
Methods to Evaluate Changes in Mitochondrial Structure and Function in Cancer
 , , ,
, , ,  , ,
, ,  and
and
Abstract
:Simple Summary
Abstract

1. Introduction
2. Structural Techniques and Parameters to Characterize Mitochondria Health
2.1. Optical Microscopy
2.2. Electron Microscopy-Based Techniques
2.3. Evaluation of mtDNA Content or Integrity Using PCR
2.4. Extracellular Vesicle (EV) Secretion and Cargo
3. Functional Parameters to Understand Mitochondrial Health
3.1. Reactive Oxygen Species (ROS) Production
3.2. Mitochondrial Membrane Potential (ΔΨm)
3.3. Calcium Retention Capacity
3.4. Mitochondrial Bioenergetics: Glycolytic and Respiratory Capacity Measurements
Measures of ETC Complex Activity and the TCA Cycle
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Szewczyk, A.; Wojtczak, L. Mitochondria as a pharmacological target. Pharmacol. Rev. 2002, 54, 101–127. [Google Scholar] [CrossRef] [PubMed]
- Gilkerson, R.W.; Selker, J.M.L.; Capaldi, R.A. The cristal membrane of mitochondria is the principal site of oxidative phosphorylation. FEBS Lett. 2003, 546, 355–358. [Google Scholar] [CrossRef] [PubMed]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.H. Mitochondria signaling to the epigenome: A novel role for an old organelle. Free Radic. Biol. Med. 2021, 170, 59–69. [Google Scholar] [CrossRef]
- Vafai, S.B.; Mootha, V.K. Mitochondrial disorders as windows into an ancient organelle. Nature 2012, 491, 374–383. [Google Scholar] [CrossRef]
- Carew, J.S.; Huang, P. Mitochondrial defects in cancer. Mol. Cancer 2002, 1, 9. [Google Scholar] [CrossRef]
- Ohta, S. Contribution of somatic mutations in the mitochondrial genome to the development of cancer and tolerance against anticancer drugs. Oncogene 2006, 25, 4768–4776. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Barbi de Moura, M.; Vincent, G.; Fayewicz, S.L.; Bateman, N.W.; Hood, B.L.; Sun, M.; Suhan, J.; Duensing, S.; Yin, Y.; Sander, C.; et al. Mitochondrial respiration—An important therapeutic target in melanoma. PLoS ONE 2012, 7, e40690. [Google Scholar] [CrossRef]
- Hirpara, J.; Eu, J.Q.; Tan, J.K.M.; Wong, A.L.; Clement, M.-V.; Kong, L.R.; Ohi, N.; Tsunoda, T.; Qu, J.; Goh, B.C.; et al. Metabolic reprogramming of oncogene-addicted cancer cells to OXPHOS as a mechanism of drug resistance. Redox Biol. 2019, 25, 101076. [Google Scholar] [CrossRef]
- Grasso, D.; Zampieri, L.X.; Capelôa, T.; Van de Velde, J.A.; Sonveaux, P. Mitochondria in cancer. Cell Stress 2020, 4, 114–146. [Google Scholar] [CrossRef]
- Andersen, J.L.; Kornbluth, S. Mcl-1 rescues a glitch in the matrix. Nat. Cell Biol. 2012, 14, 563–565. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhou, F.; Zhang, Z.; Xing, D. Mitochondrial oxidative stress causes mitochondrial fragmentation via differential modulation of mitochondrial fission-fusion proteins. Febs J. 2011, 278, 941–954. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Yu, T.; Yoon, Y. Mitochondrial clustering induced by overexpression of the mitochondrial fusion protein Mfn2 causes mitochondrial dysfunction and cell death. Eur. J. Cell Biol. 2007, 86, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Protasoni, M.; Zeviani, M. Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions. Int. J. Mol. Sci. 2021, 22, 586. [Google Scholar] [CrossRef]
- Kühlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015, 13, 89. [Google Scholar] [CrossRef]
- Singh, R.; Jain, A.; Palanichamy, J.K.; Nag, T.C.; Bakhshi, S.; Singh, A. Ultrastructural changes in cristae of lymphoblasts in acute lymphoblastic leukemia parallel alterations in biogenesis markers. Appl. Microsc. 2021, 51, 20. [Google Scholar] [CrossRef]
- Moro, L. Mitochondrial Dysfunction in Aging and Cancer. J. Clin. Med. 2019, 8, 1983. [Google Scholar] [CrossRef]
- Dar, S.; Chhina, J.; Mert, I.; Chitale, D.; Buekers, T.; Kaur, H.; Giri, S.; Munkarah, A.; Rattan, R. Bioenergetic Adaptations in Chemoresistant Ovarian Cancer Cells. Sci. Rep. 2017, 7, 8760. [Google Scholar] [CrossRef]
- Rickard, B.P.; Tan, X.; Fenton, S.E.; Rizvi, I. Select Per- and Polyfluoroalkyl Substances (PFAS) Induce Resistance to Carboplatin in Ovarian Cancer Cell Lines. Int. J. Mol. Sci. 2022, 23, 5176. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, L.; Jia, R. The role of mitochondrial dynamics in human cancers. Am. J. Cancer Res. 2020, 10, 1278–1293. [Google Scholar]
- Lozoya, O.A.; Martinez-Reyes, I.; Wang, T.; Grenet, D.; Bushel, P.; Li, J.; Chandel, N.; Woychik, R.P.; Santos, J.H. Mitochondrial nicotinamide adenine dinucleotide reduced (NADH) oxidation links the tricarboxylic acid (TCA) cycle with methionine metabolism and nuclear DNA methylation. PLoS Biol. 2018, 16, e2005707. [Google Scholar] [CrossRef] [PubMed]
- Mitra, K.; Lippincott-Schwartz, J. Analysis of Mitochondrial Dynamics and Functions Using Imaging Approaches. Curr. Protoc. Cell Biol. 2010, 46, 4.25.1–4.25.21. [Google Scholar] [CrossRef] [PubMed]
- Kessel, D.; Luguya, R.; Vicente, M.G.H. Localization and Photodynamic Efficacy of Two Cationic Porphyrins Varying in Charge Distribution. Photochem. Photobiol. 2003, 78, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Sivandzade, F.; Bhalerao, A.; Cucullo, L. Analysis of the Mitochondrial Membrane Potential Using the Cationic JC-1 Dye as a Sensitive Fluorescent Probe. Bio-protocol 2019, 9, e3128. [Google Scholar] [CrossRef]
- Jakobs, S.; Stoldt, S.; Neumann, D. Light microscopic analysis of mitochondrial heterogeneity in cell populations and within single cells. Adv. Biochem. Eng. Biotechnol. 2011, 124, 1–19. [Google Scholar]
- Egner, A.; Jakobs, S.; Hell, S.W. Fast 100-nm resolution three-dimensional microscope reveals structural plasticity of mitochondria in live yeast. Proc. Natl. Acad. Sci. USA 2002, 99, 3370–3375. [Google Scholar] [CrossRef]
- Huang, B.; Bates, M.; Zhuang, X. Super-Resolution Fluorescence Microscopy. Annu. Rev. Biochem. 2009, 78, 993–1016. [Google Scholar] [CrossRef]
- Hell, S.W. Far-Field Optical Nanoscopy. Science 2007, 316, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Schermelleh, L.; Ferrand, A.; Huser, T.; Eggeling, C.; Sauer, M.; Biehlmaier, O.; Drummen, G.P.C. Super-resolution microscopy demystified. Nat. Cell Biol. 2019, 21, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Appelhans, T.; Richter, C.P.; Wilkens, V.; Hess, S.T.; Piehler, J.; Busch, K.B. Nanoscale Organization of Mitochondrial Microcompartments Revealed by Combining Tracking and Localization Microscopy. Nano Lett. 2012, 12, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Gong, W.; Yang, Z.; Pan, W.; Verwilst, P.; Shin, J.; Yan, W.; Liu, L.; Qu, J.; Kim, J.S. STORM imaging of mitochondrial dynamics using a vicinal-dithiol-proteins-targeted probe. Biomaterials 2020, 243, 119938. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Jones, S.A.; Brandenburg, B.; Zhuang, X. Whole-cell 3D STORM reveals interactions between cellular structures with nanometer-scale resolution. Nat. Methods 2008, 5, 1047–1052. [Google Scholar] [CrossRef]
- Yang, X.; Yang, Z.; Wu, Z.; He, Y.; Shan, C.; Chai, P.; Ma, C.; Tian, M.; Teng, J.; Jin, D.; et al. Mitochondrial dynamics quantitatively revealed by STED nanoscopy with an enhanced squaraine variant probe. Nat. Commun. 2020, 11, 3699. [Google Scholar] [CrossRef]
- Vicidomini, G.; Bianchini, P.; Diaspro, A. STED super-resolved microscopy. Nat. Methods 2018, 15, 173–182. [Google Scholar] [CrossRef]
- Dong, L.-F.; Kovarova, J.; Bajzikova, M.; Bezawork-Geleta, A.; Svec, D.; Endaya, B.; Sachaphibulkij, K.; Coelho, A.R.; Sebkova, N.; Ruzickova, A.; et al. Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. eLife 2017, 6, e22187. [Google Scholar] [CrossRef]
- Samanta, S.; He, Y.; Sharma, A.; Kim, J.; Pan, W.; Yang, Z.; Li, J.; Yan, W.; Liu, L.; Qu, J.; et al. Fluorescent Probes for Nanoscopic Imaging of Mitochondria. Chem 2019, 5, 1697–1726. [Google Scholar] [CrossRef]
- Ilgen, P.; Stoldt, S.; Conradi, L.-C.; Wurm, C.A.; Rüschoff, J.; Ghadimi, B.M.; Liersch, T.; Jakobs, S. STED Super-Resolution Microscopy of Clinical Paraffin-Embedded Human Rectal Cancer Tissue. PLoS ONE 2014, 9, e101563. [Google Scholar] [CrossRef]
- Tehrani, K.F.; Pendleton, E.G.; Southern, W.M.; Call, J.A.; Mortensen, L.J. Two-photon deep-tissue spatially resolved mitochondrial imaging using membrane potential fluorescence fluctuations. Biomed. Opt. Express 2018, 9, 254–259. [Google Scholar] [CrossRef]
- Pouli, D.; Balu, M.; Alonzo, C.A.; Liu, Z.; Quinn, K.P.; Rius-Diaz, F.; Harris, R.M.; Kelly, K.M.; Tromberg, B.J.; Georgakoudi, I. Imaging mitochondrial dynamics in human skin reveals depth-dependent hypoxia and malignant potential for diagnosis. Sci. Transl. Med. 2016, 8, ra169–ra367. [Google Scholar] [CrossRef] [PubMed]
- Xylas, J.; Varone, A.; Quinn, K.P.; Pouli, D.; McLaughlin-Drubin, M.E.; Thieu, H.-T.; Garcia-Moliner, M.L.; House, M.; Hunter, M.; Munger, K.; et al. Noninvasive assessment of mitochondrial organization in three-dimensional tissues reveals changes associated with cancer development. Int. J. Cancer 2015, 136, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Pouli, D.; Alonzo, C.A.; Varone, A.; Karaliota, S.; Quinn, K.P.; Münger, K.; Karalis, K.P.; Georgakoudi, I. Mapping metabolic changes by noninvasive, multiparametric, high-resolution imaging using endogenous contrast. Sci. Adv. 2018, 4, eaap9302. [Google Scholar] [CrossRef] [PubMed]
- Stuntz, E.; Gong, Y.; Sood, D.; Liaudanskaya, V.; Pouli, D.; Quinn, K.P.; Alonzo, C.; Liu, Z.; Kaplan, D.L.; Georgakoudi, I. Endogenous Two-Photon Excited Fluorescence Imaging Characterizes Neuron and Astrocyte Metabolic Responses to Manganese Toxicity. Sci. Rep. 2017, 7, 1041. [Google Scholar] [CrossRef]
- Li, H.; Yu, J.; Zhang, R.; Li, X.; Zheng, W. Two-photon excitation fluorescence lifetime imaging microscopy: A promising diagnostic tool for digestive tract tumors. J. Innov. Opt. Health Sci. 2019, 12, 1930009. [Google Scholar] [CrossRef]
- Adur, J.; Pelegati, V.B.; Bianchi, M.; de Thomaz, A.A.; Baratti, M.O.; Carvalho, H.F.; Casco, V.H.; Cesar, C.L. Multimodal Nonlinear Optical Microscopy Used to Discriminate Human Colon Cancer. In Multiphoton Microscopy in the Biomedical Sciences XIII; SPIE: Cergy Pontoise, France, 2013; pp. 254–261. [Google Scholar]
