


The Ubiquitin–Proteasome System in Tumor Metabolism


Abstract
:Simple Summary
Abstract
1. Introduction
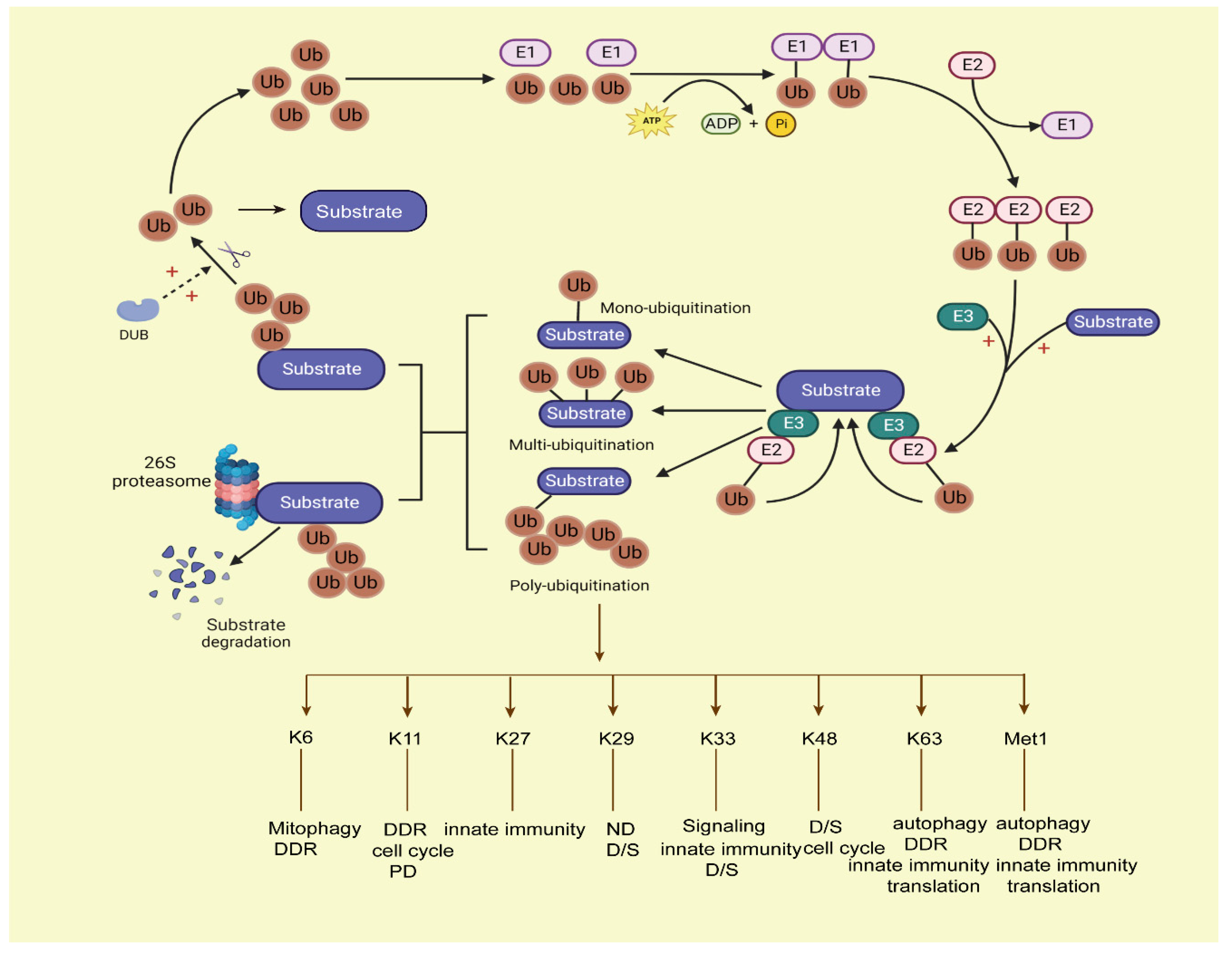
2. Ubiquitination and Deubiquitination of Metabolic Enzymes
2.1. Glucose Metabolism
2.2. FAs Metabolism
2.3. Amino Acid Metabolism
2.3.1. Glutamine Metabolism
2.3.2. Cystine Metabolism
2.3.3. Serine Metabolism
2.3.4. Arginine Metabolism
3. The UPS Links Oncogenic Signal Pathways in Cancer Metabolism
3.1. Myc Pathway
3.2. mTOR Pathway
3.3. KRAS Pathway
3.4. HIF Pathway
3.5. PI3K/AKT Pathway
3.6. Hippo Pathway
3.7. TGF-β Pathway
3.8. The Lysosome-Dependent Proteolysis Pathway
4. The Drugs Targeting UPS in Cancer Metabolism
4.1. The UPS Inhibitors in Cancer Metabolism
4.2. The Clinical Trials Targeting UPS in Cancer Metabolism
- VLX1570
- Bortezomib
- Thalidomide and its derivatives, pomalidomide and lenalidomide
- Mitoxantrone
- 2-DG
4.3. PROTAC Targeting UPS in Cancer Metabolism
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg effect: Essential part of metabolic reprogramming and central contributor to cancer progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Pant, K.; Richard, S.; Peixoto, E.; Gradilone, S.A. Role of Glucose Metabolism Reprogramming in the Pathogenesis of Cholangiocarcinoma. Front. Med. 2020, 7, 113. [Google Scholar] [CrossRef] [PubMed]
- Schworer, S.; Vardhana, S.A.; Thompson, C.B. Cancer Metabolism Drives a Stromal Regenerative Response. Cell Metab. 2019, 29, 576–591. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 2022, 21, 141–162. [Google Scholar] [CrossRef]
- Park, J.; Cho, J.; Song, E.J. Ubiquitin-proteasome system (UPS) as a target for anticancer treatment. Arch. Pharm. Res. 2020, 43, 1144–1161. [Google Scholar] [CrossRef] [PubMed]
- Adams, J. The proteasome: Structure, function, and role in the cell. Cancer Treat. Rev. 2003, 29 (Suppl. S1), 3–9. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.; Catalgol, B.; Grune, T. The proteasomal system. Mol. Asp. Med. 2009, 30, 191–296. [Google Scholar] [CrossRef]
- Ben-Nissan, G.; Sharon, M. Regulating the 20S proteasome ubiquitin-independent degradation pathway. Biomolecules 2014, 4, 862–884. [Google Scholar] [CrossRef] [PubMed]
- Vera, A.; Aris, A.; Carrio, M.; Gonzalez-Montalban, N.; Villaverde, A. Lon and ClpP proteases participate in the physiological disintegration of bacterial inclusion bodies. J. Biotechnol. 2005, 119, 163–171. [Google Scholar] [CrossRef]
- Yang, H.; Chen, X.; Li, K.; Cheaito, H.; Yang, Q.; Wu, G.; Liu, J.; Dou, Q.P. Repurposing old drugs as new inhibitors of the ubiquitin-proteasome pathway for cancer treatment. Semin. Cancer Biol. 2021, 68, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Cui, S.; Chen, Y.; Guo, S.; Chen, D. Ubiquitin specific peptidases and prostate cancer. PeerJ 2023, 11, e14799. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, B.; Akere, T.H.; Chakraborty, S.; Valsami-Jones, E.; Ali-Boucetta, H. Gold Nanoparticles Induced Size Dependent Cytotoxicity on Human Alveolar Adenocarcinoma Cells by Inhibiting the Ubiquitin Proteasome System. Pharmaceutics 2023, 15, 432. [Google Scholar] [CrossRef] [PubMed]
- Van Wijk, S.J.; Fulda, S.; Dikic, I.; Heilemann, M. Visualizing ubiquitination in mammalian cells. EMBO Rep. 2019, 20, e46520. [Google Scholar] [CrossRef]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, D.; Yao, Y.; Sun, Y.; Yao, F.; Ma, L. Monoubiquitination in Homeostasis and Cancer. Int. J. Mol. Sci. 2022, 23, 5925. [Google Scholar] [CrossRef]
- Tracz, M.; Bialek, W. Beyond K48 and K63: Non-canonical protein ubiquitination. Cell Mol. Biol. Lett. 2021, 26, 1. [Google Scholar] [CrossRef]
- Haakonsen, D.L.; Rape, M. Branching Out: Improved Signaling by Heterotypic Ubiquitin Chains. Trends Cell Biol. 2019, 29, 704–716. [Google Scholar] [CrossRef]
- Swatek, K.N.; Usher, J.L.; Kueck, A.F.; Gladkova, C.; Mevissen, T.E.T.; Pruneda, J.N.; Skern, T.; Komander, D. Insights into ubiquitin chain architecture using Ub-clipping. Nature 2019, 572, 533–537. [Google Scholar] [CrossRef]
- Schauer, N.J.; Magin, R.S.; Liu, X.; Doherty, L.M.; Buhrlage, S.J. Advances in Discovering Deubiquitinating Enzyme (DUB) Inhibitors. J. Med. Chem. 2020, 63, 2731–2750. [Google Scholar] [CrossRef]
- Lange, S.M.; Armstrong, L.A.; Kulathu, Y. Deubiquitinases: From mechanisms to their inhibition by small molecules. Mol. Cell 2022, 82, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Kukkula, A.; Ojala, V.K.; Mendez, L.M.; Sistonen, L.; Elenius, K.; Sundvall, M. Therapeutic Potential of Targeting the SUMO Pathway in Cancer. Cancers 2021, 13, 4402. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Lin, X.; Zhu, J.; Zhang, L.; Chen, S.; Yang, H.; Jia, L.; Chen, B. NEDD8-conjugating enzyme E2s: Critical targets for cancer therapy. Cell Death Discov. 2023, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Wimalarathne, M.M.; Wilkerson-Vidal, Q.C.; Hunt, E.C.; Love-Rutledge, S.T. The case for FAT10 as a novel target in fatty liver diseases. Front. Pharmacol. 2022, 13, 972320. [Google Scholar] [CrossRef]
- Xiong, T.C.; Wei, M.C.; Li, F.X.; Shi, M.; Gan, H.; Tang, Z.; Dong, H.P.; Liuyu, T.; Gao, P.; Zhong, B.; et al. The E3 ubiquitin ligase ARIH1 promotes antiviral immunity and autoimmunity by inducing mono-ISGylation and oligomerization of cGAS. Nat. Commun. 2022, 13, 5973. [Google Scholar] [CrossRef]
- Mirzalieva, O.; Juncker, M.; Schwartzenburg, J.; Desai, S. ISG15 and ISGylation in Human Diseases. Cells 2022, 11, 538. [Google Scholar] [CrossRef]
- Infantino, V.; Santarsiero, A.; Convertini, P.; Todisco, S.; Iacobazzi, V. Cancer Cell Metabolism in Hypoxia: Role of HIF-1 as Key Regulator and Therapeutic Target. Int. J. Mol. Sci. 2021, 22, 5703. [Google Scholar] [CrossRef]
- Bartman, C.R.; Weilandt, D.R.; Shen, Y.; Lee, W.D.; Han, Y.; TeSlaa, T.; Jankowski, C.S.R.; Samarah, L.; Park, N.R.; da Silva-Diz, V.; et al. Slow TCA flux and ATP production in primary solid tumours but not metastases. Nature 2023, 614, 349–357. [Google Scholar] [CrossRef]
- Jacquet, P.; Stephanou, A. Searching for the Metabolic Signature of Cancer: A Review from Warburg’s Time to Now. Biomolecules 2022, 12, 1412. [Google Scholar] [CrossRef]
- Sun, T.; Liu, Z.; Yang, Q. The role of ubiquitination and deubiquitination in cancer metabolism. Mol. Cancer 2020, 19, 146. [Google Scholar] [CrossRef]
- Chen, Y.H.; Lue, K.H.; Lin, C.B.; Chen, K.C.; Chan, S.C.; Chu, S.C.; Chang, B.S.; Chen, Y.C. Genomic and Glycolytic Entropy Are Reliable Radiogenomic Heterogeneity Biomarkers for Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2023, 24, 3988. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Bauer, C.; Newman, A.C.; Uribe, A.H.; Athineos, D.; Blyth, K.; Maddocks, O.D.K. Polyamine pathway activity promotes cysteine essentiality in cancer cells. Nat. Metab. 2020, 2, 1062–1076. [Google Scholar] [CrossRef] [PubMed]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. The pivotal roles of mitochondria in cancer: Warburg and beyond and encouraging prospects for effective therapies. Biochim. Biophys. Acta 2010, 1797, 1225–1230. [Google Scholar] [CrossRef] [PubMed]
- Mathupala, S.P.; Rempel, A.; Pedersen, P.L. Glucose catabolism in cancer cells: Identification and characterization of a marked activation response of the type II hexokinase gene to hypoxic conditions. J. Biol. Chem. 2001, 276, 43407–43412. [Google Scholar] [CrossRef]
- Garcia, S.N.; Guedes, R.C.; Marques, M.M. Unlocking the Potential of HK2 in Cancer Metabolism and Therapeutics. Curr. Med. Chem. 2019, 26, 7285–7322. [Google Scholar] [CrossRef]
- Zhao, X.; Zhou, T.; Wang, Y.; Bao, M.; Ni, C.; Ding, L.; Sun, S.; Dong, H.; Li, J.; Liang, C. Trigred motif 36 regulates neuroendocrine differentiation of prostate cancer via HK2 ubiquitination and GPx4 deficiency. Cancer Sci. 2023, 1–15. [Google Scholar] [CrossRef]
- Huang, M.; Xiong, H.; Luo, D.; Xu, B.; Liu, H. CSN5 upregulates glycolysis to promote hepatocellular carcinoma metastasis via stabilizing the HK2 protein. Exp. Cell Res. 2020, 388, 111876. [Google Scholar] [CrossRef]
