
High-Dosage NMN Promotes Ferroptosis to Suppress Lung Adenocarcinoma Growth through the NAM-Mediated SIRT1–AMPK–ACC Pathway
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Lentivirus Construction and Infection
2.3. Western Blot Assay
2.4. Cell Proliferation Assay
2.5. Cell Apoptosis Assay
2.6. Xenograft Assay in Nude Mice
2.7. Hematoxylin–Eosin (HE) Staining and Immunohistochemical (IHC) Assay
2.8. Transmission Electron Microscopy (TEM)
2.9. Cellular ROS Measurement
2.10. Lipid Peroxidation Assay
2.11. Cellular Glutathione (GSH) and Malondialdehyde (MDA) Detection
2.12. Mito-FerroGreen-Labeled Immunofluorescence Assay (Fe2+)
2.13. ELISA Assay
2.14. Statistical Analysis
3. Results
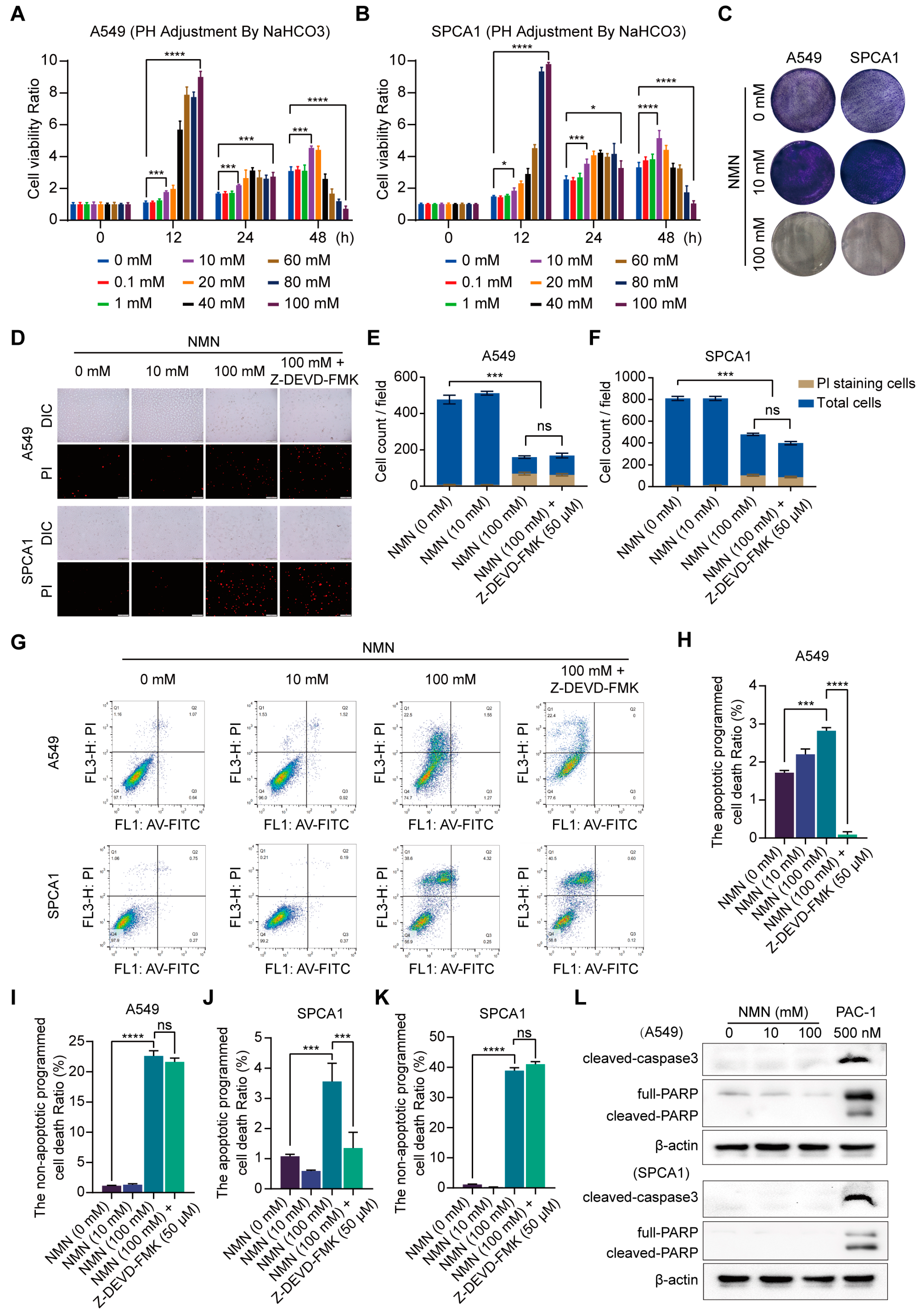
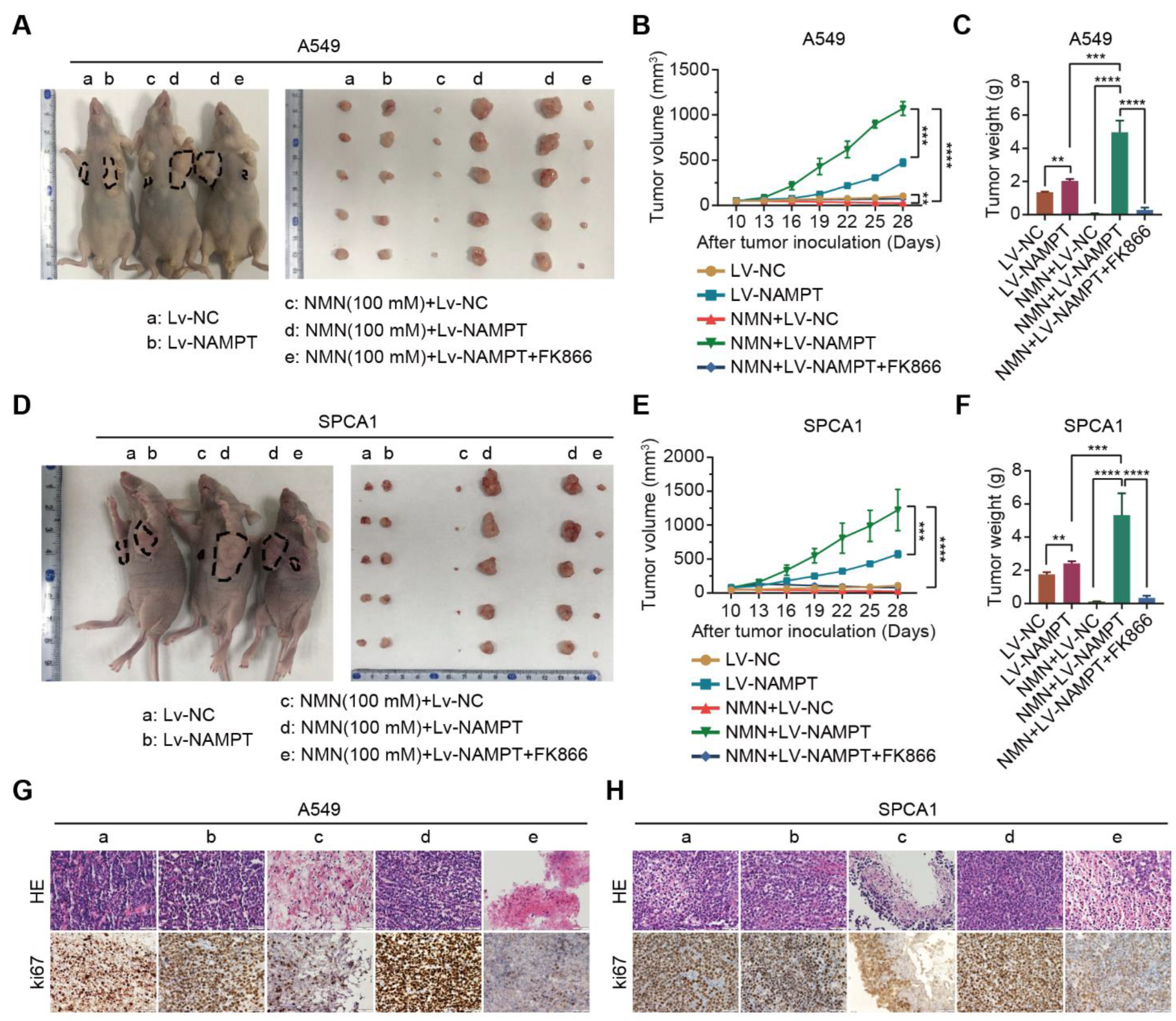
3.1. High-Dosage NMN Inhibits Lung Cancer Growth through a Non-Apoptotic, Non-Autophagy, or Non-Necrosis Program
3.2. High-Dosage NMN Inhibits Lung Cancer Growth by Inducing Ferroptosis Program
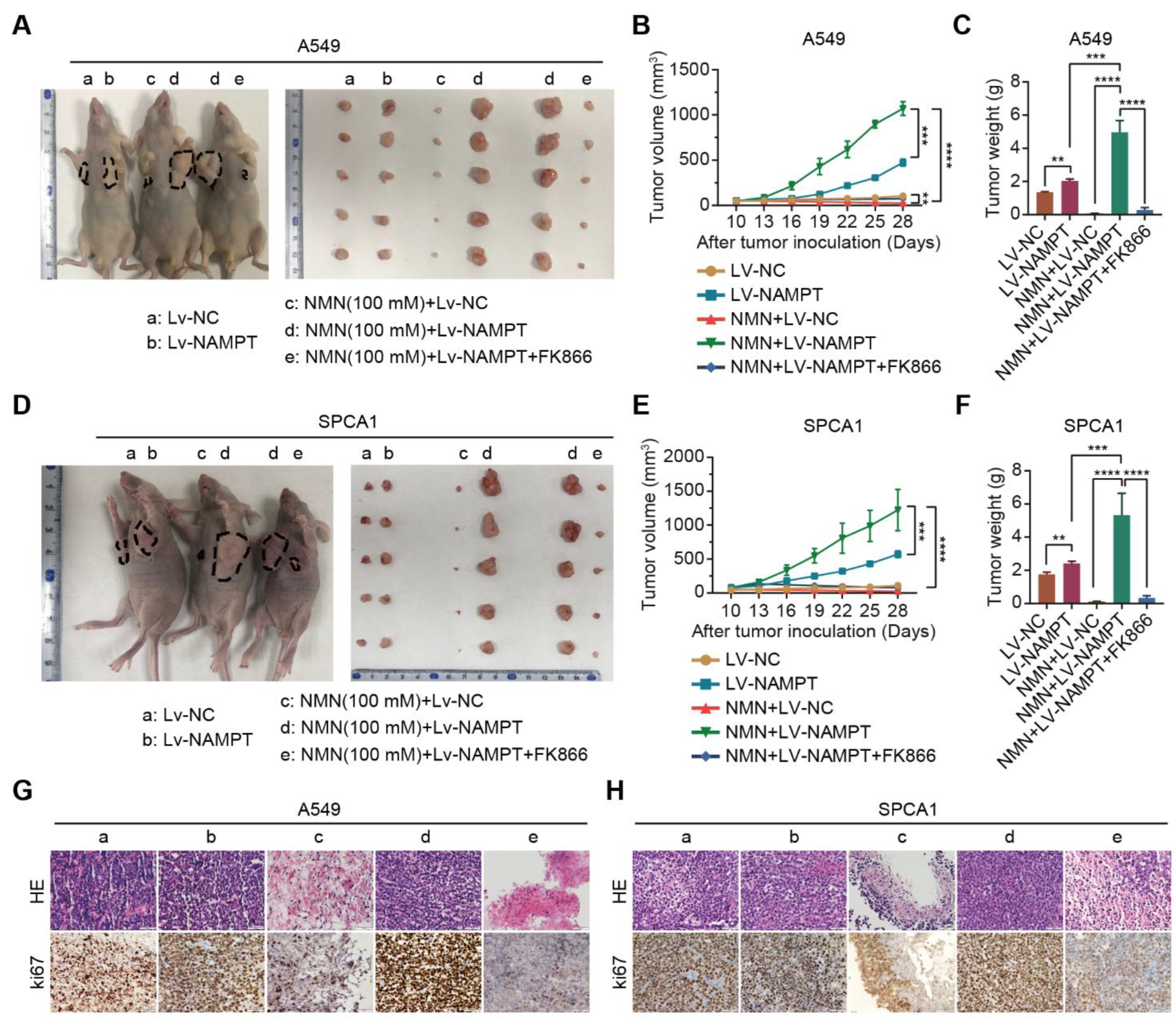
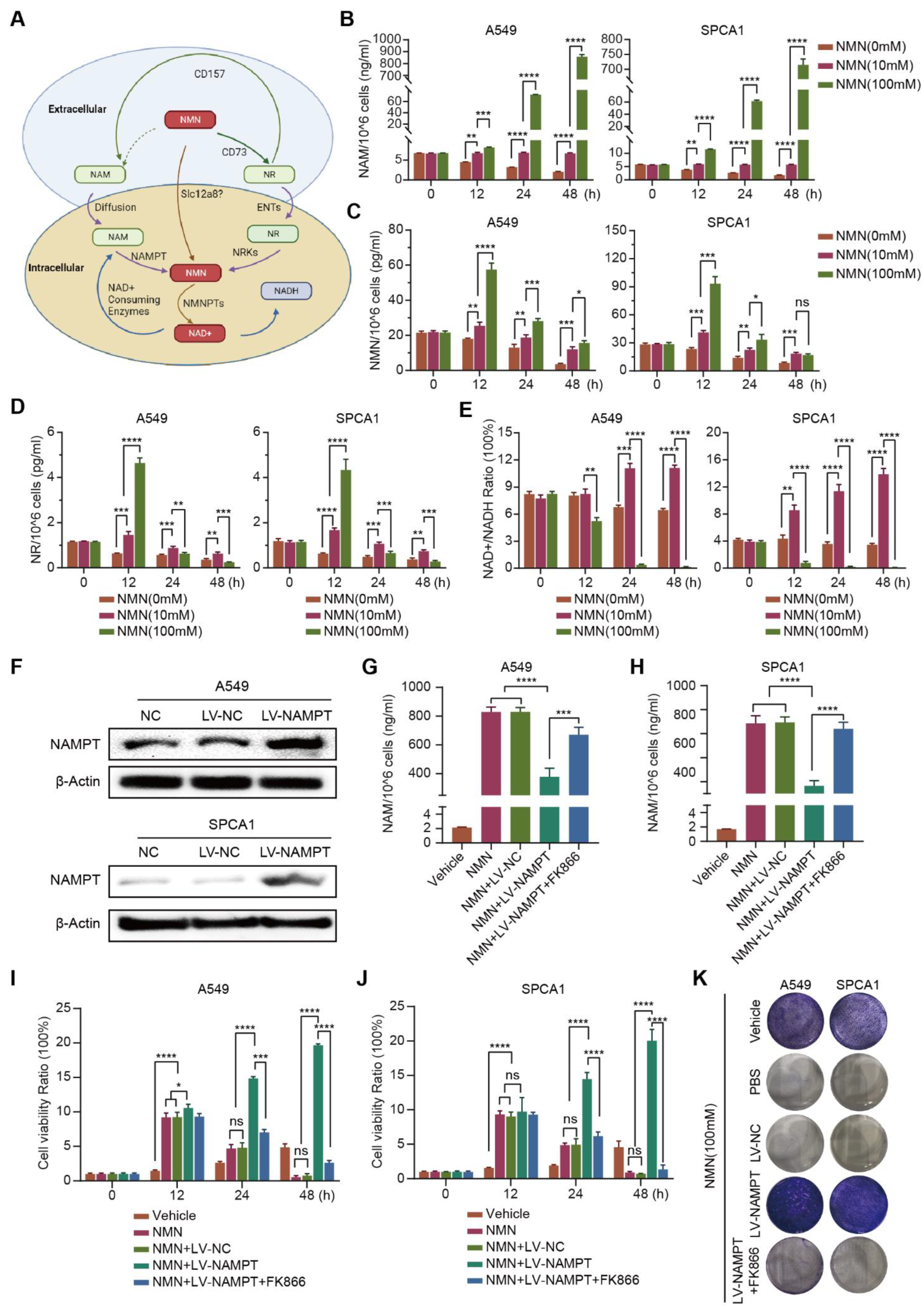
3.3. High-Dosage NMN Inhibits Lung Cancer Growth through Its Metabolite NAM
3.4. High-Dosage NMN Induces Ferroptosis via Its Metabolite NAM
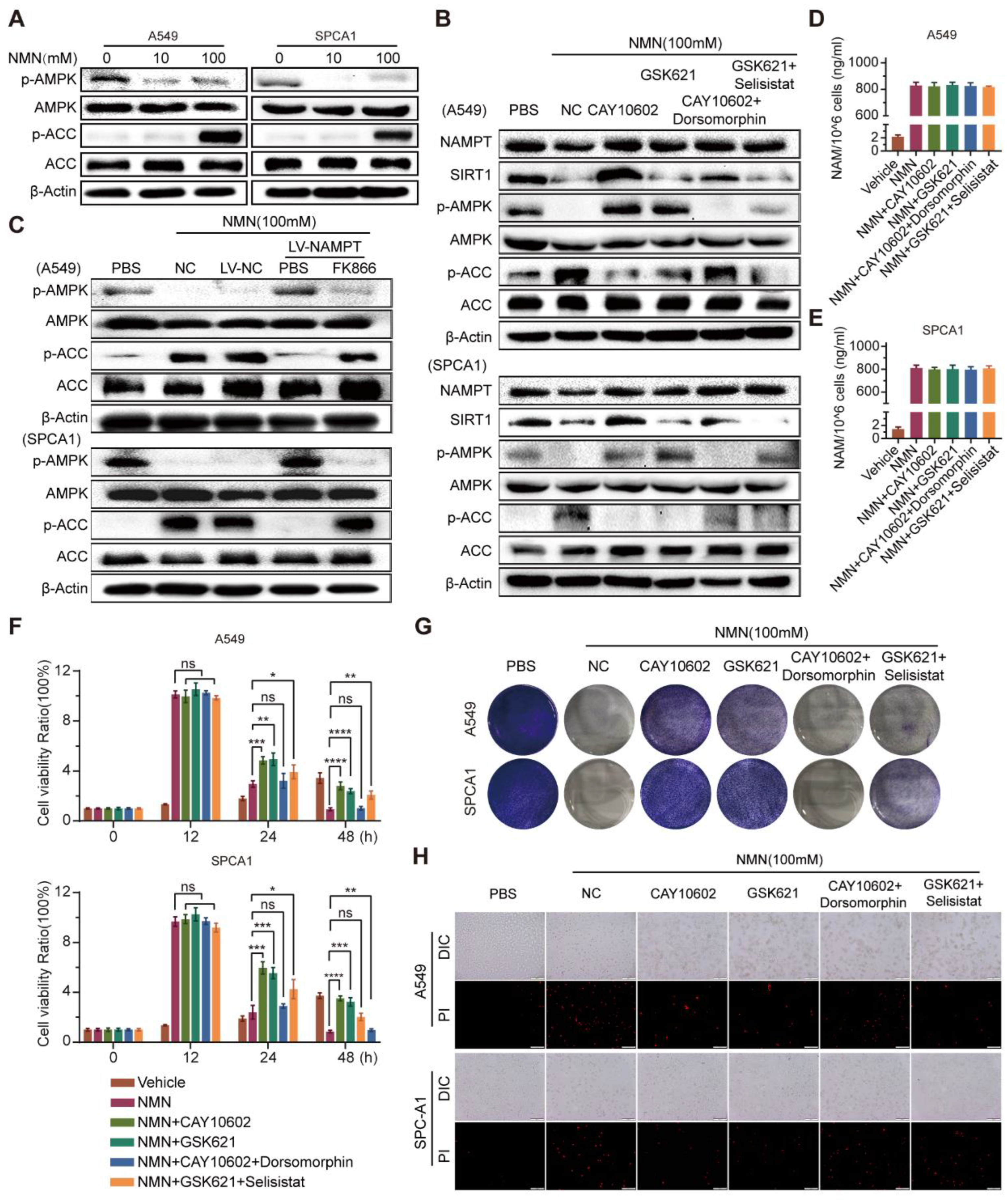
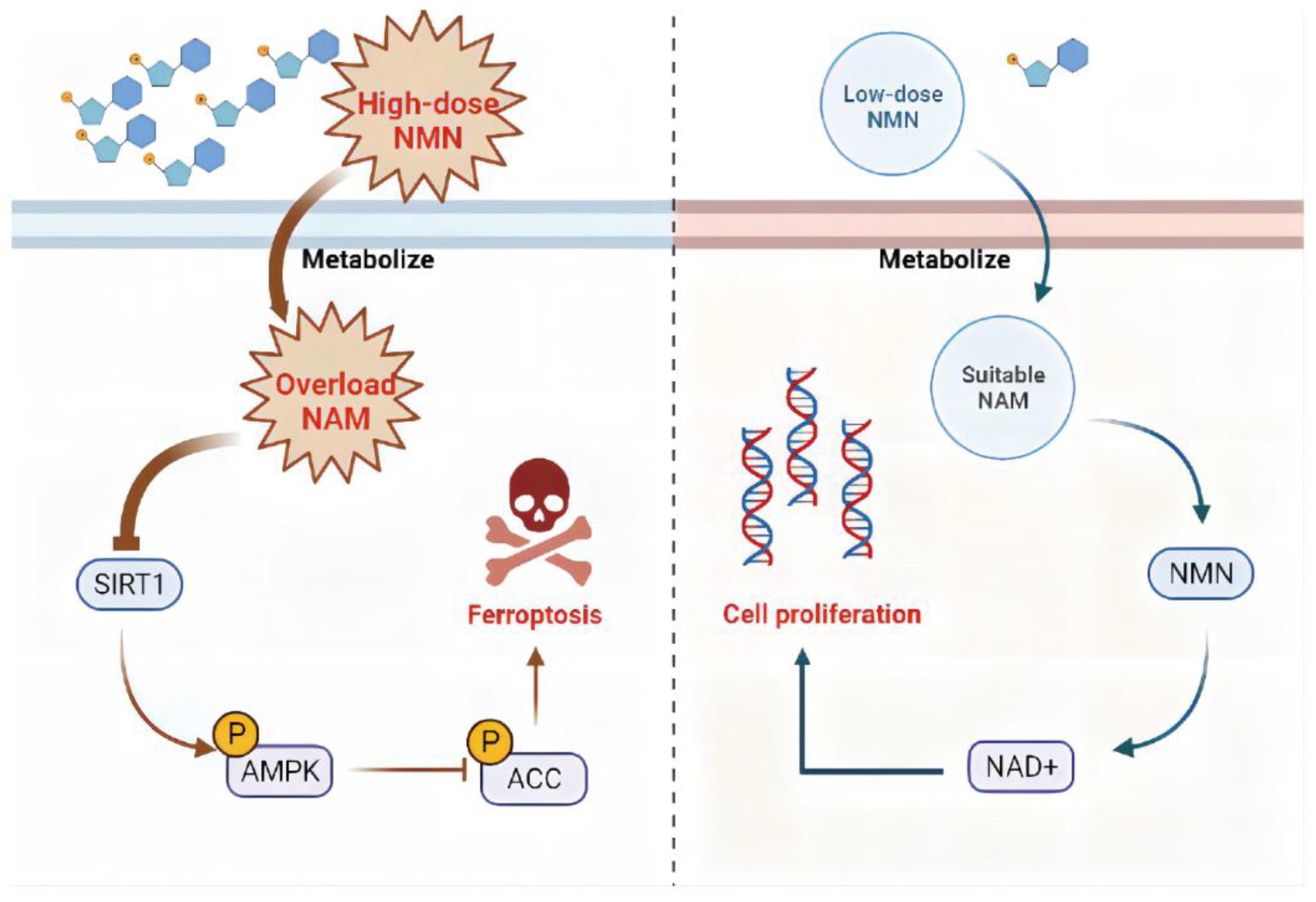