- Rück, A.; Hauser, C.; Mosch, S.; Kalinina, S. Spectrally resolved fluorescence lifetime imaging to investigate cell metabolism in malignant and nonmalignant oral mucosa cells. J. Biomed. Opt. 2014, 19, 96005. [Google Scholar] [CrossRef]
- Palade, G.E. The fine structure of mitochondria. Anat. Rec. 1952, 114, 427–451. [Google Scholar] [CrossRef]
- Hackenbrock, C.R. Ultrastructural bases for metabolically linked mechanical activity in mitochondria. I. Reversible ultrastructural changes with change in metabolic steady state in isolated liver mitochondria. J. Cell Biol. 1966, 30, 269–297. [Google Scholar] [CrossRef]
- Malka, F.; Lombès, A.; Rojo, M. Organisation et dynamique du compartiment mitochondrial. Morphologie 2004, 88, 13–18. [Google Scholar] [CrossRef]
- Benard, G.; Bellance, N.; James, D.; Parrone, P.; Fernandez, H.; Letellier, T.; Rossignol, R. Mitochondrial bioenergetics and structural network organization. J. Cell Sci. 2007, 120 Pt 5, 838–848. [Google Scholar] [CrossRef] [PubMed]
- Dimmer, K.S.; Scorrano, L. (De)constructing mitochondria: What for? Physiology 2006, 21, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Mehard, C.W.; Packer, L.; Abraham, S. Activity and ultrastructure of mitochondria from mouse mammary gland and mammary adenocarcinoma. Cancer Res. 1971, 31, 2148–2160. [Google Scholar]
- Kolosov, A.E.; Chernina, L.A.; Bychkov, V.M. Ultrastructural characteristics of endometrioid and serous ovarian adenocarcinomas. Arkh Patol. 1983, 45, 34–38. [Google Scholar] [PubMed]
- Andrews, P.A.; Albright, K.D. Mitochondrial defects in cis-diamminedichloroplatinum(II)-resistant human ovarian carcinoma cells. Cancer Res. 1992, 52, 1895–1901. [Google Scholar]
- Nicolescu, P.G.; Eskenasy, A. Electronmicroscopic observations on epidermoid (squamous cell) carcinomas of the lung. Morphol. Embryol. 1984, 30, 131–135. [Google Scholar]
- Arismendi-Morillo, G. Electron microscopy morphology of the mitochondrial network in human cancer. Int. J. Biochem. Cell Biol. 2009, 41, 2062–2068. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Ayres, M.; Taylor, J.A., 3rd. Metabolic changes in bladder cancer. Urol. Oncol. 2018, 36, 327–337. [Google Scholar] [CrossRef]
- Signorile, A.; De Rasmo, D.; Cormio, A.; Musicco, C.; Rossi, R.; Fortarezza, F.; Palese, L.L.; Loizzi, V.; Resta, L.; Scillitani, G.; et al. Human Ovarian Cancer Tissue Exhibits Increase of Mitochondrial Biogenesis and Cristae Remodeling. Cancers 2019, 11, 1350. [Google Scholar] [CrossRef]
- Ricci, F.; Corbelli, A.; Affatato, R.; Chilà, R.; Chiappa, M.; Brunelli, L.; Fruscio, R.; Pastorelli, R.; Fiordaliso, F.; Damia, G. Mitochondrial structural alterations in ovarian cancer patient-derived xenografts resistant to cisplatin. Am. J. Cancer Res. 2021, 11, 2303–2311. [Google Scholar]
- Ricci, F.; Brunelli, L.; Affatato, R.; Chilà, R.; Verza, M.; Indraccolo, S.; Falcetta, F.; Fratelli, M.; Fruscio, R.; Pastorelli, R.; et al. Overcoming platinum-acquired resistance in ovarian cancer patient-derived xenografts. Ther. Adv. Med. Oncol. 2019, 11, 1758835919839543. [Google Scholar] [CrossRef] [PubMed]
- Frank, J. Introduction: Principles of Electron Tomography. In Electron Tomography: Methods for Three-Dimensional Visualization of Structures in the Cell; Frank, J., Ed.; Springer: New York, NY, USA, 2006; pp. 1–15. [Google Scholar]
- Frey, T.G.; Renken, C.W.; Perkins, G.A. Insight into mitochondrial structure and function from electron tomography. Biochim. Biophys. Acta (BBA)—Bioenerg. 2002, 1555, 196–203. [Google Scholar] [CrossRef]
- Gan, L.; Jensen, G.J. Electron tomography of cells. Q. Rev. Biophys. 2012, 45, 27–56. [Google Scholar] [CrossRef] [PubMed]
- Perkins, G.; Renken, C.; Martone, M.E.; Young, S.J.; Ellisman, M.; Frey, T. Electron tomography of neuronal mitochondria: Three-dimensional structure and organization of cristae and membrane contacts. J. Struct. Biol. 1997, 119, 260–272. [Google Scholar] [CrossRef]
- Perkins, G.A.; Renken, C.W.; van der Klei, I.J.; Ellisman, M.H.; Neupert, W.; Frey, T.G. Electron tomography of mitochondria after the arrest of protein import associated with Tom19 depletion. Eur. J. Cell Biol. 2001, 80, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Perkins, G.A.; Song, J.Y.; Tarsa, L.; Deerinck, T.J.; Ellisman, M.H.; Frey, T.G. Electron Tomography of Mitochondria from Brown Adipocytes Reveals Crista Junctions. J. Bioenerg. Biomembr. 1998, 30, 431–442. [Google Scholar] [CrossRef]
- Sun, M.G.; Williams, J.; Munoz-Pinedo, C.; Perkins, G.A.; Brown, J.M.; Ellisman, M.H.; Green, D.R.; Frey, T.G. Correlated three-dimensional light and electron microscopy reveals transformation of mitochondria during apoptosis. Nat. Cell Biol. 2007, 9, 1057–1065. [Google Scholar] [CrossRef]
- Moscheni, C.; Malucelli, E.; Castiglioni, S.; Procopio, A.; De Palma, C.; Sorrentino, A.; Sartori, P.; Locatelli, L.; Pereiro, E.; Maier, J.A.; et al. 3D Quantitative and Ultrastructural Analysis of Mitochondria in a Model of Doxorubicin Sensitive and Resistant Human Colon Carcinoma Cells. Cancers 2019, 11, 1254. [Google Scholar] [CrossRef]
- Narayan, K.; Danielson, C.M.; Lagarec, K.; Lowekamp, B.C.; Coffman, P.; Laquerre, A.; Phaneuf, M.W.; Hope, T.J.; Subramaniam, S. Multi-resolution correlative focused ion beam scanning electron microscopy: Applications to cell biology. J. Struct. Biol. 2014, 185, 278–284. [Google Scholar] [CrossRef]
- Narayan, K.; Subramaniam, S. Focused ion beams in biology. Nat. Methods 2015, 12, 1021–1031. [Google Scholar] [CrossRef]
- Knott, G.; Rosset, S.; Cantoni, M. Focussed ion beam milling and scanning electron microscopy of brain tissue. J. Vis. Exp. 2011, 53, e2588. [Google Scholar]
- Heymann, J.A.; Hayles, M.; Gestmann, I.; Giannuzzi, L.A.; Lich, B.; Subramaniam, S. Site-specific 3D imaging of cells and tissues with a dual beam microscope. J. Struct. Biol. 2006, 155, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Schertel, A.; Snaidero, N.; Han, H.M.; Ruhwedel, T.; Laue, M.; Grabenbauer, M.; Möbius, W. Cryo FIB-SEM: Volume imaging of cellular ultrastructure in native frozen specimens. J. Struct. Biol. 2013, 184, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Bushby, A.J.; P’ng, K.M.; Young, R.D.; Pinali, C.; Knupp, C.; Quantock, A.J. Imaging three-dimensional tissue architectures by focused ion beam scanning electron microscopy. Nat. Protoc. 2011, 6, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G.E.; Lowekamp, B.C.; Zerfas, P.M.; Chandler, R.J.; Narasimha, R.; Venditti, C.P.; Subramaniam, S. Ion-abrasion scanning electron microscopy reveals distorted liver mitochondrial morphology in murine methylmalonic acidemia. J. Struct. Biol. 2010, 171, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Harwig, M.C.; Viana, M.P.; Egner, J.M.; Harwig, J.J.; Widlansky, M.E.; Rafelski, S.M.; Hill, R.B. Methods for imaging mammalian mitochondrial morphology: A prospective on MitoGraph. Anal. Biochem. 2018, 552, 81–99. [Google Scholar] [CrossRef]
- Villa, A.M.; Doglia, S.M. Mitochondria in tumor cells studied by laser scanning confocal microscopy. J. Biomed. Opt. 2004, 9, 385–394. [Google Scholar] [CrossRef]
- Jakobs, S.; Wurm, C.A. Super-resolution microscopy of mitochondria. Curr. Opin. Chem. Biol. 2014, 20, 9–15. [Google Scholar] [CrossRef]
- Miao, F.; Zhang, W.; Sun, Y.; Zhang, R.; Liu, Y.; Guo, F.; Song, G.; Tian, M.; Yu, X. Novel fluorescent probes for highly selective two-photon imaging of mitochondria in living cells. Biosens. Bioelectron. 2014, 55, 423–429. [Google Scholar] [CrossRef]
- Huang, S.; Heikal, A.A.; Webb, W.W. Two-photon fluorescence spectroscopy and microscopy of NAD(P)H and flavoprotein. Biophys. J. 2002, 82, 2811–2825. [Google Scholar] [CrossRef]
- Bartesaghi, A.; Sprechmann, P.; Liu, J.; Randall, G.; Sapiro, G.; Subramaniam, S. Classification and 3D averaging with missing wedge correction in biological electron tomography. J. Struct. Biol. 2008, 162, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Cantele, F.; Zampighi, L.; Radermacher, M.; Zampighi, G.; Lanzavecchia, S. Local refinement: An attempt to correct for shrinkage and distortion in electron tomography. J. Struct. Biol. 2007, 158, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Lučić, V.; Rigort, A.; Baumeister, W. Cryo-electron tomography: The challenge of doing structural biology in situ. J. Cell Biol. 2013, 202, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Venegas, V.; Wang, J.; Dimmock, D.; Wong, L.J. Real-time quantitative PCR analysis of mitochondrial DNA content. Curr. Protoc. Hum. Genet. 2011, 68, 19.7.1–19.7.12. [Google Scholar] [CrossRef]
- Rooney, J.P.; Ryde, I.T.; Sanders, L.H.; Howlett, E.H.; Colton, M.D.; Germ, K.E.; Mayer, G.D.; Greenamyre, J.T.; Meyer, J.N. PCR based determination of mitochondrial DNA copy number in multiple species. Methods Mol. Biol. 2015, 1241, 23–38. [Google Scholar]
- Rolo, A.P.; Palmeira, C.M. Diabetes and mitochondrial function: Role of hyperglycemia and oxidative stress. Toxicol. Appl. Pharmacol. 2006, 212, 167–178. [Google Scholar] [CrossRef]
- Yu, M. Generation, function and diagnostic value of mitochondrial DNA copy number alterations in human cancers. Life Sci. 2011, 89, 65–71. [Google Scholar] [CrossRef]
- Coskun, P.; Wyrembak, J.; Schriner, S.E.; Chen, H.W.; Marciniack, C.; Laferla, F.; Wallace, D.C. A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim. Biophys. Acta 2012, 1820, 553–564. [Google Scholar] [CrossRef]
- Furda, A.M.; Bess, A.S.; Meyer, J.N.; Van Houten, B. Analysis of DNA damage and repair in nuclear and mitochondrial DNA of animal cells using quantitative PCR. Methods Mol. Biol. 2012, 920, 111–132. [Google Scholar]
- Shanske, S.; Wong, L.J. Molecular analysis for mitochondrial DNA disorders. Mitochondrion 2004, 4, 403–415. [Google Scholar] [CrossRef]
- Hunter, S.E.; Jung, D.; Di Giulio, R.T.; Meyer, J.N. The QPCR assay for analysis of mitochondrial DNA damage, repair, and relative copy number. Methods 2010, 51, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Furda, A.; Santos, J.H.; Meyer, J.N.; Van Houten, B. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol. Biol. 2014, 1105, 419–437. [Google Scholar] [PubMed]
