
The Function of NK Cells in Tumor Metastasis and NK Cell-Based Immunotherapy


Abstract
:Simple Summary
Abstract
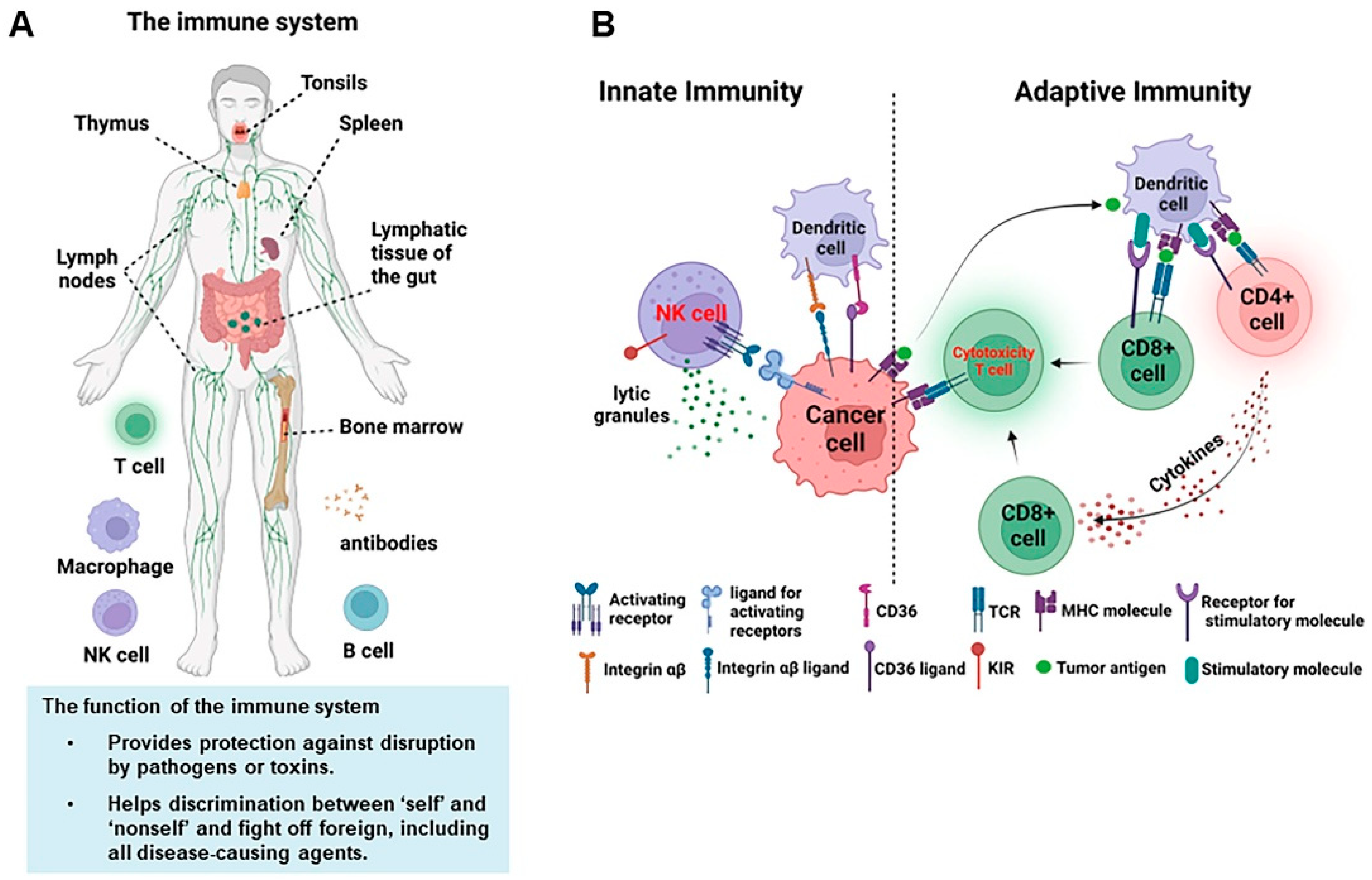
1. Introduction of the General Immune System
2. NK Cell Biology and Function
3. The Function of NK Cells in Tumor Metastasis
3.1. NK Cell Function Related to Metastasis
3.2. How NK Cells Kill Metastases
4. Immunosuppression of NK in Tumor Metastasis
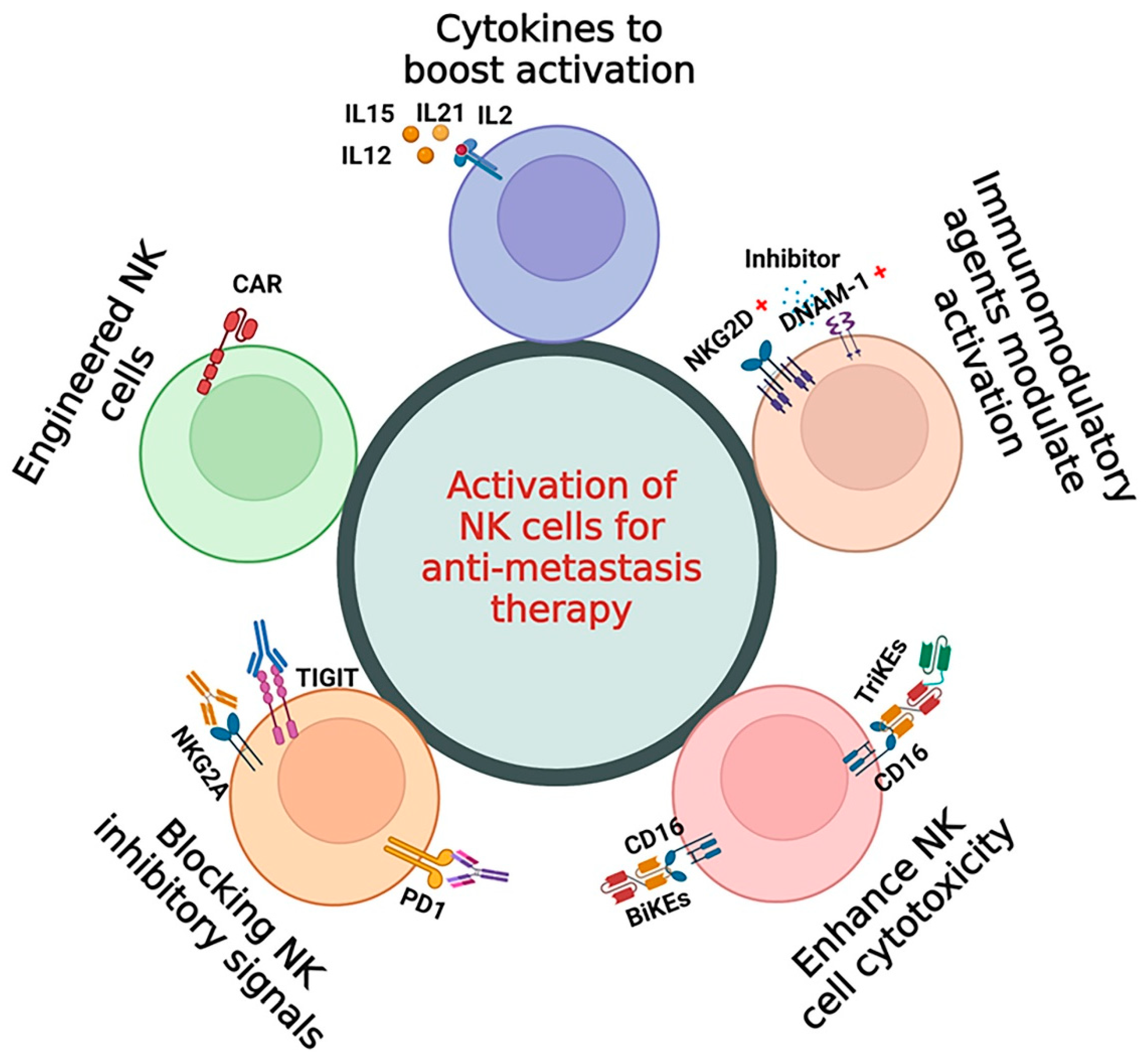
5. NK Cell-Based Immunotherapy for Metastasis Therapy
5.1. Cytokines to Boost Activation of NK Cells
5.2. Immunomodulatory Agents to Modulate the Activation of NK Cells
5.3. Enhancing NK Cell-Mediated Antibody-Dependent Cellular Cytotoxicity
5.4. Blocking (Shielding) the NK Inhibitory Receptor Signals
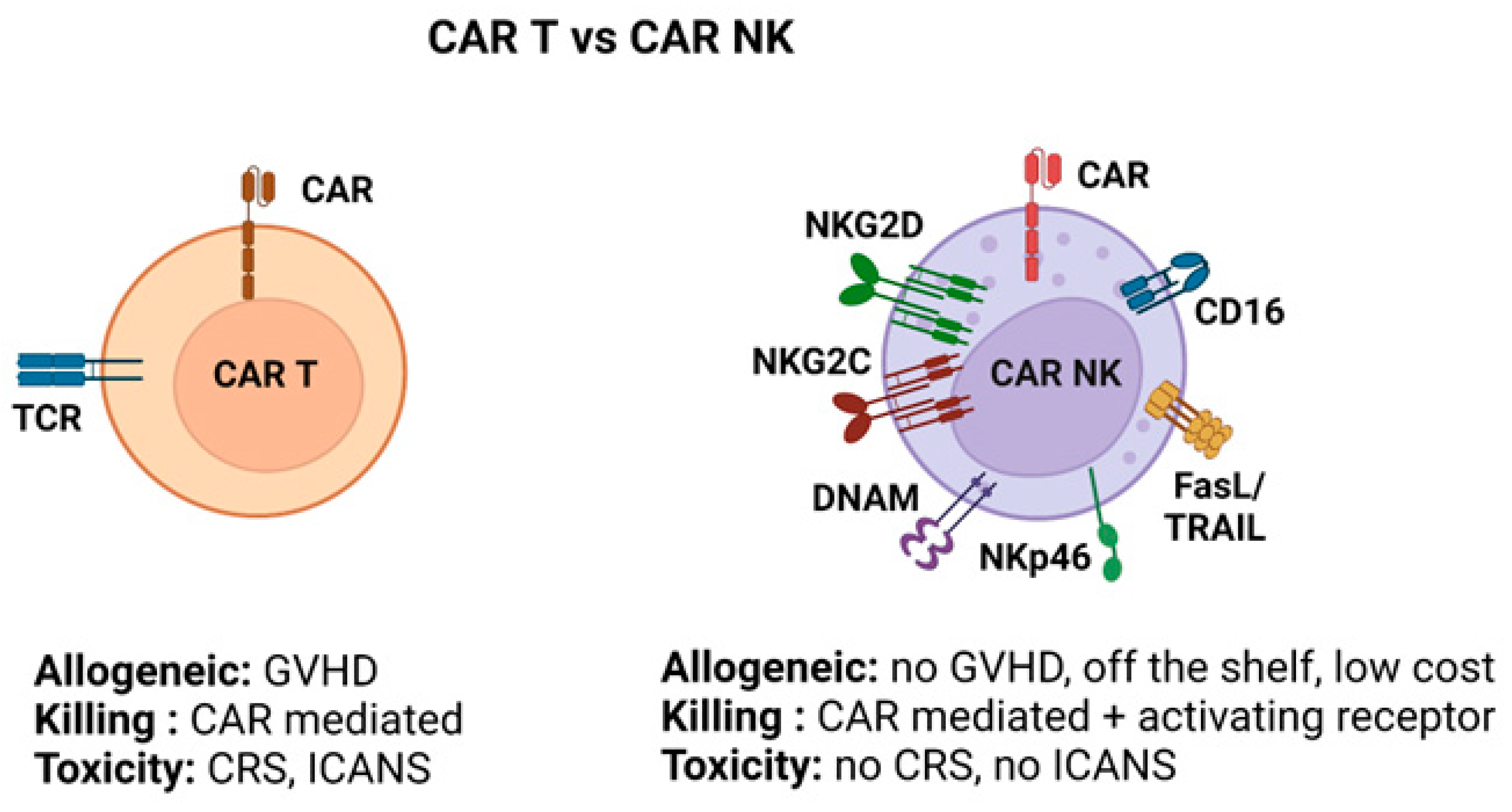
5.5. Engineered NK Cells (CAR-NK) for Metastasis Therapy
5.6. Combination of Multiple Strategies for Metastasis Therapy
6. Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Arase, H.; Mocarski, E.S.; Campbell, A.E.; Hill, A.B.; Lanier, L.L. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science 2002, 296, 1323–1326. [Google Scholar] [CrossRef]
- Bedoui, S.; Gebhardt, T.; Gasteiger, G.; Kastenmuller, W. Parallels and differences between innate and adaptive lymphocytes. Nat. Immunol. 2016, 17, 490–494. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr.; Travers, P.; Walport, M.; Shlomchik, M.J. Principles of Innate and Adaptive Immunity. Immunobiology: The Immune System in Health and Disease, 5th ed.; Garland Science: New York, NY, USA, 2001. [Google Scholar]
- Cooper, M.D.; Alder, M.N. The evolution of adaptive immune systems. Cell 2006, 124, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Goldstein, A.; Wang, H.; Ching Lo, H.; Sun Kim, I.; Welte, T.; Sheng, K.; Dobrolecki, L.E.; Zhang, X.; Putluri, N.; et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature 2017, 544, 250–254. [Google Scholar] [CrossRef]
- Ljunggren, H.G.; Glas, R.; Sandberg, J.K.; Kärre, K. Reactivity and specificity of CD8+ T cells in mice with defects in the MHC class I antigen-presenting pathway. Immunol. Rev. 1996, 151, 123–148. [Google Scholar] [CrossRef] [PubMed]
- Ljunggren, H.G.; Kärre, K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol. Today 1990, 11, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Malmberg, K.J.; Sohlberg, E.; Goodridge, J.P.; Ljunggren, H.G. Immune selection during tumor checkpoint inhibition therapy paves way for NK-cell “missing self” recognition. Immunogenetics 2017, 69, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Sonnenberg, G.F.; Hepworth, M.R. Functional interactions between innate lymphoid cells and adaptive immunity. Nature Rev. Immunol. 2019, 19, 599–613. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef]
- Driessens, G.; Kline, J.; Gajewski, T.F. Costimulatory and coinhibitory receptors in anti-tumor immunity. Immunol. Rev. 2009, 229, 126–144. [Google Scholar] [CrossRef]
- Cronin, S.J.; Penninger, J.M. From T-cell activation signals to signaling control of anti-cancer immunity. Immunol. Rev. 2007, 220, 151–168. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Rouas-Freiss, N.; Khalil-Daher, I.; Riteau, B.; Menier, C.; Paul, P.; Dausset, J.; Carosella, E.D. The immunotolerance role of HLA-G. Semin. Cancer Biol. 1999, 9, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.; Rouas-Freiss, N.; Khalil-Daher, I.; Moreau, P.; Riteau, B.; Le Gal, F.A.; Avril, M.F.; Dausset, J.; Guillet, J.G.; Carosella, E.D. HLA-G expression in melanoma: A way for tumor cells to escape from immunosurveillance. Proc. Natl. Acad. Sci. USA 1998, 95, 4510–4515. [Google Scholar] [CrossRef] [PubMed]
- O’Garra, A.; Barrat, F.J.; Castro, A.G.; Vicari, A.; Hawrylowicz, C. Strategies for use of IL-10 or its antagonists in human disease. Immunol. Rev. 2008, 223, 114–131. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef]
- Garner, H.; de Visser, K.E. Immune crosstalk in cancer progression and metastatic spread: A complex conversation. Nat. Rev. Immunol. 2020, 20, 483–497. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, X. Characteristics and Significance of the Pre-metastatic Niche. Cancer Cell 2016, 30, 668–681. [Google Scholar] [CrossRef]
- Basudan, A.M. The Role of Immune Checkpoint Inhibitors in Cancer Therapy. Clin. Pract. 2022, 13, 22–40. [Google Scholar] [CrossRef]
- Hsu, J.; Yang, Y.; Gergis, M.; Bi, X.; Yi, D.; Gergis, U. Chimeric Antigen Receptor T Cell Therapy for Solid Tumors. Hematol. Oncol. Stem Cell Ther. 2022, 15, 94–99. [Google Scholar] [CrossRef]
- Rotte, A.; Frigault, M.J.; Ansari, A.; Gliner, B.; Heery, C.; Shah, B. Dose-response correlation for CAR-T cells: A systematic review of clinical studies. J. Immunother. Cancer 2022, 10, e005678. [Google Scholar] [CrossRef]
- Beauvais, D.; Danhof, S.; Hayden, P.J.; Einsele, H.; Yakoub-Agha, I. Clinical data, limitations and perspectives on chimeric antigen receptor T-cell therapy in multiple myeloma. Curr. Opin. Oncol. 2020, 32, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Bachanova, V.; Miller, J.S. NK cells in therapy of cancer. Crit. Rev. Oncog. 2014, 19, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Bahmanyar, M.; Vakil, M.K.; Al-Awsi, G.R.L.; Kouhpayeh, S.A.; Mansoori, Y.; Mansoori, B.; Moravej, A.; Mazarzaei, A.; Ghasemian, A. Anticancer traits of chimeric antigen receptors (CARs)-Natural Killer (NK) cells as novel approaches for melanoma treatment. BMC Cancer 2022, 22, 1220. [Google Scholar] [CrossRef] [PubMed]
- Bald, T.; Krummel, M.F.; Smyth, M.J.; Barry, K.C. The NK cell–cancer cycle: Advances and new challenges in NK cell–based immunotherapies. Nat. Immunol. 2020, 21, 835–847. [Google Scholar] [CrossRef]
- Freud, A.G.; Mundy-Bosse, B.L.; Yu, J.; Caligiuri, M.A. The broad spectrum of human natural killer cell diversity. Immunity 2017, 47, 820–833. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef]
- Richards, J.O.; Chang, X.; Blaser, B.W.; Caligiuri, M.A.; Zheng, P.; Liu, Y. Tumor growth impedes natural-killer-cell maturation in the bone marrow. Blood 2006, 108, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.C.; Lanier, L.L. NK cell development, homeostasis and function: Parallels with CD8(+) T cells. Nat. Rev. Immunol. 2011, 11, 645–657. [Google Scholar] [CrossRef]
- Herberman, R.B.; Holden, H.T. Natural killer cells as antitumor effector cells. J. Natl. Cancer Inst. 1979, 62, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Freud, A.G.; Caligiuri, M.A. Location and cellular stages of natural killer cell development. Trends Immunol. 2013, 34, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Stabile, H.; Fionda, C.; Gismondi, A.; Santoni, A. Role of distinct natural killer cell subsets in anticancer response. Front. Immunol. 2017, 8, 293. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Li, B.; Lu, R.; Koelle, S.J.; Yang, Y.; Jares, A.; Krouse, A.E.; Metzger, M.; Liang, F.; Lore, K.; et al. Clonal tracking of rhesus macaque hematopoiesis highlights a distinct lineage origin for natural killer cells. Cell Stem Cell 2014, 14, 486–499. [Google Scholar] [CrossRef]
- Lotzová, E.; Savary, C.A. Human natural killer cell development from bone marrow progenitors: Analysis of phenotype, cytotoxicity and growth. Nat. Immun. 1993, 12, 209–217. [Google Scholar]
- Maskalenko, N.A.; Zhigarev, D.; Campbell, K.S. Harnessing natural killer cells for cancer immunotherapy: Dispatching the first responders. Nat. Rev. Drug Discov. 2022, 21, 559–577. [Google Scholar] [CrossRef]
- Laskowski, T.J.; Biederstädt, A.; Rezvani, K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat. Rev. Cancer 2022, 22, 557–575. [Google Scholar] [CrossRef]
- Li, Y.; Basar, R.; Wang, G.; Liu, E.; Moyes, J.S.; Li, L.; Kerbauy, L.N.; Uprety, N.; Fathi, M.; Rezvan, A.; et al. KIR-based inhibitory CARs overcome CAR-NK cell trogocytosis-mediated fratricide and tumor escape. Nat. Med. 2022, 28, 2133–2144. [Google Scholar] [CrossRef]
- Voskoboinik, I.; Whisstock, J.C.; Trapani, J.A. Perforin and granzymes: Function, dysfunction and human pathology. Nat. Rev. Immunol. 2015, 15, 388–400. [Google Scholar] [CrossRef]
- Martinez-Lostao, L.; Anel, A.; Pardo, J. How do cytotoxic lymphocytes kill cancer cells? Clin. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef]
- Martinet, L.; Smyth, M.J. Balancing natural killer cell activation through paired receptors. Nat. Rev. Immunol. 2015, 15, 243–254. [Google Scholar] [CrossRef]
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999, 285, 727–729. [Google Scholar] [CrossRef] [PubMed]
- Kriegeskorte, A.K.; Gebhardt, F.E.; Porcellini, S.; Schiemann, M.; Stemberger, C.; Franz, T.J.; Huster, K.M.; Carayannopoulos, L.N.; Yokoyama, W.M.; Colonna, M.; et al. NKG2D-independent suppression of T cell proliferation by H60 and MICA. Proc. Natl. Acad. Sci. USA 2005, 102, 11805–11810. [Google Scholar] [CrossRef]
- Lanier, L.L. NKG2D Receptor and Its Ligands in Host Defense. Cancer Immunol. Res. 2015, 3, 575–582. [Google Scholar] [CrossRef]
- Wu, J.; Song, Y.; Bakker, A.B.; Bauer, S.; Spies, T.; Lanier, L.L.; Phillips, J.H. An activating immunoreceptor complex formed by NKG2D and DAP10. Science 1999, 285, 730–732. [Google Scholar] [CrossRef]
- Bottino, C.; Castriconi, R.; Pende, D.; Rivera, P.; Nanni, M.; Carnemolla, B.; Cantoni, C.; Grassi, J.; Marcenaro, S.; Reymond, N.; et al. Identification of PVR (CD155) and Nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J. Exp. Med. 2003, 198, 557–567. [Google Scholar] [CrossRef]
