc. Natl. Acad. Sci. USA 2015, 112, 13360–13365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.P.; Coulombe-Huntington, J.; Kang, S.L.; Sheynkman, G.M.; Hao, T.; Richardson, A.; Sun, S.; Yang, F.; Shen, Y.A.; Murray, R.R.; et al. Widespread Expansion of Protein Interaction Capabilities by Alternative Splicing. Cell 2016, 164, 805–817. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Manley, J.L. Misregulation of Pre-mRNA Alternative Splicing in Cancer. Cancer Discov. 2013, 3, 1228–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prinos, P.; Garneau, D.; Lucier, J.F.; Gendron, D.; Couture, S.; Boivin, M.; Brosseau, J.P.; Lapointe, E.; Thibault, P.; Durand, M.; et al. Alternative splicing of SYK regulates mitosis and cell survival. Nat. Struct. Mol. Biol. 2011, 18, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.C. Therapeutic Applications of Targeted Alternative Splicing to Cancer Treatment. Int. J. Mol. Sci. 2018, 19, 75. [Google Scholar] [CrossRef] [Green Version]
- Miura, K.; Fujibuchi, W.; Unno, M. Splice isoforms as therapeutic targets for colorectal cancer. Carcinogenesis 2012, 33, 2311–2319. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.D.; Morini, E.; Salani, M.; Krauson, A.J.; Chekuri, A.; Sharma, N.; Ragavendran, A.; Erdin, S.; Logan, E.M.; Li, W.C.; et al. A deep learning approach to identify gene targets of a therapeutic for human splicing disorders. Nat. Commun. 2021, 12, 3332. [Google Scholar] [CrossRef]
- Koedoot, E.; Wolters, L.; van de Water, B.; Devedec, S.E.L. Splicing regulatory factors in breast cancer hallmarks and disease progression. Oncotarget 2019, 10, 6021–6037. [Google Scholar] [CrossRef] [Green Version]
- Damen, M.P.F.; van Rheenen, J.; Scheele, C.L.G.J. Targeting dormant tumor cells to prevent cancer recurrence. FEBS J. 2021, 288, 6286–6303. [Google Scholar] [CrossRef]
- Varlakhanova, N.; Hahm, J.B.; Privalsky, M.L. Regulation of SMRT corepressor dimerization and composition by MAP kinase phosphorylation. Mol. Cell. Endocrinol. 2011, 332, 180–188. [Google Scholar] [CrossRef] [Green Version]
- Leung, M.H.; Tsoi, H.; Gong, C.; Man, E.P.S.; Zona, S.; Yao, S.; Lam, E.W.F.; Khoo, U.S. A Splice Variant of NCOR2, BQ323636.1, Confers Chemoresistance in Breast Cancer by Altering the Activity of NRF2. Cancers 2020, 12, 533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsoi, H.; Tsang, W.C.; Man, E.P.S.; Leung, M.H.; You, C.P.; Chan, S.Y.; Chan, W.L.; Khoo, U.S. Checkpoint Kinase 2 Inhibition Can Reverse Tamoxifen Resistance in ER-Positive Breast Cancer. Int. J. Mol. Sci. 2022, 23, 12290. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Huang, Q.Y.; Liu, W.; Zhu, Q.; Cui, C.P.; Xu, L.; Guo, X.; Wang, P.; Liu, J.W.; Dong, G.L.; et al. Mutually exclusive acetylation and ubiquitylation of the splicing factor SRSF5 control tumor growth. Nat. Commun. 2018, 9, 2464. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Forward (5′ → 3′) | Reverse (5′ → 3′) | |
|---|---|---|
| SRSF1 | GCG GTC TGA AAA CAG AGT GG | ACA AAC TCC ACG ACA CCA GT |
| SRSF2 | TCC AAG TCC AAG TCC TCG TC | GCG ACC TGG ATT TGG ATT CC |
| SRSF3 | CGA AGT GTG TGG GTT GCT AG | AGT TCC ACT CTT ACA CGG CA |
| SRSF4 | GGA AGG TCG AGG AGA GAG TG | GCT GAC TTT GAT CTG GAG CG |
| SRSF5 | GGT GGA AGA GGT AGA GGA CG | TTT GAG ATC CTG CCA GCT GA |
| SRSF6 | CGT GCT TTG GAC AAA CTG GA | TCG AGA CCT GGA TCT GCT TC |
| SRSF7 | CGA AGA AGA AGC AGG TCA CG | GGA GAT GCT GAC CTT GAC CT |
| SRSF8 | GTC TCA CTC GAA GTC TGG GT | TAG ATG AAG ACC TGG ACC GC |
| SRSF9 | GAG AAG CTG GGG ATG TCT GT | TCC AGT TTA CGC AGG GCA TA |
| SRSF10 | ATT TCT ACA CTC GCC GTC CA | GCC GTC CAC AAA TCC ACT TT |