- Jiao, L.; Zhang, H.L.; Li, D.D.; Yang, K.L.; Tang, J.; Li, X.; Ji, J.; Yu, Y.; Wu, R.Y.; Ravichandran, S.; et al. Regulation of glycolytic metabolism by autophagy in liver cancer involves selective autophagic degradation of HK2 (hexokinase 2). Autophagy 2018, 14, 671–684. [Google Scholar] [CrossRef]
- Lee, H.J.; Li, C.F.; Ruan, D.; He, J.; Montal, E.D.; Lorenz, S.; Girnun, G.D.; Chan, C.H. Non-proteolytic ubiquitination of Hexokinase 2 by HectH9 controls tumor metabolism and cancer stem cell expansion. Nat. Commun. 2019, 10, 2625. [Google Scholar] [CrossRef]
- Gao, R.; Buechel, D.; Kalathur, R.K.R.; Morini, M.F.; Coto-Llerena, M.; Ercan, C.; Piscuoglio, S.; Chen, Q.; Blumer, T.; Wang, X.; et al. USP29-mediated HIF1alpha stabilization is associated with Sorafenib resistance of hepatocellular carcinoma cells by upregulating glycolysis. Oncogenesis 2021, 10, 52. [Google Scholar] [CrossRef]
- Xu, C.D.; Liu, Y.K.; Qiu, L.Y.; Wang, S.S.; Pan, B.Y.; Li, Y.; Wang, S.G.; Tang, B. GFAT and PFK genes show contrasting regulation of chitin metabolism in Nilaparvata lugens. Sci. Rep. 2021, 11, 5246. [Google Scholar] [CrossRef] [PubMed]
- Mor, I.; Cheung, E.C.; Vousden, K.H. Control of glycolysis through regulation of PFK1: Old friends and recent additions. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Sanchez, R.; Rodriguez-Enriquez, S.; Marin-Hernandez, A.; Saavedra, E. Energy metabolism in tumor cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Takase, S.; Nishimura, H.; Matsumoto, K.; Harada, H.; Yoshida, M. RNAi screening reveals a synthetic chemical-genetic interaction between ATP synthase and PFK1 in cancer cells. Cancer Sci. 2023, 114, 1663–1671. [Google Scholar] [CrossRef]
- Tudzarova, S.; Colombo, S.L.; Stoeber, K.; Carcamo, S.; Williams, G.H.; Moncada, S. Two ubiquitin ligases, APC/C-Cdh1 and SKP1-CUL1-F (SCF)-beta-TrCP, sequentially regulate glycolysis during the cell cycle. Proc. Natl. Acad. Sci. USA 2011, 108, 5278–5283. [Google Scholar] [CrossRef]
- Zhu, S.; Guo, Y.; Zhang, X.; Liu, H.; Yin, M.; Chen, X.; Peng, C. Pyruvate kinase M2 (PKM2) in cancer and cancer therapeutics. Cancer Lett. 2021, 503, 240–248. [Google Scholar] [CrossRef]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Suzuki, A.; Puri, S.; Leland, P.; Puri, A.; Moudgil, T.; Fox, B.A.; Puri, R.K.; Joshi, B.H. Subcellular compartmentalization of PKM2 identifies anti-PKM2 therapy response in vitro and in vivo mouse model of human non-small-cell lung cancer. PLoS ONE 2019, 14, e0217131. [Google Scholar] [CrossRef]
- Zhou, Y.; Huang, Z.; Su, J.; Li, J.; Zhao, S.; Wu, L.; Zhang, J.; He, Y.; Zhang, G.; Tao, J.; et al. Benserazide is a novel inhibitor targeting PKM2 for melanoma treatment. Int. J. Cancer 2020, 147, 139–151. [Google Scholar] [CrossRef]
- Lin, Y.; Zhai, H.; Ouyang, Y.; Lu, Z.; Chu, C.; He, Q.; Cao, X. Knockdown of PKM2 enhances radiosensitivity of cervical cancer cells. Cancer Cell Int. 2019, 19, 129. [Google Scholar] [CrossRef]
- Kim, S.R.; Kim, J.O.; Lim, K.H.; Yun, J.H.; Han, I.; Baek, K.H. Regulation of pyruvate kinase isozyme M2 is mediated by the ubiquitin-specific protease 20. Int. J. Oncol. 2015, 46, 2116–2124. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Pei, C.Z.; Park, J.H.; Kim, S.Y.; Song, S.Y.; Shin, G.J.; Baek, K.H. Protein Stability of Pyruvate Kinase Isozyme M2 Is Mediated by HAUSP. Cancers 2020, 12, 1548. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.; Wang, H.; Ni, M.; Wang, Z.; Wang, Z.; Wei, S.; Liu, M.; Wang, P.; Qiu, J.; Zhang, L.; et al. FSTL1 promotes liver fibrosis by reprogramming macrophage function through modulating the intracellular function of PKM2. Gut 2022, 71, 2539–2550. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Li, F.; Han, H.; Chen, Y.; Mao, Z.; Luo, J.; Zhao, Y.; Zheng, B.; Gu, W.; Zhao, W. Parkin Regulates the Activity of Pyruvate Kinase M2. J. Biol. Chem. 2016, 291, 10307–10317. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Zang, W.; Qiu, Y.; Liao, L.; Zheng, X. Deubiquitinase OTUB2 exacerbates the progression of colorectal cancer by promoting PKM2 activity and glycolysis. Oncogene 2022, 41, 46–56. [Google Scholar] [CrossRef]
- Sun, T.; Liu, Z.; Bi, F.; Yang, Q. Deubiquitinase PSMD14 promotes ovarian cancer progression by decreasing enzymatic activity of PKM2. Mol. Oncol. 2021, 15, 3639–3658. [Google Scholar] [CrossRef]
- Wu, H.; Jiao, Y.; Zhou, C.; Guo, X.; Wu, Z.; Lv, Q. miR-140-3p/usp36 axis mediates ubiquitination to regulate PKM2 and suppressed the malignant biological behavior of breast cancer through Warburg effect. Cell Cycle 2022, 22, 680–692. [Google Scholar] [CrossRef]
- Wu, H.; Guo, X.; Jiao, Y.; Wu, Z.; Lv, Q. TRIM35 ubiquitination regulates the expression of PKM2 tetramer and dimer and affects the malignant behaviour of breast cancer by regulating the Warburg effect. Int. J. Oncol. 2022, 61, 144. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, Z.; Guo, W.; Zhang, Z.; Zhao, F.; Zhao, Y.; Jia, D.; Ding, J.; Wang, H.; Yao, M.; et al. TRIM35 Interacts with pyruvate kinase isoform M2 to suppress the Warburg effect and tumorigenicity in hepatocellular carcinoma. Oncogene 2015, 34, 3946–3956. [Google Scholar] [CrossRef]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef]
- Chi, C.; Harth, L.; Galera, M.R.; Torrealba, M.P.; Vadivel, C.K.; Geisler, C.; Bonefeld, C.M.; Nielsen, P.R.; Bzorek, M.; Becker, J.C.; et al. Concomitant Inhibition of FASN and SREBP Provides a Promising Therapy for CTCL. Cancers 2022, 14, 4491. [Google Scholar] [CrossRef] [PubMed]
- Nakakuki, M.; Kawano, H.; Notsu, T.; Imada, K.; Mizuguchi, K.; Shimano, H. A novel processing system of sterol regulatory element-binding protein-1c regulated by polyunsaturated fatty acid. J. Biochem. 2014, 155, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yang, J.; Li, F.; Gao, F.; Zhu, L.; Hao, J. FBXW7 mediates high glucose-induced SREBP-1 expression in renal tubular cells of diabetic nephropathy under PI3K/Akt pathway regulation. Mol. Med. Rep. 2021, 23, 233. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, P.C. Acetyl-CoA Metabolism and Histone Acetylation in the Regulation of Aging and Lifespan. Antioxidants 2021, 10, 572. [Google Scholar] [CrossRef]
- Wen, J.; Min, X.; Shen, M.; Hua, Q.; Han, Y.; Zhao, L.; Liu, L.; Huang, G.; Liu, J.; Zhao, X. ACLY facilitates colon cancer cell metastasis by CTNNB1. J. Exp. Clin. Cancer Res. 2019, 38, 401. [Google Scholar] [CrossRef]
- Icard, P.; Simula, L.; Fournel, L.; Leroy, K.; Lupo, A.; Damotte, D.; Charpentier, M.C.; Durdux, C.; Loi, M.; Schussler, O.; et al. The strategic roles of four enzymes in the interconnection between metabolism and oncogene activation in non-small cell lung cancer: Therapeutic implications. Drug Resist. Updat. 2022, 63, 100852. [Google Scholar] [CrossRef]
- Noh, K.H.; Kang, H.M.; Yoo, W.; Min, Y.; Kim, D.; Kim, M.; Wang, S.; Lim, J.H.; Jung, C.R. Ubiquitination of PPAR-gamma by pVHL inhibits ACLY expression and lipid metabolism, is implicated in tumor progression. Metabolism 2020, 110, 154302. [Google Scholar] [CrossRef]
- Zhao, Z.; Liu, Y.; Liu, Q.; Wu, F.; Liu, X.; Qu, H.; Yuan, Y.; Ge, J.; Xu, Y.; Wang, H. The mRNA Expression Signature and Prognostic Analysis of Multiple Fatty Acid Metabolic Enzymes in Clear Cell Renal Cell Carcinoma. J. Cancer 2019, 10, 6599–6607. [Google Scholar] [CrossRef]
- Wei, X.; Shi, J.; Lin, Q.; Ma, X.; Pang, Y.; Mao, H.; Li, R.; Lu, W.; Wang, Y.; Liu, P. Targeting ACLY Attenuates Tumor Growth and Acquired Cisplatin Resistance in Ovarian Cancer by Inhibiting the PI3K-AKT Pathway and Activating the AMPK-ROS Pathway. Front. Oncol. 2021, 11, 642229. [Google Scholar] [CrossRef]
- Chen, Y.; Li, K.; Gong, D.; Zhang, J.; Li, Q.; Zhao, G.; Lin, P. ACLY: A biomarker of recurrence in breast cancer. Pathol. Res. Pract. 2020, 216, 153076. [Google Scholar] [CrossRef]