- Arnould, L.; Gelly, M.; Penault-Llorca, F.; Benoit, L.; Bonnetain, F.; Migeon, C.; Cabaret, V.; Fermeaux, V.; Bertheau, P.; Garnier, J.; et al. Trastuzumab-based treatment of HER2-positive breast cancer: An antibody-dependent cellular cytotoxicity mechanism? Br. J. Cancer 2006, 94, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Collins, D.M.; O’Donovan, N.; McGowan, P.M.; O’Sullivan, F.; Duffy, M.J.; Crown, J. Trastuzumab induces antibody-dependent cell-mediated cytotoxicity (ADCC) in HER-2-non-amplified breast cancer cell lines. Ann. Oncol. 2012, 23, 1788–1795. [Google Scholar] [CrossRef]
- Barrow, A.D.; Martin, C.J.; Colonna, M. The Natural Cytotoxicity Receptors in Health and Disease. Front. Immunol. 2019, 10, 909. [Google Scholar] [CrossRef] [PubMed]
- Pessino, A.; Sivori, S.; Bottino, C.; Malaspina, A.; Morelli, L.; Moretta, L.; Biassoni, R.; Moretta, A. Molecular cloning of NKp46, A novel member of the immunoglobulin superfamily involved in triggering of natural cytotoxicity. J. Exp. Med. 1998, 188, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Pende, D.; Parolini, S.; Pessino, A.; Sivori, S.; Augugliaro, R.; Morelli, L.; Marcenaro, E.; Accame, L.; Malaspina, A.; Biassoni, R.; et al. Identification and molecular characterization of NKp30, a novel triggering receptor involved in natural cytotoxicity mediated by human natural killer cells. J. Exp. Med. 1999, 190, 1505–1516. [Google Scholar] [CrossRef]
- Cantoni, C.; Bottino, C.; Vitale, M.; Pessino, A.; Augugliaro, R.; Malaspina, A.; Parolini, S.; Moretta, L.; Moretta, A.; Biassoni, R. NKp44, a triggering receptor involved in tumor cell lysis by activated human natural killer cells, is a novel member of the immunoglobulin superfamily. J. Exp. Med. 1999, 189, 787–796. [Google Scholar] [CrossRef]
- Vitale, M.; Bottino, C.; Sivori, S.; Sanseverino, L.; Castriconi, R.; Marcenaro, E.; Augugliaro, R.; Moretta, L.; Moretta, A. NKp44, a novel triggering surface molecule specifically expressed by activated natural killer cells, is involved in non-major histocompatibility complex-restricted tumor cell lysis. J. Exp. Med. 1998, 187, 2065–2072. [Google Scholar] [CrossRef] [PubMed]
- Sen Santara, S.; Lee, D.J.; Crespo, Â.; Hu, J.J.; Walker, C.; Ma, X.; Zhang, Y.; Chowdhury, S.; Meza-Sosa, K.F.; Lewandrowski, M.; et al. The NK cell receptor NKp46 recognizes ecto-calreticulin on ER-stressed cells. Nature 2023. [Google Scholar] [CrossRef]
- Raulet, D.H.; Vance, R.E. Self-tolerance of natural killer cells. Nat. Rev. Immunol. 2006, 6, 520–531. [Google Scholar] [CrossRef]
- Wieten, L.; Mahaweni, N.M.; Voorter, C.E.; Bos, G.M.; Tilanus, M.G. Clinical and immunological significance of HLA-E in stem cell transplantation and cancer. Tissue Antigens 2014, 84, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Navarro, F.; Llano, M.; Bellon, T.; Colonna, M.; Geraghty, D.E.; Lopez-Botet, M. The ILT2(LIR1) and CD94/NKG2A NK cell receptors respectively recognize HLA-G1 and HLA-E molecules co-expressed on target cells. Eur. J. Immunol. 1999, 29, 277–283. [Google Scholar] [CrossRef]
- Bi, J.; Tian, Z. NK cell dysfunction and checkpoint immunotherapy. Front. Immunol. 2019, 10, 1999. [Google Scholar] [CrossRef]
- Chan, I.S.; Knútsdóttir, H.; Ramakrishnan, G.; Padmanaban, V.; Warrier, M.; Ramirez, J.C.; Dunworth, M.; Zhang, H.; Jaffee, E.M.; Bader, J.S.; et al. Cancer cells educate natural killer cells to a metastasis-promoting cell state. J. Cell Biol. 2020, 219, e202001134. [Google Scholar] [CrossRef]
- López-Soto, A.; Gonzalez, S.; Smyth, M.J.; Galluzzi, L. Control of Metastasis by NK Cells. Cancer Cell 2017, 32, 135–154. [Google Scholar] [CrossRef] [PubMed]
- Chiossone, L.; Dumas, P.Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688. [Google Scholar] [CrossRef]
- Barlozzari, T.; Reynolds, C.W.; Herberman, R.B. In vivo role of natural killer cells: Involvement of large granular lymphocytes in the clearance of tumor cells in anti-asialo GM1-treated rats. J. Immunol. 1983, 131, 1024–1027. [Google Scholar] [CrossRef]
- Imai, K.; Matsuyama, S.; Miyake, S.; Suga, K.; Nakachi, K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: An 11-year follow-up study of a general population. Lancet 2000, 356, 1795–1799. [Google Scholar] [CrossRef]
- Balsamo, M.; Vermi, W.; Parodi, M.; Pietra, G.; Manzini, C.; Queirolo, P.; Lonardi, S.; Augugliaro, R.; Moretta, A.; Facchetti, F.; et al. Melanoma cells become resistant to NK-cell-mediated killing when exposed to NK-cell numbers compatible with NK-cell infiltration in the tumor. Eur. J. Immunol. 2012, 42, 1833–1842. [Google Scholar] [CrossRef]
- Sandel, M.H.; Speetjens, F.M.; Menon, A.G.; Albertsson, P.A.; Basse, P.H.; Hokland, M.; Nagelkerke, J.F.; Tollenaar, R.A.; van de Velde, C.J.; Kuppen, P.J. Natural killer cells infiltrating colorectal cancer and MHC class I expression. Mol. Immunol. 2005, 42, 541–546. [Google Scholar] [CrossRef]
- Esendagli, G.; Bruderek, K.; Goldmann, T.; Busche, A.; Branscheid, D.; Vollmer, E.; Brandau, S. Malignant and non-malignant lung tissue areas are differentially populated by natural killer cells and regulatory T cells in non-small cell lung cancer. Lung Cancer 2008, 59, 32–40. [Google Scholar] [CrossRef]
- Mamessier, E.; Sylvain, A.; Thibult, M.L.; Houvenaeghel, G.; Jacquemier, J.; Castellano, R.; Gonçalves, A.; André, P.; Romagné, F.; Thibault, G.; et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J. Clin. Investig. 2011, 121, 3609–3622. [Google Scholar] [CrossRef] [PubMed]
- Orange, J.S. Natural killer cell deficiency. J. Allergy Clin. Immunol. 2013, 132, 515–525. [Google Scholar] [CrossRef]
- MacFarlane, A.W.; Jillab, M.; Smith, M.R.; Alpaugh, R.K.; Cole, M.E.; Litwin, S.; Millenson, M.M.; Al-Saleem, T.; Cohen, A.D.; Campbell, K.S. NK cell dysfunction in chronic lymphocytic leukemia is associated with loss of the mature cells expressing inhibitory killer cell Ig-like receptors. OncoImmunology 2017, 6, e1330235. [Google Scholar] [CrossRef] [PubMed]
- Pazina, T.; MacFarlane, A.W.; Bernabei, L.; Dulaimi, E.; Kotcher, R.; Yam, C.; Bezman, N.A.; Robbins, M.D.; Ross, E.A.; Campbell, K.S.; et al. Alterations of NK Cell Phenotype in the Disease Course of Multiple Myeloma. Cancers 2021, 13, 226. [Google Scholar] [CrossRef]
- Vetter, C.S.; Groh, V.; Thor Straten, P.; Spies, T.; Brocker, E.B.; Becker, J.C. Expression of stress-induced MHC class I related chain molecules on human melanoma. J. Investig. Dermatol. 2002, 118, 600–605. [Google Scholar] [CrossRef]
- Lakshmikanth, T.; Burke, S.; Ali, T.H.; Kimpfler, S.; Ursini, F.; Ruggeri, L.; Capanni, M.; Umansky, V.; Paschen, A.; Sucker, A.; et al. NCRs and DNAM 1 mediate NK cell recognition and lysis of human and mouse melanoma cell lines in vitro and in vivo. J. Clin. Investig. 2009, 119, 1251–1263. [Google Scholar] [CrossRef] [PubMed]
- Erdag, G.; Schaefer, J.T.; Smolkin, M.E.; Deacon, D.H.; Shea, S.M.; Dengel, L.T.; Patterson, J.W.; Slingluff, C.L., Jr. Immuno type and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res. 2012, 72, 1070–1080. [Google Scholar] [CrossRef] [PubMed]
- Halama, N.; Braun, M.; Kahlert, C.; Spille, A.; Quack, C.; Rahbari, N.; Koch, M.; Weitz, J.; Kloor, M.; Zoernig, I.; et al. Natural killer cells are scarce in colorectal carcinoma tissue despite high levels of chemokines and cytokines. Clin. Cancer Res. 2011, 17, 678–689. [Google Scholar] [CrossRef]
- Remark, R.; Alifano, M.; Cremer, I.; Lupo, A.; Dieu-Nosjean, M.C.; Riquet, M.; Crozet, L.; Ouakrim, H.; Goc, J.; Cazes, A.; et al. Characteristics and clinical impacts of the immune environments in colorectal and renal cell carcinoma lung metastases: Influence of tumor origin. Clin. Cancer Res. 2013, 19, 4079–4091. [Google Scholar] [CrossRef] [PubMed]
- Ichise, H.; Tsukamoto, S.; Hirashima, T.; Konishi, Y.; Oki, C.; Tsukiji, S.; Iwano, S.; Miyawaki, A.; Sumiyama, K.; Terai, K.; et al. Functional visualization of NK cell-mediated killing of metastatic single tumor cells. Elife 2022, 11, e76269. [Google Scholar] [CrossRef]
- Delahaye, N.F.; Rusakiewicz, S.; Martins, I.; Ménard, C.; Roux, S.; Lyonnet, L.; Paul, P.; Sarabi, M.; Chaput, N.; Semeraro, M.; et al. Circulating CD56bright NK cells inversely correlate with survival of melanoma patients. Sci. Rep. 2019, 9, 4487. [Google Scholar]
- Semeraro, M.; Rusakiewicz, S.; Minard-Colin, V.; Delahaye, N.F.; Enot, D.; Vély, F.; Marabelle, A.; Papoular, B.; Piperoglou, C.; Ponzoni, M.; et al. Clinical impact of the NKp30/B7-H6 axis in high-risk neuroblastoma patients. Sci. Transl. Med. 2015, 7, 283ra55. [Google Scholar] [CrossRef]
- Ishigami, S.; Natsugoe, S.; Tokuda, K.; Nakajo, A.; Che, X.; Iwashige, H.; Aridome, K.; Hokita, S.; Aikou, T. Prognostic value of intratumoral natural killer cells in gastric carcinoma. Cancer 2000, 88, 577–583. [Google Scholar] [CrossRef]
- Coca, S.; Perez-Piqueras, J.; Martinez, D.; Colmenarejo, A.; Saez, M.A.; Vallejo, C.; Martos, J.A.; Moreno, M. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer 1997, 79, 2320–2328. [Google Scholar] [CrossRef]
- Donskov, F.; von der Maase, H. Impact of immune parameters on long-term survival in metastatic renal cell carcinoma. J. Clin. Oncol. 2006, 24, 1997–2005. [Google Scholar] [CrossRef]
- Gannon, P.O.; Poisson, A.O.; Delvoye, N.; Lapointe, R.; Mes-Masson, A.M.; Saad, F. Characterization of the intra-prostatic immune cell infiltration in androgen-deprived prostate cancer patients. J. Immunol. Methods 2009, 348, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Rusakiewicz, S.; Semeraro, M.; Sarabi, M.; Desbois, M.; Locher, C.; Mendez, R.; Vimond, N.; Concha, A.; Garrido, F.; Isambert, N.; et al. Immune infil trates are prognostic factors in localized gastrointestinal stromal tumors. Cancer Res. 2013, 73, 3499–3510. [Google Scholar] [CrossRef]