- Wang, W.; Esbensen, Y.; Scheffler, K.; Eide, L. Analysis of Mitochondrial DNA and RNA Integrity by a Real-Time qPCR-Based Method. In Mitochondrial Medicine: Volume I, Probing Mitochondrial Function; Weissig, V., Edeas, M., Eds.; Springer: New York, NY, USA, 2015; pp. 97–106. [Google Scholar]
- Nadalutti, C.A.; Ayala-Peña, S.; Santos, J.H. Mitochondrial DNA damage as driver of cellular outcomes. Am. J. Physiol. Cell Physiol. 2022, 322, C136–C150. [Google Scholar] [CrossRef] [PubMed]
- Mandavilli, B.S.; Santos, J.H.; Van Houten, B. Mitochondrial DNA repair and aging. Mutat. Res. 2002, 509, 127–151. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.K.; Wong, L.J. Detection and quantification of heteroplasmic mutant mitochondrial DNA by real-time amplification refractory mutation system quantitative PCR analysis: A single-step approach. Clin. Chem. 2004, 50, 996–1001. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.K.; Wong, L.J. Simultaneous detection and quantification of mitochondrial DNA deletion(s), depletion, and over-replication in patients with mitochondrial disease. J. Mol. Diagn. 2005, 7, 613–622. [Google Scholar] [CrossRef]
- Dimmock, D.; Tang, L.Y.; Schmitt, E.S.; Wong, L.J. Quantitative evaluation of the mitochondrial DNA depletion syndrome. Clin. Chem. 2010, 56, 1119–1127. [Google Scholar] [CrossRef]
- Chiang, J.L.; Shukla, P.; Pagidas, K.; Ahmed, N.S.; Karri, S.; Gunn, D.D.; Hurd, W.W.; Singh, K.K. Mitochondria in Ovarian Aging and Reproductive Longevity. Ageing Res. Rev. 2020, 63, 101168. [Google Scholar] [CrossRef]
- Shukla, P.; Singh, K.K. The mitochondrial landscape of ovarian cancer: Emerging insights. Carcinogenesis 2021, 42, 663–671. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, V.W.; Xue, W.C.; Cheung, A.N.; Ngan, H.Y. Association of decreased mitochondrial DNA content with ovarian cancer progression. Br. J. Cancer 2006, 95, 1087–1091. [Google Scholar] [CrossRef]
- Bindra, S.; McGill, M.A.; Triplett, M.K.; Tyagi, A.; Thaker, P.H.; Dahmoush, L.; Goodheart, M.J.; Ogden, R.T.; Owusu-Ansah, E.; Karan, R.K.; et al. Mitochondria in epithelial ovarian carcinoma exhibit abnormal phenotypes and blunted associations with biobehavioral factors. Sci. Rep. 2021, 11, 11595. [Google Scholar] [CrossRef] [PubMed]
- Kleih, M.; Böpple, K.; Dong, M.; Gaißler, A.; Heine, S.; Olayioye, M.A.; Aulitzky, W.E.; Essmann, F. Direct impact of cisplatin on mitochondria induces ROS production that dictates cell fate of ovarian cancer cells. Cell Death Dis. 2019, 10, 851. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Schumaker, L.M.; Egorin, M.J.; Zuhowski, E.G.; Guo, Z.; Cullen, K.J. Cisplatin Preferentially Binds Mitochondrial DNA and Voltage-Dependent Anion Channel Protein in the Mitochondrial Membrane of Head and Neck Squamous Cell Carcinoma: Possible Role in Apoptosis. Clin. Cancer Res. 2006, 12, 5817–5825. [Google Scholar] [CrossRef] [PubMed]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS ONE 2013, 8, e81162. [Google Scholar] [CrossRef]
- Park, S.Y.; Chang, I.; Kim, J.-Y.; Kang, S.W.; Park, S.-H.; Singh, K.; Lee, M.-S. Resistance of Mitochondrial DNA-depleted Cells against Cell Death: ROLE OF MITOCHONDRIAL SUPEROXIDE DISMUTASE *. J. Biol. Chem. 2004, 279, 7512–7520. [Google Scholar] [CrossRef]
- Montopoli, M.; Bellanda, M.; Lonardoni, F.; Ragazzi, E.; Dorigo, P.; Froldi, G.; Mammi, S.; Caparrotta, L. “Metabolic reprogramming” in ovarian cancer cells resistant to cisplatin. Curr. Cancer Drug Targets 2011, 11, 226–235. [Google Scholar] [CrossRef]
- Cocetta, V.; Ragazzi, E.; Montopoli, M. Mitochondrial Involvement in Cisplatin Resistance. Int. J. Mol. Sci. 2019, 20, 3384. [Google Scholar] [CrossRef]
- Mei, H.; Sun, S.; Bai, Y.; Chen, Y.; Chai, R.; Li, H. Reduced mtDNA copy number increases the sensitivity of tumor cells to chemotherapeutic drugs. Cell Death Dis. 2015, 6, e1710. [Google Scholar] [CrossRef]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hiddessen, A.L.; Legler, T.C.; et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef]
- Miotke, L.; Lau, B.T.; Rumma, R.T.; Ji, H.P. High sensitivity detection and quantitation of DNA copy number and single nucleotide variants with single color droplet digital PCR. Anal. Chem. 2014, 86, 2618–2624. [Google Scholar] [CrossRef]
- Bhat, S.; Herrmann, J.; Armishaw, P.; Corbisier, P.; Emslie, K.R. Single molecule detection in nanofluidic digital array enables accurate measurement of DNA copy number. Anal. Bioanal. Chem. 2009, 394, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Whale, A.S.; Huggett, J.F.; Cowen, S.; Speirs, V.; Shaw, J.; Ellison, S.; Foy, C.A.; Scott, D.J. Comparison of microfluidic digital PCR and conventional quantitative PCR for measuring copy number variation. Nucleic Acids Res. 2012, 40, e82. [Google Scholar] [CrossRef]
- Laurie, M.T.; Bertout, J.A.; Taylor, S.D.; Burton, J.N.; Shendure, J.A.; Bielas, J.H. Simultaneous digital quantification and fluorescence-based size characterization of massively parallel sequencing libraries. Biotechniques 2013, 55, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Memon, A.A.; Zöller, B.; Hedelius, A.; Wang, X.; Stenman, E.; Sundquist, J.; Sundquist, K. Quantification of mitochondrial DNA copy number in suspected cancer patients by a well optimized ddPCR method. BioMol. Detect. Quantif. 2017, 13, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Shen, S.; Jiang, H.; Chen, Z. Application of Digital PCR in Detecting Human Diseases Associated Gene Mutation. Cell Physiol. Biochem. 2017, 43, 1718–1730. [Google Scholar] [CrossRef] [PubMed]
- Filograna, R.; Mennuni, M.; Alsina, D.; Larsson, N.G. Mitochondrial DNA copy number in human disease: The more the better? FEBS Lett. 2021, 595, 976–1002. [Google Scholar] [CrossRef]
- O’Hara, R.; Tedone, E.; Ludlow, A.; Huang, E.; Arosio, B.; Mari, D.; Shay, J.W. Quantitative mitochondrial DNA copy number determination using droplet digital PCR with single-cell resolution. Genome Res. 2019, 29, 1878–1888. [Google Scholar] [CrossRef]
- Taylor, S.D.; Ericson, N.G.; Burton, J.N.; Prolla, T.A.; Silber, J.R.; Shendure, J.; Bielas, J.H. Targeted enrichment and high-resolution digital profiling of mitochondrial DNA deletions in human brain. Aging Cell 2014, 13, 29–38. [Google Scholar] [CrossRef]
- Li, Y.; Sundquist, K.; Wang, X.; Zhang, N.; Hedelius, A.; Sundquist, J.; Memon, A.A. Association of Mitochondrial DNA Copy Number and Telomere Length with Prevalent and Incident Cancer and Cancer Mortality in Women: A Prospective Swedish Population-Based Study. Cancers 2021, 13, 3842. [Google Scholar] [CrossRef]
- Hu, L.; Yao, X.; Shen, Y. Altered mitochondrial DNA copy number contributes to human cancer risk: Evidence from an updated meta-analysis. Sci. Rep. 2016, 6, 35859. [Google Scholar] [CrossRef]
- Chen, N.; Wen, S.; Sun, X.; Fang, Q.; Huang, L.; Liu, S.; Li, W.; Qiu, M. Elevated Mitochondrial DNA Copy Number in Peripheral Blood and Tissue Predict the Opposite Outcome of Cancer: A Meta-Analysis. Sci. Rep. 2016, 6, 37404. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A. Metabolic control of cancer cell stemness: Lessons from iPS cells. Cell Cycle 2015, 14, 3801–3811. [Google Scholar] [CrossRef] [PubMed]
- Kawada, K.; Toda, K.; Sakai, Y. Targeting metabolic reprogramming in KRAS-driven cancers. Int. J. Clin. Oncol. 2017, 22, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.S. Digital Assays Part I: Partitioning Statistics and Digital PCR. SLAS Technol. 2017, 22, 369–386. [Google Scholar] [CrossRef]
- Coulter, S.J. Mitigation of the effect of variability in digital PCR assays through use of duplexed reference assays for normalization. Biotechniques 2018, 65, 86–91. [Google Scholar] [CrossRef]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- O’Brien, C.G.; Ozen, M.O.; Ikeda, G.; Vaskova, E.; Jung, J.H.; Bayardo, N.; Santoso, M.R.; Shi, L.; Wahlquist, C.; Jiang, Z.; et al. Mitochondria-Rich Extracellular Vesicles Rescue Patient-Specific Cardiomyocytes from Doxorubicin Injury: Insights into the SENECA Trial. JACC CardioOncol. 2021, 3, 428–440. [Google Scholar] [CrossRef]
- Tian, W.; Lei, N.; Zhou, J.; Chen, M.; Guo, R.; Qin, B.; Li, Y.; Chang, L. Extracellular vesicles in ovarian cancer chemoresistance, metastasis, and immune evasion. Cell Death Dis. 2022, 13, 64. [Google Scholar] [CrossRef]
- Cocucci, E.; Meldolesi, J. Ectosomes and exosomes: Shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015, 25, 364–372. [Google Scholar] [CrossRef]
- Sinawang, P.D.; Soto, F.; Ozen, M.O.; Akin, D.; Demirci, U. Progress and challenges in biomarker enrichment for cancer early detection. Prog. Biomed. Eng. 2021, 3, 043001. [Google Scholar] [CrossRef]
- Ryu, K.J.; Lee, J.Y.; Park, C.; Cho, D.; Kim, S.J. Isolation of Small Extracellular Vesicles from Human Serum Using a Combination of Ultracentrifugation with Polymer-Based Precipitation. Ann. Lab. Med. 2020, 40, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Fortunato, D.; Giannoukakos, S.; Giménez-Capitán, A.; Hackenberg, M.; Molina-Vila, M.A.; Zarovni, N. Selective isolation of extracellular vesicles from minimally processed human plasma as a translational strategy for liquid biopsies. Biomark. Res. 2022, 10, 57. [Google Scholar] [CrossRef] [PubMed]
- Carnino, J.M.; Lee, H.; Jin, Y. Isolation and characterization of extracellular vesicles from Broncho-alveolar lavage fluid: A review and comparison of different methods. Respir. Res. 2019, 20, 240. [Google Scholar] [CrossRef] [PubMed]
- Boukouris, S.; Mathivanan, S. Exosomes in bodily fluids are a highly stable resource of disease biomarkers. Proteom. Clin. Appl. 2015, 9, 358–367. [Google Scholar] [CrossRef]
- Xu, R.; Rai, A.; Chen, M.; Suwakulsiri, W.; Greening, D.W.; Simpson, R.J. Extracellular vesicles in cancer—Implications for future improvements in cancer care. Nat. Rev. Clin. Oncol. 2018, 15, 617–638. [Google Scholar] [CrossRef]
- Nawaz, M.; Camussi, G.; Valadi, H.; Nazarenko, I.; Ekström, K.; Wang, X.; Principe, S.; Shah, N.; Ashraf, N.M.; Fatima, F.; et al. The emerging role of extracellular vesicles as biomarkers for urogenital cancers. Nat. Rev. Urol. 2014, 11, 688–701. [Google Scholar] [CrossRef]
- Lee, Y.; Ni, J.; Beretov, J.; Wasinger, V.C.; Graham, P.; Li, Y. Recent advances of small extracellular vesicle biomarkers in breast cancer diagnosis and prognosis. Mol. Cancer 2023, 22, 33. [Google Scholar] [CrossRef]