3.5. High-Dosage NMN Promotes Ferroptosis through SIRT1–AMPK–ACC Signaling with NAM Overload
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| NMN | Nicotinamide mononucleotide |
| NAM | Nicotinamide |
| NAD | Nicotinamide adenine dinucleotide |
| NAMPT | Nicotinamide phosphoribosyltransferase |
| TEM | Transmission electron microscopy |
| NR | Nicotinamide riboside |
| SIRT1 | NAD-dependent deacetylase sirtuin-1 |
| AMPK | Adenosine 5’-monophosphate (AMP)-activated protein kinase |
| ACC | Acetyl CoA carboxylase |
| Erastin | Ferroptosis inducer |
| RSL3 | GSH peroxidase 4 inhibitor |
| STR | Short Tandem Repeats |
| PARP | Poly ADP-ribose polymerase |
| Z-DEVD-FMK | Apoptosis inhibitor |
| PAC-1 | Procaspase-activating compound 1 |
| Nec-1 | Necrostatin-1 |
| CQ | Chloroquine |
| Fer-1 | Ferrostatin-1 |
| FK866 | E-Daporinad |
| CAY10602 | SIRT1 agonist |
| GSK621 | AMPK agonist |
| GSH | Glutathione |
| MDA | Malondialdehyde |
| FACS | Fluorescence-Activated Cell Sorting |
| ROS | Reactive oxygen species |
| DMEM | Dulbecco’s modified eagle medium |
| FBS | Fetal bovine serum |
| RIPA | RIPA Lysis Buffer |
| SDS-PAGE | Polyacrylamide gel electrophoresis |
| PVDF | Polyvinylidene difluoride membranes |
| PARP | Poly ADP-ribose polymerase |
| CCK8 | Cell counting kit-8 |