- Pasero, C.; Gravis, G.; Granjeaud, S.; Guerin, M.; Thomassin-Piana, J.; Rocchi, P.; Salem, N.; Walz, J.; Moretta, A.; Olive, D. Highly effective NK cells are associated with good prognosis in patients with metastatic prostate cancer. Oncotarget 2015, 6, 14360–14373. [Google Scholar] [CrossRef] [PubMed]
- Platonova, S.; Cherfils-Vicini, J.; Damotte, D.; Crozet, L.; Vieillard, V.; Validire, P.; André, P.; Dieu-Nosjean, M.C.; Alifano, M.; Régnard, J.F.; et al. Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res. 2011, 71, 5412–5422. [Google Scholar] [CrossRef]
- Pasero, C.; Gravis, G.; Guerin, M.; Granjeaud, S.; Thomassin-Piana, J.; Rocchi, P.; Paciencia-Gros, M.; Poizat, F.; Bentobji, M.; Azario-Cheillan, F.; et al. Inherent and tumor-driven immune tolerance in the prostate microenvironment impairs natural killer cell antitumor activity. Cancer Res. 2016, 76, 2153–2165. [Google Scholar] [CrossRef]
- de Andrade, L.F.; Lu, Y.; Luoma, A.; Ito, Y.; Pan, D.; Pyrdol, J.W.; Yoon, C.H.; Yuan, G.C.; Wucherpfennig, K.W. Discovery of specialized NK cell populations infiltrating human melanoma metastases. JCI Insight 2019, 4, e133103. [Google Scholar] [CrossRef]
- Couanet, D.; Gutierrez, J.C.; Nunès, J.A.; Commo, F.; Bonvalot, S.; Ibrahim, N.; Terrier, P.; Opolon, P.; Bottino, C.; Moretta, A.; et al. Alternatively spliced NKp30 isoforms affect the prognosis of gastrointestinal stromal tumors. Nat. Med. 2011, 17, 700–707. [Google Scholar]
- Ménard, C.; Blay, J.Y.; Borg, C.; Michiels, S.; Ghiringhelli, F.; Robert, C.; Nonn, C.; Chaput, N.; Taïeb, J.; Delahaye, N.F.; et al. Natural killer cell IFN-gamma levels predict long-term survival with imatinib mesylate therapy in gastrointestinal stromal tumor-bearing patients. Cancer Res. 2009, 69, 3563–3569. [Google Scholar] [CrossRef] [PubMed]
- Messaoudene, M.; Fregni, G.; Enot, D.; Jacquelot, N.; Neves, E.; Germaud, N.; Garchon, H.J.; Boukouaci, W.; Tamouza, R.; Chanal, J.; et al. NKp30 isoforms and NKp46 transcripts in metastatic melanoma patients: Unique NKp30 pattern in rare melanoma patients with favorable evolution. OncoImmunology 2016, 5, e1154251. [Google Scholar] [CrossRef]
- Correia, A.L.; Guimaraes, J.C.; Auf der Maur, P.; De Silva, D.; Trefny, M.P.; Okamoto, R.; Bruno, S.; Schmidt, A.; Mertz, K.; Volkmann, K.; et al. Hepatic stellate cells suppress NK cell-sustained breast cancer dormancy. Nature 2021, 594, 566–571. [Google Scholar] [CrossRef]
- Angka, L.; Martel, A.B.; Kilgour, M.; Jeong, A.; Sadiq, M.; de Souza, C.T.; Baker, L.; Kennedy, M.A.; Kekre, N.; Auer, R.C. Natural killer cell IFNg secretion is profoundly suppressed following colorectal cancer surgery. Ann. Surg. Oncol. 2018, 25, 3747–3754. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.; Tai, L.H.; Falls, T.; de Souza, C.T.; Bell, J.C.; Carrier, M.; Atkins, H.; Boushey, R.; Auer, R.A. Surgical stress promotes the development of cancer metastases by a coagulation-dependent mechanism involving natural killer cells in a murine model. Ann. Surg. 2013, 258, 158–168. [Google Scholar] [CrossRef]
- Angka, L.; Khan, S.; Kilgour, M.; Xu, R.; Kennedy, M.; Auer, R. Dysfunctional natural killer cells in the aftermath of cancer surgery. Int. J. Mol. Sci. 2017, 18, 1787. [Google Scholar] [CrossRef]
- Tai, L.H.; de Souza, C.T.; Bélanger, S.; Ly, L.; Alkayyal, A.A.; Zhang, J.; Rintoul, J.L.; Ananth, A.A.; Lam, T.; Breitbach, C.J.; et al. Preventing postoperative metastatic disease by inhibiting surgery-induced dysfunction in natural killer cells. Cancer Res. 2013, 73, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Angka, L.; Tanese de Souza, C.; Baxter, K.E.; Khan, S.T.; Market, M.; Martel, A.B.; Tai, L.H.; Kennedy, M.A.; Bell, J.C.; Auer, R.C. Perioperative arginine prevents metastases by accelerating natural killer cell recovery after surgery. Mol. Ther. 2022, 30, 3270–3283. [Google Scholar] [CrossRef] [PubMed]
- Koch, J.; Steinle, A.; Watzl, C.; Mandelboim, O. Activating natural cytotoxicity receptors of natural killer cells in cancer and infection. Trends Immunol. 2013, 34, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, M.L.; Idowu, M.O.; Zhao, Y.; Khalak, H.; Payne, K.K.; Wang, X.Y.; Dumur, C.I.; Bedognetti, D.; Tomei, S.; Ascierto, P.A.; et al. Molecular signatures mostly associated with NK cells are predictive of relapse free survival in breast cancer patients. J. Transl. Med. 2013, 11, 145. [Google Scholar] [CrossRef]
- Halfteck, G.G.; Elboim, M.; Gur, C.; Achdout, H.; Ghadially, H.; Mandelboim, O. Enhanced in vivo growth of lymphoma tumors in the absence of the NK-activating receptor NKp46/NCR1. J. Immunol. 2009, 182, 2221–2230. [Google Scholar] [CrossRef] [PubMed]
- Glasner, A.; Ghadially, H.; Gur, C.; Stanietsky, N.; Tsukerman, P.; Enk, J.; Mandelboim, O. Recognition and prevention of tumor metastasis by the NK receptor NKp46/NCR1. J. Immunol. 2012, 188, 2509–2515. [Google Scholar] [CrossRef]
- Glasner, A.; Levi, A.; Enk, J.; Isaacson, B.; Viukov, S.; Orlanski, S.; Scope, A.; Neuman, T.; Enk, C.D.; Hanna, J.H.; et al. NKp46 receptor-mediated interferon-γ production by natural killer cells increases fibronectin 1 to alter tumor architecture and control metastasis. Immunity 2018, 48, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Laughney, A.M.; Hu, J.; Campbell, N.R.; Bakhoum, S.F.; Setty, M.; Lavallée, V.P.; Xie, Y.; Masilionis, I.; Carr, A.J.; Kottapalli, S.; et al. Regenerative lineages and immune-mediated pruning in lung cancer metastasis. Nat. Med. 2020, 26, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.C.; Xu, Z.; Kim, I.S.; Pingel, B.; Aguirre, S.; Kodali, S.; Liu, J.; Zhang, W.; Muscarella, A.M.; Hein, S.M.; et al. Resistance to natural killer cell immunosurveillance confers a selective advantage to polyclonal metastasis. Nat. Cancer 2020, 1, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Iguchi-Manaka, A.; Kai, H.; Yamashita, Y.; Shibata, K.; Tahara-Hanaoka, S.; Honda, S.; Yasui, T.; Kikutani, H.; Shibuya, K.; Shibuya, A. Accelerated tumor growth in mice deficient in DNAM-1 receptor. J. Exp. Med. 2008, 205, 2959–2964. [Google Scholar] [CrossRef]
- Chan, C.J.; Andrews, D.M.; McLaughlin, N.M.; Yagita, H.; Gilfillan, S.; Colonna, M.; Smyth, M.J. DNAM-1/CD155 interactions promote cytokine and NK cell-mediated suppression of poorly immunogenic melanoma metastases. J. Immunol. 2010, 184, 902–911. [Google Scholar] [CrossRef] [PubMed]
- Guillerey, C.; Chow, M.T.; Miles, K.; Olver, S.; Sceneay, J.; Takeda, K.; Möller, A.; Smyth, M.J. Toll-like receptor 3 regulates NK cell responses to cytokines and controls experimental metastasis. OncoImmunology 2015, 4, e1027468. [Google Scholar] [CrossRef]
- Merzoug, L.B.; Marie, S.; Satoh-Takayama, N.; Lesjean, S.; Albanesi, M.; Luche, H.; Fehling, H.J.; Di Santo, J.P.; Vosshenrich, C.A. Conditional ablation of NKp46+ cells using a novel Ncr1(greenCre) mouse strain: NK cells are essential for protection against pulmonary B16 metastases. Eur. J. Immunol. 2014, 44, 3380–3391. [Google Scholar] [CrossRef]
- Werneck, M.B.; Lugo-Villarino, G.; Hwang, E.S.; Cantor, H.; Glimcher, L.H. T-bet plays a key role in NK-mediated control of melanoma metastatic disease. J. Immunol. 2008, 180, 8004–8010. [Google Scholar] [CrossRef]
- Malaisé, M.; Rovira, J.; Renner, P.; Eggenhofer, E.; Sabet-Baktach, M.; Lantow, M.; Lang, S.A.; Koehl, G.E.; Farkas, S.A.; Loss, M.; et al. KLRG1+ NK cells protect T-bet-deficient mice from pulmonary metastatic colorectal carcinoma. J. Immunol. 2014, 192, 1954–1961. [Google Scholar] [CrossRef]
- Chan, C.J.; Martinet, L.; Gilfillan, S.; Souza-Fonseca-Guimaraes, F.; Chow, M.T.; Town, L.; Ritchie, D.S.; Colonna, M.; Andrews, D.M.; Smyth, M.J. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat. Immunol. 2014, 15, 431–438. [Google Scholar] [CrossRef]
- Martinet, L.; Ferrari De Andrade, L.; Guillerey, C.; Lee, J.S.; Liu, J.; Souza-Fonseca-Guimaraes, F.; Hutchinson, D.S.; Kolesnik, T.B.; Nicholson, S.E.; Smyth, M.J. DNAM-1 expression marks an alternative program of NK cell maturation. Cell Rep. 2015, 11, 85–97. [Google Scholar] [CrossRef]
- de Andrade, L.F.; Ngiow, S.F.; Martinet, L.; Smyth, M.J. Natural Killer cell control of BRAFV600E mutant melanoma during targeted therapy. Oncoimmunology 2015, 4, e998119. [Google Scholar] [CrossRef]
- Paolino, M.; Choidas, A.; Wallner, S.; Pranjic, B.; Uribesalgo, I.; Loeser, S.; Jamieson, A.M.; Langdon, W.Y.; Ikeda, F.; Fededa, J.P.; et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 2014, 507, 508–512. [Google Scholar] [CrossRef]
- Ferrari de Andrade, L.; Ngiow, S.F.; Stannard, K.; Rusakiewicz, S.; Kalimutho, M.; Khanna, K.K.; Tey, S.K.; Takeda, K.; Zitvogel, L.; Martinet, L.; et al. Natural killer cells are essential for the ability of BRAF inhibitors to control BRAFV600E-mutant metastatic melanoma. Cancer Res. 2014, 74, 7298–7308. [Google Scholar] [CrossRef] [PubMed]
- Putz, E.M.; Guillerey, C.; Kos, K.; Stannard, K.; Miles, K.; Delconte, R.B.; Takeda, K.; Nicholson, S.E.; Huntington, N.D.; Smyth, M.J. Targeting cytokine signaling checkpoint CIS activates NK cells to protect from tumor initiation and metastasis. OncoImmunology 2017, 6, e1267892. [Google Scholar] [CrossRef]
- Delconte, R.B.; Kolesnik, T.B.; Dagley, L.F.; Rautela, J.; Shi, W.; Putz, E.M.; Stannard, K.; Zhang, J.G.; Teh, C.; Firth, M.; et al. CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat. Immunol. 2016, 17, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Morisaki, T.; Onishi, H.; Katano, M. Cancer immunotherapy using NKG2D and DNAM-1 systems. Anticancer Res. 2012, 32, 2241–2247. [Google Scholar] [PubMed]