- Zhang, H.; Freitas, D.; Kim, H.S.; Fabijanic, K.; Li, Z.; Chen, H.; Mark, M.T.; Molina, H.; Martin, A.B.; Bojmar, L.; et al. Identification of distinct nanoparticles and subsets of extracellular vesicles by asymmetric flow field-flow fractionation. Nat. Cell Biol. 2018, 20, 332–343. [Google Scholar] [CrossRef]
- Brennan, K.; Martin, K.; FitzGerald, S.P.; O’Sullivan, J.; Wu, Y.; Blanco, A.; Richardson, C.; Mc Gee, M.M. A comparison of methods for the isolation and separation of extracellular vesicles from protein and lipid particles in human serum. Sci. Rep. 2020, 10, 1039. [Google Scholar] [CrossRef]
- Wang, J.; Ma, P.; Kim, D.H.; Liu, B.F.; Demirci, U. Towards Microfluidic-Based Exosome Isolation and Detection for Tumor Therapy. Nano Today 2021, 37, 101066. [Google Scholar] [CrossRef]
- Liu, F.; Vermesh, O.; Mani, V.; Ge, T.J.; Madsen, S.J.; Sabour, A.; Hsu, E.C.; Gowrishankar, G.; Kanada, M.; Jokerst, J.V.; et al. The Exosome Total Isolation Chip. ACS Nano 2017, 11, 10712–10723. [Google Scholar] [CrossRef] [PubMed]
- Lobb, R.J.; Becker, M.; Wen, S.W.; Wong, C.S.; Wiegmans, A.P.; Leimgruber, A.; Möller, A. Optimized exosome isolation protocol for cell culture supernatant and human plasma. J. Extracell. Vesicles 2015, 4, 27031. [Google Scholar] [CrossRef] [PubMed]
- Lane, R.E.; Korbie, D.; Hill, M.M.; Trau, M. Extracellular vesicles as circulating cancer biomarkers: Opportunities and challenges. Clin. Transl. Med. 2018, 7, 14. [Google Scholar] [CrossRef]
- Zeng, Y.; Qiu, Y.; Jiang, W.; Shen, J.; Yao, X.; He, X.; Li, L.; Fu, B.; Liu, X. Biological Features of Extracellular Vesicles and Challenges. Front. Cell Dev. Biol. 2022, 10, 816698. [Google Scholar] [CrossRef]
- Livshits, M.A.; Khomyakova, E.; Evtushenko, E.G.; Lazarev, V.N.; Kulemin, N.A.; Semina, S.E.; Generozov, E.V.; Govorun, V.M. Isolation of exosomes by differential centrifugation: Theoretical analysis of a commonly used protocol. Sci. Rep. 2015, 5, 17319. [Google Scholar] [CrossRef] [PubMed]
- Nordin, J.Z.; Lee, Y.; Vader, P.; Mäger, I.; Johansson, H.J.; Heusermann, W.; Wiklander, O.P.; Hällbrink, M.; Seow, Y.; Bultema, J.J.; et al. Ultrafiltration with size-exclusion liquid chromatography for high yield isolation of extracellular vesicles preserving intact biophysical and functional properties. Nanomedicine 2015, 11, 879–883. [Google Scholar] [CrossRef]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef]
- Reggiardo, R.E.; Maroli, S.V.; Halasz, H.; Ozen, M.; Hrabeta-Robinson, E.; Behera, A.; Peddu, V.; Carrillo, D.; LaMontagne, E.; Whitehead, L.; et al. Mutant KRAS regulates transposable element RNA and innate immunity via KRAB zinc-finger genes. Cell Rep. 2022, 40, 111104. [Google Scholar] [CrossRef]
- Huang, X.; Yuan, T.; Liang, M.; Du, M.; Xia, S.; Dittmar, R.; Wang, D.; See, W.; Costello, B.A.; Quevedo, F.; et al. Exosomal miR-1290 and miR-375 as Prognostic Markers in Castration-resistant Prostate Cancer. Eur. Urol. 2015, 67, 33–41. [Google Scholar] [CrossRef]
- Wang, Y.; Lieberman, R.; Pan, J.; Zhang, Q.; Du, M.; Zhang, P.; Nevalainen, M.; Kohli, M.; Shenoy, N.K.; Meng, H.; et al. miR-375 induces docetaxel resistance in prostate cancer by targeting SEC23A and YAP1. Mol. Cancer 2016, 15, 70. [Google Scholar] [CrossRef]
- Zedan, A.H.; Osther, P.J.S.; Assenholt, J.; Madsen, J.S.; Hansen, T.F. Circulating miR-141 and miR-375 are associated with treatment outcome in metastatic castration resistant prostate cancer. Sci. Rep. 2020, 10, 227. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Shen, H.; Yin, X.; Yang, M.; Wei, H.; Chen, Q.; Feng, F.; Liu, Y.; Xu, W.; Li, Y. Macrophages derived exosomes deliver miR-223 to epithelial ovarian cancer cells to elicit a chemoresistant phenotype. J. Exp. Clin. Cancer Res. 2019, 38, 81. [Google Scholar] [CrossRef] [PubMed]
- Kanlikilicer, P.; Bayraktar, R.; Denizli, M.; Rashed, M.H.; Ivan, C.; Aslan, B.; Mitra, R.; Karagoz, K.; Bayraktar, E.; Zhang, X.; et al. Exosomal miRNA confers chemo resistance via targeting Cav1/p-gp/M2-type macrophage axis in ovarian cancer. EBioMedicine 2018, 38, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Weiner-Gorzel, K.; Dempsey, E.; Milewska, M.; McGoldrick, A.; Toh, V.; Walsh, A.; Lindsay, S.; Gubbins, L.; Cannon, A.; Sharpe, D.; et al. Overexpression of the microRNA miR-433 promotes resistance to paclitaxel through the induction of cellular senescence in ovarian cancer cells. Cancer Med. 2015, 4, 745–758. [Google Scholar] [CrossRef]
- Chen, W.T.; Yang, Y.J.; Zhang, Z.D.; An, Q.; Li, N.; Liu, W.; Yang, B. MiR-1307 promotes ovarian cancer cell chemoresistance by targeting the ING5 expression. J. Ovarian Res. 2017, 10, 1. [Google Scholar] [CrossRef]
- Au Yeung, C.L.; Co, N.N.; Tsuruga, T.; Yeung, T.L.; Kwan, S.Y.; Leung, C.S.; Li, Y.; Lu, E.S.; Kwan, K.; Wong, K.K.; et al. Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat. Commun. 2016, 7, 11150. [Google Scholar] [CrossRef]
- Asante, D.-B.; Calapre, L.; Ziman, M.; Meniawy, T.M.; Gray, E.S. Liquid biopsy in ovarian cancer using circulating tumor DNA and cells: Ready for prime time? Cancer Lett. 2020, 468, 59–71. [Google Scholar] [CrossRef]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar]
- Keserű, J.S.; Soltész, B.; Lukács, J.; Márton, É.; Szilágyi-Bónizs, M.; Penyige, A.; Póka, R.; Nagy, B. Detection of cell-free, exosomal and whole blood mitochondrial DNA copy number in plasma or whole blood of patients with serous epithelial ovarian cancer. J. Biotechnol. 2019, 298, 76–81. [Google Scholar] [CrossRef]
- Möhrmann, L.; Huang, H.J.; Hong, D.S.; Tsimberidou, A.M.; Fu, S.; Piha-Paul, S.A.; Subbiah, V.; Karp, D.D.; Naing, A.; Krug, A.; et al. Liquid Biopsies Using Plasma Exosomal Nucleic Acids and Plasma Cell-Free DNA Compared with Clinical Outcomes of Patients with Advanced Cancers. Clin. Cancer Res. 2018, 24, 181–188. [Google Scholar] [CrossRef]
- Kahlert, C.; Melo, S.A.; Protopopov, A.; Tang, J.; Seth, S.; Koch, M.; Zhang, J.; Weitz, J.; Chin, L.; Futreal, A.; et al. Identification of Double-stranded Genomic DNA Spanning All Chromosomes with Mutated KRAS and p53 DNA in the Serum Exosomes of Patients with Pancreatic Cancer*. J. Biol. Chem. 2014, 289, 3869–3875. [Google Scholar] [CrossRef] [PubMed]
- Thakur, B.K.; Zhang, H.; Becker, A.; Matei, I.; Huang, Y.; Costa-Silva, B.; Zheng, Y.; Hoshino, A.; Brazier, H.; Xiang, J.; et al. Double-stranded DNA in exosomes: A novel biomarker in cancer detection. Cell Res. 2014, 24, 766–769. [Google Scholar] [CrossRef] [PubMed]
- Balaj, L.; Lessard, R.; Dai, L.; Cho, Y.-J.; Pomeroy, S.L.; Breakefield, X.O.; Skog, J. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat. Commun. 2011, 2, 180. [Google Scholar] [CrossRef] [PubMed]
- Guescini, M.; Genedani, S.; Stocchi, V.; Agnati, L.F. Astrocytes and Glioblastoma cells release exosomes carrying mtDNA. J. Neural Transm. 2010, 117, 1–4. [Google Scholar] [CrossRef]
- Malkin, E.Z.; Bratman, S.V. Bioactive DNA from extracellular vesicles and particles. Cell Death Dis. 2020, 11, 584. [Google Scholar] [CrossRef]
- Ghanam, J.; Chetty, V.K.; Barthel, L.; Reinhardt, D.; Hoyer, P.-F.; Thakur, B.K. DNA in extracellular vesicles: From evolution to its current application in health and disease. Cell Biosci. 2022, 12, 37. [Google Scholar] [CrossRef]
- Sharma, A.; Johnson, A. Exosome DNA: Critical regulator of tumor immunity and a diagnostic biomarker. J. Cell. Physiol. 2020, 235, 1921–1932. [Google Scholar] [CrossRef]
- Németh, A.; Orgovan, N.; Sódar, B.W.; Osteikoetxea, X.; Pálóczi, K.; Szabó-Taylor, K.É.; Vukman, K.V.; Kittel, Á.; Turiák, L.; Wiener, Z.; et al. Antibiotic-induced release of small extracellular vesicles (exosomes) with surface-associated DNA. Sci. Rep. 2017, 7, 8202. [Google Scholar] [CrossRef]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef]
- Pérez-Treviño, P.; Velásquez, M.; García, N. Mechanisms of mitochondrial DNA escape and its relationship with different metabolic diseases. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2020, 1866, 165761. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Coelho-Junior, H.J.; Bossola, M.; Landi, F.; Bernabei, R.; Bucci, C.; Marzetti, E. Generation and Release of Mitochondrial-Derived Vesicles in Health, Aging and Disease. J. Clin. Med. 2020, 9, 1440. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, A.; Villar-Prados, A.; Oliphint, P.A.; Zhang, J.; Song, X.; De Hoff, P.; Morey, R.; Liu, J.; Roszik, J.; Clise-Dwyer, K.; et al. Mechanisms of nuclear content loading to exosomes. Sci. Adv. 2019, 5, eaax8849. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Okada, R.; Nagao, K.; Kawamata, Y.; Hanyu, A.; Yoshimoto, S.; Takasugi, M.; Watanabe, S.; Kanemaki, M.T.; Obuse, C.; et al. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat. Commun. 2017, 8, 15287. [Google Scholar] [CrossRef]
- Castillo, J.; Bernard, V.; San Lucas, F.A.; Allenson, K.; Capello, M.; Kim, D.U.; Gascoyne, P.; Mulu, F.C.; Stephens, B.M.; Huang, J.; et al. Surfaceome profiling enables isolation of cancer-specific exosomal cargo in liquid biopsies from pancreatic cancer patients. Ann. Oncol. 2018, 29, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Yoon, H.; Park, S.; Kim, J.S.; Ahn, Y.-H.; Kwon, K.; Lee, D.; Kim, K.H. Urinary Exosomal and cell-free DNA Detects Somatic Mutation and Copy Number Alteration in Urothelial Carcinoma of Bladder. Sci. Rep. 2018, 8, 14707. [Google Scholar] [CrossRef]
- NLM. Navtemadlin and Radiation Therapy in Treating Patients with Soft Tissue Sarcoma. 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03217266 (accessed on 10 February 2023).
- NLM. Contents of Circulating Extracellular Vesicles: Biomarkers in Colorectal Cancer Patients (ExoColon). 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04523389 (accessed on 10 February 2023).
- NLM. Detection of Either the EML4-ALK Gene Rearrangements or the T790M EGFR Mutation in the Plasma of Advanced NSCLC Patients. 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03236675 (accessed on 10 February 2023).
- NLM. Olmutinib Trial in T790M (+) NSCLC Patients Detected by Liquid Biopsy Using BALF Extracellular Vesicular DNA. 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03228277 (accessed on 10 February 2023).
