
Engineered Adoptive T-Cell Therapies for Breast Cancer: Current Progress, Challenges, and Potential


Abstract
:Simple Summary
Abstract
1. Introduction
2. Engineered Adoptive T-Cell Therapies for Breast Cancer
3. Identification of Suitable Tumor Targets
Remaining Challenges: Antigen Heterogeneity and On-Target/Off-Tumor Toxicity
4. Overcoming the Tumor Microenvironment
4.1. Immune Microenvironment of Breast Cancer
4.1.1. Regulatory T-Cells (Tregs)
4.1.2. Myeloid-Derived Suppressor Cells (MDSCs)
4.1.3. Tumor-Associated Macrophages (TAMs)
4.2. Non-Immune Microenvironment of Breast Cancer
4.2.1. Extracellular Matrix (ECM)
4.2.2. Cancer-Associated Fibroblasts (CAFs)
4.2.3. Endothelial Cells
4.2.4. Metabolic Conditions
5. Persistence of Adoptively Transferred Engineered Cells
5.1. Engineered Chimeric Receptors
5.2. Soluble Cytokine Production
5.2.1. Interleukin-15 (IL-15)
5.2.2. Interleukin-7 (IL-7)
5.2.3. Interleukin-18 (IL-18)
6. Cost of Autologous Therapy
6.1. Non-Viral Manufacturing Techniques
6.1.1. Transposon Systems
6.1.2. In Vitro-Transcribed (IVT) mRNA
6.2. Allogeneic (“Off-the-Shelf”) Therapies
6.2.1. Natural Killer (NK) Cells
6.2.2. Gamma Delta (ɣδ) T-Cells
7. Current Clinical Trials
7.1. Trends in E-ACT Trials for Breast Cancer
7.2. Safety and Efficacy of CAR T-Cells for Breast Cancer
7.2.1. c-Met-Specific CAR T-Cells: A Safe and Moderately Effective Target
7.2.2. ROR1-Specific CAR T-Cells: Initial Safety and Poor Intratumoral Persistence
7.2.3. Mesothelin-Specific CAR T-Cells: Emerging Results from an Ongoing Trial
7.3. Safety and Efficacy of TCR T-Cells for Breast Cancer
7.3.1. NY-ESO-1-Specific TCR T-Cells: Additional Clinical Data Needed
7.3.2. MAGE-A3-Specific TCR T-Cells: Toxicity and No Evidence of Efficacy in Breast Cancer
7.3.3. Neoantigen-Specific TCR T-Cells: Promising Results from an Ongoing Trial
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast Cancer. Nat. Rev. Dis. Prim. 2019, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Mercogliano, M.F.; Bruni, S.; Mauro, F.L.; Schillaci, R. Emerging Targeted Therapies for HER2-Positive Breast Cancer. Cancers 2023, 15, 1987. [Google Scholar] [CrossRef] [PubMed]
- Soleja, M.; Raj, G.V.; Unni, N. An Evaluation of Fulvestrant for the Treatment of Metastatic Breast Cancer. Expert Opin. Pharmacother. 2019, 20, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Rath, J.A.; Arber, C. Engineering Strategies to Enhance TCR-Based Adoptive T Cell Therapy. Cells 2020, 9, 1485. [Google Scholar] [CrossRef]
- Wei, F.; Cheng, X.X.; Xue, J.Z.; Xue, S.A. Emerging Strategies in TCR-Engineered T Cells. Front. Immunol. 2022, 13, 850358. [Google Scholar] [CrossRef]
- Yang, Y.H.; Liu, J.W.; Lu, C.; Wei, J.F. CAR-T Cell Therapy for Breast Cancer: From Basic Research to Clinical Application. Int. J. Biol. Sci. 2022, 18, 2609–2626. [Google Scholar] [CrossRef]
- Kirtane, K.; Elmariah, H.; Chung, C.H.; Abate-Daga, D. Adoptive Cellular Therapy in Solid Tumor Malignancies: Review of the Literature and Challenges Ahead. J. Immunother. Cancer 2021, 9, e002723. [Google Scholar] [CrossRef]
- Akatsuka, Y. TCR-Like CAR-T Cells Targeting MHC-Bound Minor Histocompatibility Antigens. Front. Immunol. 2020, 11, e002723. [Google Scholar] [CrossRef]
- Teppert, K.; Wang, X.; Anders, K.; Evaristo, C.; Lock, D.; Künkele, A. Joining Forces for Cancer Treatment: From “TCR versus CAR” to “TCR and CAR”. Int. J. Mol. Sci. 2022, 23, 14563. [Google Scholar] [CrossRef]
- Alnefaie, A.; Albogami, S.; Asiri, Y.; Ahmad, T.; Alotaibi, S.S.; Al-Sanea, M.M.; Althobaiti, H. Chimeric Antigen Receptor T-Cells: An Overview of Concepts, Applications, Limitations, and Proposed Solutions. Front. Bioeng. Biotechnol. 2022, 10, 797440. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Flugel, C.L.; Majzner, R.G.; Krenciute, G.; Dotti, G.; Riddell, S.R.; Wagner, D.L.; Abou-el-Enein, M. Overcoming On-Target, off-Tumour Toxicity of CAR T Cell Therapy for Solid Tumours. Nat. Rev. Clin. Oncol. 2023, 20, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Brocker, T.; Karjalainen, K. Signals through T Cell Receptor-ζ Chain Alone Are Insufficient to Prime Resting T Lymphocytes. J. Exp. Med. 1995, 181, 1653–1659. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A Phase I Study on Adoptive Immunotherapy Using Gene-Modified T Cells for Ovarian Cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T Cell Therapy: Current Limitations and Potential Strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Calderon, H.; Posey, A.D.; Maus, M.V. Engineering and Design of Chimeric Antigen Receptors. Mol. Ther. Methods Clin. Dev. 2019, 12, 145–156. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKs: The Fourth Generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKS, the Fourth-generation CAR T Cells: Current Developments and Clinical Translation. Adv. Cell Gene Ther. 2020, 3, e84. [Google Scholar] [CrossRef]
- Tomasik, J.; Jasiński, M.; Basak, G.W. Next Generations of CAR-T Cells—New Therapeutic Opportunities in Hematology? Front. Immunol. 2022, 13, 1034707. [Google Scholar] [CrossRef]
- Shafer, P.; Kelly, L.M.; Hoyos, V. Cancer Therapy with TCR-Engineered T Cells: Current Strategies, Challenges, and Prospects. Front. Immunol. 2022, 13, 835762. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Jiang, X.; Zhou, X.; Weng, J. Targeting Cancers through TCR-Peptide/MHC Interactions. J. Hematol. Oncol. 2019, 12, 139. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Liu, S.; Zhao, L.; Sun, H.X. A Comprehensive Survey of Genomic Mutations in Breast Cancer Reveals Recurrent Neoantigens as Potential Therapeutic Targets. Front. Oncol. 2022, 12, 786438. [Google Scholar] [CrossRef] [PubMed]
- Brett, J.O.; Spring, L.M.; Bardia, A.; Wander, S.A. ESR1 Mutation as an Emerging Clinical Biomarker in Metastatic Hormone Receptor-Positive Breast Cancer. Breast Cancer Res. 2021, 23, 85. [Google Scholar] [CrossRef] [PubMed]
- Mikhael, J.; Fowler, J.; Shah, N. Chimeric Antigen Receptor T-Cell Therapies: Barriers and Solutions to Access. JCO Oncol. Pract. 2022, 18, 800–807. [Google Scholar] [CrossRef] [PubMed]
- Dees, S.; Ganesan, R.; Singh, S.; Grewal, I.S. Emerging CAR-T Cell Therapy for the Treatment of Triple-Negative Breast Cancer. Mol. Cancer Ther. 2020, 19, 2409–2421. [Google Scholar] [CrossRef] [PubMed]
- Leko, V.; Rosenberg, S.A. Identifying and Targeting Human Tumor Antigens for T Cell-Based Immunotherapy of Solid Tumors. Cancer Cell 2020, 38, 454–472. [Google Scholar] [CrossRef]
- Vigneron, N. Human Tumor Antigens and Cancer Immunotherapy. BioMed Res. Int. 2015, 2015, 948501. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, S.; Fang, C.; Yang, S.; Olalere, D.; Pequignot, E.C.; Cogdill, A.P.; Li, N.; Ramones, M.; Granda, B.; et al. Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res. 2015, 75, 3596–3607. [Google Scholar] [CrossRef]
- Jakobsen, M.K.; Gjerstorff, M.F. CAR T-Cell Cancer Therapy Targeting Surface Cancer/Testis Antigens. Front. Immunol. 2020, 11, 1568. [Google Scholar] [CrossRef]
- Li, Y.; Li, J.; Wang, Y.; Zhang, Y.; Chu, J.; Sun, C.; Fu, Z.; Huang, Y.; Zhang, H.; Yuan, H.; et al. Roles of Cancer/Testis Antigens (CTAs) in Breast Cancer. Cancer Lett. 2017, 399, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Akers, S.N.; Odunsi, K.; Karpf, A.R. Regulation of Cancer Germline Antigen Gene Expression: Implications for Cancer Immunotherapy. Future Oncol. 2010, 6, 717–732. [Google Scholar] [CrossRef] [PubMed]
- Feola, S.; Chiaro, J.; Martins, B.; Cerullo, V. Uncovering the Tumor Antigen Landscape: What to Know about the Discovery Process. Cancers 2020, 12, 1660. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Shen, G.; Gao, W.; Huang, Z.; Huang, C.; Fu, L. Neoantigens: Promising Targets for Cancer Therapy. Signal Transduct. Target. Ther. 2023, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Chandran, S.S.; Ma, J.; Klatt, M.G.; Dündar, F.; Bandlamudi, C.; Razavi, P.; Wen, H.Y.; Weigelt, B.; Zumbo, P.; Fu, S.N.; et al. Immunogenicity and Therapeutic Targeting of a Public Neoantigen Derived from Mutated PIK3CA. Nat. Med. 2022, 28, 946–957. [Google Scholar] [CrossRef] [PubMed]
- Tóth, G.; Szöllősi, J.; Abken, H.; Vereb, G.; Szöőr, Á. A Small Number of HER2 Redirected CAR T Cells Significantly Improves Immune Response of Adoptively Transferred Mouse Lymphocytes against Human Breast Cancer Xenografts. Int. J. Mol. Sci. 2020, 21, 1039. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Peng, Y. Current Biological, Pathological and Clinical Landscape of HER2-Low Breast Cancer. Cancers 2023, 15, 126. [Google Scholar] [CrossRef]
- Zhang, P.F.; Huang, Y.; Liang, X.; Li, D.; Jiang, L.; Yang, X.; Zhu, M.; Gou, H.F.; Gong, Y.L.; Wei, Y.Q.; et al. Enhancement of the Antitumor Effect of HER2-Directed CAR-T Cells through Blocking Epithelial-Mesenchymal Transition in Tumor Cells. FASEB J. 2020, 34, 11185–11199. [Google Scholar] [CrossRef]
- Li, H.; Yuan, W.; Bin, S.; Wu, G.; Li, P.; Liu, M.; Yang, J.; Li, X.; Yang, K.; Gu, H. Overcome Trastuzumab Resistance of Breast Cancer Using Anti-HER2 Chimeric Antigen Receptor T Cells and PD1 Blockade. Am. J. Cancer Res. 2020, 10, 688–703. [Google Scholar]
- Globerson-Levin, A.; Waks, T.; Eshhar, Z. Elimination of Progressive Mammary Cancer by Repeated Administrations of Chimeric Antigen Receptor-Modified T Cells. Mol. Ther. 2014, 22, 1029–1038. [Google Scholar] [CrossRef]