- Spiegel, A.; Brooks, M.W.; Houshyar, S.; Reinhardt, F.; Ardolino, M.; Fessler, E.; Chen, M.B.; Krall, J.A.; DeCock, J.; Zervantonakis, I.K.; et al. Neutrophils suppress intraluminal NK cell-mediated tumor cell clearance and enhance extravasation of disseminated carcinoma cells. Cancer Discov. 2016, 6, 630–649. [Google Scholar] [CrossRef]
- Diefenbach, A.; Jensen, E.R.; Jamieson, A.M.; Raulet, D.H. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature 2001, 413, 165–171. [Google Scholar] [CrossRef]
- Malladi, S.; Macalinao, D.G.; Jin, X.; He, L.; Basnet, H.; Zou, Y.; de Stanchina, E.; Massague, J. Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell 2016, 165, 45–60. [Google Scholar] [CrossRef]
- Street, S.E.; Cretney, E.; Smyth, M.J. Perforin and interferon-gamma activities independently control tumor initiation, growth, and metastasis. Blood 2001, 97, 192–197. [Google Scholar] [CrossRef]
- Smyth, M.J.; Thia, K.Y.; Cretney, E.; Kelly, J.M.; Snook, M.B.; Forbes, C.A.; Scalzo, A.A. Perforin is a major contributor to NK cell control of tumor metastasis. J. Immunol. 1999, 162, 6658–6662. [Google Scholar] [CrossRef]
- Takeda, K.; Hayakawa, Y.; Smyth, M.J.; Kayagaki, N.; Yamaguchi, N.; Kakuta, S.; Iwakura, Y.; Yagita, H.; Okumura, K. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat. Med. 2001, 7, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Prager, I.; Watzl, C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J. Leukoc. Biol. 2019, 105, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Song, J.; Zhang, H.; Liu, X.; Zuo, F.; Zhao, Y.; Zhao, Y.; Yin, X.; Guo, X.; Wu, X.; et al. Immune ch.eckpoint HLA-E:CD94-NKG2A mediates evasion of circulating tumor cells from NK cell surveillance. Cancer Cell 2023, 41, 272–287.e9. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xing, Z.; Chen, H.; Yang, H.; Wang, Q.; Xing, T. High expression of nectin-1 indicates a poor prognosis and promotes metastasis in hepatocellular carcinoma. Front. Oncol. 2022, 12, 953529. [Google Scholar] [CrossRef]
- Benson, D.M., Jr.; Bakan, C.E.; Mishra, A.; Hofmeister, C.C.; Efebera, Y.; Becknell, B.; Baiocchi, R.A.; Zhang, J.; Yu, J.; Smith, M.K.; et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: A therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood 2010, 116, 2286–2294. [Google Scholar] [CrossRef]
- Carosella, E.D.; Rouas-Freiss, N.; Tronik-Le Roux, D.; Moreau, P.; LeMaoult, J. HLA-G: An immune checkpoint molecule. Adv. Immunol. 2015, 127, 33–144. [Google Scholar]
- Girotti, M.R.; Salatino, M.; Dalotto-Moreno, T.; Rabinovich, G.A. Sweetening the hallmarks of cancer: Galectins as multifunctional mediators of tumor progression. J. Exp. Med. 2020, 217, e20182041. [Google Scholar] [CrossRef]
- Yan, W.; Chang, Y.; Liang, X.; Cardinal, J.S.; Huang, H.; Thorne, S.H.; Monga, S.P.; Geller, D.A.; Lotze, M.T.; Tsung, A. High-mobility group box 1 activates caspase-1 and promotes hepatocellular carcinoma invasiveness and metastases. Hepatology 2012, 55, 1863–1875. [Google Scholar] [CrossRef] [PubMed]
- Moriwaka, Y.; Luo, Y.; Ohmori, H.; Fujii, K.; Tatsumoto, N.; Sasahira, T.; Kuniyasu, H. HMGB1 attenuates antimetastatic defense of the lymph nodes in colorectal cancer. Pathobiology 2010, 77, 17–23. [Google Scholar] [CrossRef]
- Chiba, S.; Baghdadi, M.; Akiba, H.; Yoshiyama, H.; Kinoshita, I.; Dosaka-Akita, H.; Fujioka, Y.; Ohba, Y.; Gorman, J.V.; Colgan, J.D.; et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat. Immunol. 2012, 13, 832–842. [Google Scholar] [CrossRef]
- Thies, A.; Mauer, S.; Fodstad, O.; Schumacher, U. Clinically proven markers of metastasis predict metastatic spread of human melanoma cells engrafted in scid mice. Br. J. Cancer 2007, 96, 609–616. [Google Scholar] [CrossRef]
- Cornel, A.M.; Mimpen, I.L.; Nierkens, S. MHC Class I downregulation in cancer: Underlying mechanisms and potential targets for cancer immunotherapy. Cancers 2020, 12, 1760. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F.; Aptsiauri, N. Cancer immune escape: MHC expression in primary tumours versus metastases. Immunology 2019, 158, 255–266. [Google Scholar] [CrossRef]
- Taylor, B.C.; Balko, J.M. Mechanisms of MHC-I downregulation and role in immunotherapy response. Front. Immunol. 2022, 13, 844866. [Google Scholar] [CrossRef]
- Armeanu, S.; Bitzer, M.; Lauer, U.M.; Venturelli, S.; Pathil, A.; Krusch, M.; Kaiser, S.; Jobst, J.; Smirnow, I.; Wagner, A.; et al. Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res. 2005, 65, 6321–6329. [Google Scholar] [CrossRef]
- Lopez-Soto, A.; Folgueras, A.R.; Seto, E.; Gonzalez, S. HDAC3 represses the expression of NKG2D ligands ULBPs in epithelial tumour cells: Potential implications for the immunosurveillance of cancer. Oncogene 2009, 28, 2370–2382. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Gowen, B.G.; Zhang, L.; Wang, L.; Lau, S.; Iannello, A.; Xu, J.; Rovis, T.L.; Xiong, N.; Raulet, D.H. Antitumor immunity. A shed NKG2D ligand that promotes natural killer cell activation and tumor rejection. Science 2015, 348, 136–139. [Google Scholar] [CrossRef]
- Chitadze, G.; Lettau, M.; Bhat, J.; Wesch, D.; Steinle, A.; Furst, D.; Mytilineos, J.; Kalthoff, H.; Janssen, O.; Oberg, H.H.; et al. Shedding of endogenous MHC class I-related chain molecules A and B from different human tumor entities: Heterogeneous involvement of the “a disintegrin and metalloproteases” 10 and 17. Int. J. Cancer 2013, 133, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Atteridge, C.L.; Wang, X.; Lundgren, A.D.; Wu, J.D. The membrane type matrix metalloproteinase MMP14 mediates constitutive shedding of MHC class I chain-related molecule A independent of A disintegrin and metalloproteinases. J. Immunol. 2010, 184, 3346–3350. [Google Scholar] [CrossRef]
- Zhang, J.; Basher, F.; Wu, J.D. NKG2D ligands in tumor immunity: Two sides of a coin. Front. Immunol. 2015, 6, 97. [Google Scholar] [CrossRef]
- Schlecker, E.; Fiegler, N.; Arnold, A.; Altevogt, P.; Rose-John, S.; Moldenhauer, G.; Sucker, A.; Paschen, A.; von Strandmann, E.P.; Textor, S.; et al. Metalloprotease-mediated tumor cell shedding of B7-H6, the ligand of the natural killer cell-activating receptor NKp30. Cancer Res. 2014, 74, 3429–3440. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Chikumi, H.; Shimizu, A.; Takata, M.; Kinoshita, N.; Hashimoto, K.; Nakamoto, M.; Matsunaga, S.; Kurai, J.; Miyake, N.; et al. Diagnostic and prognostic impact of serum-soluble UL16-binding protein 2 in lung cancer patients. Cancer Sci. 2012, 103, 1405–1413. [Google Scholar] [CrossRef]
- Paschen, A.; Sucker, A.; Hill, B.; Moll, I.; Zapatka, M.; Nguyen, X.D.; Sim, G.C.; Gutmann, I.; Hassel, J.; Becker, J.C.; et al. Differential clinical significance of individual NKG2D ligands in melanoma: Soluble ULBP2 as an indicator of poor prognosis superior to S100B. Clin. Cancer Res. 2009, 15, 5208–5215. [Google Scholar] [CrossRef]
- Maecker, H.L.; Yun, Z.; Maecker, H.T.; Giaccia, A.J. Epigenetic changes in tumor Fas levels determine immune escape and response to therapy. Cancer Cell 2002, 2, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.; Focaccetti, C.; Pagani, A.; Imperatori, A.S.; Spagnoletti, M.; Rotolo, N.; Cantelmo, A.R.; Franzi, F.; Capella, C.; Ferlazzo, G.; et al. The proangiogenic phenotype of natural killer cells in patients with non-small cell lung cancer. Neoplasia 2013, 15, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Kopp, H.G.; Placke, T.; Salih, H.R. Platelet-derived transforming growth factor-beta down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Res. 2009, 69, 7775–7783. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.M.; Zhou, S.; Meng, X.M.; Wang, Q.M.; Li, C.J.; Lian, G.Y.; Huang, X.R.; Tang, Y.J.; Guan, X.Y.; Yan, B.P.; et al. Smad3 promotes cancer progression by inhibiting E4BP4-mediated NK cell development. Nat. Commun. 2017, 8, 14677. [Google Scholar] [CrossRef]
- Pedroza-Pacheco, I.; Madrigal, A.; Saudemont, A. Interaction between natural killer cells and regulatory T cells: Perspectives for immunotherapy. Cell Mol. Immunol. 2013, 10, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Han, Y.; Guo, Q.; Zhang, M.; Cao, X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane- bound TGF-beta 1. J. Immunol. 2009, 182, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Sceneay, J.; Chow, M.T.; Chen, A.; Halse, H.M.; Wong, C.S.; Andrews, D.M.; Sloan, E.K.; Parker, B.S.; Bowtell, D.D.; Smyth, M.J.; et al. Primary tumor hypoxia recruits CD11b+/Ly6Cmed/Ly6G+ immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche. Cancer Res. 2012, 72, 3906–3911. [Google Scholar] [CrossRef] [PubMed]
- Della Chiesa, M.; Carlomagno, S.; Frumento, G.; Balsamo, M.; Cantoni, C.; Conte, R.; Moretta, L.; Moretta, A.; Vitale, M. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood 2006, 108, 4118–4125. [Google Scholar] [CrossRef]
- Bidwell, B.N.; Slaney, C.Y.; Withana, N.P.; Forster, S.; Cao, Y.; Loi, S.; Andrews, D.; Mikeska, T.; Mangan, N.E.; Samarajiwa, S.A.; et al. Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat. Med. 2012, 18, 1224–1231. [Google Scholar] [CrossRef]
- Mlecnik, B.; Bindea, G.; Angell, H.K.; Sasso, M.S.; Obenauf, A.C.; Fredriksen, T.; Lafontaine, L.; Bilocq, A.M.; Kirilovsky, A.; Tosolini, M.; et al. Functional network pipeline reveals genetic determinants associated with in situ lymphocyte proliferation and survival of cancer patients. Sci. Transl. Med. 2014, 6, 228ra37. [Google Scholar] [CrossRef]