- Elzanowska, J.; Berrocal, L.; García-Peláez, B.; Vives-Usano, M.; Sebo, B.P.; Maia, J.; Batista, S.; Teppo, J.; Varjosalo, M.; Moraes, M.C.S.; et al. Defining Optimal Conditions for Tumor Extracellular Vesicle DNA Extraction for Mutation Profiling. Cancers 2022, 14, 3258. [Google Scholar] [CrossRef]
- Liu, H.; Tian, Y.; Xue, C.; Niu, Q.; Chen, C.; Yan, X. Analysis of extracellular vesicle DNA at the single-vesicle level by nano-flow cytometry. J. Extracell. Vesicles 2022, 11, e12206. [Google Scholar] [CrossRef]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Chandel, N.S. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014, 2, 17. [Google Scholar] [CrossRef]
- Duanghathaipornsuk, S.; Farrell, E.J.; Alba-Rubio, A.C.; Zelenay, P.; Kim, D.S. Detection Technologies for Reactive Oxygen Species: Fluorescence and Electrochemical Methods and Their Applications. Biosensors 2021, 11, 30. [Google Scholar] [CrossRef]
- Luo, Y.; Wang, D.; Abbruzzese, J.L.; Lu, W. Measurement of Reactive Oxygen Species by Fluorescent Probes in Pancreatic Cancer Cells. Methods Mol. Biol. 2019, 1882, 207–219. [Google Scholar] [PubMed]
- Kalyanaraman, B.; Cheng, G.; Hardy, M.; Ouari, O.; Bennett, B.; Zielonka, J. Teaching the basics of reactive oxygen species and their relevance to cancer biology: Mitochondrial reactive oxygen species detection, redox signaling, and targeted therapies. Redox Biol. 2018, 15, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xu, X.; Che, D.; Fan, R.; Gao, M.; Cao, Y.; Ge, C.; Feng, Y.; Li, J.; Xie, S.; et al. Reactive Oxygen Species Mediate 6c-Induced Mitochondrial and Lysosomal Dysfunction, Autophagic Cell Death, and DNA Damage in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 10987. [Google Scholar] [CrossRef] [PubMed]
- Tavsan, Z.; Kayali, H.A. Flavonoids showed anticancer effects on the ovarian cancer cells: Involvement of reactive oxygen species, apoptosis, cell cycle and invasion. Biomed. Pharmacother. 2019, 116, 109004. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, S.A.; Gordon, L.I. Inflammation and cancer: Role of phagocyte-generated oxidants in carcinogenesis. Blood 1990, 76, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Babior, B.M.; Woodman, R.C. Chronic granulomatous disease. Semin. Hematol. 1990, 27, 247–259. [Google Scholar] [PubMed]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef] [PubMed]
- Stadtman, E.R. Oxidation of free amino acids and amino acid residues in proteins by radiolysis and by metal-catalyzed reactions. Annu. Rev. Biochem. 1993, 62, 797–821. [Google Scholar] [CrossRef]
- Davies, K.J.; Delsignore, M.E.; Lin, S.W. Protein damage and degradation by oxygen radicals. II. Modification of amino acids. J. Biol. Chem. 1987, 262, 9902–9907. [Google Scholar] [CrossRef]
- Uchida, K.; Kawakishi, S. Site-specific oxidation of angiotensin I by copper(II) and L-ascorbate: Conversion of histidine residues to 2-imidazolones. Arch. Biochem. Biophys. 1990, 283, 20–26. [Google Scholar] [CrossRef]
- Heinecke, J.W.; Li, W.; Daehnke, H.L., 3rd; Goldstein, J.A. Dityrosine, a specific marker of oxidation, is synthesized by the myeloperoxidase-hydrogen peroxide system of human neutrophils and macrophages. J. Biol. Chem. 1993, 268, 4069–4077. [Google Scholar] [CrossRef]
- Climent, I.; Tsai, L.; Levine, R.L. Derivatization of gamma-glutamyl semialdehyde residues in oxidized proteins by fluoresceinamine. Anal. Biochem. 1989, 182, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L. Oxidative modification of glutamine synthetase. I. Inactivation is due to loss of one histidine residue. J. Biol. Chem. 1983, 258, 11823–11827. [Google Scholar] [CrossRef] [PubMed]
- Oliver, C.N. Inactivation of enzymes and oxidative modification of proteins by stimulated neutrophils. Arch. Biochem. Biophys. 1987, 253, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Pognonec, P.; Kato, H.; Roeder, R.G. The helix-loop-helix/leucine repeat transcription factor USF can be functionally regulated in a redox-dependent manner. J. Biol. Chem. 1992, 267, 24563–24567. [Google Scholar] [CrossRef]
- Rivett, A.J. Regulation of intracellular protein turnover: Covalent modification as a mechanism of marking proteins for degradation. Curr. Top Cell Regul. 1986, 28, 291–337. [Google Scholar]
- Wolff, S.P.; Dean, R.T. Fragmentation of proteins by free radicals and its effect on their susceptibility to enzymic hydrolysis. Biochem. J. 1986, 234, 399–403. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M. Role of free radicals and catalytic metal ions in human disease: An overview. Methods EnzyMol. 1990, 186, 1–85. [Google Scholar]
- Mehrabi, S.; Partridge, E.E.; Seffens, W.; Yao, X.; Aikhionbare, F.O. Oxidatively modified proteins in the serous subtype of ovarian carcinoma. Biomed. Res. Int. 2014, 2014, 585083. [Google Scholar] [CrossRef]
- Lardy, H.A.; Ferguson, S.M. Oxidative phosphorylation in mitochondria. Annu. Rev. Biochem. 1969, 38, 991–1034. [Google Scholar] [CrossRef]
- Hatefi, Y. ATP synthesis in mitochondria. Eur. J. Biochem. 1993, 218, 759–767. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.V.; Walsh, M.L.; Bockus, B.J.; Chen, L.B. Monitoring of relative mitochondrial membrane potential in living cells by fluorescence microscopy. J. Cell Biol. 1981, 88, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J.; Ramshesh, V.K. Imaging of mitochondrial polarization and depolarization with cationic fluorophores. Methods Cell Biol. 2007, 80, 283–295. [Google Scholar] [PubMed]
- Liu, X.; Yang, L.; Long, Q.; Weaver, D.; Hajnóczky, G. Choosing proper fluorescent dyes, proteins, and imaging techniques to study mitochondrial dynamics in mammalian cells. Biophys. Rep. 2017, 3, 64–72. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Ward, M.W. Mitochondrial membrane potential and neuronal glutamate excitotoxicity: Mortality and millivolts. Trends Neurosci. 2000, 23, 166–174. [Google Scholar] [CrossRef]
- Solaini, G.; Sgarbi, G.; Lenaz, G.; Baracca, A. Evaluating mitochondrial membrane potential in cells. Biosci. Rep. 2007, 27, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G. Simultaneous monitoring of ionophore- and inhibitor-mediated plasma and mitochondrial membrane potential changes in cultured neurons. J. Biol. Chem. 2006, 281, 14864–14874. [Google Scholar] [CrossRef]
- Perry, S.W.; Norman, J.P.; Barbieri, J.; Brown, E.B.; Gelbard, H.A. Mitochondrial membrane potential probes and the proton gradient: A practical usage guide. Biotechniques 2011, 50, 98–115. [Google Scholar] [CrossRef]
- Badrinath, N.; Yoo, S.Y. Mitochondria in cancer: In the aspects of tumorigenesis and targeted therapy. Carcinogenesis 2018, 39, 1419–1430. [Google Scholar] [CrossRef]
- Mani, S.; Swargiary, G.; Singh, K.K. Natural Agents Targeting Mitochondria in Cancer. Int. J. Mol. Sci. 2020, 21, 6992. [Google Scholar] [CrossRef]
- Zhang, B.B.; Wang, D.G.; Guo, F.F.; Xuan, C. Mitochondrial membrane potential and reactive oxygen species in cancer stem cells. Fam. Cancer 2015, 14, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Forrest, M.D. Why cancer cells have a more hyperpolarised mitochondrial membrane potential and emergent prospects for therapy. bioRxiv 2015. [Google Scholar] [CrossRef]
- Grieco, J.P.; Allen, M.E.; Perry, J.B.; Wang, Y.; Song, Y.; Rohani, A.; Compton, S.L.E.; Smyth, J.W.; Swami, N.S.; Brown, D.A.; et al. Progression-Mediated Changes in Mitochondrial Morphology Promotes Adaptation to Hypoxic Peritoneal Conditions in Serous Ovarian Cancer. Front Oncol. 2020, 10, 600113. [Google Scholar] [CrossRef] [PubMed]
- Heerdt, B.G.; Houston, M.A.; Wilson, A.J.; Augenlicht, L.H. The Intrinsic Mitochondrial Membrane Potential (Δψm) Is Associated with Steady-State Mitochondrial Activity and the Extent to Which Colonic Epithelial Cells Undergo Butyrate-mediated Growth Arrest and Apoptosis1. Cancer Res. 2003, 63, 6311–6319. [Google Scholar] [PubMed]
- MacDonald, J.A.; Kura, N.; Sussman, C.; Woods, D.C. Mitochondrial membrane depolarization enhances TRAIL-induced cell death in adult human granulosa tumor cells, KGN, through inhibition of BIRC5. J. Ovarian Res. 2018, 11, 89. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, Y.; Wang, M.; Lin, X.; Zhang, Y.; Laurent, I.; Zhong, Y.; Li, J. Ampelopsin Inhibits Breast Cancer Cell Growth through Mitochondrial Apoptosis Pathway. Biol. Pharm. Bull. 2021, 44, 1738–1745. [Google Scholar] [CrossRef]
- Liu, Z.; Zhao, L.; Song, Y. Eya2 Is Overexpressed in Human Prostate Cancer and Regulates Docetaxel Sensitivity and Mitochondrial Membrane Potential through AKT/Bcl-2 Signaling. Biomed. Res. Int. 2019, 2019, 3808432. [Google Scholar] [CrossRef]