- Wen, H.; Lee, S.; Zhu, W.G.; Lee, O.J.; Yun, S.J.; Kim, J.; Park, S. Glucose-derived acetate and ACSS2 as key players in cisplatin resistance in bladder cancer. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Zhu, Y.; Lin, X.; Lu, B.; Zhou, X.; Zhou, F.; Zhao, Q.; Prochownik, E.V.; Li, Y. The IKKbeta-USP30-ACLY Axis Controls Lipogenesis and Tumorigenesis. Hepatology 2021, 73, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Torrini, C.; Nguyen, T.T.T.; Shu, C.; Mela, A.; Humala, N.; Mahajan, A.; Seeley, E.H.; Zhang, G.; Westhoff, M.A.; Karpel-Massler, G.; et al. Lactate is an epigenetic metabolite that drives survival in model systems of glioblastoma. Mol. Cell 2022, 82, 3061–3076.e3066. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Zhang, K.; Wang, H.; Wu, Y.; Chen, N.; Chen, J.; Qiu, C.; Cai, P.; Li, M.; Liang, X.; et al. Hrd1-mediated ACLY ubiquitination alleviate NAFLD in db/db mice. Metabolism 2021, 114, 154349. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Hao, F.; Jin, X.; Sun, X.; Jiang, Y.; Wang, Y.; Li, D.; Chang, T.; Zou, Y.; Peng, P.; et al. ACLY ubiquitination by CUL3-KLHL25 induces the reprogramming of fatty acid metabolism to facilitate iTreg differentiation. Elife 2021, 10, e62394. [Google Scholar] [CrossRef]
- Chen, L.; Duan, Y.; Wei, H.; Ning, H.; Bi, C.; Zhao, Y.; Qin, Y.; Li, Y. Acetyl-CoA carboxylase (ACC) as a therapeutic target for metabolic syndrome and recent developments in ACC1/2 inhibitors. Expert Opin. Investig. Drugs 2019, 28, 917–930. [Google Scholar] [CrossRef]
- Qi, L.; Heredia, J.E.; Altarejos, J.Y.; Screaton, R.; Goebel, N.; Niessen, S.; Macleod, I.X.; Liew, C.W.; Kulkarni, R.N.; Bain, J.; et al. TRB3 links the E3 ubiquitin ligase COP1 to lipid metabolism. Science 2006, 312, 1763–1766. [Google Scholar] [CrossRef]
- Ito, H.; Nakamae, I.; Kato, J.Y.; Yoneda-Kato, N. Stabilization of fatty acid synthesis enzyme acetyl-CoA carboxylase 1 suppresses acute myeloid leukemia development. J. Clin. Investig. 2021, 131, e141529. [Google Scholar] [CrossRef]
- Jin, Y.; Chen, Z.; Dong, J.; Wang, B.; Fan, S.; Yang, X.; Cui, M. SREBP1/FASN/cholesterol axis facilitates radioresistance in colorectal cancer. FEBS Open Bio 2021, 11, 1343–1352. [Google Scholar] [CrossRef]
- McClellan, B.; Pham, T.; Harlow, B.; Lee, G.; Quach, D.; Jolly, C.; Brenner, A.; deGraffenried, L. Modulation of Breast Cancer Cell FASN Expression by Obesity-Related Systemic Factors. Breast Cancer 2022, 16, 11782234221111374. [Google Scholar] [CrossRef]
- Raab, S.; Gadault, A.; Very, N.; Decourcelle, A.; Baldini, S.; Schulz, C.; Mortuaire, M.; Lemaire, Q.; Hardiville, S.; Dehennaut, V.; et al. Dual regulation of fatty acid synthase (FASN) expression by O-GlcNAc transferase (OGT) and mTOR pathway in proliferating liver cancer cells. Cell Mol. Life Sci. 2021, 78, 5397–5413. [Google Scholar] [CrossRef] [PubMed]
- Tao, T.; Su, Q.; Xu, S.; Deng, J.; Zhou, S.; Zhuang, Y.; Huang, Y.; He, C.; He, S.; Peng, M.; et al. Down-regulation of PKM2 decreases FASN expression in bladder cancer cells through AKT/mTOR/SREBP-1c axis. J. Cell. Physiol. 2019, 234, 3088–3104. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, W.; Zheng, Y.; Dai, W.; Ji, J.; Wu, L.; Cheng, Z.; Zhang, J.; Li, J.; Xu, X.; et al. Targeting fatty acid synthase modulates sensitivity of hepatocellular carcinoma to sorafenib via ferroptosis. J. Exp. Clin. Cancer Res. 2023, 42, 6. [Google Scholar] [CrossRef] [PubMed]
- El-Saudi, A.M.; Altouhamy, M.A.; Shaaban, S.; Badria, F.A.; Youssef, M.M.; El-Senduny, F.F. Down regulation of fatty acid synthase via inhibition of PI3K/AKT/mTOR in ovarian cancer cell line by novel organoselenium pseudopeptide. Curr. Res. Pharmacol. Drug Discov. 2022, 3, 100134. [Google Scholar] [CrossRef]
- Yu, J.; Deng, R.; Zhu, H.H.; Zhang, S.S.; Zhu, C.; Montminy, M.; Davis, R.; Feng, G.S. Modulation of fatty acid synthase degradation by concerted action of p38 MAP kinase, E3 ligase COP1, and SH2-tyrosine phosphatase Shp2. J. Biol. Chem. 2013, 288, 3823–3830. [Google Scholar] [CrossRef]
- Tao, B.B.; He, H.; Shi, X.H.; Wang, C.L.; Li, W.Q.; Li, B.; Dong, Y.; Hu, G.H.; Hou, L.J.; Luo, C.; et al. Up-regulation of USP2a and FASN in gliomas correlates strongly with glioma grade. J. Clin. Neurosci. 2013, 20, 717–720. [Google Scholar] [CrossRef]
- Liu, B.; Jiang, S.; Li, M.; Xiong, X.; Zhu, M.; Li, D.; Zhao, L.; Qian, L.; Zhai, L.; Li, J.; et al. Proteome-wide analysis of USP14 substrates revealed its role in hepatosteatosis via stabilization of FASN. Nat. Commun. 2018, 9, 4770. [Google Scholar] [CrossRef]
- Hu, Y.; He, W.; Huang, Y.; Xiang, H.; Guo, J.; Che, Y.; Cheng, X.; Hu, F.; Hu, M.; Ma, T.; et al. Fatty Acid Synthase-Suppressor Screening Identifies Sorting Nexin 8 as a Therapeutic Target for NAFLD. Hepatology 2021, 74, 2508–2525. [Google Scholar] [CrossRef]
- Gang, X.; Xuan, L.; Zhao, X.; Lv, Y.; Li, F.; Wang, Y.; Wang, G. Speckle-type POZ protein suppresses lipid accumulation and prostate cancer growth by stabilizing fatty acid synthase. Prostate 2019, 79, 864–871. [Google Scholar] [CrossRef]
- Mevissen, T.E.T.; Prasad, A.V.; Walter, J.C. TRIM21-dependent target protein ubiquitination mediates cell-free Trim-Away. Cell Rep. 2023, 42, 112125. [Google Scholar] [CrossRef]
- Andrilenas, K.K.; Ramlall, V.; Kurland, J.; Leung, B.; Harbaugh, A.G.; Siggers, T. DNA-binding landscape of IRF3, IRF5 and IRF7 dimers: Implications for dimer-specific gene regulation. Nucleic Acids Res. 2018, 46, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.A.; Sun, Y.; Jiang, Y.P.; Bott, A.J.; Jaber, N.; Dou, Z.; Yang, B.; Chen, J.S.; Catanzaro, J.M.; Du, C.; et al. TRIM21 Ubiquitylates SQSTM1/p62 and Suppresses Protein Sequestration to Regulate Redox Homeostasis. Mol. Cell 2016, 61, 720–733. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.P.; Cheng, Z.L.; He, R.Y.; Song, L.; Tian, M.X.; Zhou, L.S.; Groh, B.S.; Liu, W.R.; Ji, M.B.; Ding, C.; et al. Destabilization of Fatty Acid Synthase by Acetylation Inhibits De Novo Lipogenesis and Tumor Cell Growth. Cancer Res. 2016, 76, 6924–6936. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Peng, Z.; Chen, Y.; Li, H.; Du, M.; Tan, Y.; Zhang, X.; Lu, Z.; Cui, C.P.; Liu, C.H.; et al. Neddylation of PTEN regulates its nuclear import and promotes tumor development. Cell Res. 2021, 31, 291–311. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Zhu, Y.; Lin, X.; Tan, X.; Lu, B.; Li, Y. Stabilization of FASN by ACAT1-mediated GNPAT acetylation promotes lipid metabolism and hepatocarcinogenesis. Oncogene 2020, 39, 2437–2449. [Google Scholar] [CrossRef]
- Vettore, L.; Westbrook, R.L.; Tennant, D.A. New aspects of amino acid metabolism in cancer. Br. J. Cancer 2020, 122, 150–156. [Google Scholar] [CrossRef]
- Cruzat, V.; Macedo Rogero, M.; Noel Keane, K.; Curi, R.; Newsholme, P. Glutamine: Metabolism and Immune Function, Supplementation and Clinical Translation. Nutrients 2018, 10, 1564. [Google Scholar] [CrossRef]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef]
- Yoo, H.C.; Park, S.J.; Nam, M.; Kang, J.; Kim, K.; Yeo, J.H.; Kim, J.K.; Heo, Y.; Lee, H.S.; Lee, M.Y.; et al. A Variant of SLC1A5 Is a Mitochondrial Glutamine Transporter for Metabolic Reprogramming in Cancer Cells. Cell Metab. 2020, 31, 267–283.e212. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, T.; Li, Z.; Wang, L.; Yuan, S.; Sun, L. The role of ASCT2 in cancer: A review. Eur. J. Pharmacol. 2018, 837, 81–87. [Google Scholar] [CrossRef]
- van Geldermalsen, M.; Wang, Q.; Nagarajah, R.; Marshall, A.D.; Thoeng, A.; Gao, D.; Ritchie, W.; Feng, Y.; Bailey, C.G.; Deng, N.; et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene 2016, 35, 3201–3208. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Tao, W.; Zhang, F.; Jie, Q.; He, Y.; Zhu, W.; Tan, J.; Shen, W.; Li, L.; Yang, Y.; et al. Lobetyolin induces apoptosis of colon cancer cells by inhibiting glutamine metabolism. J. Cell. Mol. Med. 2020, 24, 3359–3369. [Google Scholar] [CrossRef] [PubMed]