- Whitley, M.J.; Suwanpradid, J.; Lai, C.; Jiang, S.W.; Cook, J.L.; Zelac, D.E.; Rudolph, R.; Corcoran, D.L.; Degan, S.; Spasojevic, I.; et al. ENTPD1 (CD39) expression inhibits UVR-induced DNA damage repair through purinergic signaling and is associated with metastasis in human cutaneous squamous cell carcinoma. J. Investig. Dermatol. 2021, 141, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Young, A.; Mittal, D.; Stagg, J.; Smyth, M.J. Targeting cancer derived adenosine: New therapeutic approaches. Cancer Discov. 2014, 4, 879–888. [Google Scholar] [CrossRef]
- Beavis, P.A.; Divisekera, U.; Paget, C.; Chow, M.T.; John, L.B.; Devaud, C.; Dwyer, K.; Stagg, J.; Smyth, M.J.; Darcy, P.K. Blockade of A2A receptors potently suppresses the metastasis of CD73+ tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 14711–14716. [Google Scholar] [CrossRef]
- Cekic, C.; Day, Y.J.; Sag, D.; Linden, J. Myeloid expression of adenosine A2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer Res. 2014, 74, 7250–7259. [Google Scholar] [CrossRef]
- Young, A.; Ngiow, S.F.; Barkauskas, D.S.; Sult, E.; Hay, C.; Blake, S.J.; Huang, Q.; Liu, J.; Takeda, K.; Teng, M.W.; et al. Co-inhibition of CD73 and A2AR adenosine signaling improves anti-tumor immune responses. Cancer Cell 2016, 30, 391–403. [Google Scholar] [CrossRef]
- Berchem, G.; Noman, M.Z.; Bosseler, M.; Paggetti, J.; Baconnais, S.; Le Cam, E.; Nanbakhsh, A.; Moussay, E.; Mami-Chouaib, F.; Janji, B.; et al. Hypoxic tumor-derived microvesicles negatively regulate NK cell function by a mechanism involving TGF-beta and miR23a transfer. Oncoimmunology 2016, 5, e1062968. [Google Scholar] [CrossRef] [PubMed]
- Baginska, J.; Viry, E.; Berchem, G.; Poli, A.; Noman, M.Z.; van Moer, K.; Medves, S.; Zimmer, J.A.; Niclou, S.P.; Bleackley, R.C.; et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc. Natl. Acad. Sci. USA 2013, 110, 17450–17455. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Schreiber, T.H.; Belikoff, B.; Abbott, R.; Sethumadhavan, S.; Philbrook, P.; Ko, K.; Cannici, R.; et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci. Transl. Med. 2015, 7, 277ra230. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wu, Y.; Gao, W.; Enjyoji, K.; Csizmadia, E.; Muller, C.E.; Murakami, T.; Robson, S.C. CD39/ENTPD1 expression by CD4+Foxp3+ regulatory T cells promotes hepatic metastatic tumor growth in mice. Gastroenterology 2010, 139, 1030–1040. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming, and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Sheppard, S.; Santosa, E.K.; Lau, C.M.; Violante, S.; Giovanelli, P.; Kim, H.; Cross, J.R.; Li, M.O.; Sun, J.C. Lactate dehydrogenase A-dependent aerobic glycolysis promotes natural killer cell anti-viral and anti-tumor function. Cell Rep. 2021, 35, 109210. [Google Scholar] [CrossRef]
- Chow, M.T.; Sceneay, J.; Paget, C.; Wong, C.S.; Duret, H.; Tschopp, J.; Moller, A.; Smyth, M.J. NLRP3 suppresses NK cell-mediated responses to carcinogen-induced tumors and metastases. Cancer Res. 2012, 72, 5721–5732. [Google Scholar] [CrossRef] [PubMed]
- Dupaul-Chicoine, J.; Arabzadeh, A.; Dagenais, M.; Douglas, T.; Champagne, C.; Morizot, A.; Rodrigue-Gervais, I.G.; Breton, V.; Colpitts, S.L.; Beauchemin, N.; et al. The Nlrp3 inflammasome suppresses colorectal cancer metastatic growth in the liver by promoting natural killer cell tumoricidal activity. Immunity 2015, 43, 751–763. [Google Scholar] [CrossRef]
- Shabani, N.; Mylonas, I.; Kunert-Keil, C.; Briese, V.; Janni, W.; Gerber, B.; Friese, K.; Jeschke, U. Expression of glycodelin in human breast cancer: Immunohistochemical analysis in mammary carcinoma in situ, invasive carcinomas and their lymph node metastases. Anticancer Res. 2005, 25, 1761–1764. [Google Scholar]
- Okamoto, N.; Uchida, A.; Takakura, K.; Kariya, Y.; Kanzaki, H.; Riittinen, L.; Koistinen, R.; Seppälä, M.; Mori, T. Suppression by human placental protein 14 of natural killer cell activity. Am. J. Reprod. Immunol. 1991, 26, 137–142. [Google Scholar] [CrossRef]
- Phan, T.G.; Croucher, P.I. The dormant cancer cell life cycle. Nat. Rev. Cancer 2020, 20, 398–411. [Google Scholar] [CrossRef]
- Wang, B.; Wang, Q.; Wang, Z.; Jiang, J.; Yu, S.C.; Ping, Y.F.; Yang, J.; Xu, S.L.; Ye, X.Z.; Xu, C.; et al. Metastatic consequences of immune escape from NK cell cytotoxicity by human breast cancer stem cells. Cancer Res. 2014, 74, 5746–5757. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Best, S.A.; Hess, J.B.; Souza-Fonseca-Guimaraes, F.; Cursons, J.; Kersbergen, A.; Dong, X.; Rautela, J.; Hyslop, S.R.; Ritchie, M.E.; Davis, M.J.; et al. Harnessing natural killer immunity in metastatic SCLC. J. Thorac. Oncol. 2020, 15, 1507–1521. [Google Scholar] [CrossRef] [PubMed]
- Blake, S.J.; Stannard, K.; Liu, J.; Allen, S.; Yong, M.C.; Mittal, D.; Aguilera, A.R.; Miles, J.J.; Lutzky, V.P.; de Andrade, L.F.; et al. Suppression of metastases using a new lymphocyte checkpoint target for cancer immunotherapy. Cancer Discov. 2016, 6, 446–459. [Google Scholar] [CrossRef]
- Ni, J.; Miller, M.; Stojanovic, A.; Garbi, N.; Cerwenka, A. Sustained effector function of IL-12/15/18-preactivated NK cells against established tumors. J. Exp. Med. 2012, 209, 2351–2365. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Voronov, E.; Dvorkin, T.; Fima, E.; Cagnano, E.; Benharroch, D.; Shendler, Y.; Bjorkdahl, O.; Segal, S.; Dinarello, C.A.; et al. Differential effects of IL-1 alpha and IL-1 beta on tumorigenicity patterns and invasiveness. J. Immunol. 2003, 171, 6448–6456. [Google Scholar] [CrossRef]
- Sun, R.; Gao, D.S.; Shoush, J.; Lu, B. The IL-1 family in tumorigenesis and antitumor immunity. Semin. Cancer Biol. 2022, 86 Pt 2, 280–295. [Google Scholar] [CrossRef]
- Robertson, M.J.; Mier, J.W.; Logan, T.; Atkins, M.; Koon, H.; Koch, K.M.; Kathman, S.; Pandite, L.N.; Oei, C.; Kirby, L.C.; et al. Clinical and biological effects of recombinant human interleukin-18 administered by intravenous infusion to patients with advanced cancer. Clin. Cancer Res. 2006, 12 Pt 1, 4265–4273. [Google Scholar] [CrossRef]
- Swann, J.B.; Hayakawa, Y.; Zerafa, N.; Sheehan, K.C.; Scott, B.; Schreiber, R.D.; Hertzog, P.; Smyth, M.J. Type I IFN contributes to NK cell homeostasis, activation, and antitumor function. J. Immunol. 2007, 178, 7540–7549. [Google Scholar] [CrossRef]
- Rosenberg, S. Lymphokine-activated killer cells: A new approach to immunotherapy of cancer. J. Natl. Cancer Inst. 1985, 75, 595–603. [Google Scholar]
- Yamauchi, A.; Taga, K.; Mostowski, H.S.; Bloom, E.T. Target cell-induced apoptosis of interleukin-2-activated human natural killer cells: Roles of cell surface molecules and intracellular events. Blood 1996, 87, 5127–5135. [Google Scholar] [CrossRef]
- Woan, K.V.; Miller, J.S. Harnessing natural killer cell antitumor immunity: From the bench to bedside. Cancer Immunol. Res. 2019, 7, 1742–1747. [Google Scholar] [CrossRef]
- Escudier, B.; Farace, F.; Angevin, E.; Charpentier, F.; Nitenberg, G.; Triebel, F.; Hercend, T. Immunotherapy with interleukin-2 (IL2) and lymphokine-activated natural killer cells: Improvement of clinical responses in metastatic renal cell carcinoma patients previously treated with IL2. Eur. J. Cancer 1994, 30, 1078–1083. [Google Scholar] [CrossRef]
- Schmidt-Wolf, I.; Lefterova, P.; Mehta, B.A.; Fernandez, L.P.; Huhn, D.; Blume, K.G.; Weissman, I.L.; Negrin, R.S. Phenotypic characterization and identification of effector cells involved in tumor cell recognition of cytokine-induced killer cells. Exp. Hematol. 1993, 21, 1673–1679. [Google Scholar] [PubMed]
- Grudzien, M.; Rapak, A. Effect of Natural Compounds on NK Cell Activation. J. Immunol. Res. 2018, 2018, 4868417. [Google Scholar] [CrossRef]
- Thiery, J.; Keefe, D.; Saffarian, S.; Martinvalet, D.; Walch, M.; Boucrot, E.; Kirchhausen, T.; Lieberman, J. Perforin activates clathrin- and dynamin-dependent endocytosis, which is required for plasma membrane repair and delivery of granzyme B for granzyme-mediated apoptosis. Blood 2010, 115, 1582–1593. [Google Scholar] [CrossRef] [PubMed]
- Bachanova, V.; Cooley, S.; Defor, T.E.; Verneris, M.R.; Zhang, B.; McKenna, D.H.; Curtsinger, J.; Panoskaltsis-Mortari, A.; Lewis, D.; Hippen, K.; et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood 2014, 123, 3855–3863. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.M.; Bates, D.L.; Ring, A.M.; Krieg, C.; Lin, J.T.; Su, L.; Moraga, I.; Raeber, M.E.; Bowman, G.R.; Novick, P.; et al. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature 2012, 484, 529–533. [Google Scholar] [CrossRef]
- Sim, G.C.; Radvanyi, L. The IL-2 cytokine family in cancer immunotherapy. Cytokine Growth Factor Rev. 2014, 25, 377–390. [Google Scholar] [CrossRef]
- Carson, W.E.; Giri, J.G.; Lindemann, M.J.; Linett, M.L.; Ahdieh, M.; Paxton, R.; Anderson, D.; Eisenmann, J.; Grabstein, K.; Caligiuri, M.A. Interleukin (IL) 15 is a novel cytokine that activates human natural killer cells via components of the IL-2 receptor. J. Exp. Med. 1994, 180, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Carson, W.E.; Ross, M.E.; Baiocchi, R.A.; Marien, M.J.; Boiani, N.; Grabstein, K.; Caligiuri, M.A. Endogenous production of interleukin 15 by activated human monocytes is critical for optimal production of interferon-gamma by natural killer cells in vitro. J. Clin. Investig. 1995, 96, 2578–2582. [Google Scholar] [CrossRef]
- Vignali, D.A.; Kuchroo, V.K. IL-12 family cytokines: Immunological playmakers. Nat. Immunol. 2012, 13, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Gollob, J.A.; Mier, J.W.; Veenstra, K.; McDermott, D.F.; Clancy, D.; Clancy, M.; Atkins, M.B. Phase I trial of twice-weekly intravenous interleukin 12 in patients with metastatic renal cell cancer or malignant melanoma: Ability to maintain IFN-gamma induction is associated with clinical response. Clin. Cancer Res. 2000, 6, 1678–1692. [Google Scholar]