| PI | Propidium iodide |
| HE | Hematoxylin-eosin stain |
| IHC | Immunohistochemical assay |
| ki67 | Proliferative-cell-associated antigen |
| PBS | Phosphate buffer solution |
| DAB | 3′-diaminobenzidine chromogen |
| GSH | Glutathione |
| MDA | Malondialdehyde |
References
- Zhang, W.; Qu, J.; Liu, G.H.; Belmonte, J.C.I. The ageing epigenome and its rejuvenation. Nat. Rev. Mol. Cell Biol. 2020, 21, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Saez-Atienzar, S.; Masliah, E. Cellular senescence and Alzheimer disease: The egg and the chicken scenario. Nat. Rev. Neurosci. 2020, 21, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Soukas, A.A.; Hao, H.; Wu, L. Metformin as Anti-Aging Therapy: Is It for Everyone? Trends Endocrinol. Metab. 2019, 30, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; Lebrasseur, N.K.; Childs, B.G.; Van De Sluis, B.; Kirkland, J.L.; Van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Yoshino, J.; Baur, J.A.; Imai, S.I. NAD(+) Intermediates: The Biology and Therapeutic Potential of NMN and NR. Cell Metab. 2018, 27, 513–528. [Google Scholar] [CrossRef]
- Nadeeshani, H.; Li, J.; Ying, T.; Zhang, B.; Lu, J. Nicotinamide mononucleotide (NMN) as an anti-aging health product—Promises and safety concerns. J. Adv. Res. 2022, 37, 267–278. [Google Scholar] [CrossRef]