- Schönfeld, K.; Sahm, C.; Zhang, C.; Naundorf, S.; Brendel, C.; Odendahl, M.; Nowakowska, P.; Bönig, H.; Köhl, U.; Kloess, S.; et al. Selective Inhibition of Tumor Growth by Clonal NK Cells Expressing an ErbB2/HER2-Specific Chimeric Antigen Receptor. Mol. Ther. 2015, 23, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Szöőr, Á.; Tóth, G.; Zsebik, B.; Szabó, V.; Eshhar, Z.; Abken, H.; Vereb, G. Trastuzumab Derived HER2-Specific CARs for the Treatment of Trastuzumab-Resistant Breast Cancer: CAR T Cells Penetrate and Eradicate Tumors That Are Not Accessible to Antibodies. Cancer Lett. 2020, 484, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Priceman, S.J.; Tilakawardane, D.; Jeang, B.; Aguilar, B.; Murad, J.P.; Park, A.K.; Chang, W.-C.; Ostberg, J.R.; Neman, J.; Jandial, R.; et al. Regional Delivery of Chimeric Antigen Receptor–Engineered T Cells Effectively Targets HER2+ Breast Cancer Metastasis to the Brain. Clin. Cancer Res. 2018, 24, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Shi, H.; Liu, C.; Liu, J.; Liu, X.; Sun, Y. Construction and Evaluation of a Novel Humanized HER2-Specific Chimeric Receptor. Breast Cancer Res. 2014, 16, R61. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Yang, X.; Tian, W.; Gou, S.; Huang, W.; Zhao, W. Increased Expression of C-Met Is Associated with Chemotherapy-Resistant Breast Cancer and Poor Clinical Outcome. Med. Sci. Monit. 2018, 24, 8239–8249. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Qu, J.; Hui, Y.; Zhang, H.; Sun, Y.; Liu, X.; Zhao, X.; Zhao, Z.; Yang, Q.; Wang, F.; et al. Clinicopathological and Prognostic Significance of C-Met Overexpression in Breast Cancer. Oncotarget 2017, 8, 56758–56767. [Google Scholar] [CrossRef] [PubMed]
- Tchou, J.; Zhao, Y.; Levine, B.L.; Zhang, P.J.; Davis, M.M.; Melenhorst, J.J.; Kulikovskaya, I.; Brennan, A.L.; Liu, X.; Lacey, S.F.; et al. Safety and Efficacy of Intratumoral Injections of Chimeric Antigen Receptor (CAR) T Cells in Metastatic Breast Cancer. Cancer Immunol. Res. 2017, 5, 1152–1161. [Google Scholar] [CrossRef] [PubMed]
- Ho-Yen, C.M.; Jones, J.L.; Kermorgant, S. The Clinical and Functional Significance of C-Met in Breast Cancer: A Review. Breast Cancer Res. 2015, 17, 52. [Google Scholar] [CrossRef]
- Nath, S.; Mukherjee, P. MUC1: A Multifaceted Oncoprotein with a Key Role in Cancer Progression. Trends Mol. Med. 2014, 20, 332–342. [Google Scholar] [CrossRef]
- Rakha, E.A.; Boyce, R.W.G.; El-Rehim, D.A.; Kurien, T.; Green, A.R.; Paish, E.C.; Robertson, J.F.R.; Ellis, I.O. Expression of Mucins (MUC1, MUC2, MUC3, MUC4, MUC5AC and MUC6) and Their Prognostic Significance in Human Breast Cancer. Mod. Pathol. 2005, 18, 1295–1304. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdanifar, M.; Roy, L.D.; Whilding, L.M.; Gavrill, A.; Maher, J.; Mukherjee, P. CAR T Cells Targeting the Tumor MUC1 Glycoprotein Reduce Triple-Negative Breast Cancer Growth. Front. Immunol. 2019, 10, 1149. [Google Scholar] [CrossRef] [PubMed]
- Nalawade, S.A.; Shafer, P.; Bajgain, P.; McKenna, M.K.; Ali, A.; Kelly, L.; Joubert, J.; Gottschalk, S.; Watanabe, N.; Leen, A.; et al. Selectively Targeting Myeloid-Derived Suppressor Cells through TRAIL Receptor 2 to Enhance the Efficacy of CAR T Cell Therapy for Treatment of Breast Cancer. J. Immunother. Cancer 2021, 9, e003237. [Google Scholar] [CrossRef] [PubMed]
- Bajgain, P.; Tawinwung, S.; D’Elia, L.; Sukumaran, S.; Watanabe, N.; Hoyos, V.; Lulla, P.; Brenner, M.K.; Leen, A.M.; Vera, J.F. CAR T Cell Therapy for Breast Cancer: Harnessing the Tumor Milieu to Drive T Cell Activation. J. Immunother. Cancer 2018, 6, 34. [Google Scholar] [CrossRef] [PubMed]
- Tozbikian, G.; Brogi, E.; Kadota, K.; Catalano, J.; Akram, M.; Patil, S.; Ho, A.Y.; Reis-Filho, J.S.; Weigelt, B.; Norton, L.; et al. Mesothelin Expression in Triple Negative Breast Carcinomas Correlates Significantly with Basal-like Phenotype, Distant Metastases and Decreased Survival. PLoS ONE 2014, 9, e114900. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.R.; Xian, R.R.; Ziober, A.; Conejo-Garcia, J.; Perales-Puchalt, A.; June, C.H.; Zhang, P.J.; Tchou, J. Mesothelin Expression Is Associated with Poor Outcomes in Breast Cancer. Breast Cancer Res. Treat. 2014, 147, 675–684. [Google Scholar] [CrossRef]
- Suzuki, T.; Yamagishi, Y.; Einama, T.; Koiwai, T.; Yamasaki, T.; Fukumura-Koga, M.; Ishibashi, Y.; Takihata, Y.; Shiraishi, T.; Miyata, Y.; et al. Membrane Mesothelin Expression Positivity Is Associated with Poor Clinical Outcome of Luminal-Type Breast Cancer. Oncol. Lett. 2020, 20, 193. [Google Scholar] [CrossRef]
- Tchou, J.; Wang, L.-C.; Selven, B.; Zhang, H.; Conejo-Garcia, J.; Borghaei, H.; Kalos, M.; Vondeheide, R.H.; Albelda, S.M.; June, C.H.; et al. Mesothelin, a Novel Immunotherapy Target for Triple Negative Breast Cancer. Breast Cancer Res. Treat. 2012, 133, 799–804. [Google Scholar] [CrossRef]
- Yang, M.; Guan, T.; Chen, C.F.; He, L.F.; Wu, H.M.; Zhang, R.D.; Li, Y.; Lin, Y.C.; Zeng, H.; Wu, J.D. Mesothelin-Targeted CAR-NK Cells Derived From Induced Pluripotent Stem Cells Have a High Efficacy in Killing Triple-Negative Breast Cancer Cells as Shown in Several Preclinical Models. J. Immunother. 2023, 46, 285–294. [Google Scholar] [CrossRef]
- Li, Y.; Xiao, F.; Zhang, A.; Zhang, D.; Nie, W.; Xu, T.; Han, B.; Seth, P.; Wang, H.; Yang, Y.; et al. Oncolytic Adenovirus Targeting TGF-β Enhances Anti-Tumor Responses of Mesothelin-Targeted Chimeric Antigen Receptor T Cell Therapy against Breast Cancer. Cell. Immunol. 2020, 348, 104041. [Google Scholar] [CrossRef]
- Soysal, S.D.; Muenst, S.; Barbie, T.; Fleming, T.; Gao, F.; Spizzo, G.; Oertli, D.; Viehl, C.T.; Obermann, E.C.; Gillanders, W.E. EpCAM Expression Varies Significantly and Is Differentially Associated with Prognosis in the Luminal B HER2+, Basal-like, and HER2 Intrinsic Subtypes of Breast Cancer. Br. J. Cancer 2013, 108, 1480–1487. [Google Scholar] [CrossRef]
- Gao, G.; Liao, W.; Shu, P.; Ma, Q.; He, X.; Zhang, B.; Qin, D.; Wang, Y. Targeting Sphingosine 1-Phosphate Receptor 3 Inhibits T-Cell Exhaustion and Regulates Recruitment of Proinflammatory Macrophages to Improve Antitumor Efficacy of CAR-T Cells against Solid Tumor. J. Immunother. Cancer 2023, 11, e006343. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; McCloskey, J.E.; Yang, H.; Puc, J.; Alcaina, Y.; Vedvyas, Y.; Gomez Gallegos, A.A.; Ortiz-Sánchez, E.; de Stanchina, E.; Min, I.M.; et al. Bispecific CAR T Cells against EpCAM and Inducible ICAM-1 Overcome Antigen Heterogeneity and Generate Superior Antitumor Responses. Cancer Immunol. Res. 2021, 9, 1158–1174. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Chen, L.; Cui, B.; Chuang, H.Y.; Yu, J.; Wang-Rodriguez, J.; Tang, L.; Chen, G.; Basak, G.W.; Kipps, T.J. ROR1 Is Expressed in Human Breast Cancer and Associated with Enhanced Tumor-Cell Growth. PLoS ONE 2012, 7, e31127. [Google Scholar] [CrossRef] [PubMed]
- Irmer, B.; Efing, J.; Reitnauer, L.E.; Angenendt, A.; Heinrichs, S.; Schubert, A.; Schulz, M.; Binder, C.; Tio, J.; Hansen, U.; et al. Extracellular Vesicle-Associated Tyrosine Kinase-like Orphan Receptors ROR1 and ROR2 Promote Breast Cancer Progression. Cell Commun. Signal. 2023, 21, 171. [Google Scholar] [CrossRef] [PubMed]
- Nadanaka, S.; Tamura, J.I.; Kitagawa, H. Chondroitin Sulfates Control Invasiveness of the Basal-Like Breast Cancer Cell Line MDA-MB-231 Through ROR1. Front. Oncol. 2022, 12, 914838. [Google Scholar] [CrossRef] [PubMed]
- Chien, H.P.; Ueng, S.H.; Chen, S.C.; Chang, Y.S.; Lin, Y.C.; Lo, Y.F.; Chang, H.K.; Chuang, W.Y.; Huang, Y.T.; Cheung, Y.C.; et al. Expression of ROR1 Has Prognostic Significance in Triple Negative Breast Cancer. Virchows Arch. 2016, 468, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, A.; Goodpaster, T.; Randolph-Habecker, J.; Hoffstrom, B.G.; Jalikis, F.G.; Koch, L.K.; Berger, C.; Kosasih, P.L.; Rajan, A.; Sommermeyer, D.; et al. Analysis of ROR1 Protein Expression in Human Cancer and Normal Tissues. Clin. Cancer Res. 2017, 23, 3061–3071. [Google Scholar] [CrossRef]
- Stüber, T.; Monjezi, R.; Wallstabe, L.; Kühnemundt, J.; Nietzer, S.L.; Dandekar, G.; Wöckel, A.; Einsele, H.; Wischhusen, J.; Hudecek, M. Inhibition of TGF-β-Receptor Signaling Augments the Antitumor Function of ROR1-Specific CAR T-Cells against Triple-Negative Breast Cancer. J. Immunother. Cancer 2020, 8, e000676. [Google Scholar] [CrossRef]
- Wallstabe, L.; Göttlich, C.; Nelke, L.C.; Kühnemundt, J.; Schwarz, T.; Nerreter, T.; Einsele, H.; Walles, H.; Dandekar, G.; Nietzer, S.L.; et al. ROR1-CAR T Cells Are Effective against Lung and Breast Cancer in Advanced Microphysiologic 3D Tumor Models. JCI Insight 2019, 4, e126345. [Google Scholar] [CrossRef]
- Shao, Y.; Sun, X.; He, Y.; Liu, C.; Liu, H. Elevated Levels of Serum Tumor Markers CEA and CA15-3 Are Prognostic Parameters for Different Molecular Subtypes of Breast Cancer. PLoS ONE 2015, 10, e0133830. [Google Scholar] [CrossRef]
- Guadagni, F.; Ferroni, P.; Carlini, S.; Mariotti, S.; Spila, A.; Aloe, S.; D’Alessandro, R.; Carone, M.D.; Cicchetti, A.; Ricciotti, A.; et al. A Re-Evaluation of Carcinoembryonic Antigen (CEA) as a Serum Marker for Breast Cancer: A Prospective Longitudinal Study. Clin. Cancer Res. 2001, 7, 2357–2362. [Google Scholar] [PubMed]