- Bothwell, P.J.; Kron, C.D.; Wittke, E.F.; Czerniak, B.N.; Bode, B.P. Targeted Suppression and Knockout of ASCT2 or LAT1 in Epithelial and Mesenchymal Human Liver Cancer Cells Fail to Inhibit Growth. Int. J. Mol. Sci. 2018, 19, 2093. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Lin, W.; Wang, C.; Sun, F.; Ju, S.; Chen, Q.; Wang, Y.; Chen, Y.; Li, H.; Wang, L.; et al. Neddylation inhibition induces glutamine uptake and metabolism by targeting CRL3(SPOP) E3 ligase in cancer cells. Nat. Commun. 2022, 13, 3034. [Google Scholar] [CrossRef]
- Ma, H.; Wu, Z.; Peng, J.; Li, Y.; Huang, H.; Liao, Y.; Zhou, M.; Sun, L.; Huang, N.; Shi, M.; et al. Inhibition of SLC1A5 sensitizes colorectal cancer to cetuximab. Int. J. Cancer 2018, 142, 2578–2588. [Google Scholar] [CrossRef]
- Lee, D.E.; Yoo, J.E.; Kim, J.; Kim, S.; Kim, S.; Lee, H.; Cheong, H. NEDD4L downregulates autophagy and cell growth by modulating ULK1 and a glutamine transporter. Cell Death Dis. 2020, 11, 38. [Google Scholar] [CrossRef]
- Yuan, S.; Tanzeel, Y.; Tian, X.; Zheng, D.; Wajeeha, N.; Xu, J.; Ke, Y.; Zhang, Z.; Peng, X.; Lu, L.; et al. Global analysis of HBV-mediated host proteome and ubiquitylome change in HepG2.2.15 human hepatoblastoma cell line. Cell Biosci. 2021, 11, 75. [Google Scholar] [CrossRef]
- Penugurti, V.; Khumukcham, S.S.; Padala, C.; Dwivedi, A.; Kamireddy, K.R.; Mukta, S.; Bhopal, T.; Manavathi, B. HPIP protooncogene differentially regulates metabolic adaptation and cell fate in breast cancer cells under glucose stress via AMPK and RNF2 dependent pathways. Cancer Lett. 2021, 518, 243–255. [Google Scholar] [CrossRef]
- Greene, K.S.; Lukey, M.J.; Wang, X.; Blank, B.; Druso, J.E.; Lin, M.J.; Stalnecker, C.A.; Zhang, C.; Negron Abril, Y.; Erickson, J.W.; et al. SIRT5 stabilizes mitochondrial glutaminase and supports breast cancer tumorigenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 26625–26632. [Google Scholar] [CrossRef]
- Zhao, S.; Wang, J.M.; Yan, J.; Zhang, D.L.; Liu, B.Q.; Jiang, J.Y.; Li, C.; Li, S.; Meng, X.N.; Wang, H.Q. BAG3 promotes autophagy and glutaminolysis via stabilizing glutaminase. Cell Death Dis. 2019, 10, 284. [Google Scholar] [CrossRef]
- Legendre, F.; MacLean, A.; Appanna, V.P.; Appanna, V.D. Biochemical pathways to alpha-ketoglutarate, a multi-faceted metabolite. World J. Microbiol. Biotechnol. 2020, 36, 123. [Google Scholar] [CrossRef] [PubMed]
- Bodineau, C.; Tome, M.; Murdoch, P.D.S.; Duran, R.V. Glutamine, MTOR and autophagy: A multiconnection relationship. Autophagy 2022, 18, 2749–2750. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Shi, T.; Yu, H.; Ding, Y.; Li, L.; Wang, X.; Wang, X. Cytosolic GDH1 degradation restricts protein synthesis to sustain tumor cell survival following amino acid deprivation. EMBO J. 2022, 41, e110306. [Google Scholar] [CrossRef]
- Guo, S.; Zhao, W.; Zhang, W.; Li, S.; Teng, G.; Liu, L. Vitamin D Promotes Ferroptosis in Colorectal Cancer Stem Cells via SLC7A11 Downregulation. Oxid. Med. Cell. Longev. 2023, 2023, 4772134. [Google Scholar] [CrossRef]
- Badgley, M.A.; Kremer, D.M.; Maurer, H.C.; DelGiorno, K.E.; Lee, H.J.; Purohit, V.; Sagalovskiy, I.R.; Ma, A.; Kapilian, J.; Firl, C.E.M.; et al. Cysteine depletion induces pa.ancreatic tumor ferroptosis in mice. Science 2020, 368, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Olszewski, K.; Zhang, Y.; Lim, E.W.; Shi, J.; Zhang, X.; Zhang, J.; Lee, H.; Koppula, P.; Lei, G.; et al. Cystine transporter regulation of pentose phosphate pathway dependency and disulfide stress exposes a targetable metabolic vulnerability in cancer. Nat. Cell Biol. 2020, 22, 476–486. [Google Scholar] [CrossRef]
- Koppula, P.; Zhuang, L.; Gan, B. Cystine transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 2021, 12, 599–620. [Google Scholar] [CrossRef]
- Chen, Q.; Zheng, W.; Guan, J.; Liu, H.; Dan, Y.; Zhu, L.; Song, Y.; Zhou, Y.; Zhao, X.; Zhang, Y.; et al. SOCS2-enhanced ubiquitination of SLC7A11 promotes ferroptosis and radiosensitization in hepatocellular carcinoma. Cell Death Differ. 2022, 30, 137–151. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, C.; Huang, M.; Lin, J.; Fan, X.; Ni, T. TRIM26 Induces Ferroptosis to Inhibit Hepatic Stellate Cell Activation and Mitigate Liver Fibrosis through Mediating SLC7A11 Ubiquitination. Front. Cell Dev. Biol. 2021, 9, 644901. [Google Scholar] [CrossRef]
- Zhang, Y.; Koppula, P.; Gan, B. Regulation of H2A ubiquitination and SLC7A11 expression by BAP1 and PRC1. Cell Cycle 2019, 18, 773–783. [Google Scholar] [CrossRef]
- Liu, T.; Jiang, L.; Tavana, O.; Gu, W. The Deubiquitylase OTUB1 Mediates Ferroptosis via Stabilization of SLC7A11. Cancer Res. 2019, 79, 1913–1924. [Google Scholar] [CrossRef]
- Li, A.M.; Ye, J. Reprogramming of serine, glycine and one-carbon metabolism in cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165841. [Google Scholar] [CrossRef]
- Zhao, J.Y.; Feng, K.R.; Wang, F.; Zhang, J.W.; Cheng, J.F.; Lin, G.Q.; Gao, D.; Tian, P. A retrospective overview of PHGDH and its inhibitors for regulating cancer metabolism. Eur. J. Med. Chem. 2021, 217, 113379. [Google Scholar] [CrossRef]
- Rossi, M.; Altea-Manzano, P.; Demicco, M.; Doglioni, G.; Bornes, L.; Fukano, M.; Vandekeere, A.; Cuadros, A.M.; Fernandez-Garcia, J.; Riera-Domingo, C.; et al. PHGDH heterogeneity potentiates cancer cell dissemination and metastasis. Nature 2022, 605, 747–753. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Wu, H.; Sun, X.X.; Li, Y.; Huang, S.; Yue, X.; Lu, S.E.; Shen, Z.; Su, X.; et al. Parkin ubiquitinates phosphoglycerate dehydrogenase to suppress serine synthesis and tumor progression. J. Clin. Investig. 2020, 130, 3253–3269. [Google Scholar] [CrossRef]
- Gao, X.; Wang, Y.; Lu, F.; Chen, X.; Yang, D.; Cao, Y.; Zhang, W.; Chen, J.; Zheng, L.; Wang, G.; et al. Extracellular vesicles derived from oesophageal cancer containing P4HB promote muscle wasting via regulating PHGDH/Bcl-2/caspase-3 pathway. J. Extracell. Vesicles 2021, 10, e12060. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yu, H.; Zhang, J.; Gao, H.; Wang, S.; Li, S.; Wei, P.; Liang, J.; Yu, G.; Wang, X.; et al. Cul4A-DDB1-mediated monoubiquitination of phosphoglycerate dehydrogenase promotes colorectal cancer metastasis via increased S-adenosylmethionine. J. Clin. Investig. 2021, 131, e146187. [Google Scholar] [CrossRef]
- Chen, H.; Liu, C.; Wang, Q.; Xiong, M.; Zeng, X.; Yang, D.; Xie, Y.; Su, H.; Zhang, Y.; Huang, Y.; et al. Renal UTX-PHGDH-serine axis regulates metabolic disorders in the kidney and liver. Nat. Commun. 2022, 13, 3835. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wan, X.; Yu, T.; Huang, Z.; Shen, C.; Qi, Q.; Xiang, S.; Chen, X.; Arbely, E.; Ling, Z.Q.; et al. Acetylation Stabilizes Phosphoglycerate Dehydrogenase by Disrupting the Interaction of E3 Ligase RNF5 to Promote Breast Tumorigenesis. Cell Rep. 2020, 32, 108021. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Z.X.; Wang, J.G.; Li, L.H.; Shen, W.L.; Dang, X.W. Deubiquitinating enzyme Josephin-2 stabilizes PHGDH to promote a cancer stem cell phenotype in hepatocellular carcinoma. Genes Genom. 2023, 45, 215–224. [Google Scholar] [CrossRef]
- Chen, C.L.; Hsu, S.C.; Chung, T.Y.; Chu, C.Y.; Wang, H.J.; Hsiao, P.W.; Yeh, S.D.; Ann, D.K.; Yen, Y.; Kung, H.J. Arginine is an epigenetic regulator targeting TEAD4 to modulate OXPHOS in prostate cancer cells. Nat. Commun. 2021, 12, 2398. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Yang, Y.; Li, M.; Chu, Y.; Song, H.; Zhang, J.; Zhang, D.; Zhang, Q.; Xu, Y.; Wang, J.; et al. Snail enhances arginine synthesis by inhibiting ubiquitination-mediated degradation of ASS1. EMBO Rep. 2021, 22, e51780. [Google Scholar] [CrossRef]
- Wu, X.; Sun, X.; Sharma, S.; Lu, Q.; Yegambaram, M.; Hou, Y.; Wang, T.; Fineman, J.R.; Black, S.M. Arginine recycling in endothelial cells is regulated BY HSP90 and the ubiquitin proteasome system. Nitric Oxide 2021, 108, 12–19. [Google Scholar] [CrossRef]
- Sha, Y.; Pandit, L.; Zeng, S.; Eissa, N.T. A critical role for CHIP in the aggresome pathway. Mol. Cell Biol. 2009, 29, 116–128. [Google Scholar] [CrossRef]
- Dang, C.V.; Le, A.; Gao, P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin. Cancer Res. 2009, 15, 6479–6483. [Google Scholar] [CrossRef]