- Cai, J.; Zhong, Y.Y.; Tian, S. Naturally occurring davanone terpenoid exhibits anticancer potential against ovarian cancer cells by inducing programmed cell death, by inducing caspase-dependent apoptosis, loss of mitochondrial membrane potential, inhibition of cell migration and invasion and targeting PI3K/AKT/MAPK signaling pathway. J. Buon. 2020, 25, 2301–2307. [Google Scholar]
- Singh, T.; Sharma, S.D.; Katiyar, S.K. Grape proanthocyanidins induce apoptosis by loss of mitochondrial membrane potential of human non-small cell lung cancer cells in vitro and in vivo. PLoS ONE 2011, 6, e27444. [Google Scholar] [CrossRef]
- Rickard, B.P.; Overchuk, M.; Obaid, G.; Ruhi, M.K.; Demirci, U.; Fenton, S.E.; Santos, J.H.; Kessel, D.; Rizvi, I. Photochemical Targeting of Mitochondria to Overcome Chemoresistance in Ovarian Cancer †. Photochem. Photobiol. 2022, 99, 448–468. [Google Scholar] [CrossRef] [PubMed]
- Rickard, B.P.; Tan, X.; Fenton, S.E.; Rizvi, I. Photodynamic Priming Overcomes Per- and Polyfluoroalkyl Substance ( PFAS )-Induced Platinum Resistance in Ovarian Cancer †. Photochem. Photobiol. 2022, 99, 793–813. [Google Scholar] [CrossRef]
- Mahalingam, S.M.; Ordaz, J.D.; Low, P.S. Targeting of a Photosensitizer to the Mitochondrion Enhances the Potency of Photodynamic Therapy. ACS Omega 2018, 3, 6066–6074. [Google Scholar] [CrossRef] [PubMed]
- Chiu, S.M.; Oleinick, N.L. Dissociation of mitochondrial depolarization from cytochrome c release during apoptosis induced by photodynamic therapy. Br. J. Cancer 2001, 84, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Kessel, D.; Reiners, J.J. Enhanced efficacy of photodynamic therapy via a sequential targeting protocol. Photochem. Photobiol. 2014, 90, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Kessel, D. Reversible effects of photodamage directed toward mitochondria. Photochem. Photobiol. 2014, 90, 1211–1213. [Google Scholar] [CrossRef]
- Roy, S.S.; Hajnóczky, G. Calcium, mitochondria and apoptosis studied by fluorescence measurements. Methods 2008, 46, 213–223. [Google Scholar] [CrossRef]
- Szalai, G.; Krishnamurthy, R.; Hajnóczky, G. Apoptosis driven by IP3-linked mitochondrial calcium signals. EMBO J. 1999, 18, 6349–6361. [Google Scholar] [CrossRef]
- Jang, S.; Chapa-Dubocq, X.R.; Fossati, S.; Javadov, S. Analysis of Mitochondrial Calcium Retention Capacity in Cultured Cells: Permeabilized Cells Versus Isolated Mitochondria. Front. Physiol. 2021, 12, 773839. [Google Scholar] [CrossRef]
- Halestrap, A.P. Regulation of mitochondrial metabolism through changes in matrix volume. Biochem. Soc. Trans. 1994, 22, 522–529. [Google Scholar] [CrossRef]
- Tarasov, A.I.; Griffiths, E.J.; Rutter, G.A. Regulation of ATP production by mitochondrial Ca2+. Cell Calcium 2012, 52, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Ichas, F.; Mazat, J.-P. From calcium signaling to cell death: Two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochim. Biophys. Acta (BBA)-Bioenerg. 1998, 1366, 33–50. [Google Scholar] [CrossRef]
- Kwong, J.Q.; Molkentin, J.D. Physiological and Pathological Roles of the Mitochondrial Permeability Transition Pore in the Heart. Cell Metab. 2015, 21, 206–214. [Google Scholar] [CrossRef]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium–apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Rasola, A.; Forte, M.; Lippe, G.; Zorov, D.B.; Juhaszova, M.; Sollott, S.J.; Szabo, I.; Zoratti, M.; Delmotte, P.; et al. Mitochondrial Transport of Cations: Channels, Exchangers, and Permeability Transition. Physiol. Rev. 1999, 79, 1127–1155. [Google Scholar] [CrossRef]
- Kim, J.-S.; He, L.; Lemasters, J.J. Mitochondrial permeability transition: A common pathway to necrosis and apoptosis. Biochem. Biophys. Res. Commun. 2003, 304, 463–470. [Google Scholar] [CrossRef]
- Green, D.R.; Kroemer, G. The Pathophysiology of Mitochondrial Cell Death. Science 2004, 305, 626–629. [Google Scholar] [CrossRef]
- Hajnóczky, G.; Robb-Gaspers, L.D.; Seitz, M.B.; Thomas, A.P. Decoding of cytosolic calcium oscillations in the mitochondria. Cell 1995, 82, 415–424. [Google Scholar] [CrossRef]
- Roy, S.S.; Hajnóczky, G. Fluorometric Methods for Detection of Mitochondrial Membrane Permeabilization in Apoptosis. In Apoptosis; Humana: Totowa, NJ, USA, 2009; Volume 559, pp. 173–190. [Google Scholar] [CrossRef]
- Liao, P.-C.; Bergamini, C.; Fato, R.; Pon, L.A.; Pallotti, F. Isolation of mitochondria from cells and tissues. Methods Cell Biol. 2020, 155, 3–31. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Veksler, V.; Gellerich, F.N.; Saks, V.; Margreiter, R.; Kunz, W.S. Analysis of mitochondrial function in situ in permeabilized muscle fibers, tissues and cells. Nat. Protoc. 2008, 3, 965–976. [Google Scholar] [CrossRef]
- Dedkova, E.N.; Blatter, L.A. Measuring mitochondrial function in intact cardiac myocytes. J. Mol. Cell. Cardiol. 2012, 52, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Salabei, J.K.; A Gibb, A.; Hill, B.G. Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nat. Protoc. 2014, 9, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Monteith, G.R.; Davis, F.; Roberts-Thomson, S. Calcium Channels and Pumps in Cancer: Changes and Consequences. J. Biol. Chem. 2012, 287, 31666–31673. [Google Scholar] [CrossRef] [PubMed]
- Prevarskaya, N.; Ouadid-Ahidouch, H.; Skryma, R.; Shuba, Y. Remodelling of Ca2+ transport in cancer: How it contributes to cancer hallmarks? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130097. [Google Scholar] [CrossRef] [PubMed]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Targeting Ca2+transport in cancer: Close reality or long perspective? Expert Opin. Ther. Targets 2013, 17, 225–241. [Google Scholar] [CrossRef] [PubMed]
- Caravia, L.; Staicu, C.E.; Radu, B.M.; Condrat, C.E.; Crețoiu, D.; Bacalbașa, N.; Suciu, N.; Crețoiu, S.M.; Voinea, S.C. Altered Organelle Calcium Transport in Ovarian Physiology and Cancer. Cancers 2020, 12, 2232. [Google Scholar] [CrossRef] [PubMed]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion channels and the hallmarks of cancer. Trends Mol. Med. 2010, 16, 107–121. [Google Scholar] [CrossRef]
- Giorgi, C.; Baldassari, F.; Bononi, A.; Bonora, M.; De Marchi, E.; Marchi, S.; Missiroli, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; et al. Mitochondrial Ca2+ and apoptosis. Cell Calcium 2012, 52, 36–43. [Google Scholar] [CrossRef]
- Hoyerhansen, M.; Jaattela, M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 2007, 14, 1576–1582. [Google Scholar] [CrossRef]
- Li, W.; Zhang, S.-L.; Wang, N.; Zhang, B.-B.; Li, M. Blockade of T-Type Ca2+Channels Inhibits Human Ovarian Cancer Cell Proliferation. Cancer Investig. 2011, 29, 339–346. [Google Scholar] [CrossRef]
- Monteith, G.R.; Prevarskaya, N.; Roberts-Thomson, S.J. The calcium–cancer signalling nexus. Nat. Rev. Cancer 2017, 17, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Selstam, G.; Rosberg, S.; Liljekvist, J.; Grönquist, L.; Perklev, T.; Ahrén, K. Differences in action of lh and fsh on the formation of cyclic amp in the prepubertal rat ovary. Eur. J. Endocrinol. 1976, 81, 150–164. [Google Scholar] [CrossRef] [PubMed]
- Casarini, L.; Crépieux, P. Molecular Mechanisms of Action of FSH. Front. Endocrinol. 2019, 10, 305. [Google Scholar] [CrossRef] [PubMed]
- Dier, U.; Shin, D.-H.; Hemachandra, L.P.M.P.; Uusitalo, L.M.; Hempel, N. Bioenergetic Analysis of Ovarian Cancer Cell Lines: Profiling of Histological Subtypes and Identification of a Mitochondria-Defective Cell Line. PLOS ONE 2014, 9, e98479. [Google Scholar] [CrossRef]
- Andrzejewski, S.; Klimcakova, E.; Johnson, R.M.; Tabariès, S.; Annis, M.G.; McGuirk, S.; Northey, J.J.; Chénard, V.; Sriram, U.; Papadopoli, D.J.; et al. PGC-1α Promotes Breast Cancer Metastasis and Confers Bioenergetic Flexibility against Metabolic Drugs. Cell Metab. 2017, 26, 778–787. [Google Scholar] [CrossRef]
- Li, W.; Xu, M.; Li, Y.; Huang, Z.; Zhou, J.; Zhao, Q.; Le, K.; Dong, F.; Wan, C.; Yi, P. Comprehensive analysis of the association between tumor glycolysis and immune/inflammation function in breast cancer. J. Transl. Med. 2020, 18, 1–12. [Google Scholar] [CrossRef]
- Wu, Z.; Zuo, M.; Zeng, L.; Cui, K.; Liu, B.; Yan, C.; Chen, L.; Dong, J.; Shangguan, F.; Hu, W.; et al. OMA1 reprograms metabolism under hypoxia to promote colorectal cancer development. EMBO Rep. 2021, 22, e50827. [Google Scholar] [CrossRef]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.G.; Wikman, H.; Pantel, K.; Haigis, M.C.; De Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef]
- Martínez, J.; Tarallo, D.; Martínez-Palma, L.; Victoria, S.; Bresque, M.; Rodríguez-Bottero, S.; Marmisolle, I.; Escande, C.; Cassina, P.; Casanova, G.; et al. Mitofusins modulate the increase in mitochondrial length, bioenergetics and secretory phenotype in therapy-induced senescent melanoma cells. Biochem. J. 2019, 476, 2463–2486. [Google Scholar] [CrossRef]