- De Kruijf, E.M.; Sajet, A.; Van Nes, J.G.H.; Putter, H.; Smit, V.T.; Eagle, R.A.; Jafferji, I.; Trowsdale, J.; Liefers, G.J.; Van De Velde, C.J.H.; et al. NKG2D Ligand Tumor Expression and Association with Clinical Outcome in Early Breast Cancer Patients: An Observational Study. BMC Cancer 2012, 12, 24. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Xie, W.; Song, D.G.; Powell, D.J. Control of Triple-Negative Breast Cancer Using Ex Vivo Self-Enriched, Costimulated NKG2D CAR T Cells. J. Hematol. Oncol. 2018, 11, 92. [Google Scholar] [CrossRef] [PubMed]
- Hsu, N.C.; Nien, P.Y.; Yokoyama, K.K.; Chu, P.Y.; Hou, M.F. High Chondroitin Sulfate Proteoglycan 4 Expression Correlates with Poor Outcome in Patients with Breast Cancer. Biochem. Biophys. Res. Commun. 2013, 441, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Geldres, C.; Savoldo, B.; Hoyos, V.; Caruana, I.; Zhang, M.; Yvon, E.; Del Vecchio, M.; Creighton, C.J.; Ittmann, M.; Ferrone, S.; et al. T Lymphocytes Redirected against the Chondroitin Sulfate Proteoglycan-4 Control the Growth of Multiple Solid Tumors Both in Vitro and in Vivo. Clin. Cancer Res. 2014, 20, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Beard, R.E.; Zheng, Z.; Lagisetty, K.H.; Burns, W.R.; Tran, E.; Hewitt, S.M.; Abate-Daga, D.; Rosati, S.F.; Fine, H.A.; Ferrone, S.; et al. Multiple Chimeric Antigen Receptors Successfully Target Chondroitin Sulfate Proteoglycan 4 in Several Different Cancer Histologies and Cancer Stem Cells. J. Immunother. Cancer 2014, 2, 25. [Google Scholar] [CrossRef]
- O’Shannessy, D.J.; Somers, E.B.; Maltzman, J.; Smale, R.; Fu, Y.S. Folate Receptor Alpha (FRA) Expression in Breast Cancer: Identification of a New Molecular Subtype and Association with Triple Negative Disease. Springerplus 2012, 1, 22. [Google Scholar] [CrossRef]
- Hartmann, L.C.; Keeney, G.L.; Lingle, W.L.; Christianson, T.J.H.; Varghese, B.; Hillman, D.; Oberg, A.L.; Low, P.S. Folate Receptor Overexpression Is Associated with Poor Outcome in Breast Cancer. Int. J. Cancer 2007, 121, 938–942. [Google Scholar] [CrossRef]
- Song, D.; Ye, Q.; Poussin, M.; Chacon, J.A.; Figini, M.; Powell, D.J., Jr. Effective Adoptive Immunotherapy of Triple-Negative Breast Cancer by Folate Receptor-Alpha Redirected CAR T Cells Is Influenced by Surface Antigen Expression Level. J. Hematol. Oncol. 2016, 9, 56. [Google Scholar] [CrossRef]
- Chuangchot, N.; Jamjuntra, P.; Yangngam, S.; Luangwattananun, P.; Thongchot, S.; Junking, M.; Thuwajit, P.; Yenchitsomanus, P.T.; Thuwajit, C. Enhancement of PD-L1-Attenuated CAR-T Cell Function through Breast Cancer-Associated Fibroblasts-Derived IL-6 Signaling via STAT3/AKT Pathways. Breast Cancer Res. 2023, 25, 86. [Google Scholar] [CrossRef]
- Battula, V.L.; Shi, Y.; Evans, K.W.; Wang, R.Y.; Spaeth, E.L.; Jacamo, R.O.; Guerra, R.; Sahin, A.A.; Marini, F.C.; Hortobagyi, G.; et al. Ganglioside GD2 Identifies Breast Cancer Stem Cells and Promotes Tumorigenesis. J. Clin. Investig. 2012, 122, 2066–2078. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Anand, V.; Andreeff, M.; Battula, V.L. Ganglioside GD2: A Novel Therapeutic Target in Triple-Negative Breast Cancer. Ann. N. Y. Acad. Sci. 2022, 1508, 35–53. [Google Scholar] [CrossRef] [PubMed]
- Zhong, E.; Brogi, E.; D’Alfonso, T.M.; Wen, H.; Frosina, D.; Cheung, N.K.; Jungbluth, A.A.; Ross, D.S. Expression Analysis of GD2 by Immunohistochemistry in Invasive Breast Carcinoma: Clinical and Pathologic Correlation. Appl. Immunohistochem. Mol. Morphol. 2022, 30, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Seitz, C.M.; Schroeder, S.; Knopf, P.; Krahl, A.C.; Hau, J.; Schleicher, S.; Martella, M.; Quintanilla-Martinez, L.; Kneilling, M.; Pichler, B.; et al. GD2-Targeted Chimeric Antigen Receptor T Cells Prevent Metastasis Formation by Elimination of Breast Cancer Stem-like Cells. Oncoimmunology 2020, 9, 1683345. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhou, Y.; Huang, K.H.; Li, Y.; Fang, X.; An, L.; Wang, F.; Chen, Q.; Zhang, Y.; Shi, A.; et al. EGFR-Specific CAR-T Cells Trigger Cell Lysis in EGFR-Positive TNBC. Aging 2019, 11, 11054–11072. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Zheng, Z.Z.; Liu, J.Y.; Chen, Y.J.; Ding, J.C.; Xia, N.S.; Luo, W.X.; Liu, W. EGFR-Targeted CAR-T Cells Are Potent and Specific in Suppressing Triple-Negative Breast Cancer Both in Vitro and in Vivo. Clin. Transl. Immunol. 2020, 9, e1135. [Google Scholar] [CrossRef] [PubMed]
- Li, R.H.; Huang, W.H.; Wu, J.D.; Du, C.W.; Zhang, G.J. EGFR Expression Is Associated with Cytoplasmic Staining of CXCR4 and Predicts Poor Prognosis in Triple-Negative Breast Carcinomas. Oncol. Lett. 2017, 13, 695–703. [Google Scholar] [CrossRef]
- Park, H.S.; Jang, M.H.; Kim, E.J.; Kim, H.J.; Lee, H.J.; Kim, Y.J.; Kim, J.H.; Kang, E.; Kim, S.W.; Kim, I.A.; et al. High EGFR Gene Copy Number Predicts Poor Outcome in Triple-Negative Breast Cancer. Mod. Pathol. 2014, 27, 1212–1222. [Google Scholar] [CrossRef]
- Chen, M.; Wu, C.; Fu, Z.; Liu, S. ICAM1 Promotes Bone Metastasis via Integrin-Mediated TGF-β/EMT Signaling in Triple-Negative Breast Cancer. Cancer Sci. 2022, 113, 3751–3765. [Google Scholar] [CrossRef]
- Guo, P.; Huang, J.; Wang, L.; Jia, D.; Yang, J.; Dillon, D.A.; Zurakowski, D.; Mao, H.; Moses, M.A.; Auguste, D.T.; et al. ICAM-1 as a Molecular Target for Triple Negative Breast Cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 14710–14715. [Google Scholar] [CrossRef]
- Wei, H.; Wang, Z.; Kuang, Y.; Wu, Z.; Zhao, S.; Zhang, Z.; Li, H.; Zheng, M.; Zhang, N.; Long, C.; et al. Intercellular Adhesion Molecule-1 as Target for CAR-T-Cell Therapy of Triple-Negative Breast Cancer. Front. Immunol. 2020, 11, 573823. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Yu, F.; Yao, Z.; Ding, X.; Xu, H.; Zhang, J. CD24 Is a Novel Target of Chimeric Antigen Receptor T Cells for the Treatment of Triple Negative Breast Cancer. Cancer Immunol. Immunother. 2023, 72, 3191–3202. [Google Scholar] [CrossRef] [PubMed]
- Baba, S.A.; Sun, Q.; Mugisha, S.; Labhsetwar, S.; Klemke, R.; Desgrosellier, J.S. Breast Cancer Stem Cells Tolerate Chromosomal Instability during Tumor Progression via C-Jun/AXL Stress Signaling. Heliyon 2023, 9, e20182. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Ding, Y.; Kong, Y.; Fang, M.; Yu, X.; Lai, X.; Gu, Q. Triple-negative Breast Cancer Cells That Survive Ionizing Radiation Exhibit an Axl-dependent Aggressive Radioresistant Phenotype. Exp. Ther. Med. 2023, 26, 448. [Google Scholar] [CrossRef] [PubMed]
- Gjerdrum, C.; Tiron, C.; Høiby, T.; Stefansson, I.; Haugen, H.; Sandal, T.; Collett, K.; Li, S.; McCormack, E.; Gjertsen, B.T.; et al. Axl Is an Essential Epithelial-to-Mesenchymal Transition-Induced Regulator of Breast Cancer Metastasis and Patient Survival. Proc. Natl. Acad. Sci. USA 2010, 107, 1124–1129. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Chen, H.; You, S.; Tian, Z.; Wang, Z.; Liu, F.; Hu, W.; Zhang, H.; Zhang, G.; Zhao, H.; et al. AXL Upregulates C-Myc Expression through AKT and ERK Signaling Pathways in Breast Cancers. Mol. Clin. Oncol. 2023, 18, 22. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Sun, H.; Zhang, A.; Wu, X.; Li, Y.; Liu, J.; Duan, Y.; Xiao, F.; Wang, H.; Lv, M.; et al. A Novel AXL Chimeric Antigen Receptor Endows T Cells with Anti-Tumor Effects against Triple Negative Breast Cancers. Cell. Immunol. 2018, 331, 49–58. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, Y.; Liu, W.; Li, X. Engineered IL-7 Receptor Enhances the Therapeutic Effect of AXL-CAR-T Cells on Triple-Negative Breast Cancer. BioMed Res. Int. 2020, 2020, 4795171. [Google Scholar] [CrossRef]
- Tessari, A.; Pilla, L.; Silvia, D.; Duca, M.; Paolini, B.; Carcangiu, M.L.; Mariani, L.; de Braud, F.G.; Cresta, S. Expression of NY-ESO-1, MAGE-A3, PRAME and WT1 in Different Subgroups of Breast Cancer: An Indication to Immunotherapy? Breast 2018, 42, 68–73. [Google Scholar] [CrossRef]
- Raghavendra, A.; Kalita-de Croft, P.; Vargas, A.C.; Smart, C.E.; Simpson, P.T.; Saunus, J.M.; Lakhani, S.R. Expression of MAGE-A and NY-ESO-1 Cancer/Testis Antigens Is Enriched in Triple-Negative Invasive Breast Cancers. Histopathology 2018, 73, 68–80. [Google Scholar] [CrossRef]
- Liu, X.; Xu, Y.; Xiong, W.; Yin, B.; Huang, Y.; Chu, J.; Xing, C.; Qian, C.; Du, Y.; Duan, T.; et al. Development of a TCR-like Antibody and Chimeric Antigen Receptor against NY-ESO-1/HLA-A2 for Cancer Immunotherapy. J. Immunother. Cancer 2022, 10, e004035. [Google Scholar] [CrossRef] [PubMed]
- Ademuyiwa, F.O.; Bshara, W.; Attwood, K.; Morrison, C.; Edge, S.B.; Ambrosone, C.B.; O’Connor, T.L.; Levine, E.G.; Miliotto, A.; Ritter, E.; et al. NY-ESO-1 Cancer Testis Antigen Demonstrates High Immunogenicity in Triple Negative Breast Cancer. PLoS ONE 2012, 7, e38783. [Google Scholar] [CrossRef]
- Oh, C.; Kim, H.-R.; Oh, S.; Ko, J.Y.; Kim, Y.; Kang, K.; Yang, Y.; Kim, J.; Park, J.H.; Roe, J.-S.; et al. Epigenetic Upregulation of MAGE-A Isoforms Promotes Breast Cancer Cell Aggressiveness. Cancers 2021, 13, 3176. [Google Scholar] [CrossRef] [PubMed]