- Cheng, W.; Li, G.; Ye, Z.; Hu, J.; Gao, L.; Jia, X.; Zhao, S.; Wang, Y.; Zhou, Q. NEDD4L inhibits cell viability, cell cycle progression, and glutamine metabolism in esophageal squamous cell carcinoma via ubiquitination of c-Myc. Acta Biochim. Biophys. Sin. 2022, 54, 716–724. [Google Scholar] [CrossRef]
- Popov, N.; Schulein, C.; Jaenicke, L.A.; Eilers, M. Ubiquitylation of the amino terminus of Myc by SCF(beta-TrCP) antagonizes SCF(Fbw7)-mediated turnover. Nat. Cell Biol. 2010, 12, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ding, C.; Chen, Y.; Hu, W.; Lu, Y.; Wu, W.; Zhang, Y.; Yang, B.; Wu, H.; Peng, C.; et al. ACSL4 promotes hepatocellular carcinoma progression via c-Myc stability mediated by ERK/FBW7/c-Myc axis. Oncogenesis 2020, 9, 42. [Google Scholar] [CrossRef]
- Ruiz, E.J.; Pinto-Fernandez, A.; Turnbull, A.P.; Lan, L.; Charlton, T.M.; Scott, H.C.; Damianou, A.; Vere, G.; Riising, E.M.; Da Costa, C.; et al. USP28 deletion and small-molecule inhibition destabilizes c-MYC and elicits regression of squamous cell lung carcinoma. Elife 2021, 10, e71596. [Google Scholar] [CrossRef] [PubMed]
- Cui, B.; Luo, Y.; Tian, P.; Peng, F.; Lu, J.; Yang, Y.; Su, Q.; Liu, B.; Yu, J.; Luo, X.; et al. Stress-induced epinephrine enhances lactate dehydrogenase A and promotes breast cancer stem-like cells. J. Clin. Investig. 2019, 129, 1030–1046. [Google Scholar] [CrossRef]
- Zhang, J.; Ren, P.; Xu, D.; Liu, X.; Liu, Z.; Zhang, C.; Li, Y.; Wang, L.; Du, X.; Xing, B. Human UTP14a promotes colorectal cancer progression by forming a positive regulation loop with c-Myc. Cancer Lett. 2019, 440–441, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.X.; He, X.; Yin, L.; Komada, M.; Sears, R.C.; Dai, M.S. The nucleolar ubiquitin-specific protease USP36 deubiquitinates and stabilizes c-Myc. Proc. Natl. Acad. Sci. USA 2015, 112, 3734–3739. [Google Scholar] [CrossRef]
- Nicklas, S.; Hillje, A.L.; Okawa, S.; Rudolph, I.M.; Collmann, F.M.; van Wuellen, T.; Del Sol, A.; Schwamborn, J.C. A complex of the ubiquitin ligase TRIM32 and the deubiquitinase USP7 balances the level of c-Myc ubiquitination and thereby determines neural stem cell fate specification. Cell Death Differ. 2019, 26, 728–740. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Zhou, W.; Wu, Q.; Huang, Z.; Shi, Y.; Yang, K.; Chen, C.; Xie, Q.; Mack, S.C.; Wang, X.; et al. Deubiquitinase USP13 maintains glioblastoma stem cells by antagonizing FBXL14-mediated Myc ubiquitination. J. Exp. Med. 2017, 214, 245–267. [Google Scholar] [CrossRef] [PubMed]
- Old, J.B.; Kratzat, S.; Hoellein, A.; Graf, S.; Nilsson, J.A.; Nilsson, L.; Nakayama, K.I.; Peschel, C.; Cleveland, J.L.; Keller, U.B. Skp2 directs Myc-mediated suppression of p27Kip1 yet has modest effects on Myc-driven lymphomagenesis. Mol. Cancer Res. 2010, 8, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Hong, A.; Park, H.I.; Shin, W.H.; Yoo, L.; Jeon, S.J.; Chung, K.C. Deubiquitinating enzyme USP22 positively regulates c-Myc stability and tumorigenic activity in mammalian and breast cancer cells. J. Cell Physiol. 2017, 232, 3664–3676. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef]
- Wang, B.; Jie, Z.; Joo, D.; Ordureau, A.; Liu, P.; Gan, W.; Guo, J.; Zhang, J.; North, B.J.; Dai, X.; et al. TRAF2 and OTUD7B govern a ubiquitin-dependent switch that regulates mTORC2 signalling. Nature 2017, 545, 365–369. [Google Scholar] [CrossRef]
- Linares, J.F.; Duran, A.; Yajima, T.; Pasparakis, M.; Moscat, J.; Diaz-Meco, M.T. K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol. Cell 2013, 51, 283–296. [Google Scholar] [CrossRef]
- Wang, L.; Li, D.; Su, X.; Zhao, Y.; Huang, A.; Li, H.; Li, J.; Xia, W.; Jia, T.; Zhang, H.; et al. AGO4 suppresses tumor growth by modulating autophagy and apoptosis via enhancing TRIM21-mediated ubiquitination of GRP78 in a p53-independent manner. Oncogene 2022, 42, 62–77. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Chen, L.; Zhao, L.; Xu, Y.; Peng, X.; Wang, X.; Ding, L.; Jin, J.; Teng, H.; Wang, Y.; et al. Ubiquitination of Rheb governs growth factor-induced mTORC1 activation. Cell Res. 2019, 29, 136–150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Shao, Y.; Quan, F.; Liu, L.; Yang, J. FBP1 enhances the radiosensitivity by suppressing glycolysis via the FBXW7/mTOR axis in nasopharyngeal carcinoma cells. Life Sci. 2021, 283, 119840. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kim, H.J.; Kim, H.J.; Kim, C.H. Non-Thermal Plasma Induces Antileukemic Effect through mTOR Ubiquitination. Cells 2020, 9, 595. [Google Scholar] [CrossRef] [PubMed]
- Nanayakkara, D.M.; Nguyen, M.N.; Wood, S.A. Deubiquitylating enzyme, USP9X, regulates proliferation of cells of head and neck cancer lines. Cell Prolif. 2016, 49, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Song, S.; Li, Y.; Fan, Y.; Wang, L.; Wang, R.; Huo, L.; Scott, A.; Xu, Y.; Pizzi, M.P.; et al. Loss of ARID1A activates mTOR signaling and SOX9 in gastric adenocarcinoma-rationale for targeting ARID1A deficiency. Gut 2022, 71, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhou, Y.F.; Cao, J.; Burley, S.K.; Wang, H.Y.; Zheng, X.F.S. mTORC1 Promotes ARID1A Degradation and Oncogenic Chromatin Remodeling in Hepatocellular Carcinoma. Cancer Res. 2021, 81, 5652–5665. [Google Scholar] [CrossRef]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef]
- Zhu, G.; Pei, L.; Xia, H.; Tang, Q.; Bi, F. Role of oncogenic KRAS in the prognosis, diagnosis and treatment of colorectal cancer. Mol. Cancer 2021, 20, 143. [Google Scholar] [CrossRef]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.; Markowitz, S.; Zhou, S.; et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009, 325, 1555–1559. [Google Scholar] [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Carbone, D.P.; Garassino, M.; Barlesi, F. Targeting KRAS in non-small-cell lung cancer: Recent progress and new approaches. Ann. Oncol. 2021, 32, 1101–1110. [Google Scholar] [CrossRef]
- Abe, T.; Umeki, I.; Kanno, S.I.; Inoue, S.I.; Niihori, T.; Aoki, Y. LZTR1 facilitates polyubiquitination and degradation of RAS-GTPases. Cell Death Differ. 2020, 27, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, G.A.; Baker, N.M.; Miermont, A.M.; Thurman, R.D.; Pierobon, M.; Tran, T.H.; Anderson, A.O.; Waters, A.M.; Diehl, J.N.; Papke, B.; et al. Atypical KRAS(G12R) Mutant Is Impaired in PI3K Signaling and Macropinocytosis in Pancreatic Cancer. Cancer Discov. 2020, 10, 104–123. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.; Wilkerson, E.M.; Sumita, K.; Isom, D.G.; Sasaki, A.T.; Dohlman, H.G.; Campbell, S.L. Differences in the regulation of K-Ras and H-Ras isoforms by monoubiquitination. J. Biol. Chem. 2013, 288, 36856–36862. [Google Scholar] [CrossRef]
- Zeng, T.; Wang, Q.; Fu, J.; Lin, Q.; Bi, J.; Ding, W.; Qiao, Y.; Zhang, S.; Zhao, W.; Lin, H.; et al. Impeded Nedd4-1-mediated Ras degradation underlies Ras-driven tumorigenesis. Cell Rep. 2014, 7, 871–882. [Google Scholar] [CrossRef]
- Hsu, C.Y.; Huang, J.W.; Huang, W.R.; Chen, I.C.; Chen, M.S.; Liao, T.L.; Chang, Y.K.; Munir, M.; Liu, H.J. Oncolytic Avian Reovirus sigmaA-Modulated Upregulation of the HIF-1alpha/C-myc/glut1 Pathway to Produce More Energy in Different Cancer Cell Lines Benefiting Virus Replication. Viruses 2023, 15, 523. [Google Scholar] [CrossRef]
- Dang, C.V. The interplay between MYC and HIF in the Warburg effect. Ernst Scher. Found. Symp Proc. 2007, 4, 35–53. [Google Scholar] [CrossRef]
- Kim, J.A.; Choi, D.K.; Min, J.S.; Kang, I.; Kim, J.C.; Kim, S.; Ahn, J.K. VBP1 represses cancer metastasis by enhancing HIF-1alpha degradation induced by pVHL. FEBS J. 2018, 285, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wen, Y.; Zhang, N. (1)H, (13)C and (15)N backbone and side-chain resonance assignments of the ZnF-UBP domain of USP20/VDU2. Biomol. NMR Assign. 2017, 11, 91–93. [Google Scholar] [CrossRef]