- Smyth, M.J.; Taniguchi, M.; Street, S.E. The anti-tumor activity of IL-12, mechanisms of innate immunity that are model and dose dependent. J. Immunol. 2000, 165, 2665–2670. [Google Scholar] [CrossRef] [PubMed]
- Mittal, D.; Vijayan, D.; Putz, E.M.; Aguilera, A.R.; Markey, K.A.; Straube, J.; Kazakoff, S.; Nutt, S.L.; Takeda, K.; Hill, G.R.; et al. Interleukin-12 from CD103+ Batf3-dependent dendritic cells required for NK-cell suppression of metastasis. Cancer Immunol. Res. 2017, 5, 1098–1108. [Google Scholar] [CrossRef]
- Bajetta, E.; Del Vecchio, M.; Mortarini, R.; Nadeau, R.; Rakhit, A.; Rimassa, L.; Fowst, C.; Borri, A.; Anichini, A.; Parmiani, G. Pilot study of subcutaneous recombinant human interleukin 12 in metastatic melanoma. Clin. Cancer Res. 1998, 4, 75–85. [Google Scholar]
- Leonard, J.P.; Sherman, M.L.; Fisher, G.L.; Buchanan, L.J.; Larsen, G.; Atkins, M.B.; Sosman, J.A.; Dutcher, J.P.; Vogelzang, N.J.; Ryan, J.L. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood 1997, 90, 2541–2548. [Google Scholar]
- Lind, H.; Gameiro, S.R.; Jochems, C.; Donahue, R.N.; Strauss, J.; Gulley, J.L.; Palena, C.; Schlom, J. Dual targeting of TGF-β and PD-L1 via a bifunctional anti-PD-L1/TGF-βRII agent: Status of preclinical and clinical advances. J. Immunother. Cancer 2020, 8, e000433. [Google Scholar] [CrossRef]
- Davis, I.D.; Skrumsager, B.K.; Cebon, J.; Nicholaou, T.; Barlow, J.W.; Moller, N.P.; Skak, K.; Lundsgaard, D.; Frederiksen, K.S.; Thygesen, P.; et al. An open-label, two arm, phase I trial of recombinant human interleukin-21 in patients with metastatic melanoma. Clin. Cancer Res. 2007, 13, 3630–3636. [Google Scholar] [CrossRef] [PubMed]
- Steele, N.; Anthony, A.; Saunders, M.; Esmarck, B.; Ehrnrooth, E.; Kristjansen, P.E.; Nihlen, A.; Hansen, L.T.; Cassidy, J. A phase 1 trial of recombinant human IL-21 in combination with cetuximab in patients with metastatic colorectal cancer. Br. J. Cancer 2012, 106, 793–798. [Google Scholar] [CrossRef]
- Davis, L.D.; Brady, B.; Kefford, R.F.; Millward, M.; Cebon, J.; Skrumsager, B.K.; Mouritzen, U.; Hansen, L.T.; Skak, K.; Lundsgaard, D.; et al. Clinical and biological efficacy of recombinant human interleukin-21 in patients with stage IV malignant melanoma without prior treatment: A phase IIa trial. Clin. Cancer Res. 2009, 15, 2123–2129. [Google Scholar] [CrossRef]
- Rahimi Kalateh Shah Mohammad, G.; Ghahremanloo, A.; Soltani, A.; Fathi, E.; Hashemy, S.I. Cytokines as potential combination agents with PD-1/PD-L1 blockade for cancer treatment. J. Cell Physiol. 2020, 235, 5449–5460. [Google Scholar] [CrossRef]
- Waldmann, T.A. Cytokines in Cancer Immunotherapy. Cold Spring Harb. Perspect. Biol. 2018, 10, a028472. [Google Scholar] [CrossRef]
- Shaw, S.G.; Maung, A.A.; Steptoe, R.J.; Thomson, A.W.; Vujanovic, N.L. Expansion of functional NK cells in multiple tissue compartments of mice treated with Flt3-ligand: Implications for anti-cancer and anti-viral therapy. J. Immunol. 1998, 161, 2817–2824. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, A.; Nagayoshi, K.; Nakamura, K.; Nakauchi, H. Lymphokine requirement for the generation of natural killer cells from CD34+ hematopoietic progenitor cells. Blood 1995, 85, 3538–3546. [Google Scholar] [CrossRef]
- Neal, Z.C.; Sondel, P.M.; Bates, M.K.; Gillies, S.D.; Herweijer, H. Flt3-L gene therapy enhances immunocytokine-mediated antitumor effects and induces long-term memory. Cancer Immunol. Immunother. 2007, 56, 1765–1774. [Google Scholar] [CrossRef]
- Long, S.; Gu, Y.; An, Y.; Lin, X.; Chen, X.; Wang, X.; Liao, C.; Ouyang, W.; Wang, N.; He, Z.; et al. Reovirus enhances cytotoxicity of natural killer cells against colorectal cancer via TLR3 pathway. J. Transl. Med. 2021, 19, 185. [Google Scholar] [CrossRef] [PubMed]
- Le Noci, V.; Sommariva, M.; Tortoreto, M.; Zaffaroni, N.; Campiglio, M.; Tagliabue, E.; Balsari, A.; Sfondrini, L. Reprogramming the lung microenvironment by inhaled immunotherapy fosters immune destruction of tumor. OncoImmunology 2016, 5, e1234571. [Google Scholar] [CrossRef]
- Yang, J.C.; Sherry, R.M.; Steinberg, S.M.; Topalian, S.L.; Schwartzentruber, D.J.; Hwu, P.; Seipp, C.A.; Rogers-Freezer, L.; Morton, K.E.; White, D.E.; et al. Randomized study of high-dose and low-dose interleukin-2 in patients with metastatic renal cancer. J. Clin. Oncol. 2003, 21, 3127–3132. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lundqvist, A. Immunomodulatory effects of IL-2 and IL-15; implications for cancer immunotherapy. Cancers 2020, 12, 3586. [Google Scholar] [CrossRef]
- Peng, L.S.; Penichet, M.L.; Morrison, S.L. A single-chain IL-12 IgG3 antibody fusion protein retains antibody specificity and IL-12 bioactivity and demonstrates antitumor activity. J. Immunol. 1999, 163, 250–258. [Google Scholar] [CrossRef]
- Young, P.A.; Morrison, S.L.; Timmerman, J.M. Antibody-cytokine fusion proteins for treatment of cancer: Engineering cytokines for improved efficacy and safety. Semin. Oncol. 2014, 41, 623–636. [Google Scholar] [CrossRef]
- Robertson, M.J. Role of chemokines in the biology of natural killer cells. J. Leukoc. Biol. 2002, 71, 173–183. [Google Scholar] [CrossRef]
- Lucas, M.; Schachterle, W.; Oberle, K.; Aichele, P.; Diefenbach, A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity 2007, 26, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, J.S.; Lee, H.K.; Kim, H.S.; Park, E.J.; Choi, J.E.; Choi, Y.J.; Shin, B.R.; Kim, E.Y.; Hong, J.T.; et al. CXCR3-deficient natural killer cells fail to migrate to B16F10 melanoma cells. Int. Immunopharmacol. 2018, 63, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Melaiu, O.; Chierici, M.; Lucarini, V.; Jurman, G.; Conti, L.A.; De Vito, R.; Boldrini, R.; Cifaldi, L.; Castellano, A.; Furlanello, C.; et al. Cellular and gene signatures of tumor-infiltrating dendritic cells and natural-killer cells predict prognosis of neuroblastoma. Nat. Commun. 2020, 11, 5992. [Google Scholar] [CrossRef]
- Fernandez, N.C.; Lozier, A.; Flament, C.; Ricciardi-Castagnoli, P.; Bellet, D.; Suter, M.; Perricaudet, M.; Tursz, T.; Maraskovsky, E.; Zitvogel, L. Dendritic cells directly trigger NK cell functions: Cross-talk relevant in innate anti-tumor immune responses in vivo. Nat. Med. 1999, 5, 405–411. [Google Scholar] [CrossRef]
- Biber, G.; Sabag, B.; Raiff, A.; Ben-Shmuel, A.; Puthenveetil, A.; Benichou, J.I.C.; Jubany, T.; Levy, M.; Killner, S.; Barda-Saad, M. Modulation of intrinsic inhibitory checkpoints using nano-carriers to unleash NK cell activity. EMBO Mol. Med. 2022, 14, e14073. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, Y.; Lew, E.D.; Seelige, R.; Tindall, E.A.; Walsh, C.; Fagan, P.C.; Lee, J.Y.; Nevarez, R.; Oh, J.; Tucker, K.D.; et al. Immuno-oncological efficacy of RXDX-106, a novel TAM (TYRO3, AXL, MER) family small-molecule kinase inhibitor. Cancer Res. 2019, 79, 1996–2008. [Google Scholar] [CrossRef]
- Sanchez-Correa, B.; Lopez-Sejas, N.; Duran, E.; Labella, F.; Alonso, C.; Solana, R.; Tarazona, R. Modulation of NK cells with checkpoint inhibitors in the context of cancer immunotherapy. Cancer Immunol. Immunother. 2019, 68, 861–870. [Google Scholar] [CrossRef]
- Semeraro, M.; Galluzzi, L. Novel insights into the mechanism of action of lenalidomide. Oncoimmunology 2014, 3, e28386. [Google Scholar] [CrossRef]
- Sehgal, K.; Das, R.; Zhang, L.; Verma, R.; Deng, Y.; Kocoglu, M.; Vasquez, J.; Koduru, S.; Ren, Y.; Wang, M.; et al. Clinical and pharmacodynamic analysis of pomalidomide dosing strategies in myeloma: Impact of immune activation and cereblon targets. Blood 2015, 125, 4042–4051. [Google Scholar] [CrossRef]
- Lin, D.J.; Ng, J.C.; Huang, L.; Robinson, M.; O’Hara, J.; Wilson, J.A.; Mellor, A.L. The immunotherapeutic role of indoleamine 2,3-dioxygenase in head and neck squamous cell carcinoma: A systematic review. Clin. Otolaryngol. 2021, 46, 919–934. [Google Scholar] [CrossRef]
- Lemos, H.; Huang, L.; Prendergast, G.C.; Mellor, A.L. Immune control by amino acid catabolism during tumorigenesis and therapy. Nat. Rev. Cancer 2019, 19, 162–175. [Google Scholar] [CrossRef]
- Lim, C.M.; Liou, A.; Poon, M.; Koh, L.P.; Tan, L.K.; Loh, K.S.; Petersson, B.F.; Ting, E.; Campana, D.; Goh, B.C.; et al. Phase I study of expanded natural killer cells in combination with cetuximab for recurrent/metastatic nasopharyngeal carcinoma. Cancer Immunol. Immunother. 2022, 71, 2277–2286. [Google Scholar] [CrossRef]
- Weiner, L.M.; Surana, R.; Wang, S. Monoclonal antibodies: Versatile platforms for cancer immunotherapy. Nat. Rev. Immunol. 2010, 10, 317–327. [Google Scholar] [CrossRef]
- Collins, D.M.; Madden, S.F.; Gaynor, N.; AlSultan, D.; Le Gal, M.; Eustace, A.J.; Gately, K.A.; Hughes, C.; Davies, A.M.; Mahgoub, T.; et al. Effects of HER family-targeting tyrosine kinase inhibitors on antibody-dependent cell-mediated cytotoxicity in HER2-expressing breast cancer. Clin. Cancer Res. 2021, 27, 807–818. [Google Scholar] [CrossRef]
- Cho, H.; Kim, K.H.; Lee, H.; Kim, C.G.; Chung, H.; Choi, Y.S.; Park, S.H.; Cheong, J.W.; Min, Y.H.; Shin, E.C.; et al. Adaptive Natural Killer Cells Facilitate Effector Functions of Daratumumab in Multiple Myeloma. Clin. Cancer Res. 2021, 27, 2947–2958. [Google Scholar] [CrossRef]
- Rosario, M.; Liu, B.; Kong, L.; Collins, L.I.; Schneider, S.E.; Chen, X.; Han, K.; Jeng, E.K.; Rhode, P.R.; Leong, J.W.; et al. The IL-15-based ALT-803 complex enhances FcγRIIIa-triggered NK cell responses and in vivo clearance of B cell lymphomas. Clin. Cancer Res. 2016, 22, 596–608. [Google Scholar] [CrossRef]