- Chinnasamy, N.; Wargo, J.A.; Yu, Z.; Rao, M.; Frankel, T.L.; Riley, J.P.; Hong, J.J.; Parkhurst, M.R.; Feldman, S.A.; Schrump, D.S.; et al. A TCR Targeting the HLA-A*0201–Restricted Epitope of MAGE-A3 Recognizes Multiple Epitopes of the MAGE-A Antigen Superfamily in Several Types of Cancer. J. Immunol. 2011, 186, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Matković, B.; Juretić, A.; Spagnoli, G.C.; Šeparović, V.; Gamulin, M.; Šeparović, R.; Šarić, N.; Bašić-Koretić, M.; Novosel, I.; Krušlin, B. Expression of MAGE-A and NY-ESO-1 Cancer/Testis Antigens in Medullary Breast Cancer: Retrospective Immunohistochemical Study. Croat. Med. J. 2011, 52, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Bu, J.; Zhang, Y.; Wu, S.; Li, H.; Sun, L.; Liu, Y.; Zhu, X.; Qiao, X.; Ma, Q.; Liu, C.; et al. KK-LC-1 as a Therapeutic Target to Eliminate ALDH+ Stem Cells in Triple Negative Breast Cancer. Nat. Commun. 2023, 14, 2602. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Bu, J.; Zhu, T.; Jiang, Y. Targeting KK-LC-1 Inhibits Malignant Biological Behaviors of Triple-Negative Breast Cancer. J. Transl. Med. 2023, 21, 184. [Google Scholar] [CrossRef]
- Kondo, Y.; Fukuyama, T.; Yamamura, R.; Futawatari, N.; Ichiki, Y.; Tanaka, Y.; Nishi, Y.; Takahashi, Y.; Yamazaki, H.; Kobayashi, N.; et al. Detection of KK-LC-1 Protein, a Cancer/Testis Antigen, in Patients with Breast Cancer. Anticancer Res. 2018, 38, 5923–5928. [Google Scholar] [CrossRef]
- Keraite, I.; Alvarez-Garcia, V.; Garcia-Murillas, I.; Beaney, M.; Turner, N.C.; Bartos, C.; Oikonomidou, O.; Kersaudy-Kerhoas, M.; Leslie, N.R. PIK3CA Mutation Enrichment and Quantitation from Blood and Tissue. Sci. Rep. 2020, 10, 17082. [Google Scholar] [CrossRef]
- Martínez-Saéz, O.; Chic, N.; Pascual, T.; Adamo, B.; Vidal, M.; González-Farré, B.; Sanfeliu, E.; Schettini, F.; Conte, B.; Brasó-Maristany, F.; et al. Frequency and Spectrum of PIK3CA Somatic Mutations in Breast Cancer. Breast Cancer Res. 2020, 22, 45. [Google Scholar] [CrossRef]
- Reinhardt, K.; Stückrath, K.; Hartung, C.; Kaufhold, S.; Uleer, C.; Hanf, V.; Lantzsch, T.; Peschel, S.; John, J.; Pöhler, M.; et al. PIK3CA-Mutations in Breast Cancer. Breast Cancer Res. Treat. 2022, 196, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Bindal, N.; Bamford, S.; Cole, C.; Kok, C.Y.; Beare, D.; Jia, M.; Shepherd, R.; Leung, K.; Menzies, A.; et al. COSMIC: Mining Complete Cancer Genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011, 39, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.J.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant P53 Drives Invasion by Promoting Integrin Recycling. Cell 2009, 139, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Capaci, V.; Bascetta, L.; Fantuz, M.; Beznoussenko, G.V.; Sommaggio, R.; Cancila, V.; Bisso, A.; Campaner, E.; Mironov, A.A.; Wiśniewski, J.R.; et al. Mutant P53 Induces Golgi Tubulo-Vesiculation Driving a Prometastatic Secretome. Nat. Commun. 2020, 11, 3945. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Lim, B.; Wang, Y.; Krishnamurthy, S.; Woodward, W.; Alvarez, R.H.; Lucci, A.; Valero, V.; Reuben, J.M.; Meric-Bernstam, F.; et al. Identification of Frequent Somatic Mutations in Inflammatory Breast Cancer. Breast Cancer Res. Treat. 2017, 163, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.P.; Vale, N.R.; Zacharakis, N.; Krishna, S.; Yu, Z.; Gasmi, B.; Gartner, J.J.; Sindiri, S.; Malekzadeh, P.; Deniger, D.C.; et al. Adoptive Cellular Therapy with Autologous Tumor-Infiltrating Lymphocytes and T-Cell Receptor-Engineered T Cells Targeting Common P53 Neoantigens in Human Solid Tumors. Cancer Immunol. Res. 2022, 10, 932–946. [Google Scholar] [CrossRef] [PubMed]
- Walerych, D.; Napoli, M.; Collavin, L.; Del Sal, G. The Rebel Angel: Mutant P53 as the Driving Oncogene in Breast Cancer. Carcinogenesis 2012, 33, 2007–2017. [Google Scholar] [CrossRef]
- Chen, N.; Li, X.; Chintala, N.K.; Tano, Z.E.; Adusumilli, P.S. Driving CARs on the Uneven Road of Antigen Heterogeneity in Solid Tumors. Curr. Opin. Immunol. 2018, 51, 103–110. [Google Scholar] [CrossRef]
- Hou, Y.; Nitta, H.; Li, Z. HER2 Intratumoral Heterogeneity in Breast Cancer, an Evolving Concept. Cancers 2023, 15, 2664. [Google Scholar] [CrossRef]
- Anurathapan, U.; Chan, R.C.; Hindi, H.F.; Mucharla, R.; Bajgain, P.; Hayes, B.C.; Fisher, W.E.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; et al. Kinetics of Tumor Destruction by Chimeric Antigen Receptor-Modified T Cells. Mol. Ther. 2014, 22, 623–633. [Google Scholar] [CrossRef]
- Kailayangiri, S.; Altvater, B.; Lesch, S.; Balbach, S.; Göttlich, C.; Kühnemundt, J.; Mikesch, J.-H.; Schelhaas, S.; Jamitzky, S.; Meltzer, J.; et al. EZH2 Inhibition in Ewing Sarcoma Upregulates GD2 Expression for Targeting with Gene-Modified T Cells. Mol. Ther. 2019, 27, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Driouk, L.; Gicobi, J.; Kamihara, Y.; Rutherford, K.; Dranoff, G.; Ritz, J.; Baumeister, S.H.C. Chimeric Antigen Receptor T Cells Targeting NKG2D-Ligands Show Robust Efficacy Against Acute Myeloid Leukemia and T-Cell Acute Lymphoblastic Leukemia. Blood 2019, 134, 1930. [Google Scholar] [CrossRef]
- Ruella, M.; Barrett, D.M.; Kenderian, S.S.; Shestova, O.; Hofmann, T.J.; Perazzelli, J.; Klichinsky, M.; Aikawa, V.; Nazimuddin, F.; Kozlowski, M.; et al. Dual CD19 and CD123 Targeting Prevents Antigen-Loss Relapses after CD19-Directed Immunotherapies. J. Clin. Investig. 2016, 126, 3814–3826. [Google Scholar] [CrossRef] [PubMed]
- Fousek, K.; Watanabe, J.; Joseph, S.K.; George, A.; An, X.; Byrd, T.T.; Morris, J.S.; Luong, A.; Martínez-Paniagua, M.A.; Sanber, K.; et al. CAR T-Cells That Target Acute B-Lineage Leukemia Irrespective of CD19 Expression. Leukemia 2021, 35, 75–89. [Google Scholar] [CrossRef]
- Wilkie, S.; Van Schalkwyk, M.C.I.; Hobbs, S.; Davies, D.M.; Van Der Stegen, S.J.C.; Pereira, A.C.P.; Burbridge, S.E.; Box, C.; Eccles, S.A.; Maher, J. Dual Targeting of ErbB2 and MUC1 in Breast Cancer Using Chimeric Antigen Receptors Engineered to Provide Complementary Signaling. J. Clin. Immunol. 2012, 32, 1059–1070. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.S.; Hong, J.A.; Irvine, K.R.; Chen, G.A.; Spiess, P.J.; Liu, Y.; Zeng, G.; Wunderlich, J.R.; Nguyen, D.M.; Restifo, N.P.; et al. De Novo Induction of a Cancer/Testis Antigen by 5-Aza-2′-Deoxycytidine Augments Adoptive Immunotherapy in a Murine Tumor Model. Cancer Res. 2006, 66, 1105–1113. [Google Scholar] [CrossRef] [PubMed]
- Tousley, A.M.; Rotiroti, M.C.; Labanieh, L.; Rysavy, L.W.; Kim, W.J.; Lareau, C.; Sotillo, E.; Weber, E.W.; Rietberg, S.P.; Dalton, G.N.; et al. Co-Opting Signalling Molecules Enables Logic-Gated Control of CAR T Cells. Nature 2023, 615, 507–516. [Google Scholar] [CrossRef]
- Soysal, S.D.; Tzankov, A.; Muenst, S.E. Role of the Tumor Microenvironment in Breast Cancer. Pathobiology 2015, 82, 142–152. [Google Scholar] [CrossRef]
- Li, J.J.; Tsang, J.Y.; Tse, G.M. Tumor Microenvironment in Breast Cancer—Updates on Therapeutic Implications and Pathologic Assessment. Cancers 2021, 13, 4233. [Google Scholar] [CrossRef]
- Wu, S.Z.; Al-Eryani, G.; Roden, D.L.; Junankar, S.; Harvey, K.; Andersson, A.; Thennavan, A.; Wang, C.; Torpy, J.R.; Bartonicek, N.; et al. A Single-Cell and Spatially Resolved Atlas of Human Breast Cancers. Nat. Genet. 2021, 53, 1334–1347. [Google Scholar] [CrossRef]
- Tan, Z.; Kan, C.; Sun, M.; Yang, F.; Wong, M.; Wang, S.; Zheng, H. Mapping Breast Cancer Microenvironment Through Single-Cell Omics. Front. Immunol. 2022, 13, 868813. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Deng, W.; Deng, X.; Liang, J.Y.; Tang, Y.; Huang, J.; Tang, H.; Zou, Y.; Zhou, H.; Xie, X. Single-Cell Histone Chaperones Patterns Guide Intercellular Communication of Tumor Microenvironment That Contribute to Breast Cancer Metastases. Cancer Cell Int. 2023, 23, 311. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Yang, C.; Peng, A.; Sun, T.; Ji, X.; Mi, J.; Wei, L.; Shen, S.; Feng, Q. Pan-Cancer Spatially Resolved Single-Cell Analysis Reveals the Crosstalk between Cancer-Associated Fibroblasts and Tumor Microenvironment. Mol. Cancer 2023, 22, 170. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Xu, M.; Xu, S.; Guan, Q.; Geng, S.; Wang, J.; Wei, W.; Xu, H.; Liu, Y.; Meng, Y.; et al. Single-Cell RNA Reveals a Tumorigenic Microenvironment in the Interface Zone of Human Breast Tumors. Breast Cancer Res. 2023, 25, 100. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhen, F.; Sun, Y.; Han, B.; Wang, H.; Zhang, Y.; Zhang, H.; Hu, J. Single-Cell RNA Sequencing Reveals Small Extracellular Vesicles Derived from Malignant Cells That Contribute to Angiogenesis in Human Breast Cancers. J. Transl. Med. 2023, 21, 570. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Luo, Y.; Wu, N. TCL1A+ B Cells Predict Prognosis in Triple-Negative Breast Cancer through Integrative Analysis of Single-Cell and Bulk Transcriptomic Data. Open Life Sci. 2023, 18, 20220707. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.A.E.; Oda, J.M.M.; Amarante, M.K.; Cesar Voltarelli, J. Regulatory T Cells and Breast Cancer: Implications for Immunopathogenesis. Cancer Metastasis Rev. 2010, 29, 569–579. [Google Scholar] [CrossRef]
- Hashemi, V.; Maleki, L.A.; Esmaily, M.; Masjedi, A.; Ghalamfarsa, G.; Namdar, A.; Yousefi, M.; Yousefi, B.; Jadidi-Niaragh, F. Regulatory T Cells in Breast Cancer as a Potent Anti-Cancer Therapeutic Target. Int. Immunopharmacol. 2020, 78, 106087. [Google Scholar] [CrossRef]