- Troilo, A.; Alexander, I.; Muehl, S.; Jaramillo, D.; Knobeloch, K.P.; Krek, W. HIF1alpha deubiquitination by USP8 is essential for ciliogenesis in normoxia. EMBO Rep. 2014, 15, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Du, S.C.; Zhu, L.; Wang, Y.X.; Liu, J.; Zhang, D.; Chen, Y.L.; Peng, Q.; Liu, W.; Liu, B. SENP1-mediated deSUMOylation of USP28 regulated HIF-1alpha accumulation and activation during hypoxia response. Cancer Cell Int. 2019, 19, 4. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Lin, T.C.; Bi, X.; Lu, G.; Dawson, B.C.; Miranda, R.; Medeiros, L.J.; McNiece, I.; McCarty, N. TRIM44 promotes quiescent multiple myeloma cell occupancy and survival in the osteoblastic niche via HIF-1alpha stabilization. Leukemia 2019, 33, 469–486. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.T.; Kuo, Y.C.; Hung, J.J.; Huang, C.H.; Chen, W.Y.; Chou, T.Y.; Chen, Y.; Chen, Y.J.; Chen, Y.J.; Cheng, W.C.; et al. K63-polyubiquitinated HAUSP deubiquitinates HIF-1alpha and dictates H3K56 acetylation promoting hypoxia-induced tumour progression. Nat. Commun. 2016, 7, 13644. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Zhao, Y.; Yue, X.; Wu, H.; Huang, S.; Chen, J.; Tomsky, K.; Xie, H.; Khella, C.A.; et al. Parkin targets HIF-1alpha for ubiquitination and degradation to inhibit breast tumor progression. Nat. Commun. 2017, 8, 1823. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Xu, H.; Li, W.; Ji, J.; Jin, Q.; Chen, L.; Gan, Q.; Tao, Q.; Chai, Y. MDM2 promotes cancer cell survival through regulating the expression of HIF-1alpha and pVHL in retinoblastoma. Pathol. Oncol. Res. 2023, 29, 1610801. [Google Scholar] [CrossRef]
- Sun, H.; Li, X.B.; Meng, Y.; Fan, L.; Li, M.; Fang, J. TRAF6 upregulates expression of HIF-1alpha and promotes tumor angiogenesis. Cancer Res. 2013, 73, 4950–4959. [Google Scholar] [CrossRef]
- Sun, L. F-box and WD repeat domain-containing 7 (FBXW7) mediates the hypoxia inducible factor-1alpha (HIF-1alpha)/vascular endothelial growth factor (VEGF) signaling pathway to affect hypoxic-ischemic brain damage in neonatal rats. Bioengineered 2022, 13, 560–572. [Google Scholar] [CrossRef]
- Ju, U.I.; Park, J.W.; Park, H.S.; Kim, S.J.; Chun, Y.S. FBXO11 represses cellular response to hypoxia by destabilizing hypoxia-inducible factor-1alpha mRNA. Biochem. Biophys. Res. Commun. 2015, 464, 1008–1015. [Google Scholar] [CrossRef]
- Jafari, M.; Ghadami, E.; Dadkhah, T.; Akhavan-Niaki, H. PI3k/AKT signaling pathway: Erythropoiesis and beyond. J. Cell Physiol. 2019, 234, 2373–2385. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.L.; Wang, J.; Chan, C.H.; Lee, S.W.; Campos, A.D.; Lamothe, B.; Hur, L.; Grabiner, B.C.; Lin, X.; Darnay, B.G.; et al. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science 2009, 325, 1134–1138. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Hu, Y.; Xu, T.; Yan, K.; Zhang, T.; Li, Q.; Chang, F.; Guo, X.; Peng, J.; Li, M.; et al. RNF8-mediated regulation of Akt promotes lung cancer cell survival and resistance to DNA damage. Cell Rep. 2021, 37, 109854. [Google Scholar] [CrossRef]
- Zhu, Z.; Cao, C.; Zhang, D.; Zhang, Z.; Liu, L.; Wu, D.; Sun, J. UBE2T-mediated Akt ubiquitination and Akt/beta-catenin activation promotes hepatocellular carcinoma development by increasing pyrimidine metabolism. Cell Death Dis. 2022, 13, 154. [Google Scholar] [CrossRef]
- Kuang, X.; Xiong, J.; Lu, T.; Wang, W.; Zhang, Z.; Wang, J. Inhibition of USP1 induces apoptosis via ID1/AKT pathway in B-cell acute lymphoblastic leukemia cells. Int. J. Med. Sci. 2021, 18, 245–255. [Google Scholar] [CrossRef]
- Cilenti, L.; Di Gregorio, J.; Ambivero, C.T.; Andl, T.; Liao, R.; Zervos, A.S. Mitochondrial MUL1 E3 ubiquitin ligase regulates Hypoxia Inducible Factor (HIF-1alpha) and metabolic reprogramming by modulating the UBXN7 cofactor protein. Sci. Rep. 2020, 10, 1609. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, S.Y.; Kim, D.H.; Park, J.S.; Jeong, S.H.; Choi, Y.W.; Kim, C.H. Crosstalk between HSPA5 arginylation and sequential ubiquitination leads to AKT degradation through autophagy flux. Autophagy 2021, 17, 961–979. [Google Scholar] [CrossRef]
- Yang, W.H.; Lin, C.C.; Wu, J.; Chao, P.Y.; Chen, K.; Chen, P.H.; Chi, J.T. The Hippo Pathway Effector YAP Promotes Ferroptosis via the E3 Ligase SKP2. Mol. Cancer Res. 2021, 19, 1005–1014. [Google Scholar] [CrossRef]
- Zhang, X.; Qiao, Y.; Wu, Q.; Chen, Y.; Zou, S.; Liu, X.; Zhu, G.; Zhao, Y.; Chen, Y.; Yu, Y.; et al. The essential role of YAP O-GlcNAcylation in high-glucose-stimulated liver tumorigenesis. Nat. Commun. 2017, 8, 15280. [Google Scholar] [CrossRef]
- Koo, J.H.; Guan, K.L. Interplay between YAP/TAZ and Metabolism. Cell Metab. 2018, 28, 196–206. [Google Scholar] [CrossRef]
- Zhou, X.; Li, Y.; Wang, W.; Wang, S.; Hou, J.; Zhang, A.; Lv, B.; Gao, C.; Yan, Z.; Pang, D.; et al. Regulation of Hippo/YAP signaling and Esophageal Squamous Carcinoma progression by an E3 ubiquitin ligase PARK2. Theranostics 2020, 10, 9443–9457. [Google Scholar] [CrossRef]
- Qian, M.; Yan, F.; Wang, W.; Du, J.; Yuan, T.; Wu, R.; Zhao, C.; Wang, J.; Lu, J.; Zhang, B.; et al. Deubiquitinase JOSD2 stabilizes YAP/TAZ to promote cholangiocarcinoma progression. Acta Pharm. Sin. B 2021, 11, 4008–4019. [Google Scholar] [CrossRef] [PubMed]
- Yao, F.; Zhou, Z.; Kim, J.; Hang, Q.; Xiao, Z.; Ton, B.N.; Chang, L.; Liu, N.; Zeng, L.; Wang, W.; et al. SKP2- and OTUD1-regulated non-proteolytic ubiquitination of YAP promotes YAP nuclear localization and activity. Nat. Commun. 2018, 9, 2269. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Deng, J. Ubiquitination-deubiquitination in the Hippo signaling pathway (Review). Oncol. Rep. 2019, 41, 1455–1475. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.A.; Kim, D.W.; Cho, J.Y. Neural precursor cell-expressed, developmentally down-regulated 4 (NEDD4) regulates hydrogen peroxide-induced cell proliferation and death through inhibition of Hippo signaling. FASEB J. 2019, 33, 14772–14783. [Google Scholar] [CrossRef]
- Takahashi, H.; Alves, C.R.R.; Stanford, K.I.; Middelbeek, R.J.W.; Nigro, P.; Ryan, R.E.; Xue, R.; Sakaguchi, M.; Lynes, M.D.; So, K.; et al. TGF-beta2 is an exercise-induced adipokine that regulates glucose and fatty acid metabolism. Nat. Metab. 2019, 1, 291–303. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, F.; Garcia de Vinuesa, A.; de Kruijf, E.M.; Mesker, W.E.; Hui, L.; Drabsch, Y.; Li, Y.; Bauer, A.; Rousseau, A.; et al. TRAF4 promotes TGF-beta receptor signaling and drives breast cancer metastasis. Mol. Cell 2013, 51, 559–572. [Google Scholar] [CrossRef]
- Sorrentino, A.; Thakur, N.; Grimsby, S.; Marcusson, A.; von Bulow, V.; Schuster, N.; Zhang, S.; Heldin, C.H.; Landstrom, M. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat. Cell Biol. 2008, 10, 1199–1207. [Google Scholar] [CrossRef]
- Gudey, S.K.; Sundar, R.; Mu, Y.; Wallenius, A.; Zang, G.; Bergh, A.; Heldin, C.H.; Landstrom, M. TRAF6 stimulates the tumor-promoting effects of TGFbeta type I receptor through polyubiquitination and activation of presenilin 1. Sci. Signal. 2014, 7, ra2. [Google Scholar] [CrossRef]
- Mu, Y.; Sundar, R.; Thakur, N.; Ekman, M.; Gudey, S.K.; Yakymovych, M.; Hermansson, A.; Dimitriou, H.; Bengoechea-Alonso, M.T.; Ericsson, J.; et al. TRAF6 ubiquitinates TGFbeta type I receptor to promote its cleavage and nuclear translocation in cancer. Nat. Commun. 2011, 2, 330. [Google Scholar] [CrossRef]
- Chen, J.; Li, W.; Cui, K.; Ji, K.; Xu, S.; Xu, Y. Artemisitene suppresses tumorigenesis by inducing DNA damage through deregulating c-Myc-topoisomerase pathway. Oncogene 2018, 37, 5079–5087. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, L.; Pan, H.; Wang, Y.; Shi, M.; Yu, H.; Wang, C.; Pan, X.; Chen, Z. Exosomes Derived From Macrophages Enhance Aerobic Glycolysis and Chemoresistance in Lung Cancer by Stabilizing c-Myc via the Inhibition of NEDD4L. Front. Cell Dev. Biol. 2020, 8, 620603. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, W.; Ko, C.; Ryu, W.S. Hepatitis B virus X protein enhances Myc stability by inhibiting SCF(Skp2) ubiquitin E3 ligase-mediated Myc ubiquitination and contributes to oncogenesis. Oncogene 2016, 35, 1857–1867. [Google Scholar] [CrossRef]