- Gardell, S.J.; Hopf, M.; Khan, A.; Dispagna, M.; Sessions, E.H.; Falter, R.; Kapoor, N.; Brooks, J.; Culver, J.; Petucci, C.; et al. Boosting NAD(+) with a small molecule that activates NAMPT. Nat. Commun. 2019, 10, 3241. [Google Scholar] [CrossRef] [PubMed]
- Nacarelli, T.; Lau, L.; Fukumoto, T.; Zundell, J.; Fatkhutdinov, N.; Wu, S.; Aird, K.M.; Iwasaki, O.; Kossenkov, A.V.; Schultz, D.; et al. NAD(+) metabolism governs the proinflammatory senescence-associated secretome. Nat. Cell Biol. 2019, 21, 397–407. [Google Scholar] [CrossRef]
- Drapela, S.; Ilter, D.; Gomes, A.P. Metabolic reprogramming: A bridge between aging and tumorigenesis. Mol. Oncol. 2022, 16, 3295–3318. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Thai, A.A.; Solomon, B.J.; Sequist, L.V.; Gainor, J.F.; Heist, R.S. Lung cancer. Lancet 2021, 398, 535–554. [Google Scholar] [CrossRef] [PubMed]
- Fane, M.; Weeraratna, A.T. How the ageing microenvironment influences tumour progression. Nat. Rev. Cancer 2020, 20, 89–106. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.H.; Chen, Z.; Chen, K.; Liao, F.T.; Chung, C.E.; Liu, X.; Lin, Y.C.; Keohavong, P.; Leikauf, G.D.; Di, Y.P. Lipopolysaccharide-Mediated Chronic Inflammation Promotes Tobacco Carcinogen-Induced Lung Cancer and Determines the Efficacy of Immunotherapy. Cancer Res. 2021, 81, 144–157. [Google Scholar] [CrossRef]