- Kos, K.; De Visser, K.E. The Multifaceted Role of Regulatory T Cells in Breast Cancer. Annu. Rev. Cancer Biol. 2020, 5, 291–310. [Google Scholar] [CrossRef]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T Cells in Cancer Immunosuppression—Implications for Anticancer Therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef]
- Lucca, L.E.; Dominguez-Villar, M. Modulation of Regulatory T Cell Function and Stability by Co-Inhibitory Receptors. Nat. Rev. Immunol. 2020, 20, 680–693. [Google Scholar] [CrossRef] [PubMed]
- Bohling, S.D.; Allison, K.H. Immunosuppressive Regulatory T Cells Are Associated with Aggressive Breast Cancer Phenotypes: A Potential Therapeutic Target. Mod. Pathol. 2008, 21, 1527–1532. [Google Scholar] [CrossRef] [PubMed]
- Stenström, J.; Hedenfalk, I.; Hagerling, C. Regulatory T Lymphocyte Infiltration in Metastatic Breast Cancer—An Independent Prognostic Factor That Changes with Tumor Progression. Breast Cancer Res. 2021, 23, 27. [Google Scholar] [CrossRef] [PubMed]
- Plitas, G.; Konopacki, C.; Wu, K.; Bos, P.D.; Morrow, M.; Putintseva, E.V.; Chudakov, D.M.; Rudensky, A.Y. Regulatory T Cells Exhibit Distinct Features in Human Breast Cancer. Immunity 2016, 45, 1122–1134. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.J.; Zhao, J.W.; Zhang, D.H.; Zheng, A.H.; Wu, G.Q. Immunotherapy of Cancer by Targeting Regulatory T Cells. Int. Immunopharmacol. 2022, 104, 108469. [Google Scholar] [CrossRef]
- Tanaka, A.; Sakaguchi, S. Regulatory T Cells in Cancer Immunotherapy. Cell Res. 2017, 27, 109–118. [Google Scholar] [CrossRef]
- Taylor, N.A.; Vick, S.C.; Iglesia, M.D.; Brickey, W.J.; Midkiff, B.R.; McKinnon, K.P.; Reisdorf, S.; Anders, C.K.; Carey, L.A.; Parker, J.S.; et al. Treg Depletion Potentiates Checkpoint Inhibition in Claudin-Low Breast Cancer. J. Clin. Investig. 2017, 127, 3472–3483. [Google Scholar] [CrossRef]
- Terranova-Barberio, M.; Thomas, S.; Ali, N.; Pawlowska, N.; Park, J.; Krings, G.; Rosenblum, M.D.; Budillon, A.; Munster, P.N. HDAC Inhibition Potentiates Immunotherapy in Triple Negative Breast Cancer. Oncotarget 2017, 8, 114156–114172. [Google Scholar] [CrossRef]
- Ge, Y.; Domschke, C.; Stoiber, N.; Schott, S.; Heil, J.; Rom, J.; Blumenstein, M.; Thum, J.; Sohn, C.; Schneeweiss, A.; et al. Metronomic Cyclophosphamide Treatment in Metastasized Breast Cancer Patients: Immunological Effects and Clinical Outcome. Cancer Immunol. Immunother. 2012, 61, 353–362. [Google Scholar] [CrossRef]
- Generali, D.; Bates, G.; Berruti, A.; Brizzi, M.P.; Campo, L.; Bonardi, S.; Bersiga, A.; Allevi, G.; Milani, M.; Aguggini, S.; et al. Immunomodulation of FOXP3+ Regulatory T Cells by the Aromatase Inhibitor Letrozole in Breast Cancer Patients. Clin. Cancer Res. 2009, 15, 1046–1051. [Google Scholar] [CrossRef]
- Rech, A.J.; Mick, R.; Recio, A.; DeMichele, A.; Tweed, C.K.; Fox, K.R.; Domchek, S.M.; Vonderheide, R.H. Phase I Study of Anti-CD25 Mab Daclizumab to Deplete Regulatory T Cells Prior to Telomerase/Survivin Peptide Vaccination in Patients (Pts) with Metastatic Breast Cancer (MBC) (Meeting Abstract). J. Clin. Oncol. 2010, 28, 2508. [Google Scholar] [CrossRef]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Law, A.M.K.; Valdes-Mora, F.; Gallego-Ortega, D. Myeloid-Derived Suppressor Cells as a Therapeutic Target for Cancer. Cells 2020, 9, 561. [Google Scholar] [CrossRef] [PubMed]
- Cassetta, L.; Baekkevold, E.S.; Brandau, S.; Bujko, A.; Cassatella, M.A.; Dorhoi, A.; Krieg, C.; Lin, A.; Loré, K.; Marini, O.; et al. Deciphering Myeloid-Derived Suppressor Cells: Isolation and Markers in Humans, Mice and Non-Human Primates. Cancer Immunol. Immunother. 2019, 68, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Brandau, S.; Chen, S.-H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for Myeloid-Derived Suppressor Cell Nomenclature and Characterization Standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [PubMed]
- De Cicco, P.; Ercolano, G.; Ianaro, A. The New Era of Cancer Immunotherapy: Targeting Myeloid-Derived Suppressor Cells to Overcome Immune Evasion. Front. Immunol. 2020, 11, 1680. [Google Scholar] [CrossRef] [PubMed]
- Bergenfelz, C.; Larsson, A.M.; Von Stedingk, K.; Gruvberger-Saal, S.; Aaltonen, K.; Jansson, S.; Jernström, H.; Janols, H.; Wullt, M.; Bredberg, A.; et al. Systemic Monocytic-MDSCs Are Generated from Monocytes and Correlate with Disease Progression in Breast Cancer Patients. PLoS ONE 2015, 10, e0127028. [Google Scholar] [CrossRef]
- Bergenfelz, C.; Roxå, A.; Mehmeti, M.; Leandersson, K.; Larsson, A.M. Clinical Relevance of Systemic Monocytic-MDSCs in Patients with Metastatic Breast Cancer. Cancer Immunol. Immunother. 2020, 69, 435–448. [Google Scholar] [CrossRef]
- Di, S.; Zhou, M.; Pan, Z.; Sun, R.; Chen, M.; Jiang, H.; Shi, B.; Luo, H.; Li, Z. Combined Adjuvant of Poly I:C Improves Antitumor Effects of CAR-T Cells. Front. Oncol. 2019, 9, 241. [Google Scholar] [CrossRef]
- Bauer, R.; Udonta, F.; Wroblewski, M.; Ben-Batalla, I.; Santos, I.M.; Taverna, F.; Kuhlencord, M.; Gensch, V.; Päsler, S.; Vinckier, S.; et al. Blockade of Myeloid-Derived Suppressor Cell Expansion with All-Trans Retinoic Acid Increases the Efficacy of Antiangiogenic Therapy. Cancer Res. 2018, 78, 3220–3232. [Google Scholar] [CrossRef]
- Christmas, B.J.; Rafie, C.I.; Hopkins, A.C.; Scott, B.A.; Ma, H.S.; Cruz, K.A.; Woolman, S.; Armstrong, T.D.; Connolly, R.M.; Azad, N.A.; et al. Entinostat Converts Immune-Resistant Breast and Pancreatic Cancers into Checkpoint-Responsive Tumors by Reprogramming Tumor-Infiltrating MDSCs. Cancer Immunol. Res. 2018, 6, 1561–1577. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Luo, H.; Su, J.; Di, S.; Zhou, M.; Shi, B.; Sun, Y.; Du, G.; Zhang, H.; Jiang, H.; et al. Olaparib Suppresses MDSC Recruitment via SDF1α/CXCR4 Axis to Improve the Anti-Tumor Efficacy of CAR-T Cells on Breast Cancer in Mice. Mol. Ther. 2021, 29, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Kumar, V.; Ramachandran, I.R.; Youn, J.I.; Celis, E.; Finnberg, N.; El-Deiry, W.S.; Winograd, R.; Vonderheide, R.H.; English, N.R.; et al. ER Stress Regulates Myeloid-Derived Suppressor Cell Fate through TRAIL-R-Mediated Apoptosis. J. Clin. Investig. 2014, 124, 2626–2639. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, G.A.; Condamine, T.; Mony, S.; Hashimoto, A.; Wang, F.; Liu, Q.; Forero, A.; Bendell, J.; Witt, R.; Hockstein, N.; et al. Selective Targeting of Myeloid-Derived Suppressor Cells in Cancer Patients Using DS-8273a, an Agonistic TRAIL-R2 Antibody. Clin. Cancer Res. 2017, 23, 2942–2950. [Google Scholar] [CrossRef] [PubMed]
- Cha, Y.J.; Koo, J.S. Role of Tumor-Associated Myeloid Cells in Breast Cancer. Cells 2020, 9, 1785. [Google Scholar] [CrossRef] [PubMed]
- Cendrowicz, E.; Sas, Z.; Bremer, E.; Rygiel, T.P. The Role of Macrophages in Cancer Development and Therapy. Cancers 2021, 13, 1946. [Google Scholar] [CrossRef] [PubMed]
- Bozorgi, A.; Bozorgi, M.; Khazaei, M. Immunotherapy and Immunoengineering for Breast Cancer; a Comprehensive Insight into CAR-T Cell Therapy Advancements, Challenges and Prospects. Cell. Oncol. 2022, 45, 755–777. [Google Scholar] [CrossRef]
- Zhang, Y.; Cheng, S.; Zhang, M.; Zhen, L.; Pang, D.; Zhang, Q.; Li, Z. High-Infiltration of Tumor-Associated Macrophages Predicts Unfavorable Clinical Outcome for Node-Negative Breast Cancer. PLoS ONE 2013, 8, e76147. [Google Scholar] [CrossRef]
- Mehta, A.K.; Kadel, S.; Townsend, M.G.; Oliwa, M.; Guerriero, J.L. Macrophage Biology and Mechanisms of Immune Suppression in Breast Cancer. Front. Immunol. 2021, 12, 643771. [Google Scholar] [CrossRef]
- Ye, X.; Li, Y.; Stawicki, S.; Couto, S.; Eastham-Anderson, J.; Kallop, D.; Weimer, R.; Wu, Y.; Pei, L. An Anti-Axl Monoclonal Antibody Attenuates Xenograft Tumor Growth and Enhances the Effect of Multiple Anticancer Therapies. Oncogene 2010, 29, 5254–5264. [Google Scholar] [CrossRef]
- Cha, J.H.; Chan, L.C.; Wang, Y.N.; Chu, Y.Y.; Wang, C.H.; Lee, H.H.; Xia, W.; Shyu, W.C.; Liu, S.P.; Yao, J.; et al. Ephrin Receptor A10 Monoclonal Antibodies and the Derived Chimeric Antigen Receptor T Cells Exert an Antitumor Response in Mouse Models of Triple-Negative Breast Cancer. J. Biol. Chem. 2022, 298, 101817. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.Y.; Nguyen, A.V.; Russell, R.G.; Pollard, J.W. Colony-Stimulating Factor 1 Promotes Progression of Mammary Tumors to Malignancy. J. Exp. Med. 2001, 193, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Goswami, S.; Sahai, E.; Wyckoff, J.B.; Cammer, M.; Cox, D.; Pixley, F.J.; Stanley, E.R.; Segall, J.E.; Condeelis, J.S. Macrophages Promote the Invasion of Breast Carcinoma Cells via a Colony-Stimulating Factor-1/Epidermal Growth Factor Paracrine Loop. Cancer Res. 2005, 65, 5278–5283. [Google Scholar] [CrossRef] [PubMed]
- Strachan, D.C.; Ruffell, B.; Oei, Y.; Bissell, M.J.; Coussens, L.M.; Pryer, N.; Daniel, D. CSF1R Inhibition Delays Cervical and Mammary Tumor Growth in Murine Models by Attenuating the Turnover of Tumor-Associated Macrophages and Enhancing Infiltration by CD8+ T Cells. Oncoimmunology 2013, 2, e26968. [Google Scholar] [CrossRef]
- Gomez-Roca, C.A.; Italiano, A.; Le Tourneau, C.; Cassier, P.A.; Toulmonde, M.; D’Angelo, S.P.; Campone, M.; Weber, K.L.; Loirat, D.; Cannarile, M.A.; et al. Phase i Study of Emactuzumab Single Agent or in Combination with Paclitaxel in Patients with Advanced/Metastatic Solid Tumors Reveals Depletion of Immunosuppressive M2-like Macrophages. Ann. Oncol. 2019, 30, 1381–1392. [Google Scholar] [CrossRef]