- The MULE/HUWE1 E3 ubiquitin ligase is a tumor suppressor. Cancer Discov. 2013, 3, OF32. [CrossRef] [PubMed]
- Choi, S.H.; Wright, J.B.; Gerber, S.A.; Cole, M.D. Myc protein is stabilized by suppression of a novel E3 ligase complex in cancer cells. Genes Dev. 2010, 24, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, N.; Zheng, Y.; Yang, B.; Liu, P.; Zhang, F.; Li, M.; Song, J.; Chang, X.; Wang, Z. Caveolin-1 inhibits breast cancer stem cells via c-Myc-mediated metabolic reprogramming. Cell Death Dis. 2020, 11, 450. [Google Scholar] [CrossRef] [PubMed]
- Paul, I.; Ahmed, S.F.; Bhowmik, A.; Deb, S.; Ghosh, M.K. The ubiquitin ligase CHIP regulates c-Myc stability and transcriptional activity. Oncogene 2013, 32, 1284–1295. [Google Scholar] [CrossRef] [PubMed]
- Popov, N.; Wanzel, M.; Madiredjo, M.; Zhang, D.; Beijersbergen, R.; Bernards, R.; Moll, R.; Elledge, S.J.; Eilers, M. The ubiquitin-specific protease USP28 is required for MYC stability. Nat. Cell Biol. 2007, 9, 765–774. [Google Scholar] [CrossRef]
- Linares, J.F.; Duran, A.; Reina-Campos, M.; Aza-Blanc, P.; Campos, A.; Moscat, J.; Diaz-Meco, M.T. Amino Acid Activation of mTORC1 by a PB1-Domain-Driven Kinase Complex Cascade. Cell Rep. 2015, 12, 1339–1352. [Google Scholar] [CrossRef]
- Mao, J.H.; Kim, I.J.; Wu, D.; Climent, J.; Kang, H.C.; DelRosario, R.; Balmain, A. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science 2008, 321, 1499–1502. [Google Scholar] [CrossRef]
- Agrawal, P.; Chen, Y.T.; Schilling, B.; Gibson, B.W.; Hughes, R.E. Ubiquitin-specific peptidase 9, X-linked (USP9X) modulates activity of mammalian target of rapamycin (mTOR). J. Biol. Chem. 2012, 287, 21164–21175. [Google Scholar] [CrossRef]
- Dorr, C.; Janik, C.; Weg, M.; Been, R.A.; Bader, J.; Kang, R.; Ng, B.; Foran, L.; Landman, S.R.; O’Sullivan, M.G.; et al. Transposon Mutagenesis Screen Identifies Potential Lung Cancer Drivers and CUL3 as a Tumor Suppressor. Mol. Cancer Res. 2015, 13, 1238–1247. [Google Scholar] [CrossRef]
- Thirusangu, P.; Vigneshwaran, V.; Prashanth, T.; Vijay Avin, B.R.; Malojirao, V.H.; Rakesh, H.; Khanum, S.A.; Mahmood, R.; Prabhakar, B.T. BP-1T, an antiangiogenic benzophenone-thiazole pharmacophore, counteracts HIF-1 signalling through p53/MDM2-mediated HIF-1alpha proteasomal degradation. Angiogenesis 2017, 20, 55–71. [Google Scholar] [CrossRef]
- Flugel, D.; Gorlach, A.; Kietzmann, T. GSK-3beta regulates cell growth, migration, and angiogenesis via Fbw7 and USP28-dependent degradation of HIF-1alpha. Blood 2012, 119, 1292–1301. [Google Scholar] [CrossRef]
- Park, J.J.; Yun, J.H.; Baek, K.H. Polyclonal and monoclonal antibodies specific for ubiquitin-specific protease 20. Monoclon. Antib. Immunodiagn. Immunother. 2013, 32, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Yang, R.; Huang, M.L.; Kong, Y.G.; Sheng, J.F.; Tao, Z.Z.; Gao, L.; Chen, S.M. NOTCH2 negatively regulates metastasis and epithelial-Mesenchymal transition via TRAF6/AKT in nasopharyngeal carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 456. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Chen, Q.; Liu, Q.; Li, Y.; Sun, X.; Hong, L.; Ji, S.; Liu, C.; Geng, J.; Zhang, W.; et al. Hippo Signaling Suppresses Cell Ploidy and Tumorigenesis through Skp2. Cancer Cell 2017, 31, 669–684 e667. [Google Scholar] [CrossRef]
- Grattarola, M.; Cucci, M.A.; Roetto, A.; Dianzani, C.; Barrera, G.; Pizzimenti, S. Post-translational down-regulation of Nrf2 and YAP proteins, by targeting deubiquitinases, reduces growth and chemoresistance in pancreatic cancer cells. Free Radic. Biol. Med. 2021, 174, 202–210. [Google Scholar] [CrossRef]
- Yeung, B.; Ho, K.C.; Yang, X. WWP1 E3 ligase targets LATS1 for ubiquitin-mediated degradation in breast cancer cells. PLoS ONE 2013, 8, e61027. [Google Scholar] [CrossRef] [PubMed]
- Thakur, N.; Sorrentino, A.; Heldin, C.H.; Landstrom, M. TGF-beta uses the E3-ligase TRAF6 to turn on the kinase TAK1 to kill prostate cancer cells. Future Oncol. 2009, 5, 1–3. [Google Scholar] [CrossRef]
- Eichhorn, P.J.; Rodon, L.; Gonzalez-Junca, A.; Dirac, A.; Gili, M.; Martinez-Saez, E.; Aura, C.; Barba, I.; Peg, V.; Prat, A.; et al. USP15 stabilizes TGF-beta receptor I and promotes oncogenesis through the activation of TGF-beta signaling in glioblastoma. Nat. Med. 2012, 18, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Ying, Y. The Post-translational Modifications of Smurf2 in TGF-beta Signaling. Front. Mol. Biosci. 2020, 7, 128. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Popelka, H.; Lei, Y.; Yang, Y.; Klionsky, D.J. The Roles of Ubiquitin in Mediating Autophagy. Cells 2020, 9, 2025. [Google Scholar] [CrossRef]
- Lin, P.W.; Chu, M.L.; Liu, H.S. Autophagy and metabolism. Kaohsiung J. Med. Sci. 2021, 37, 12–19. [Google Scholar] [CrossRef]
- Grumati, P.; Dikic, I. Ubiquitin signaling and autophagy. J. Biol. Chem. 2018, 293, 5404–5413. [Google Scholar] [CrossRef]
- Wang, Y.T.; Liu, T.Y.; Shen, C.H.; Lin, S.Y.; Hung, C.C.; Hsu, L.C.; Chen, G.C. K48/K63-linked polyubiquitination of ATG9A by TRAF6 E3 ligase regulates oxidative stress-induced autophagy. Cell Rep. 2022, 38, 110354. [Google Scholar] [CrossRef]
- Feng, X.; Jia, Y.; Zhang, Y.; Ma, F.; Zhu, Y.; Hong, X.; Zhou, Q.; He, R.; Zhang, H.; Jin, J.; et al. Ubiquitination of UVRAG by SMURF1 promotes autophagosome maturation and inhibits hepatocellular carcinoma growth. Autophagy 2019, 15, 1130–1149. [Google Scholar] [CrossRef]
- Chen, R.H.; Chen, Y.H.; Huang, T.Y. Ubiquitin-mediated regulation of autophagy. J. Biomed. Sci. 2019, 26, 80. [Google Scholar] [CrossRef]
- Moon, S.; Muniyappan, S.; Lee, S.B.; Lee, B.H. Small-Molecule Inhibitors Targeting Proteasome-Associated Deubiquitinases. Int. J. Mol. Sci. 2021, 22, 6213. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Liu, X.; Jiang, X.; Zhang, K.; Wang, Y.; Li, X.; Jiang, S.; Han, T. USP10 as a Potential Therapeutic Target in Human Cancers. Genes 2022, 13, 831. [Google Scholar] [CrossRef]
- Kim, S.; Woo, S.M.; Min, K.J.; Seo, S.U.; Lee, T.J.; Kubatka, P.; Kim, D.E.; Kwon, T.K. WP1130 Enhances TRAIL-Induced Apoptosis through USP9X-Dependent miR-708-Mediated Downregulation of c-FLIP. Cancers 2019, 11, 344. [Google Scholar] [CrossRef]
- Mao, Y. Structure, Dynamics and Function of the 26S Proteasome. Subcell. Biochem. 2021, 96, 1–151. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, T.; Yang, Q.; Cheng, S.; Liu, F.; Yang, G.; Wang, F.; Wang, R.; Yang, D.; Zhou, M.; et al. Proteasomal deubiquitylase activity enhances cell surface recycling of the epidermal growth factor receptor in non-small cell lung cancer. Cell Oncol. 2022, 45, 951–965. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Liao, Y.; Guo, Z.; Li, Y.; Jiang, L.; Zhang, F.; Huang, C.; Liu, Y.; Wang, X.; Liu, N.; et al. Targeting proteasome-associated deubiquitinases as a novel strategy for the treatment of estrogen receptor-positive breast cancer. Oncogenesis 2018, 7, 75. [Google Scholar] [CrossRef] [PubMed]
- Hillert, E.K.; Brnjic, S.; Zhang, X.; Mazurkiewicz, M.; Saei, A.A.; Mofers, A.; Selvaraju, K.; Zubarev, R.; Linder, S.; D’Arcy, P. Proteasome inhibitor b-AP15 induces enhanced prote.eotoxicity by inhibiting cytoprotective aggresome formation. Cancer Lett. 2019, 448, 70–83. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Chen, Y.; He, D.; He, S. USP14-mediated deubiquitination of SIRT1 in macrophage promotes fatty acid oxidation amplification and M2 phenotype polarization. Biochem. Biophys. Res. Commun. 2023, 646, 19–29. [Google Scholar] [CrossRef]
- Li, J.; Yakushi, T.; Parlati, F.; Mackinnon, A.L.; Perez, C.; Ma, Y.; Carter, K.P.; Colayco, S.; Magnuson, G.; Brown, B.; et al. Capzimin is a potent and specific inhibitor of proteasome isopeptidase Rpn11. Nat. Chem. Biol. 2017, 13, 486–493. [Google Scholar] [CrossRef]
- Guo, W.; Ding, Y.; Pu, C.; Wang, Z.; Deng, W.; Jin, X. Curcumin inhibits pancreatic cancer cell proliferation by regulating Beclin1 expression and inhibiting the hypoxia-inducible factor-1alpha-mediated glycolytic pathway. J. Gastrointest. Oncol. 2022, 13, 3254–3262. [Google Scholar] [CrossRef]