- Margolin, K.; Morishima, C.; Velcheti, V.; Miller, J.S.; Lee, S.M.; Silk, A.W.; Holtan, S.G.; Lacroix, A.M.; Fling, S.P.; Kaiser, J.C.; et al. Phase I trial of ALT-803, a novel recombinant IL15 complex, in patients with advanced solid tumors. Clin. Cancer Res. 2018, 24, 5552–5561. [Google Scholar] [CrossRef]
- Junttila, T.T.; Parsons, K.; Olsson, C.; Lu, Y.; Xin, Y.; Theriault, J.; Crocker, L.; Pabonan, O.; Baginski, T.; Meng, G.; et al. Superior in vivo efficacy of afucosylated trastuzumab in the treatment of HER2-amplified breast cancer. Cancer Res. 2010, 70, 4481–4489. [Google Scholar] [CrossRef]
- Au, K.M.; Park, S.I.; Wang, A.Z. Trispecifc natural killer cell nanoengagers for targeted chemoimmunotherapy. Sci. Adv. 2020, 6, eaba8564. [Google Scholar] [CrossRef]
- Davis, Z.B.; Vallera, D.A.; Miller, J.S.; Felices, M. Natural killer cells unleashed: Checkpoint receptor blockade and BiKE/TriKE utilization in NK-mediated anti-tumor immunotherapy. Semin. Immunol. 2017, 31, 64–75. [Google Scholar] [CrossRef]
- Li, J.; Piskol, R.; Ybarra, R.; Chen, Y.-J.J.; Li, J.; Slaga, D.; Hristopoulos, M.; Clark, R.; Modrusan, Z.; Totpal, K.; et al. CD3 bispecific antibody–induced cytokine release is dispensable for cytotoxic T cell activity. Sci. Trans. Med. 2019, 11, eaax8861. [Google Scholar] [CrossRef]
- Felices, M.; Lenvik, T.R.; Davis, Z.B.; Miller, J.S.; Vallera, D.A. Generation of BiKEs and TriKEs to improve NK cell-mediated targeting of tumor cells. Methods Mol. Biol. 2016, 1441, 333–346. [Google Scholar]
- Sarhan, D.; Brandt, L.; Felices, M.; Guldevall, K.; Lenvik, T.; Hinderlie, P.; Curtsinger, J.; Warlick, E.; Spellman, S.R.; Blazar, B.R.; et al. 161533 TriKE stimulates NK-cell function to overcome myeloid-derived suppressor cells in MDS. Blood Adv. 2018, 2, 1459–1469. [Google Scholar] [CrossRef]
- Wingert, S.; Reusch, U.; Knackmuss, S.; Kluge, M.; Damrat, M.; Pahl, J.; Schniegler-Mattox, U.; Mueller, T.; Fucek, I.; Ellwanger, K.; et al. Preclinical evaluation of AFM24, a novel CD16A-specific innate immune cell engager targeting EGFR-positive tumors. MAbs 2021, 13, 1950264. [Google Scholar] [CrossRef]
- Vallera, D.A.; Felices, M.; McElmurry, R.; McCullar, V.; Zhou, X.; Schmohl, J.U.; Zhang, B.; Lenvik, A.J.; Panoskaltsis-Mortari, A.; Verneris, M.R.; et al. IL15 trispecific killer engagers (TriKE) make natural killer cells specific to CD33+ targets while also inducing persistence, in vivo expansion, and enhanced function. Clin. Cancer Res. 2016, 22, 3440–3450. [Google Scholar] [CrossRef]
- Reusing, S.B.; Vallera, D.A.; Manser, A.R.; Vatrin, T.; Bhatia, S.; Felices, M.; Miller, J.S.; Uhrberg, M.; Babor, F. CD16xCD33 bispecifc killer cell engager (BiKE) as potential immunotherapeutic in pediatric patients with AML and biphenotypic ALL. Cancer Immunol. Immunother. 2021, 70, 3701–3708. [Google Scholar] [CrossRef]
- Arvindam, U.S.; van Hauten, P.M.M.; Schirm, D.; Schaap, N.; Hobo, W.; Blazar, B.R.; Vallera, D.A.; Dolstra, H.; Felices, M.; Miller, J.S. A trispecific killer engager molecule against CLEC12A effectively induces NK-cell mediated killing of AML cells. Leukemia 2021, 35, 1586–1596. [Google Scholar] [CrossRef]
- Groh, V.; Wu, J.; Yee, C.; Spies, T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 2002, 419, 734–738. [Google Scholar] [CrossRef]
- Lopez, K.J.; Cross-Najafi, A.A.; Farag, K.; Obando, B.; Thadasina, D.; Isidan, A.; Park, Y.; Zhang, W.; Ekser, B.; Li, P. Strategies to induce natural killer cell tolerance in xenotransplantation. Front. Immunol. 2022, 13, 941880. [Google Scholar] [CrossRef]
- Andre, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell 2018, 175, 1731–1743. [Google Scholar] [CrossRef]
- van Hall, T.; André, P.; Horowitz, A.; Ruan, D.F.; Borst, L.; Zerbib, R.; Narni-Mancinelli, E.; van der Burg, S.H.; Vivier, E. Monalizumab: Inhibiting the novel immune checkpoint NKG2A. J. Immunother. Cancer 2019, 7, 263. [Google Scholar] [CrossRef]
- Benson, D.M., Jr.; Bakan, C.E.; Zhang, S.; Collins, S.M.; Liang, J.; Srivastava, S.; Hofmeister, C.C.; Efebera, Y.; Andre, P.; Romagne, F.; et al. IPH2101, a novel anti-inhibitory KIR antibody, and lenalidomide combine to enhance the natural killer cell versus multiple myeloma effect. Blood 2011, 118, 6387–6391. [Google Scholar] [CrossRef]
- Kohrt, H.E.; Thielens, A.; Marabelle, A.; Sagiv-Barfi, I.; Sola, C.; Chanuc, F.; Fuseri, N.; Bonnafous, C.; Czerwinski, D.; Rajapaksa, A.; et al. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood 2014, 123, 678–686. [Google Scholar] [CrossRef]
- Ellwanger, K.; Reusch, U.; Fucek, I.; Wingert, S.; Ross, T.; Müller, T.; Schniegler-Mattox, U.; Haneke, T.; Rajkovic, E.; Koch, J.; et al. Redirected optimized cell killing (ROCK®): A highly versatile multispecific fit-for-purpose antibody platform for engaging innate immunity. MAbs 2019, 11, 899–918. [Google Scholar] [CrossRef]
- Benson, D.M., Jr.; Hofmeister, C.C.; Padmanabhan, S.; Suvannasankha, A.; Jagannath, S.; Abonour, R.; Bakan, C.; Andre, P.; Efebera, Y.; Tiollier, J.; et al. A phase 1 trial of the anti-KIR antibody IPH2101 in patients with relapsed/refractory multiple myeloma. Blood 2012, 120, 4324–4333. [Google Scholar] [CrossRef]
- Armand, P.; Lesokhin, A.; Borrello, I.; Timmerman, J.; Gutierrez, M.; Zhu, L.; Popa McKiver, M.; Ansell, S.M. A phase 1b study of dual PD-1 and CTLA-4 or KIR blockade in patients with relapsed/refractory lymphoid malignancies. Leukemia 2021, 35, 777–786. [Google Scholar] [CrossRef]
- Benson, D.M., Jr.; Cohen, A.D.; Jagannath, S.; Munshi, N.C.; Spitzer, G.; Hofmeister, C.C.; Efebera, Y.A.; Andre, P.; Zerbib, R.; Caligiuri, M.A. A phase I trial of the anti-KIR antibody IPH2101 and lenalidomide in patients with relapsed/refractory multiple myeloma. Clin. Cancer Res. 2015, 21, 4055–4061. [Google Scholar] [CrossRef]
- Esen, F.; Deniz, G.; Aktas, E.C. PD-1, CTLA-4, LAG-3, and TIGIT: The roles of immune checkpoint receptors on the regulation of human NK cell phenotype and functions. Immunol. Lett. 2021, 240, 15–23. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory receptors with specialized functions in immune regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef]
- Chauvin, J.M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef]
- Cho, B.C.; Abreu, D.R.; Hussein, M.; Cobo, M.; Patel, A.J.; Secen, N.; Lee, K.H.; Massuti, B.; Hiret, S.; Yang, J.C.H.; et al. Tiragolumab plus atezolizumab versus placebo plus atezolizumab as a first-line treatment for PD-L1-selected non-small-cell lung cancer (CITYSCAPE): Primary and follow-up analyses of a randomised, double-blind, phase 2 study. Lancet Oncol. 2022, 23, 781–792. [Google Scholar] [CrossRef]
- Thibaudin, M.; Limagne, E.; Hampe, L.; Ballot, E.; Truntzer, C.; Ghiringhelli, F. Targeting PD-L1 and TIGIT could restore intratumoral CD8 T cell function in human colorectal cancer. Cancer Immunol. Immunother. 2022, 71, 2549–2563. [Google Scholar] [CrossRef]
- Hsu, J.; Hodgins, J.J.; Marathe, M.; Nicolai, C.J.; Bourgeois-Daigneault, M.C.; Trevino, T.N.; Azimi, C.S.; Scheer, A.K.; Randolph, H.E.; Thompson, T.W.; et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Investig. 2018, 128, 4654–4668. [Google Scholar] [CrossRef]
- Igarashi, T.; Wynberg, J.; Srinivasan, R.; Becknell, B.; McCoy, J.P., Jr.; Takahashi, Y.; Suffredini, D.A.; Linehan, W.M.; Caligiuri, M.A.; Childs, R.W. Enhanced cytotoxicity of allogeneic NK cells with killer immunoglobulin-like receptor ligand incompatibility against melanoma and renal cell carcinoma cells. Blood 2004, 104, 170–177. [Google Scholar] [CrossRef]
- Delgado, D.; Webster, D.E.; DeSantes, K.B.; Durkin, E.T.; Shaaban, A.F. KIR receptor-ligand incompatibility predicts killing of osteosarcoma cell lines by allogeneic NK cells. Pediatr. Blood Cancer 2010, 55, 1300–1305. [Google Scholar] [CrossRef]
- Grote, S.; Ureña-Bailén, G.; Chan, K.C.; Baden, C.; Mezger, M.; Handgretinger, R.; Schleicher, S. In vitro evaluation of CD276-CAR NK-92 functionality, migration and invasion potential in the presence of immune Inhibitory factors of the tumor microenvironment. Cells 2021, 10, 1020. [Google Scholar] [CrossRef]
- Soltantoyeh, T.; Akbari, B.; Karimi, A.; Mahmoodi Chalbatani, G.; Ghahri-Saremi, N.; Hadjati, J.; Hamblin, M.R.; Mirzaei, H.R. Chimeric antigen receptor (CAR) T cell therapy for metastatic melanoma: Challenges and road ahead. Cells 2021, 10, 1450. [Google Scholar] [CrossRef]
- Daher, M.; Rezvani, K. Outlook for new CAR-based therapies with a focus on CAR NK cells: What lies beyond CAR-engineered T cells in the race against cancer. Cancer Discov. 2020, 11, 45–58. [Google Scholar] [CrossRef]
- Azoulay, E.; Shimabukuro-Vornhagen, A.; Darmon, M.; von Bergwelt-Baildon, M. Critical care management of chimeric antigen receptor T cell–related toxicity. Be aware and prepared. Am. J. Respir. Crit. Care Med. 2019, 200, 20–23. [Google Scholar] [CrossRef]
- Daher, M.; Melo Garcia, L.; Li, Y.; Rezvani, K. CAR-NK cells: The next wave of cellular therapy for cancer. Clin. Transl. Immunol. 2021, 10, e1274. [Google Scholar] [CrossRef]
- Siegler, E.L.; Zhu, Y.; Wang, P.; Yang, L. Of-the-shelf CAR-NK cells for cancer immunotherapy. Cell Stem Cell 2018, 23, 160–161. [Google Scholar] [CrossRef]
- Klingemann, H.; Boissel, L.; Toneguzzo, F. Natural killer cells for immunotherapy–advantages of the NK-92 cell line over blood NK cells. Front. Immunol. 2016, 7, 91. [Google Scholar] [CrossRef]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell 2018, 23, 181–192.e5. [Google Scholar] [CrossRef]
- Gong, Y.; Klein Wolterink, R.G.J.; Wang, J.; Bos, G.M.J.; Germeraad, W.T.V. Chimeric antigen receptor natural killer (CAR-NK) cell design and engineering for cancer therapy. J. Hematol. Oncol. 2021, 14, 73. [Google Scholar] [CrossRef]