- Guerriero, J.L.; Sotayo, A.; Ponichtera, H.E.; Castrillon, J.A.; Pourzia, A.L.; Schad, S.; Johnson, S.F.; Carrasco, R.D.; Lazo, S.; Bronson, R.T.; et al. Class IIa HDAC Inhibition Reduces Breast Tumours and Metastases through Anti-Tumour Macrophages. Nature 2017, 543, 428–432. [Google Scholar] [CrossRef]
- Georgoudaki, A.M.; Prokopec, K.E.; Boura, V.F.; Hellqvist, E.; Sohn, S.; Östling, J.; Dahan, R.; Harris, R.A.; Rantalainen, M.; Klevebring, D.; et al. Reprogramming Tumor-Associated Macrophages by Antibody Targeting Inhibits Cancer Progression and Metastasis. Cell Rep. 2016, 15, 2000–2011. [Google Scholar] [CrossRef]
- Insua-Rodríguez, J.; Oskarsson, T. The Extracellular Matrix in Breast Cancer. Adv. Drug Deliv. Rev. 2016, 97, 41–55. [Google Scholar] [CrossRef]
- Oskarsson, T. Extracellular Matrix Components in Breast Cancer Progression and Metastasis. Breast 2013, 22, S66–S72. [Google Scholar] [CrossRef]
- Papanicolaou, M.; Parker, A.L.; Yam, M.; Filipe, E.C.; Wu, S.Z.; Chitty, J.L.; Wyllie, K.; Tran, E.; Mok, E.; Nadalini, A.; et al. Temporal Profiling of the Breast Tumour Microenvironment Reveals Collagen XII as a Driver of Metastasis. Nat. Commun. 2022, 13, 4587. [Google Scholar] [CrossRef]
- Acerbi, I.; Cassereau, L.; Dean, I.; Shi, Q.; Au, A.; Park, C.; Chen, Y.Y.; Liphardt, J.; Hwang, E.S.; Weaver, V.M. Human Breast Cancer Invasion and Aggression Correlates with ECM Stiffening and Immune Cell Infiltration. Integr. Biol. 2015, 7, 1120–1134. [Google Scholar] [CrossRef] [PubMed]
- Jena, M.K.; Janjanam, J. Role of Extracellular Matrix in Breast Cancer Development: A Brief Update. F1000Research 2018, 7, 274. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wu, B.; Chiang, H.-C.; Deng, H.; Zhang, X.; Xiong, W.; Liu, J.; Rozeboom, A.M.; Harris, B.T.; Blommaert, E.; et al. Tumour DDR1 Promotes Collagen Fibre Alignment to Instigate Immune Exclusion. Nature 2021, 599, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Zhang, W.; Sun, T. DDR1 Promotes Breast Tumor Growth by Suppressing Antitumor Immunity. Oncol. Rep. 2019, 42, 2844–2854. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, L.; Su, H.F.; Liu, Q.; Shen, J.; Dai, H.; Zheng, W.; Lu, Y.; Zhang, W.; Bei, Y.; et al. Chimeric Antigen Receptor Macrophage Therapy for Breast Tumours Mediated by Targeting the Tumour Extracellular Matrix. Br. J. Cancer 2019, 121, 837–845. [Google Scholar] [CrossRef]
- Hu, D.; Li, Z.; Zheng, B.; Lin, X.; Pan, Y.; Gong, P.; Zhuo, W.; Hu, Y.; Chen, C.; Chen, L.; et al. Cancer-Associated Fibroblasts in Breast Cancer: Challenges and Opportunities. Cancer Commun. 2022, 42, 401–434. [Google Scholar] [CrossRef]
- Bughda, R.; Dimou, P.; D’Souza, R.R.; Klampatsa, A. Fibroblast Activation Protein (FAP)-Targeted CAR-T Cells: Launching an Attack on Tumor Stroma. ImmunoTargets Ther. 2021, 10, 313–323. [Google Scholar] [CrossRef]
- Elwakeel, E.; Weigert, A. Breast Cancer Cafs: Spectrum of Phenotypes and Promising Targeting Avenues. Int. J. Mol. Sci. 2021, 22, 11636. [Google Scholar] [CrossRef]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal Fibroblasts Present in Invasive Human Breast Carcinomas Promote Tumor Growth and Angiogenesis through Elevated SDF-1/CXCL12 Secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef]
- Chen, B.; Sang, Y.; Song, X.; Zhang, D.; Wang, L.; Zhao, W.; Liang, Y.; Zhang, N.; Yang, Q. Exosomal MiR-500a-5p Derived from Cancer-Associated Fibroblasts Promotes Breast Cancer Cell Proliferation and Metastasis through Targeting USP28. Theranostics 2021, 11, 3932–3947. [Google Scholar] [CrossRef]
- Tran, E.; Chinnasamy, D.; Yu, Z.; Morgan, R.A.; Lee, C.C.R.; Restifo, N.P.; Rosenberg, S.A. Immune Targeting of Fibroblast Activation Protein Triggers Recognition of Multipotent Bone Marrow Stromal Cells and Cachexia. J. Exp. Med. 2013, 210, 1065–1068. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-C.S.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting Fibroblast Activation Protein in Tumor Stroma with Chimeric Antigen Receptor T Cells Can Inhibit Tumor Growth and Augment Host Immunity without Severe Toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Kakarla, S.; Chow, K.K.H.; Mata, M.; Shaffer, D.R.; Song, X.T.; Wu, M.F.; Liu, H.; Wang, L.L.; Rowley, D.R.; Pfizenmaier, K.; et al. Antitumor Effects of Chimeric Receptor Engineered Human T Cells Directed to Tumor Stroma. Mol. Ther. 2013, 21, 1611–1620. [Google Scholar] [CrossRef] [PubMed]
- Xing, H.; Yang, X.; Xu, Y.; Tang, K.; Tian, Z.; Chen, Z.; Zhang, Y.; Xue, Z.; Rao, Q.; Wang, M.; et al. Anti-Tumor Effects of Vascular Endothelial Growth Factor/Vascular Endothelial Growth Factor Receptor Binding Domain-Modified Chimeric Antigen Receptor T Cells. Cytotherapy 2021, 23, 810–819. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, F.C.; Longatto Filho, A.; Lopes, J.M. Angiogenesis and Breast Cancer. J. Oncol. 2010, 2010, 576384. [Google Scholar] [CrossRef]
- Akbari, P.; Katsarou, A.; Daghighian, R.; van Mil, L.W.H.G.; Huijbers, E.J.M.; Griffioen, A.W.; van Beijnum, J.R. Directing CAR T Cells towards the Tumor Vasculature for the Treatment of Solid Tumors. Biochim. Biophys. Acta-Rev. Cancer 2022, 1877, 188701. [Google Scholar] [CrossRef]
- Ghiabi, P.; Jiang, J.; Pasquier, J.; Maleki, M.; Abu-Kaoud, N.; Halabi, N.; Guerrouahen, B.S.; Rafii, S.; Rafii, A. Breast Cancer Cells Promote a Notch-Dependent Mesenchymal Phenotype in Endothelial Cells Participating to a pro-Tumoral Niche. J. Transl. Med. 2015, 13, 27. [Google Scholar] [CrossRef]
- Byrd, T.T.; Fousek, K.; Pignata, A.; Szot, C.; Samaha, H.; Seaman, S.; Dobrolecki, L.; Salsman, V.S.; Oo, H.Z.; Bielamowicz, K.; et al. TEM8/ANTXR1-Specific CAR T Cells as a Targeted Therapy for Triple-Negative Breast Cancer. Cancer Res. 2018, 78, 489–500. [Google Scholar] [CrossRef]
- Duan, Z.; Li, Z.; Wang, Z.; Chen, C.; Luo, Y. Chimeric Antigen Receptor Macrophages Activated through TLR4 or IFN-γ Receptors Suppress Breast Cancer Growth by Targeting VEGFR2. Cancer Immunol. Immunother. 2023, 72, 3243–3257. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, L.; Li, X.F. Hypoxia and the Tumor Microenvironment. Technol. Cancer Res. Treat. 2021, 20, 15330338211036304. [Google Scholar] [CrossRef]
- Schurich, A.; Magalhaes, I.; Mattsson, J. Metabolic Regulation of CAR T Cell Function by the Hypoxic Microenvironment in Solid Tumors. Immunotherapy 2019, 11, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.D.; Sowell, R.T.; Kaech, S.M.; Pearce, E.L. Metabolic Instruction of Immunity. Cell 2017, 169, 570–586. [Google Scholar] [CrossRef] [PubMed]
- Lourdes Mora-García, M.; García-Rocha, R.; Morales-Ramírez, O.; Montesinos, J.J.; Weiss-Steider, B.; Hernández-Montes, J.; Ávila-Ibarra, L.R.; Don-López, C.A.; Velasco-Velázquez, M.A.; Gutiérrez-Serrano, V.; et al. Mesenchymal Stromal Cells Derived from Cervical Cancer Produce High Amounts of Adenosine to Suppress Cytotoxic T Lymphocyte Functions. J. Transl. Med. 2016, 14, 302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ertl, H.C.J. Depletion of FAP+ Cells Reduces Immunosuppressive Cells and Improves Metabolism and Functions CD8+T Cells within Tumors. Oncotarget 2016, 7, 23282–23299. [Google Scholar] [CrossRef] [PubMed]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of Antitumor Immunity by Stromal Cells Expressing Fibroblast Activation Protein–α. Science 2010, 330, 827–830. [Google Scholar] [CrossRef]
- Lo, A.; Wang, L.-C.S.; Scholler, J.; Monslow, J.; Avery, D.; Newick, K.; O’Brien, S.; Evans, R.A.; Bajor, D.J.; Clendenin, C.; et al. Tumor-Promoting Desmoplasia Is Disrupted by Depleting FAP-Expressing Stromal Cells. Cancer Res. 2015, 75, 2800–2810. [Google Scholar] [CrossRef]
- Leone, R.D.; Sun, I.M.; Oh, M.H.; Sun, I.H.; Wen, J.; Englert, J.; Powell, J.D. Inhibition of the Adenosine A2a Receptor Modulates Expression of T Cell Coinhibitory Receptors and Improves Effector Function for Enhanced Checkpoint Blockade and ACT in Murine Cancer Models. Cancer Immunol. Immunother. 2018, 67, 1271–1284. [Google Scholar] [CrossRef]
- Zohair, B.; Chraa, D.; Rezouki, I.; Benthami, H.; Razzouki, I.; Elkarroumi, M.; Olive, D.; Karkouri, M.; Badou, A. The Immune Checkpoint Adenosine 2A Receptor Is Associated with Aggressive Clinical Outcomes and Reflects an Immunosuppressive Tumor Microenvironment in Human Breast Cancer. Front. Immunol. 2023, 14, 1201632. [Google Scholar] [CrossRef]
- Giuffrida, L.; Sek, K.; Henderson, M.A.; Lai, J.; Chen, A.X.Y.; Meyran, D.; Todd, K.L.; Petley, E.V.; Mardiana, S.; Mølck, C.; et al. CRISPR/Cas9 Mediated Deletion of the Adenosine A2A Receptor Enhances CAR T Cell Efficacy. Nat. Commun. 2021, 12, 3236. [Google Scholar] [CrossRef]
- Leen, A.M.; Sukumaran, S.; Watanabe, N.; Mohammed, S.; Keirnan, J.; Yanagisawa, R.; Anurathapan, U.; Rendon, D.; Heslop, H.E.; Rooney, C.M.; et al. Reversal of Tumor Immune Inhibition Using a Chimeric Cytokine Receptor. Mol. Ther. 2014, 22, 1211–1220. [Google Scholar] [CrossRef]