| SRSF11 | GGG GCT CCT ACT CTT GAT CC | TGA AAC GAG ACC AGC AGC TA |
| SRSF12 | AAT AGG AGG CGG TCA GAC AG | GTT CTT GAC TGC CTT GCT GA |
| Actin | ATC GTG CGT GAC ATT AAG GAG AAG | AGG AAG GAA GGC TGG AAG AGT G |
| BQ | GGA GCG CAT GCA GAG AAC C | CTG GCG GTC TTT GTA CAC CT |
| NCOR2 | GGT GGA GCG CAT CGA GAA C | CCC GCT GGC CCA CCC TCT GCA TG |
| NCOR2-BQ splicing | GAG CAGA AGT TCT GCC AGC GC | TTC TCC CGG AAG GTC TCC TTC |
| Number of Cases | Percentage (%) | ||
|---|---|---|---|
| ER +ve breast cancer | 137 | 100 | |
| Age | <56 | 65 | 47.4 |
| ≥56 | 52.6 | 52.6 | |
| T stage | I, II | 41 | 29.9 |
| III, IV | 10 | 7.3 | |
| Lymph node status | Positive | 67 | 48.9 |
| Negative | 56 | 40.9 | |
| Tumour grade | 1, 2 | 49 | 35.8 |
| 3 | 74 | 54.0 | |
| Tumour size | <2 cm | 14 | 10.2 |
| ≥2 cm | 79 | 57.7 | |
| ER status | Positive | 73 | 53.3 |
| Negative | 25 | 18.2 | |
| PR status | Positive | 49 | 35.8 |
| Negative | 37 | 27.0 | |
| HER2 receptor status | Positive | 35 | 25.5 |
| Negative | 37 | 27.0 | |
| Triple negative status | Yes | 13 | 9.5 |
| No | 71 | 51.8 |
| Overall Survival | Disease-Specific Survival | ||||
|---|---|---|---|---|---|
| Clinical–Pathological Parameters | No. of Cases | RR (95% CI) | p-Value | RR (95% CI) | p-Value |
| Age | 71 | 1.736 (0.811, 3.718) | 0.155 | 1.421 (0.564, 3.584) | 0.456 |
| T-stage | 32 | 5.897 (1.311, 26.531) | 0.021 | 3.931 (0.714, 21.647) | 0.116 |
| Lymph node involvement | 65 | 0.948 (0.423, 2.214) | 0.896 | 1.323 (0.478, 3.662) | 0.589 |
| Tumour grade | 70 | 1.336 (0.613, 2.913) | 0.466 | 3.319 (1.092, 10.087) | 0.034 |
| Histological type | 71 | 1.342 (0.317, 5.689) | 0.690 | 0.791 (0.182, 3.447) | 0.755 |
| PR status | 59 | 0.237 (0.096, 0.582) | 0.002 | 0.111 (0.038, 0.330) | <0.001 |
| HER2 status | 49 | 1.110 (0.427, 2.888) | 0.830 | 1.715 (0.499, 5.893) | 0.392 |
| Tumour size | 54 | 1.691 (0.390, 7.327) | 0.482 | 26.528 (0.035, 20173.302) | 0.333 |
| Cases with high SRSF5 | 58 | 0.182 (0.071, 0.466) | <0.001 | 0.142 (0.039, 0.519) | 0.003 |
| Cases with high BQ | 64 | 7.450 (2.559, 21.692) | <0.001 | 21.188 (2.802, 160.238) | 0.003 |
| Either high SRSF5/low BQ or low SRSF5/high BQ | 40 | 0.122 (0.035, 0.421) | 0.001 | 0.060 (0.008, 0.466) | 0.007 |
| Overall Survival | Disease-Specific Survival | ||||
|---|---|---|---|---|---|
| Clinical–Pathological Parameters | Cases | RR (95% CI) | p-Value | RR (95% CI) | p-Value |
| T-stage | 21 | 12.931 (0.708, 236.235) | 0.084 | 2.583 (0.537, 12.436) | 0.237 |
| PR status | 21 | 0.361 (0.053, 2.437) | 0.296 | 0.288 (0.084, 0.986) | 0.047 |
| Cases with high SRSF5 | 21 | 0.087 (0.008, 0.901) | 0.041 | 0.178 (0.035, 0.920) | 0.039 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsoi, H.; Fung, N.N.-C.; Man, E.P.S.; Leung, M.-H.; You, C.-P.; Chan, W.-L.; Chan, S.-Y.; Khoo, U.-S. SRSF5 Regulates the Expression of BQ323636.1 to Modulate Tamoxifen Resistance in ER-Positive Breast Cancer. Cancers 2023, 15, 2271. https://doi.org/10.3390/cancers15082271
Tsoi H, Fung NN-C, Man EPS, Leung M-H, You C-P, Chan W-L, Chan S-Y, Khoo U-S. SRSF5 Regulates the Expression of BQ323636.1 to Modulate Tamoxifen Resistance in ER-Positive Breast Cancer. Cancers. 2023; 15(8):2271. https://doi.org/10.3390/cancers15082271
Chicago/Turabian StyleTsoi, Ho, Nicholas Nok-Ching Fung, Ellen P. S. Man, Man-Hong Leung, Chan-Ping You, Wing-Lok Chan, Sum-Yin Chan, and Ui-Soon Khoo. 2023. "SRSF5 Regulates the Expression of BQ323636.1 to Modulate Tamoxifen Resistance in ER-Positive Breast Cancer" Cancers 15, no. 8: 2271. https://doi.org/10.3390/cancers15082271