- Papkovsky, D.B.; Zhdanov, A.V. Cell Energy Budget Platform for Multiparametric Assessment of Cell and Tissue Metabolism. In Mitochondrial Medicine; Springer: New York, NY, USA, 2021; Volume 2276, pp. 305–324. [Google Scholar] [CrossRef]
- Zhang, J. Using Seahorse Machine to Measure OCR and ECAR in Cancer Cells. In Cancer Metabolism; Humana: New York, NY, USA, 2019; Volume 1928, pp. 353–363. [Google Scholar] [CrossRef]
- Smolina, N.; Bruton, J.; Kostareva, A.; Sejersen, T. Assaying Mitochondrial Respiration as an Indicator of Cellular Metabolism and Fitness. In Cell Viability Assays; Humana: New York, NY, USA, 2017; Volume 1601, pp. 79–87. [Google Scholar] [CrossRef]
- Iuso, A.; Repp, B.; Biagosch, C.; Terrile, C.; Prokisch, H. Assessing Mitochondrial Bioenergetics in Isolated Mitochondria from Various Mouse Tissues Using Seahorse XF96 Analyzer. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2017; Volume 1567, pp. 217–230. [Google Scholar]
- Shao, Y.; Ye, G.; Ren, S.; Piao, H.-L.; Zhao, X.; Lu, X.; Wang, F.; Ma, W.; Li, J.; Yin, P.; et al. Metabolomics and transcriptomics profiles reveal the dysregulation of the tricarboxylic acid cycle and related mechanisms in prostate cancer. Int. J. Cancer 2018, 143, 396–407. [Google Scholar] [CrossRef]
- Anderson, N.M.; Mucka, P.; Kern, J.G.; Feng, H. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell 2018, 9, 216–237. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Sato, K.; Yamaguchi, M.; Mitamura, K.; Taga, A. Development of simultaneous quantitative analysis of tricarboxylic acid cycle metabolites to identify specific metabolites in cancer cells by targeted metabolomic approach. Biochem. Biophys. Res. Commun. 2021, 584, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Gnaiger, E. Bioenergetics at low oxygen: Dependence of respiration and phosphorylation on oxygen and adenosine diphosphate supply. Respir. Physiol. 2001, 128, 277–297. [Google Scholar] [CrossRef]
- Pesta, D.; Gnaiger, E. High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol. Biol. 2012, 810, 25–58. [Google Scholar]
- Long, Q.; Huang, L.; Huang, K.; Yang, Q. Assessing Mitochondrial Bioenergetics in Isolated Mitochondria from Mouse Heart Tissues Using Oroboros 2k-Oxygraph. In Nuclear Receptors; Humana: New York, NY, USA, 2019; Volume 1966, pp. 237–246. [Google Scholar] [CrossRef]
- Doerrier, C.; Garcia-Souza, L.F.; Krumschnabel, G.; Wohlfarter, Y.; Mészáros, A.T.; Gnaiger, E. High-Resolution Fluorespirometry and Oxphos Protocols for Human Cells, Permeabilized Fibers from Small Biopsies of Muscle, and Isolated Mitochondria. In Mitochondrial Bioenergetics; Humana: New York, NY, USA, 2018; Volume 1782, pp. 31–70. [Google Scholar] [CrossRef] [PubMed]
- Schöpf, B.; Weissensteiner, H.; Schäfer, G.; Fazzini, F.; Charoentong, P.; Naschberger, A.; Rupp, B.; Fendt, L.; Bukur, V.; Giese, I.; et al. OXPHOS remodeling in high-grade prostate cancer involves mtDNA mutations and increased succinate oxidation. Nat. Commun. 2020, 11, 1487. [Google Scholar] [CrossRef] [PubMed]
- Chuang, K.-C.; Chang, C.-R.; Chang, S.-H.; Huang, S.-W.; Chuang, S.-M.; Li, Z.-Y.; Wang, S.-T.; Kao, J.-K.; Chen, Y.-J.; Shieh, J.-J. Imiquimod-induced ROS production disrupts the balance of mitochondrial dynamics and increases mitophagy in skin cancer cells. J. Dermatol. Sci. 2020, 98, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.C.; Carey, K.T.; McKenzie, M. Anti-cancer analogues ME-143 and ME-344 exert toxicity by directly inhibiting mitochondrial NADH: Ubiquinone oxidoreductase (Complex I). Am. J. Cancer Res. 2015, 5, 689–701. [Google Scholar] [PubMed]
- Silva, A.M.; Oliveira, P.J. Evaluation of Respiration with Clark-Type Electrode in Isolated Mitochondria and Permeabilized Animal Cells. In Mitochondrial Bioenergetics; Humana: New York, NY, USA, 2018; Volume 1782, pp. 7–29. [Google Scholar] [CrossRef]
- Li, Z.; Graham, B.H. Measurement of Mitochondrial Oxygen Consumption Using a Clark Electrode. In Mitochondrial Disorders; Humana: Totowa, NJ, USA, 2012; Volume 837, pp. 63–72. [Google Scholar] [CrossRef]
- Divakaruni, A.S.; Rogers, G.W.; Murphy, A.N. Measuring Mitochondrial Function in Permeabilized Cells Using the Seahorse XF Analyzer or a Clark-Type Oxygen Electrode. Curr. Protoc. Toxicol. 2014, 60, 25.2.1–25.2.16. [Google Scholar] [CrossRef]
- Elbaz, H.A.; Lee, I.; Antwih, D.A.; Liu, J.; Hüttemann, M.; Zielske, S.P. Epicatechin Stimulates Mitochondrial Activity and Selectively Sensitizes Cancer Cells to Radiation. PLoS ONE 2014, 9, e88322. [Google Scholar] [CrossRef]
- Ghilardi, C.; Moreira-Barbosa, C.; Brunelli, L.; Ostano, P.; Panini, N.; Lupi, M.; Anastasia, A.; Fiordaliso, F.; Salio, M.; Formenti, L.; et al. PGC1α/β Expression Predicts Therapeutic Response to Oxidative Phosphorylation Inhibition in Ovarian Cancer. Cancer Res. 2022, 82, 1423–1434. [Google Scholar] [CrossRef] [PubMed]
- Chiappetta, G.; Gamberi, T.; Faienza, F.; Limaj, X.; Rizza, S.; Messori, L.; Filomeni, G.; Modesti, A.; Vinh, J. Redox proteome analysis of auranofin exposed ovarian cancer cells (A2780). Redox Biol. 2022, 52, 102294. [Google Scholar] [CrossRef]
- Ahmad, R.; Kuppusamy, P. Theory, Instrumentation, and Applications of Electron Paramagnetic Resonance Oximetry. Chem. Rev. 2010, 110, 3212–3236. [Google Scholar] [CrossRef] [PubMed]
- Subczynski, W.K.; Lukiewicz, S.; Hyde, J.S. Murinein vivo L-band ESR spin-label oximetry with a loop-gap resonator. Magn. Reson. Med. 1986, 3, 747–754. [Google Scholar] [CrossRef]
- Li, Y.; Xu, S.; Cai, M. PO2-based biodosimetry evaluation using an EPR technique acts as a sensitive index for chemotherapy. Oncol. Lett. 2018, 16, 2167–2174. [Google Scholar] [CrossRef]
- Ilangovan, G.; Liebgott, T.; Kutala, V.K.; Petryakov, S.; Zweier, J.L.; Kuppusamy, P. EPR oximetry in the beating heart: Myocardial oxygen consumption rate as an index of postischemic recovery. Magn. Reson. Med. 2004, 51, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Angelos, M.G.; Kutala, V.K.; Torres, C.A.; He, G.; Stoner, J.D.; Mohammad, M.; Kuppusamy, P. Hypoxic reperfusion of the ischemic heart and oxygen radical generation. Am. J. Physiol. Circ. Physiol. 2006, 290, H341–H347. [Google Scholar] [CrossRef] [PubMed]
- Selvendiran, K.; Bratasz, A.; Kuppusamy, M.L.; Tazi, M.F.; Rivera, B.K.; Kuppusamy, P. Hypoxia induces chemoresistance in ovarian cancer cells by activation of signal transducer and activator of transcription 3. Int. J. Cancer 2009, 125, 2198–2204. [Google Scholar] [CrossRef]
- Diepart, C.; Verrax, J.; Calderon, P.B.; Feron, O.; Jordan, B.F.; Gallez, B. Comparison of methods for measuring oxygen consumption in tumor cells in vitro. Anal. Biochem. 2010, 396, 250–256. [Google Scholar] [CrossRef]
- Horan, M.P.; Pichaud, N.; Ballard, J.W.O. Review: Quantifying Mitochondrial Dysfunction in Complex Diseases of Aging. Journals Gerontol. Ser. A 2012, 67, 1022–1035. [Google Scholar] [CrossRef]
- Djafarzadeh, S.; Jakob, S.M. High-resolution Respirometry to Assess Mitochondrial Function in Permeabilized and Intact Cells. J. Vis. Exp. 2017, e54985. [Google Scholar] [CrossRef]
- Wolfbeis, O.S. Luminescent sensing and imaging of oxygen: Fierce competition to the Clark electrode. Bioessays 2015, 37, 921–928. [Google Scholar] [CrossRef]
- Melnikov, P.V.; Alexandrovskaya, A.Y.; Naumova, A.O.; Arlyapov, V.A.; Kamanina, O.A.; Popova, N.M.; Zaitsev, N.K.; Yashtulov, N.A. Optical Oxygen Sensing and Clark Electrode: Face-to-Face in a Biosensor Case Study. Sensors 2022, 22, 7626. [Google Scholar] [CrossRef]
- Jha, P.; Wang, X.; Auwerx, J. Analysis of Mitochondrial Respiratory Chain Supercomplexes Using Blue Native Polyacrylamide Gel Electrophoresis (BN-PAGE). Curr. Protoc. Mouse Biol. 2016, 6, 1–14. [Google Scholar] [CrossRef]
- Beutner, G.; Porter, G.A., Jr. Analyzing Supercomplexes of the Mitochondrial Electron Transport Chain with Native Electrophoresis, In-gel Assays, and Electroelution. J. Vis. Exp. 2017, 124, e55738. [Google Scholar] [CrossRef]
- Fernandez-Vizarra, E.; Zeviani, M. Blue-Native Electrophoresis to Study the OXPHOS Complexes. In Mitochondrial Gene Expression; Humana: New York, NY, USA, 2020; Volume 2192, pp. 287–311. [Google Scholar] [CrossRef]
- Nadtochiy, S.M.; Ngai, M.; Brookes, P.S. Modified Blue Native Gel Approach for Analysis of Respiratory Supercomplexes. In Mitochondrial Medicine; Springer: New York, NY, USA, 2021; Volume 2276, pp. 227–234. [Google Scholar] [CrossRef]
- Schagger, H.; Cramer, W.A.; Vonjagow, G. Analysis of Molecular Masses and Oligomeric States of Protein Complexes by Blue Native Electrophoresis and Isolation of Membrane Protein Complexes by Two-Dimensional Native Electrophoresis. Anal. Biochem. 1994, 217, 220–230. [Google Scholar] [CrossRef]