- Stoiber, S.; Cadilha, B.L.; Benmebarek, M.R.; Lesch, S.; Endres, S.; Kobold, S. Limitations in the design of chimeric antigen receptors for cancer therapy. Cells 2019, 8, 472. [Google Scholar] [CrossRef]
- Liu, S.; Galat, V.; Galat, Y.; Lee, Y.K.A.; Wainwright, D.; Wu, J. NK cell-based cancer immunotherapy: From basic biology to clinical development. J. Hematol. Oncol. 2021, 14, 7. [Google Scholar] [CrossRef]
- Marof, F.; Abdul-Rasheed, O.F.; Rahman, H.S.; Budi, H.S.; Jalil, A.T.; Yumashev, A.V.; Hassanzadeh, A.; Yazdanifar, M.; Motavalli, R.; Chartrand, M.S.; et al. CAR-NK cell in cancer immunotherapy; A promising frontier. Cancer Sci. 2021, 112, 3427–3436. [Google Scholar] [CrossRef]
- Matosevic, S. Viral and nonviral engineering of natural killer cells as emerging adoptive cancer immunotherapies. J. Immunol. Res. 2018, 2018, 4054815. [Google Scholar] [CrossRef]
- Rafei, H.; Daher, M.; Rezvani, K. Chimeric antigen receptor (CAR) natural killer (NK)-cell therapy: Leveraging the power of innate immunity. Br. J. Haematol. 2021, 193, 216–230. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, L.; Virone-Oddos, A.; Beninga, J.; Rossi, B.; Nicolazzi, C.; Amara, C.; Blanchard-Alvarez, A.; Gourdin, N.; Courta, J.; Basset, A.; et al. Control of acute myeloid leukemia by a trifunctional NKp46-CD16a-NK cell engager targeting CD123. Nat. Biotechnol. 2023. [Google Scholar] [CrossRef]
- Yanakieva, D.; Pekar, L.; Evers, A.; Fleischer, M.; Keller, S.; Mueller-Pompalla, D.; Toleikis, L.; Kolmar, H.; Zielonka, S.; Krah, S. Beyond bispecificity: Controlled Fab arm exchange for the generation of antibodies with multiple specificities. mAbs 2022, 14, 2018960. [Google Scholar] [CrossRef]
- Schmohl, J.U.; Felices, M.; Todhunter, D.; Taras, E.; Miller, J.S.; Vallera, D.A. Tetraspecific scFv construct provides NK cell mediated ADCC and self-sustaining stimuli via insertion of IL-15 as a cross-linker. Oncotarget 2016, 7, 73830–73844. [Google Scholar] [CrossRef]
- Hamieh, M.; Dobrin, A.; Cabriolu, A.; van der Stegen, S.J.C.; Giavridis, T.; Mansilla-Soto, J.; Eyquem, J.; Zhao, Z.; Whitlock, B.M.; Miele, M.M.; et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature 2019, 568, 112–116. [Google Scholar] [CrossRef]
- Li, H.S.; Wong, N.M.; Tague, E.; Ngo, J.T.; Khalil, A.S.; Wong, W.W. High-performance multiplex drug-gated CAR circuits. Cancer Cell 2022, 40, 1294–1305.e4. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | NK Cell | T Cells |
|---|---|---|
| Type of immune response | Innate immunity, Adaptive Immunity | Adaptive immunity |
| Cell marker | CD56+CD3-CD16+ | CD3+CD4+ or CD3+CD8+ |
| Development | Differentiate in the BM | Differentiate in the thymus |
| pre-stimulation | No antigen priming | Antigen priming required |
| Location | Primarily in blood and tissue resident | Antigen specific tumor site |
| Side effect of overactivation | No/low risk of GVHD | Allogeneic T cells induce GVHD |
| Mechanism of activation | Recognition of abnormal or missing self-molecules on the surface of cancer cells through the complex array of receptors | Antigen-specific recognition of of cancer cells through TCR |
| MHC dependence | Do not require MHC matching for antigen recognition and activation | Require MHC matching for antigen recognition and activation |
| Cytotoxicity | Directly kill cancer cells through the release of cytotoxic granules, greater cytotoxicity | Directly kill cancer cells through the release of cytotoxic granules |
| Cytokine secretion | Secrete cytokines to stimulate other immune cells to attack cancer cells | Secrete cytokines that stimulate other immune cells to attack cancer cells |
| Target | Target various cancer cells that have lost MHC expression or express stress-induced molecules | Target cancer cells that express specific antigens on their surface |
| ADCC | mediated antibody-dependent cellular cytotoxicity of cancer cells | No |
| Anti-metastatic activity | Highly effective against dissimilating tumor cells and distant site tumor cells | Have limited efficacy against metastatic tumor cells |
| Name | Target | Format | Mechanism | Disease | Status |
|---|---|---|---|---|---|
| AFM-13 | CD30/CD16 | scFv-scFv (BiKE) | CD30 inhibitor CD16 regulator | relapsed or refractory MM | phase II |
| AFM-24 | EGFR/CD16 | scFv-scFv (BiKE) | EGFR blockers CD16 regulator | advanced solid tumors | phase II |
| AFM26 | BCMA/CD16 | scFv-scFv (BiKE) | BCMA blocker CD16 regulator | relapsed or refractory MM | phase I/II |
| 6MW3411 | PD-L1/CD16 | scFv-scFv (BiKE) | PD-L1 inhibitors CD16 NK cell recruitment agent | solid tumor | pre-clinical |
| HRS-3/A9 or Anti-CD16/CD30 BiMAB | CD16/CD30 | scFv-scFv (BiKE) | CD30 regulator CD16 regulator | hodgkin’s disease | pre-clinical |
| NKp46 NKCE | NCR1/CD16 | scFv-scFv (BiKE) | NCR1 inhibitor CD16 NK cell recruitment agent | tumor | pre-clinical |
| 161533/GTB3550/OXS3550 | CD16/IL15/CD33 | scFv-IL15-scFv (TriKE) | fusion protein trifunctional, NK cell stimulant, CD16 regulator, CD33 inhibitor | High -risk MDS, relapsed or refractory AML | phase II |
| cam161533 TriKE | CD16/IL15/CD33 | scFv-IL15-scFv (TriKE) | CD33 regulator IL15 regulator CD16 regulator | High -risk MDS, relapsed or refractory AML | pre-clinical |
| GTB-3650 (humanized CD16scFv) | CD16/IL-15/CD33 | scFv-IL15-scFv (TriKE) | fusion protein trifunctional, NK cell stimulant, CD16 regulator, CD33 inhibitor | AML, MDS | pre-clinical |
| CD16-IL15-CLEC12A | CD16/IL15/CLEC12A | scFv-IL15-scFv (TriKE) | NK cell stimulant ADCC effect | acute myeloid leukemia, Leukemic stem cells | pre-clinical |
| triplebody | NKG2D/CD19/CD33 | ULPB2-scFv-scFv (TriKE) | activating NK cell, CD19 and CD33 inhibitors | mixed lineage leukemia (MLL) | pre-clinical |
| triplebody | CD33/CD16/CD19 | scFv-scFv-scFv (TriKE) | CD16 regulator, CD19 and CD33 inhibitors ADCC | MLL | pre-clinical |
| sctb | CD123/CD16/CD33 | scFv-scFv-scFv (TriKE) | CD16 regulator, CD123 and CD133 inhibitors, ADCC | AML | pre-clinical |
| SPM-2 | CD33/CD16/CD123 | scFv-scFv-scFv (TriKE) | CD16 regulator, CD19 and CD133 inhibitors, ADCC | AML | pre-clinical |
| TriKE | CD16/CD22/CD19 | scFv-scFv-scFv (TriKE) | CD16 regulator, CD19 and CD22 inhibitors, ADCC | B-ALL, B-CLL, AML | pre-clinical |
| ATriFlex | BCMA/CD200/CD16A | scFv-diabody-scFv (TriKE) | CD16 regulator, CD200 and BCMA inhibitors, ADCC | MM | pre-clinical |
| SAR443579 (ANKET) | CD123/CD16/NKp46 | NKp46-Fc-CD123 (TriKE) | fusion protein trifunctional, NK cell stimulant, CD16 regulator, CD123 inhibitor | AML, MDS | pre-clinical |
| 1615EpCAM | CD16/IL-15/EpCAM | scFv-IL15-scFv (TriKE) | fusion protein trifunctional, NK cell stimulant, CD16 regulator, EpCAM inhibitor | Various carcinomas | pre-clinical |
| 1615133 | CD16/IL-15/CD133 | scFv-IL15-scFv (TriKE) | fusion protein trifunctional, NK cell stimulant, CD16 regulator, CD133 inhibitor | Cancer stem cells | pre-clinical |
| 161519 | CD16/IL-15/CD19 | scFv-IL15-scFv (TriKE) | fusion protein trifunctional, NK cell stimulant, CD16 regulator, CD19 inhibitor | B-CLL | pre-clinical |
| cam1615B7H3 | CD16/IL-15/B7H3 | VHH-IL15-scFv (TriKE) | fusion protein trifunctional, NK cell stimulant, CD16 regulator, B7H3 inhibitor | Ovarian cancer | pre-clinical |
| cam1615HER2 | CD16/IL-15/HER2 | VHH-IL15-scFv (TriKE) | fusion protein trifunctional, NK cell stimulant, CD16 regulator, HER2 inhibitor | Ovarian cancer | pre-clinical |
| cam1615TEM8 | CD16/IL-15/TEM8 | VHH-IL15-scFv (TriKE) | fusion protein trifunctional, NK cell stimulant, CD16 regulator, TEM8 inhibitor | NSCLC, solid tumor | pre-clinical |
| TetraKE (TtsAb) | CD16/IL-5/EpCAM/CD133 | scFv-IL15-scFv-scFv (tetraKE) | fusion protein trifunctional, NK cell stimulant, CD16 regulator, CD133 and EpCAM inhibitor | CRC | pre-clinical |
| SEEDbody | EGFR/HER2/NKG2D | IgG-like VHH-based (NKCE) | activating NK cell, EGFR and HER2inhibitors | Breast cancer | pre-clinical |
| TsAb | EGFR/CD16a/PD-L1 | Bs IgG-Fab (NKCE) | EGFR blocker, PD-L1 blocker and CD16 regulator, ADCC | Epidermoid carcinoma | pre-clinical |
| ANKET | NKp46/CD16/CD19-CD20-EGFR | Fab-Fc-Fab (NKCE) | activating NK cell, CD16 regulator CD19 and CD20 EGFR inhibitors | LNH | pre-clinical |
| ANKET | NKp46-NKp30/CD16/CD19-CD20 | Fab-Fc-Fab(NKCE) | activating NK cell, CD16 regulator CD19 and CD20 inhibitors | B-ALL | pre-clinical |
| ANKET4 | NKp46/CD16/CD20/IL-2v | Fab-Fc-Fab (NKCE) | activating NK cell, CD16 regulator CD20 inhibitors | B-ALL | pre-clinical |
| HLE-nano-BiKE | CD38/CD16/HSA | VHH-VHH-VHH (nano) | fusion protein trifunctional, NK cell stimulant, CD16 regulator, CD38 inhibitor | multiple myeloma | pre-clinical |
| DuoBody (DB)-VHH (TtsAb) | HER2/cMET/EGFR-IL6R-NKG2D | bs IgG-VHH-VHH (NKCE) | fusion protein trifunctional, NK cell stimulant, HER2, cMet, EGFR inhibitor | Breast cancer | pre-clinical |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, Y. The Function of NK Cells in Tumor Metastasis and NK Cell-Based Immunotherapy. Cancers 2023, 15, 2323. https://doi.org/10.3390/cancers15082323
Yu Y. The Function of NK Cells in Tumor Metastasis and NK Cell-Based Immunotherapy. Cancers. 2023; 15(8):2323. https://doi.org/10.3390/cancers15082323
Chicago/Turabian StyleYu, Yanlin. 2023. "The Function of NK Cells in Tumor Metastasis and NK Cell-Based Immunotherapy" Cancers 15, no. 8: 2323. https://doi.org/10.3390/cancers15082323
APA StyleYu, Y. (2023). The Function of NK Cells in Tumor Metastasis and NK Cell-Based Immunotherapy. Cancers, 15(8), 2323. https://doi.org/10.3390/cancers15082323