- Conticello, C.; Pedini, F.; Zeuner, A.; Patti, M.; Zerilli, M.; Stassi, G.; Messina, A.; Peschle, C.; De Maria, R. IL-4 Protects Tumor Cells from Anti-CD95 and Chemotherapeutic Agents via Up-Regulation of Antiapoptotic Proteins. J. Immunol. 2004, 172, 5467–5477. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.-C.; Hwang, Y.-S.; Chen, Y.-Y.; Liu, C.-L.; Shen, C.-N.; Hong, W.-H.; Lo, S.-M.; Shen, C.-R. Interleukin-4 Supports the Suppressive Immune Responses Elicited by Regulatory T Cells. Front. Immunol. 2017, 8, 1508. [Google Scholar] [CrossRef] [PubMed]
- Shum, T.; Omer, B.; Tashiro, H.; Kruse, R.L.; Wagner, D.L.; Parikh, K.; Yi, Z.; Sauer, T.; Liu, D.; Parihar, R.; et al. Constitutive Signaling from an Engineered IL7 Receptor Promotes Durable Tumor Elimination by Tumor-Redirected T Cells. Cancer Discov. 2017, 7, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, E.R.; D’Souza, R.R.; Klampatsa, A. Armored CAR T-Cells: The Next Chapter in T-Cell Cancer Immunotherapy. Biol. Targets Ther. 2021, 15, 95–105. [Google Scholar] [CrossRef]
- Zhou, Y.; Husman, T.; Cen, X.; Tsao, T.; Brown, J.; Bajpai, A.; Li, M.; Zhou, K.; Yang, L. Interleukin 15 in Cell-Based Cancer Immunotherapy. Int. J. Mol. Sci. 2022, 23, 7311. [Google Scholar] [CrossRef] [PubMed]
- Makkouk, A.; Yang, X.C.; Barca, T.; Lucas, A.; Turkoz, M.; Wong, J.T.S.; Nishimoto, K.P.; Brodey, M.M.; Tabrizizad, M.; Gundurao, S.R.Y.; et al. Off-the-Shelf Vδ 1 Gamma Delta T Cells Engineered with Glypican-3 (GPC-3)-Specific Chimeric Antigen Receptor (CAR) and Soluble IL-15 Display Robust Antitumor Efficacy against Hepatocellular Carcinoma. J. Immunother. Cancer 2021, 9, e003441. [Google Scholar] [CrossRef]
- Batra, S.A.; Rathi, P.; Guo, L.; Courtney, A.N.; Fleurence, J.; Balzeau, J.; Shaik, R.S.; Nguyen, T.P.; Wu, M.F.; Bulsara, S.; et al. Glypican-3-Specific CAR T Cells Coexpressing IL15 and IL21 Have Superior Expansion and Antitumor Activity against Hepatocellular Carcinoma. Cancer Immunol. Res. 2020, 8, 309–320. [Google Scholar] [CrossRef]
- Ruixin, S.; Yifan, L.; Chuanlong, W.; Min, Z.; Hong, L.; Guoxiu, D.; Zhengyang, L.; Yansha, S.; Yiwei, D.; Jingwen, S.; et al. Expressing IL-15/IL-18 and CXCR2 Improve Infiltration and Survival of EGFRvIII-Targeting CAR-T Cells in Breast Cancer. Biochem. Pharmacol. 2023, 212, 115536. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, C.; Landoni, E.; Metelitsa, L.; Dotti, G.; Savoldo, B. Eradication of Neuroblastoma by T Cells Redirected with an Optimized GD2-Specific Chimeric Antigen Receptor and Interleukin-15. Clin. Cancer Res. 2019, 25, 2915–2924. [Google Scholar] [CrossRef]
- Gargett, T.; Ebert, L.M.; Truong, N.T.H.; Kollis, P.M.; Sedivakova, K.; Yu, W.; Yeo, E.C.F.; Wittwer, N.L.; Gliddon, B.L.; Tea, M.N.; et al. GD2-Targeting CAR-T Cells Enhanced by Transgenic IL-15 Expression Are an Effective and Clinically Feasible Therapy for Glioblastoma. J. Immunother. Cancer 2022, 10, e005187. [Google Scholar] [CrossRef]
- Lanitis, E.; Rota, G.; Kosti, P.; Ronet, C.; Spill, A.; Seijo, B.; Romero, P.; Dangaj, D.; Coukos, G.; Irving, M. Optimized Gene Engineering of Murine CAR-T Cells Reveals the Beneficial Effects of IL-15 Coexpression. J. Exp. Med. 2021, 218, e20192203. [Google Scholar] [CrossRef] [PubMed]
- Krenciute, G.; Prinzing, B.L.; Yi, Z.; Wu, M.-F.; Liu, H.; Dotti, G.; Balyasnikova, I.V.; Gottschalk, S. Transgenic Expression of IL15 Improves Antiglioma Activity of IL13Rα2-CAR T Cells but Results in Antigen Loss Variants. Cancer Immunol. Res. 2017, 5, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Perna, S.K.; Pagliara, D.; Mahendravada, A.; Liu, H.; Brenner, M.K.; Savoldo, B.; Dotti, G. Interleukin-7 Mediates Selective Expansion of Tumor-Redirected Cytotoxic T Lymphocytes (CTLs) without Enhancement of Regulatory T-Cell Inhibition. Clin. Cancer Res. 2014, 20, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Bradley, L.M.; Haynes, L.; Swain, S.L. IL-7: Maintaining T-Cell Memory and Achieving Homeostasis. Trends Immunol. 2005, 26, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Su, J.; Sun, R.; Sun, Y.; Wang, Y.; Dong, Y.; Shi, B.; Jiang, H.; Li, Z. Coexpression of IL7 and CCL21 Increases Efficacy of CAR-T Cells in Solid Tumors without Requiring Preconditioned Lymphodepletion. Clin. Cancer Res. 2020, 26, 5494–5505. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Deng, X.; Wen, J.; Li, Y.; Zhang, M.; Cai, Z.; Liu, G.; Wang, H.; Cai, J. Folate Receptor-Alpha Targeted 7x19 CAR-Γδ T Suppressed Triple-Negative Breast Cancer Xenograft Model in Mice. J. Oncol. 2022, 2022, 2112898. [Google Scholar] [CrossRef] [PubMed]
- Swan, S.L.; Mehta, N.; Ilich, E.; Shen, S.H.; Wilkinson, D.S.; Anderson, A.R.; Segura, T.; Sanchez-Perez, L.; Sampson, J.H.; Bellamkonda, R.V. IL7 and IL7 Flt3L Co-Expressing CAR T Cells Improve Therapeutic Efficacy in Mouse EGFRvIII Heterogeneous Glioblastoma. Front. Immunol. 2023, 14, 1085547. [Google Scholar] [CrossRef]
- Li, G.; Zhang, Q.; Han, Z.; Zhu, Y.; Shen, H.; Liu, Z.; Zhou, Z.; Ding, W.; Han, S.; He, J.; et al. IL-7 and CCR2b Co-Expression-Mediated Enhanced CAR-T Survival and Infiltration in Solid Tumors. Front. Oncol. 2021, 11, 734593. [Google Scholar] [CrossRef]
- Xiong, X.; Xi, J.; Liu, Q.; Wang, C.; Jiang, Z.; Yue, S.; Shi, L.; Rong, Y. Co-expression of IL-7 and PH20 Promote Anti-GPC3 CAR-T Tumour Suppressor Activity in Vivo and in Vitro. Liver Int. 2021, 41, 1033–1043. [Google Scholar] [CrossRef]
- Yasuda, K.; Nakanishi, K.; Tsutsui, H. Interleukin-18 in Health and Disease. Int. J. Mol. Sci. 2019, 20, 649. [Google Scholar] [CrossRef]
- Hu, B.; Ren, J.; Luo, Y.; Keith, B.; Young, R.M.; Scholler, J.; Zhao, Y.; June, C.H. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep. 2017, 20, 3025–3033. [Google Scholar] [CrossRef] [PubMed]
- Carroll, R.G.; Carpenito, C.; Shan, X.; Danet-Desnoyers, G.; Liu, R.; Jiang, S.; Albelda, S.M.; Golovina, T.; Coukos, G.; Riley, J.L.; et al. Distinct Effects of IL-18 on the Engraftment and Function of Human Effector CD8 T Cells and Regulatory T Cells. PLoS ONE 2008, 3, e3289. [Google Scholar] [CrossRef] [PubMed]
- Jaspers, J.E.; Khan, J.F.; Godfrey, W.D.; Lopez, A.V.; Ciampricotti, M.; Rudin, C.M.; Brentjens, R.J. IL-18-Secreting CAR T Cells Targeting DLL3 Are Highly Effective in Small Cell Lung Cancer Models. J. Clin. Investig. 2023, 133, e166028. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Abken, H. CAR T Cells Releasing IL-18 Convert to T-Bethigh FoxO1low Effectors That Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep. 2017, 21, 3205–3219. [Google Scholar] [CrossRef] [PubMed]
- Glienke, W.; Dragon, A.C.; Zimmermann, K.; Martyniszyn-Eiben, A.; Mertens, M.; Abken, H.; Rossig, C.; Altvater, B.; Aleksandrova, K.; Arseniev, L.; et al. GMP-Compliant Manufacturing of TRUCKs: CAR T Cells Targeting GD2 and Releasing Inducible IL-18. Front. Immunol. 2022, 13, 839783. [Google Scholar] [CrossRef] [PubMed]
- Choi, G.; Shin, G.; Bae, S.J. Price and Prejudice? The Value of Chimeric Antigen Receptor (CAR) T-Cell Therapy. Int. J. Environ. Res. Public Health 2022, 19, 12366. [Google Scholar] [CrossRef] [PubMed]
- Fiorenza, S.; Ritchie, D.S.; Ramsey, S.D.; Turtle, C.J.; Roth, J.A. Value and Affordability of CAR T-Cell Therapy in the United States. Bone Marrow Transplant. 2020, 55, 1706–1715. [Google Scholar] [CrossRef]
- Hernandez, I.; Prasad, V.; Gellad, W.F. Total Costs of Chimeric Antigen Receptor T-Cell Immunotherapy. JAMA Oncol. 2018, 4, 994. [Google Scholar] [CrossRef]
- Vormittag, P.; Gunn, R.; Ghorashian, S.; Veraitch, F.S. A Guide to Manufacturing CAR T Cell Therapies. Curr. Opin. Biotechnol. 2018, 53, 164–181. [Google Scholar] [CrossRef]
- Billingsley, M.M.; Singh, N.; Ravikumar, P.; Zhang, R.; June, C.H.; Mitchell, M.J. Ionizable Lipid Nanoparticle-Mediated MRNA Delivery for Human CAR T Cell Engineering. Nano Lett. 2020, 20, 1578–1589. [Google Scholar] [CrossRef]
- Nakazawa, Y.; Huye, L.E.; Dotti, G.; Foster, A.E.; Vera, J.F.; Manuri, P.R.; June, C.H.; Rooney, C.M.; Wilson, M.H. Optimization of the PiggyBac Transposon System for the Sustained Genetic Modification of Human T Lymphocytes. J. Immunother. 2009, 32, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Vargas, J.E.; Chicaybam, L.; Stein, R.T.; Tanuri, A.; Delgado-Cañedo, A.; Bonamino, M.H. Retroviral Vectors and Transposons for Stable Gene Therapy: Advances, Current Challenges and Perspectives. J. Transl. Med. 2016, 14, 288. [Google Scholar] [CrossRef] [PubMed]
- Ivics, Z.N.; Hackett, P.B.; Plasterk, R.H.; Izsvá, Z. Molecular Reconstruction of Sleeping Beauty, a Tc1-like Transposon from Fish, and Its Transposition in Human Cells. Cell 1997, 91, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Moretti, A.; Ponzo, M.; Nicolette, C.A.; Tcherepanova, I.Y.; Biondi, A.; Magnani, C.F. The Past, Present, and Future of Non-Viral CAR T Cells. Front. Immunol. 2022, 13, 867013. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.B.; Barrett, D.M.; Karikó, K. The Emerging Role of In Vitro-Transcribed MRNA in Adoptive T Cell Immunotherapy. Mol. Ther. 2019, 27, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Soundara Rajan, T.; Gugliandolo, A.; Bramanti, P.; Mazzon, E. In Vitro-Transcribed MRNA Chimeric Antigen Receptor T Cell (IVT MRNA CAR T) Therapy in Hematologic and Solid Tumor Management: A Preclinical Update. Int. J. Mol. Sci. 2020, 21, 6514. [Google Scholar] [CrossRef] [PubMed]