- Miyamoto, C.; Kojo, S.; Yamashita, M.; Moro, K.; Lacaud, G.; Shiroguchi, K.; Taniuchi, I.; Ebihara, T. Runx/Cbfbeta complexes protect group 2 innate lymphoid cells from exhausted-like hyporesponsiveness during allergic airway inflammation. Nat. Commun. 2019, 10, 447. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef]
- Liao, P.; Wang, W.; Wang, W.; Kryczek, I.; Li, X.; Bian, Y.; Sell, A.; Wei, S.; Grove, S.; Johnson, J.K.; et al. CD8(+) T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell 2022, 40, 365–378.e6. [Google Scholar] [CrossRef]
- Park, E.; Chung, S.W. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019, 10, 822. [Google Scholar] [CrossRef]
- Posavec Marjanović, M.; Hurtado-Bagès, S.; Lassi, M.; Valero, V.; Malinverni, R.; Delage, H.; Navarro, M.; Corujo, D.; Guberovic, I.; Douet, J.; et al. MacroH2A1.1 regulates mitochondrial respiration by limiting nuclear NAD(+) consumption. Nat. Struct. Mol. Biol. 2017, 24, 902–910. [Google Scholar] [CrossRef]
- Cui, J.; Shu, C.; Xu, J.; Chen, D.; Li, J.; Ding, K.; Chen, M.; Li, A.; He, J.; Shu, Y.; et al. JP1 suppresses proliferation and metastasis of melanoma through MEK1/2 mediated NEDD4L-SP1-Integrin alphavbeta3 signaling. Theranostics 2020, 10, 8036–8050. [Google Scholar] [CrossRef]