- Zhou, B.; Zuo, Y.; Li, B.; Wang, H.; Liu, H.; Wang, X.; Qiu, X.; Hu, Y.; Wen, S.; Du, J.; et al. Deubiquitinase inhibition of 19S regulatory particles by 4-arylidene curcumin analog AC17 causes NF-kappaB inhibition and p53 reactivation in human lung cancer cells. Mol. Cancer Ther. 2013, 12, 1381–1392. [Google Scholar] [CrossRef]
- Wang, J.; Du, T.; Lu, Y.; Lv, Y.; Du, Y.; Wu, J.; Ma, R.; Xu, C.; Feng, J. VLX1570 regulates the proliferation and apoptosis of human lung cancer cells through modulating ER stress and the AKT pathway. J. Cell Mol. Med. 2022, 26, 108–122. [Google Scholar] [CrossRef]
- Ambrosio, F.A.; Costa, G.; Gallo Cantafio, M.E.; Torcasio, R.; Trapasso, F.; Alcaro, S.; Viglietto, G.; Amodio, N. Natural Agents as Novel Potential Source of Proteasome Inhibitors with Anti-Tumor Activity: Focus on Multiple Myeloma. Molecules 2023, 28, 1438. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Xia, J.; Zhang, J.; Zhu, Y.; Wu, Y.; Guo, J.; Chen, S.; Lei, Q.; Meng, B.; Kuang, C.; et al. Phosphoglycerate dehydrogenase promotes proliferation and bortezomib resistance through increasing reduced glutathione synthesis in multiple myeloma. Br. J. Haematol. 2020, 190, 52–66. [Google Scholar] [CrossRef]
- Lindner, S.; Kronke, J. The molecular mechanism of thalidomide analogs in hematologic malignancies. J. Mol. Med. 2016, 94, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Hesterberg, R.S.; Beatty, M.S.; Han, Y.; Fernandez, M.R.; Akuffo, A.A.; Goodheart, W.E.; Yang, C.; Chang, S.; Colin, C.M.; Alontaga, A.Y.; et al. Cereblon harnesses Myc-dependent bioenergetics and activity of CD8+ T lymphocytes. Blood 2020, 136, 857–870. [Google Scholar] [CrossRef]
- Chen, S.; Liu, Y.; Zhou, H. Advances in the Development Ubiquitin-Specific Peptidase (USP) Inhibitors. Int. J. Mol. Sci. 2021, 22, 4546. [Google Scholar] [CrossRef] [PubMed]
- Pajak, B.; Siwiak, E.; Soltyka, M.; Priebe, A.; Zielinski, R.; Fokt, I.; Ziemniak, M.; Jaskiewicz, A.; Borowski, R.; Domoradzki, T.; et al. 2-Deoxy-d-Glucose and Its Analogs: From Diagnostic to Therapeutic Agents. Int. J. Mol. Sci. 2019, 21, 234. [Google Scholar] [CrossRef]
- Luh, L.M.; Scheib, U.; Juenemann, K.; Wortmann, L.; Brands, M.; Cromm, P.M. Prey for the Proteasome: Targeted Protein Degradation-A Medicinal Chemist’s Perspective. Angew. Chem. Int. Ed. Engl. 2020, 59, 15448–15466. [Google Scholar] [CrossRef]
- Jiang, H.; Xiong, H.; Gu, S.X.; Wang, M. E3 ligase ligand optimization of Clinical PROTACs. Front. Chem. 2023, 11, 1098331. [Google Scholar] [CrossRef]
- Lebraud, H.; Wright, D.J.; Johnson, C.N.; Heightman, T.D. Protein Degradation by In-Cell Self-Assembly of Proteolysis Targeting Chimeras. ACS Cent. Sci. 2016, 2, 927–934. [Google Scholar] [CrossRef]
- Zhou, H.; Bai, L.; Xu, R.; Zhao, Y.; Chen, J.; McEachern, D.; Chinnaswamy, K.; Wen, B.; Dai, L.; Kumar, P.; et al. Structure-Based Discovery of SD-36 as a Potent, Selective, and Efficacious PROTAC Degrader of STAT3 Protein. J. Med. Chem. 2019, 62, 11280–11300. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Kim, J.W.; Choi, H.I.; Maeng, H.J.; Koo, T.S. Development of an LC-MS/MS Method for ARV-110, a PROTAC Molecule, and Applications to Pharmacokinetic Studies. Molecules 2022, 27, 1977. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, Y.; Wu, Y.; Xing, D. Developments of CRBN-based PROTACs as potential therapeutic agents. Eur. J. Med. Chem. 2021, 225, 113749. [Google Scholar] [CrossRef]
- Li, X.; Song, Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J. Hematol. Oncol. 2020, 13, 50. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tandon, I.; Heelan, W.; Wang, Y.; Tang, W.; Hu, Q. Proteolysis-targeting chimera (PROTAC) delivery system: Advancing protein degraders towards clinical translation. Chem. Soc. Rev. 2022, 51, 5330–5350. [Google Scholar] [CrossRef]
- Zhang, C.; Zeng, Z.; Cui, D.; He, S.; Jiang, Y.; Li, J.; Huang, J.; Pu, K. Semiconducting polymer nano-PROTACs for activatable photo-immunometabolic cancer therapy. Nat. Commun. 2021, 12, 2934. [Google Scholar] [CrossRef]
- Schapira, M.; Calabrese, M.F.; Bullock, A.N.; Crews, C.M. Targeted protein degradation: Expanding the toolbox. Nat. Rev. Drug Discov. 2019, 18, 949–963. [Google Scholar] [CrossRef]
- Xue, G.; Wang, K.; Zhou, D.; Zhong, H.; Pan, Z. Light-Induced Protein Degradation with Photocaged PROTACs. J. Am. Chem. Soc. 2019, 141, 18370–18374. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Target | E3 Ligase/DUB | Disease Association | Tumor | Refs | Site |
|---|---|---|---|---|---|
| c-Myc | NEDD4L | ESCC | Suppressor | [136,201] | |
| Lung cancer | Carcinogenesis | [202] | |||
| Skp2 | Hepatoma carcinoma | Suppressor | [203] | ||
| Fbxw7 | Hepatoma carcinoma | Suppressor | [203] | ||
| HUWE1 | Skin tumorigenesis | Suppressor | [204] | Lys48 | |
| CUL4 | Many cancer types | Suppressor | [205] | ||
| VHL | Breast cancer stem | Suppressor | [206] | ||
| FBXL14 | Glioblastoma stem | Suppressor | [144] | ||
| CHIP | Glioma | Suppressor | [207] | ||
| USP13 | Glioblastoma stem cell | Carcinogenesis | [144] | ||
| USP22 | Breast cancer | Carcinogenesis | [146] | ||
| USP28 | Breast and colon cancer | Carcinogenesis | [208] | ||
| USP36 | Lung/breast cancer | Carcinogenesis | [142] | ||
| mTOR | TRAF2 | Lung/melanoma cancer | Suppressor | [149] | Lys 63 |
| TRAF6 | Prostate cancer | Suppressor | [149,209] | Lys 63 | |
| RNF152 | Colorectal cancer | Suppressor | [152] | ||
| TRIM21 | Lung cancer | Suppressor | [151] | ||
| FBXW7 | Breast cancer | Suppressor | [210] | ||
| RNF126 | Myeloid leukemia cells | Suppressor | [154] | ||
| OTUD7B | Lung/melanoma cancer | Carcinogenesis | [149] | ||
| USP9X | Head and neck cancer | Suppressor | [155,211] | ||
| KRAS | NEDD4L | Many cancers | Suppressor | [166] | |
| CUL3 | Lung cancer | Suppressor | [163,212] | Lys 48, 63, 33 | |
| HIF-a | pVHL | ccRCC | Suppressor | [171] | |
| MDM2 | Solid tumors | Suppressor | [213] | ||
| FBXW7 | Many cancers | Suppressor | [214] | ||
| FBXO11 | Many cancers | Suppressor | [179] | ||
| Parkin | Breast cancer | Suppressor | [175] | Lys477 | |
| TRAF6 | Colon and cervix cancer | Suppressor | [177] | Lys 63 | |
| USP7 | Lung cancer | Carcinogenesis | [174] | Lys 63 | |
| USP8 | ccRCC | Carcinogenesis | [171] | ||
| USP20 | Many cancer types | Carcinogenesis | [215] | ||
| USP28 | Many cancer types | Carcinogenesis | [214] | ||
| TRIM44 | Myeloma | Carcinogenesis | [173] | ||
| PI3K/AKT | TRAF6 | Nasopharyngeal carcinoma | Carcinogenesis | [216] | Lys 63 |
| RNF8 | Lung cancer | Carcinogenesis | [183] | Lys 63 | |
| UBE2T | Hepatoma carcinoma | Carcinogenesis | [184] | Lys63 | |
| USP1 | Leukemia | Carcinogenesis | [185] | ||
| Hippo pathway | PARK2 | ESCC | Carcinogenesis | [191] | Lys48 |
| SCF/Skp2 | Hepatoma carcinoma | Carcinogenesis | [217] | Lys63 | |
| OTUD1 | Pancreatic | Suppressor | [218] | ||
| WWP1 | Breast cancer | Carcinogenesis | [194,219] | ||
| TGF-β pathway | TRAF4 | Breast cancer | Carcinogenesis | [197] | |
| TRAF6 | Prostate cancer | Suppressor | [220] | ||
| USP15 | Glioblastoma | Carcinogenesis | [221] | ||
| SMURF2 | Glioma | Suppressor | [222] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Xiang, Y.; Fan, M.; Fang, S.; Hua, Q. The Ubiquitin–Proteasome System in Tumor Metabolism. Cancers 2023, 15, 2385. https://doi.org/10.3390/cancers15082385
Wang J, Xiang Y, Fan M, Fang S, Hua Q. The Ubiquitin–Proteasome System in Tumor Metabolism. Cancers. 2023; 15(8):2385. https://doi.org/10.3390/cancers15082385
Chicago/Turabian StyleWang, Jie, Yuandi Xiang, Mengqi Fan, Shizhen Fang, and Qingquan Hua. 2023. "The Ubiquitin–Proteasome System in Tumor Metabolism" Cancers 15, no. 8: 2385. https://doi.org/10.3390/cancers15082385
APA StyleWang, J., Xiang, Y., Fan, M., Fang, S., & Hua, Q. (2023). The Ubiquitin–Proteasome System in Tumor Metabolism. Cancers, 15(8), 2385. https://doi.org/10.3390/cancers15082385