- D’Aurelio, M.; Gajewski, C.D.; Lenaz, G.; Manfredi, G. Respiratory chain supercomplexes set the threshold for respiration defects in human mtDNA mutant cybrids. Hum. Mol. Genet. 2006, 15, 2157–2169. [Google Scholar] [CrossRef]
- Lenaz, G.; Genova, M.L. Structure and Organization of Mitochondrial Respiratory Complexes: A New Understanding of an Old Subject. Antioxidants Redox Signal. 2010, 12, 961–1008. [Google Scholar] [CrossRef]
- Chaban, Y.; Boekema, E.J.; Dudkina, N.V. Structures of mitochondrial oxidative phosphorylation supercomplexes and mechanisms for their stabilisation. Biochim. Biophys. Acta (BBA)-Bioenerg. 2014, 1837, 418–426. [Google Scholar] [CrossRef]
- Lamantea, E.; Carrara, F.; Mariotti, C.; Morandi, L.; Tiranti, V.; Zeviani, M. A novel nonsense mutation (Q352X) in the mitochondrial cytochrome b gene associated with a combined deficiency of complexes I and III. Neuromuscul. Disord. 2002, 12, 49–52. [Google Scholar] [CrossRef]
- Budde, S.M.S.; van den Heuvel, L.P.W.J.; Janssen, A.J.; Smeets, R.J.P.; Buskens, C.A.F.; DeMeirleir, L.; Van Coster, R.; Baethmann, M.; Voit, T.; Trijbels, J.M.F.; et al. Combined Enzymatic Complex I and III Deficiency Associated with Mutations in the Nuclear Encoded NDUFS4 Gene. Biochem. Biophys. Res. Commun. 2000, 275, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Saada, A.; Edvardson, S.; Shaag, A.; Chung, W.K.; Segel, R.; Miller, C.; Jalas, C.; Elpeleg, O. Combined OXPHOS complex I and IV defect, due to mutated complex I assembly factor C20ORF7. J. Inherit. Metab. Dis. 2011, 35, 125–131. [Google Scholar] [CrossRef]
- Wittig, I.; Karas, M.; Schägger, H. High Resolution Clear Native Electrophoresis for In-gel Functional Assays and Fluorescence Studies of Membrane Protein Complexes. Mol. Cell. Proteom. 2007, 6, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
- Rai, N.K.; Mathur, S.; Singh, S.K.; Tiwari, M.; Singh, V.K.; Haque, R.; Tiwari, S.; Sharma, L.K. Differential regulation of mitochondrial complex I and oxidative stress based on metastatic potential of colorectal cancer cells. Oncol. Lett. 2020, 20, 1. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Horie-Inoue, K.; Suzuki, T.; Hobo, R.; Nakasato, N.; Takeda, S.; Inoue, S. Mitochondrial supercomplex assembly promotes breast and endometrial tumorigenesis by metabolic alterations and enhanced hypoxia tolerance. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- De Luise, M.; Sollazzo, M.; Lama, E.; Coadă, C.A.; Bressi, L.; Iorio, M.; Cavina, B.; D’angelo, L.; Milioni, S.; Marchio, L.; et al. Inducing respiratory complex I impairment elicits an increase in PGC1α in ovarian cancer. Sci. Rep. 2022, 12, 1–12. [Google Scholar] [CrossRef]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms Controlling Mitochondrial Biogenesis and Respiration through the Thermogenic Coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Bartman, C.R.; Weilandt, D.R.; Shen, Y.; Lee, W.D.; Han, Y.; TeSlaa, T.; Jankowski, C.S.R.; Samarah, L.; Park, N.R.; da Silva-Diz, V.; et al. Slow TCA flux and ATP production in primary solid tumours but not metastases. Nature 2023, 614, 349–357. [Google Scholar] [CrossRef]
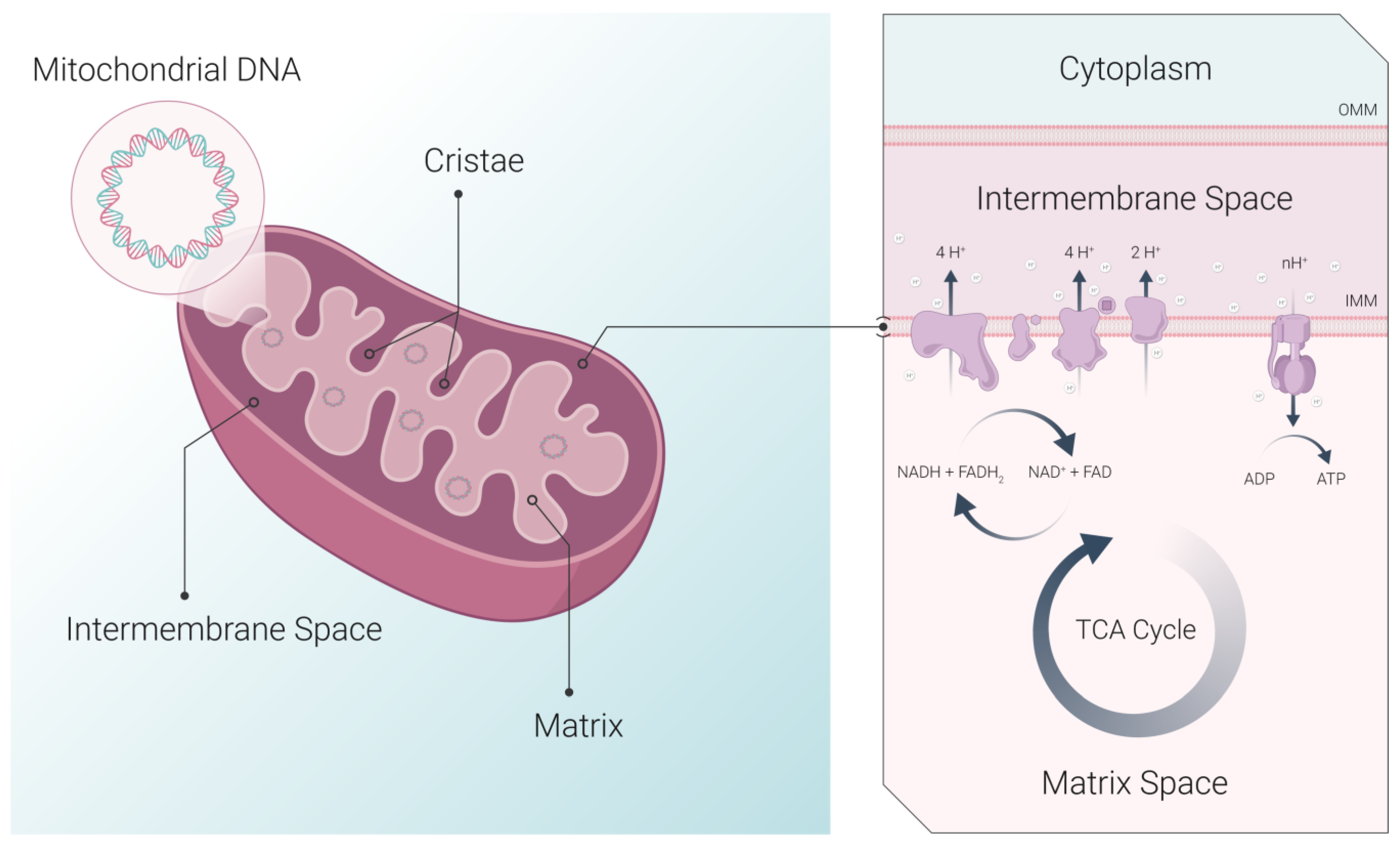
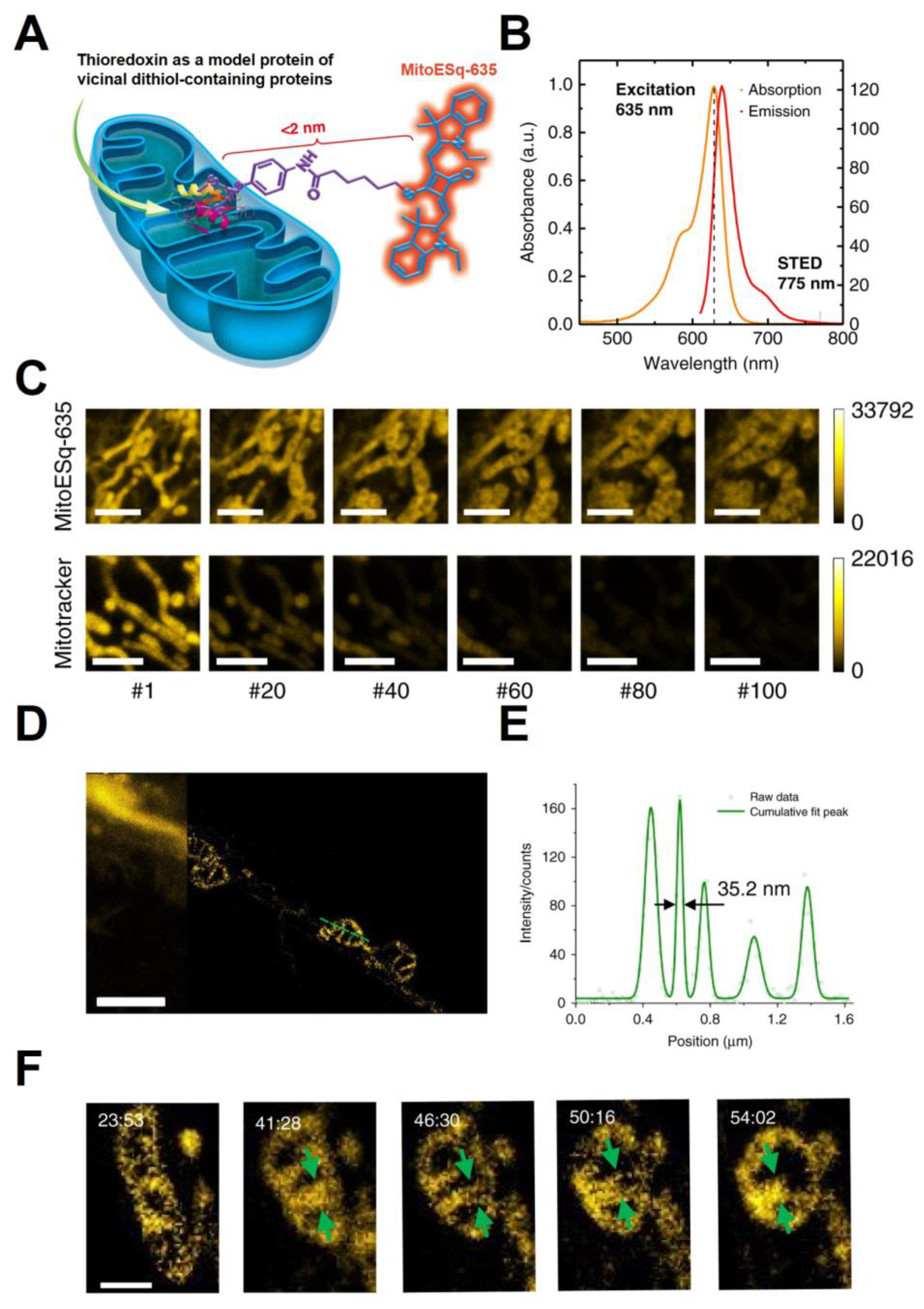
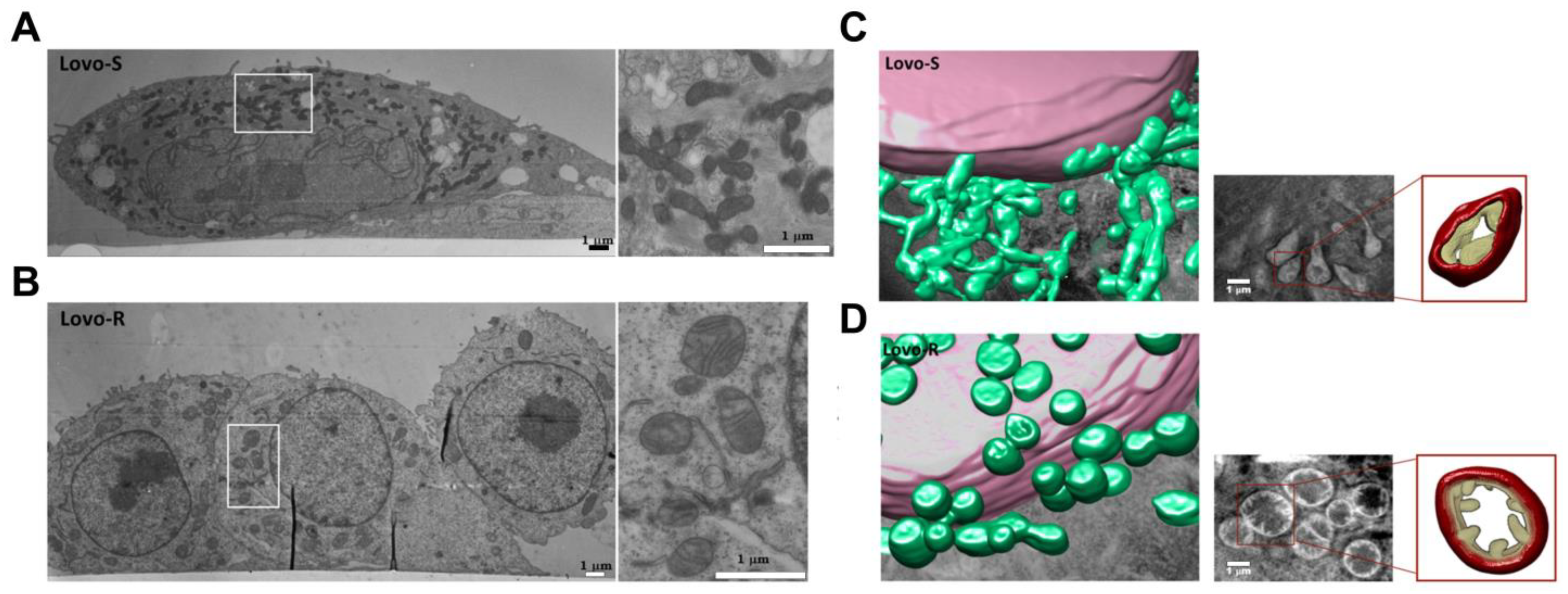


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Applications | Strengths | Limitations | Ref |
|---|---|---|---|---|
| Widefield fluorescence and confocal microscopy | Mitochondrial morphology and dynamics, ΔΨm | Live-cell and time-lapse imaging Visualization of fluorescent proteins, dyes, and immunofluorescence staining | Resolution limited by diffraction (~1/2 λ) Fluorophore photobleaching | [25,78,79] |
| Super-resolution microscopy (STED, FPALM, STORM, etc.) | Fine details of mitochondrial morphology and dynamics (e.g., cristae shape, width, etc.) | Superior resolution compared to the confocal Live-cell and time-lapse imaging | Requires specific fluorophores Often needs high laser powers | [33,34,35,39,80] |
| Two-photon excitation fluorescence (TPEF) and fluorescence lifetime imaging | Mitochondrial dynamics, morphology and label-free redox state measurements, in vivo applications | Low background Superior light penetration depth Live-cell and time-lapse imaging Visualization of external probes or endogenous metabolites | Limited number of external fluorophores suitable for two-photon excitation | [44,45,81,82] |
| Electron microscopy (EM) | Ultrastructural changes in mitochondrial shape, size, and components | Sub-nanometer resolution | Requires extensive fixation Limited ability to visualize markers Prone to artifacts | [58] |
| Electron tomography | 3D information on mitochondrial ultrastructure | 3D structural information with high resolution | Requires extensive fixation “Missing wedge” because of the restricted tilt range Sample shrinkage due to high electron dose Limited ability to visualize markers | [65,83,84,85] |
| Focused ion beam scanning electron microscopy (FIB-SEM) | 3D structural and compositional analysis | Label-free Resolution approaching EM | Not suitable for live cells Long image acquisition time (up to 60 h) | [71,72,73,74,75,76,77] |
| Method | Strengths | Limitations | Refs |
|---|---|---|---|
| Quantitative PCR | High throughput High sensitivity and specificity Established methods Simply and widely used Real-time monitoring of target Versatile | Reliant on standard curves or normalization to reference gene Prone to PCR efficiency bias Sensitive to PCR inhibitors Sample quality requirements | [117,118,119] |
| Digital PCR | High throughput Highly sensitive Absolute quantification Improved reproducibility Reduced PCR efficiency bias and PCR inhibition Highly accurate for low target concentrations No need for reference genes | Specialized equipment needed Cost prohibitive Extensive optimization required Sample quality requirements Narrow dynamic range Less accurate for high target concentrations | [117,118,127,128] |
| Method | Strengths | Limitations | Refs |
|---|---|---|---|
| Seahorse Extracellular Flux Analyzer | High throughput Requires small quantities of cells Automated process/injections Compatible with broad range of cell and tissue types | Costly equipment setup Moderate optimization required Significant plate and reagents costs per experiments | [271,284,295] |
| MitoXpress-Xtra® | Microplate-based assay High throughput Compatible with broad range of in vitro models Compatible with many plate readers | Moderate optimization required Significant plate and reagent costs per experiment Lower sensitivity Less well-established | [268,294] |
| Orosboros-2k Oxygraph | Relatively cheap Low sample volumes required Reduced oxygen leakage from device compared to other electrodes Increased sensitivity compared to other electrodes | Measurements are not automated Labor-intensive Low throughput Time-intensive (hour-long sample reads) Lacks background controls | [277,296] |
| Clark Electrode | Well-established method Reliable Mechanically robust Relatively low cost | Consumes oxygen Interference with various gases Labor-intensive maintenance | [294,297,298] |
| EPR Oximetry | Higher sensitivity Relatively non-invasive Enables 3D oxygen mapping Minimal interference issues Spin probes are non-toxic & stable | Poor signal-to-noise ratio Motion artifacts Requires exogenous probe Longer acquisition times Limited penetration depth | [288,294] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rickard, B.P.; Overchuk, M.; Chappell, V.A.; Kemal Ruhi, M.; Sinawang, P.D.; Nguyen Hoang, T.T.; Akin, D.; Demirci, U.; Franco, W.; Fenton, S.E.; et al. Methods to Evaluate Changes in Mitochondrial Structure and Function in Cancer. Cancers 2023, 15, 2564. https://doi.org/10.3390/cancers15092564
Rickard BP, Overchuk M, Chappell VA, Kemal Ruhi M, Sinawang PD, Nguyen Hoang TT, Akin D, Demirci U, Franco W, Fenton SE, et al. Methods to Evaluate Changes in Mitochondrial Structure and Function in Cancer. Cancers. 2023; 15(9):2564. https://doi.org/10.3390/cancers15092564
Chicago/Turabian StyleRickard, Brittany P., Marta Overchuk, Vesna A. Chappell, Mustafa Kemal Ruhi, Prima Dewi Sinawang, Tina Thuy Nguyen Hoang, Demir Akin, Utkan Demirci, Walfre Franco, Suzanne E. Fenton, and et al. 2023. "Methods to Evaluate Changes in Mitochondrial Structure and Function in Cancer" Cancers 15, no. 9: 2564. https://doi.org/10.3390/cancers15092564
APA StyleRickard, B. P., Overchuk, M., Chappell, V. A., Kemal Ruhi, M., Sinawang, P. D., Nguyen Hoang, T. T., Akin, D., Demirci, U., Franco, W., Fenton, S. E., Santos, J. H., & Rizvi, I. (2023). Methods to Evaluate Changes in Mitochondrial Structure and Function in Cancer. Cancers, 15(9), 2564. https://doi.org/10.3390/cancers15092564