- Lee, H.; Zandkarimi, F.; Zhang, Y.; Meena, J.K.; Kim, J.; Zhuang, L.; Tyagi, S.; Ma, L.; Westbrook, T.F.; Steinberg, G.R.; et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 2020, 22, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Nikas, I.P.; Paschou, S.A.; Ryu, H.S. The Role of Nicotinamide in Cancer Chemoprevention and Therapy. Biomolecules 2020, 10, 477. [Google Scholar] [CrossRef] [PubMed]
- Garten, A.; Schuster, S.; Penke, M.; Gorski, T.; de Giorgis, T.; Kiess, W. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat. Rev. Endocrinol. 2015, 11, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular Senescence: Aging, Cancer, and Injury. Physiol. Rev. 2019, 99, 1047–1078. [Google Scholar] [CrossRef] [PubMed]
- Pan, F.; Kang, S.; Zhao, Y.; Dai, L.; Shao, Q.; Yang, Y.; Chen, Q.; Zhu, J.; Cui, L. Effect of β-nicotinamide mononucleotide on tumor formation and growth in a lung cancer mouse model. Mater. Chem. Front. 2021, 5, 995–1002. [Google Scholar] [CrossRef]
- Lin, G.L.; Wilson, K.M.; Ceribelli, M.; Stanton, B.Z.; Woo, P.J.; Kreimer, S.; Qin, E.Y.; Zhang, X.; Lennon, J.; Nagaraja, S.; et al. Therapeutic strategies for diffuse midline glioma from high-throughput combination drug screening. Sci. Transl. Med. 2019, 11, eaaw0064. [Google Scholar] [CrossRef]
- Hong, S.M.; Lee, A.-Y.; Hwang, S.-M.; Ha, Y.-J.; Kim, M.-J.; Min, S.; Hwang, W.; Yoon, G.; Kwon, S.M.; Woo, H.G.; et al. NAMPT mitigates colitis severity by supporting redox-sensitive activation of phagocytosis in inflammatory macrophages. Redox Biol. 2022, 50, 102237. [Google Scholar] [CrossRef]
- Grozio, A.; Mills, K.F.; Yoshino, J.; Bruzzone, S.; Sociali, G.; Tokizane, K.; Lei, H.C.; Cunningham, R.; Sasaki, Y.; Migaud, M.E.; et al. Slc12a8 is a nicotinamide mononucleotide transporter. Nat. Metab. 2019, 1, 47–57. [Google Scholar] [CrossRef]
- Okur, M.N.; Fang, E.F.; Fivenson, E.M.; Tiwari, V.; Croteau, D.L.; Bohr, V.A. Cockayne syndrome proteins CSA and CSB maintain mitochondrial homeostasis through NAD(+) signaling. Aging Cell 2020, 19, e13268. [Google Scholar] [CrossRef]
- Segatto, M.; Szokoll, R.; Fittipaldi, R.; Bottino, C.; Nevi, L.; Mamchaoui, K.; Filippakopoulos, P.; Caretti, G. BETs inhibition attenuates oxidative stress and preserves muscle integrity in Duchenne muscular dystrophy. Nat. Commun. 2020, 11, 6108. [Google Scholar] [CrossRef]
- Turner, J.; Licollari, A.; Mihalcea, E.; Tan, A. Safety Evaluation for Restorin(R) NMN, a NAD+ Precursor. Front. Pharmacol. 2021, 12, 749727. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Cui, J.; Chen, H.; Wang, Y.; Kuai, X.; Sun, S.; Tang, Q.; Zong, F.; Chen, Q.; Wu, J.; et al. High-Dosage NMN Promotes Ferroptosis to Suppress Lung Adenocarcinoma Growth through the NAM-Mediated SIRT1–AMPK–ACC Pathway. Cancers 2023, 15, 2427. https://doi.org/10.3390/cancers15092427
Zhang M, Cui J, Chen H, Wang Y, Kuai X, Sun S, Tang Q, Zong F, Chen Q, Wu J, et al. High-Dosage NMN Promotes Ferroptosis to Suppress Lung Adenocarcinoma Growth through the NAM-Mediated SIRT1–AMPK–ACC Pathway. Cancers. 2023; 15(9):2427. https://doi.org/10.3390/cancers15092427
Chicago/Turabian StyleZhang, Mingjiong, Jiahua Cui, Haoyan Chen, Yu Wang, Xingwang Kuai, Sibo Sun, Qi Tang, Feng Zong, Qiaoyu Chen, Jianqing Wu, and et al. 2023. "High-Dosage NMN Promotes Ferroptosis to Suppress Lung Adenocarcinoma Growth through the NAM-Mediated SIRT1–AMPK–ACC Pathway" Cancers 15, no. 9: 2427. https://doi.org/10.3390/cancers15092427
APA StyleZhang, M., Cui, J., Chen, H., Wang, Y., Kuai, X., Sun, S., Tang, Q., Zong, F., Chen, Q., Wu, J., & Wu, S. (2023). High-Dosage NMN Promotes Ferroptosis to Suppress Lung Adenocarcinoma Growth through the NAM-Mediated SIRT1–AMPK–ACC Pathway. Cancers, 15(9), 2427. https://doi.org/10.3390/cancers15092427