- Schaft, N.; Dörrie, J.; Müller, I.; Beck, V.; Baumann, S.; Schunder, T.; Kämpgen, E.; Schuler, G. A New Way to Generate Cytolytic Tumor-Specific T Cells: Electroporation of RNA Coding for a T Cell Receptor into T Lymphocytes. Cancer Immunol. Immunother. 2006, 55, 1132–1141. [Google Scholar] [CrossRef]
- Zhao, Y.; Zheng, Z.; Cohen, C.J.; Gattinoni, L.; Palmer, D.C.; Restifo, N.P.; Rosenberg, S.A.; Morgan, R.A. High-Efficiency Transfection of Primary Human and Mouse T Lymphocytes Using RNA Electroporation. Mol. Ther. 2006, 13, 151–159. [Google Scholar] [CrossRef]
- Billingsley, M.M.; Hamilton, A.G.; Mai, D.; Patel, S.K.; Swingle, K.L.; Sheppard, N.C.; June, C.H.; Mitchell, M.J. Orthogonal Design of Experiments for Optimization of Lipid Nanoparticles for MRNA Engineering of CAR T Cells. Nano Lett. 2022, 22, 533–542. [Google Scholar] [CrossRef]
- Parayath, N.N.; Stephan, S.B.; Koehne, A.L.; Nelson, P.S.; Stephan, M.T. In Vitro-Transcribed Antigen Receptor MRNA Nanocarriers for Transient Expression in Circulating T Cells in Vivo. Nat. Commun. 2020, 11, 6080. [Google Scholar] [CrossRef]
- Caldwell, K.J.; Gottschalk, S.; Talleur, A.C. Allogeneic CAR Cell Therapy—More Than a Pipe Dream. Front. Immunol. 2021, 11, 618427. [Google Scholar] [CrossRef] [PubMed]
- Berrien-Elliott, M.M.; Jacobs, M.T.; Fehniger, T.A. Allogeneic Natural Killer Cell Therapy. Blood 2023, 141, 856–868. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.A.; Büning, H.; Sauer, M.; Schambach, A. Use of Cell and Genome Modification Technologies to Generate Improved “Off-the-Shelf” CAR T and CAR NK Cells. Front. Immunol. 2020, 11, 1965. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Hamano, Y.; Shirane, S.; Kinoshita, S.; Azusawa, Y.; Ando, J.; Nakauchi, H.; Ando, M. Advances in Allogeneic Cancer Cell Therapy and Future Perspectives on “Off-the-Shelf” T Cell Therapy Using IPSC Technology and Gene Editing. Cells 2022, 11, 269. [Google Scholar] [CrossRef] [PubMed]
- Saetersmoen, M.L.; Hammer, Q.; Valamehr, B.; Kaufman, D.S.; Malmberg, K.J. Off-the-Shelf Cell Therapy with Induced Pluripotent Stem Cell-Derived Natural Killer Cells. Semin. Immunopathol. 2019, 41, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Heipertz, E.L.; Zynda, E.R.; Stav-Noraas, T.E.; Hungler, A.D.; Boucher, S.E.; Kaur, N.; Vemuri, M.C. Current Perspectives on “Off-The-Shelf” Allogeneic NK and CAR-NK Cell Therapies. Front. Immunol. 2021, 12, 732135. [Google Scholar] [CrossRef] [PubMed]
- Daher, M.; Melo Garcia, L.; Li, Y.; Rezvani, K. CAR-NK Cells: The next Wave of Cellular Therapy for Cancer. Clin. Transl. Immunol. 2021, 10, e1274. [Google Scholar] [CrossRef]
- Liu, H.; Yang, B.; Sun, T.; Lin, L.; Hu, Y.; Deng, M.; Yang, J.; Liu, T.; Li, J.; Sun, S.; et al. Specific Growth Inhibition of ErbB2-Expressing Human Breast Cancer Cells by Genetically Modified NK-92 Cells. Oncol. Rep. 2015, 33, 95–102. [Google Scholar] [CrossRef]
- Chen, X.; Han, J.; Chu, J.; Zhang, L.; Zhang, J.; Chen, C.; Chen, L.; Wang, Y.; Wang, H.; Yi, L.; et al. A Combinational Therapy of EGFR-CAR NK Cells and Oncolytic Herpes Simplex Virus 1 for Breast Cancer Brain Metastases. Oncotarget 2016, 7, 27764–27777. [Google Scholar] [CrossRef]
- Hu, Z. Tissue Factor as a New Target for CAR-NK Cell Immunotherapy of Triple-Negative Breast Cancer. Sci. Rep. 2020, 10, 2815. [Google Scholar] [CrossRef]
- Uherek, C.; Tonn, T.; Uherek, B.; Becker, S.; Schnierle, B.; Klingemann, H.G.; Wels, W. Retargeting of Natural Killer-Cell Cytolytic Activity to ErbB2-Expressing Cancer Cells Results in Efficient and Selective Tumor Cell Destruction. Blood 2002, 100, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Sahm, C.; Schönfeld, K.; Wels, W.S. Expression of IL-15 in NK Cells Results in Rapid Enrichment and Selective Cytotoxicity of Gene-Modified Effectors That Carry a Tumor-Specific Antigen Receptor. Cancer Immunol. Immunother. 2012, 61, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-Z.; Lee, C.-C.; Cho, D.-Y.; Wang, Y.-L.; Chen, C.-Y.; Weng, C.-Y.; Chiu, S.-C.; Hung, M.-C.; Wang, S.-C. Suppression of Breast Cancer Cells Resistant to a Pure Anti-Estrogen with CAR-Transduced Natural Killer Cells. Am. J. Cancer Res. 2021, 11, 4455–4469. [Google Scholar] [PubMed]
- Liu, Y.; Zhou, Y.; Huang, K.H.; Fang, X.; Li, Y.; Wang, F.; An, L.; Chen, Q.; Zhang, Y.; Shi, A.; et al. Targeting Epidermal Growth Factor-Overexpressing Triple-Negative Breast Cancer by Natural Killer Cells Expressing a Specific Chimeric Antigen Receptor. Cell Prolif. 2020, 53, e12858. [Google Scholar] [CrossRef] [PubMed]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-Shelf’ Allogeneic CAR T Cells: Development and Challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Yin, H. Gamma Delta (Γδ) T Cells in Cancer Immunotherapy; Where It Comes from, Where It Will Go? Eur. J. Pharmacol. 2022, 919, 174803. [Google Scholar] [CrossRef] [PubMed]
- Capietto, A.-H.; Martinet, L.; Fournié, J.-J. Stimulated Γδ T Cells Increase the In Vivo Efficacy of Trastuzumab in HER-2+ Breast Cancer. J. Immunol. 2011, 187, 1031–1038. [Google Scholar] [CrossRef]
- Dhar, S.; Chiplunkar, S.V. Lysis of Aminobisphosphonate-Sensitized MCF-7 Breast Tumor Cells by Vγ9Vδ2 T Cells. Cancer Immun. 2010, 10, 1–10. [Google Scholar]
- Chen, H.; Joalland, N.; Bridgeman, J.S.; Alchami, F.S.; Jarry, U.; Khan, M.W.A.; Piggott, L.; Shanneik, Y.; Li, J.; Herold, M.J.; et al. Synergistic Targeting of Breast Cancer Stem-like Cells by Human Γδ T Cells and CD8+ T Cells. Immunol. Cell Biol. 2017, 95, 620–629. [Google Scholar] [CrossRef]
- Klebanoff, C.A.; Khong, H.T.; Antony, P.A.; Palmer, D.C.; Restifo, N.P. Sinks, Suppressors and Antigen Presenters: How Lymphodepletion Enhances T Cell-Mediated Tumor Immunotherapy. Trends Immunol. 2005, 26, 111–117. [Google Scholar] [CrossRef]
- Hu, W.; Zi, Z.; Jin, Y.; Li, G.; Shao, K.; Cai, Q.; Ma, X.; Wei, F. CRISPR/Cas9-Mediated PD-1 Disruption Enhances Human Mesothelin-Targeted CAR T Cell Effector Functions. Cancer Immunol. Immunother. 2019, 68, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.D.; Huang, A.C.; Xu, X.; Orlowski, R.; Amaravadi, R.K.; Schuchter, L.M.; Zhang, P.; Tchou, J.; Matlawski, T.; Cervini, A.; et al. Phase I Trial of Autologous RNA-Electroporated CMET-Directed CAR T Cells Administered Intravenously in Patients with Melanoma and Breast Carcinoma. Cancer Res. Commun. 2023, 3, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Furlan, S.N.; Jaeger-Ruckstuhl, C.A.; Sarvothama, M.; Berger, C.; Smythe, K.S.; Garrison, S.M.; Specht, J.M.; Lee, S.M.; Amezquita, R.A.; et al. Immunogenic Chemotherapy Enhances Recruitment of CAR-T Cells to Lung Tumors and Improves Antitumor Efficacy When Combined with Checkpoint Blockade. Cancer Cell 2021, 39, 193–208.e10. [Google Scholar] [CrossRef] [PubMed]
- Adusumilli, P.S.; Zauderer, M.G.; Rivière, I.; Solomon, S.B.; Rusch, V.W.; O’Cearbhaill, R.E.; Zhu, A.; Cheema, W.; Chintala, N.K.; Halton, E.; et al. A Phase I Trial of Regional Mesothelin-Targeted CAR T-Cell Therapy in Patients with Malignant Pleural Disease, in Combination with the Anti–PD-1 Agent Pembrolizumab. Cancer Discov. 2021, 11, 2748–2763. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.C.; Parker, L.L.; Lu, T.; Zheng, Z.; Toomey, M.A.; White, D.E.; Yao, X.; Li, Y.F.; Robbins, P.F.; Feldman, S.A.; et al. Treatment of Patients with Metastatic Cancer Using a Major Histocompatibility Complex Class II-Restricted T-Cell Receptor Targeting the Cancer Germline Antigen MAGE-A3. J. Clin. Oncol. 2017, 35, 3322–3329. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, M.; Kitano, S.; Kageyama, S.; Miyahara, Y.; Yamamoto, N.; Kato, H.; Mishima, H.; Hattori, H.; Funakoshi, T.; Kojima, T.; et al. NY-ESO-1-Specific Redirected T Cells with Endogenous TCR Knockdown Mediate Tumor Response and Cytokine Release Syndrome. J. Immunother. Cancer 2022, 10, e003811. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Salter, A.I.; Liggitt, D.; Yechan-Gunja, S.; Sarvothama, M.; Cooper, K.; Smythe, K.S.; Dudakov, J.A.; Pierce, R.H.; Rader, C.; et al. Logic-Gated ROR1 Chimeric Antigen Receptor Expression Rescues T Cell-Mediated Toxicity to Normal Tissues and Enables Selective Tumor Targeting. Cancer Cell 2019, 35, 489–503.e8. [Google Scholar] [CrossRef]
- Mhaidly, R.; Verhoeyen, E. Humanized Mice Are Precious Tools for Preclinical Evaluation of Car t and Car Nk Cell Therapies. Cancers 2020, 12, 1915. [Google Scholar] [CrossRef]
- Wang, M.; Yao, L.C.; Cheng, M.; Cai, D.; Martinek, J.; Pan, C.X.; Shi, W.; Ma, A.H.; De Vere White, R.W.; Airhart, S.; et al. Humanized Mice in Studying Efficacy and Mechanisms of PD-1-Targeted Cancer Immunotherapy. FASEB J. 2018, 32, 1537–1549. [Google Scholar] [CrossRef]
- Scherer, S.D.; Riggio, A.I.; Haroun, F.; DeRose, Y.S.; Ekiz, H.A.; Fujita, M.; Toner, J.; Zhao, L.; Li, Z.; Oesterreich, S.; et al. An Immune-Humanized Patient-Derived Xenograft Model of Estrogen-Independent, Hormone Receptor Positive Metastatic Breast Cancer. Breast Cancer Res. 2021, 23, 100. [Google Scholar] [CrossRef]
- Capasso, A.; Lang, J.; Pitts, T.M.; Jordan, K.R.; Lieu, C.H.; Davis, S.L.; Diamond, J.R.; Kopetz, S.; Barbee, J.; Peterson, J.; et al. Characterization of Immune Responses to Anti-PD-1 Mono and Combination Immunotherapy in Hematopoietic Humanized Mice Implanted with Tumor Xenografts. J. Immunother. Cancer 2019, 7, 37. [Google Scholar] [CrossRef] [PubMed]



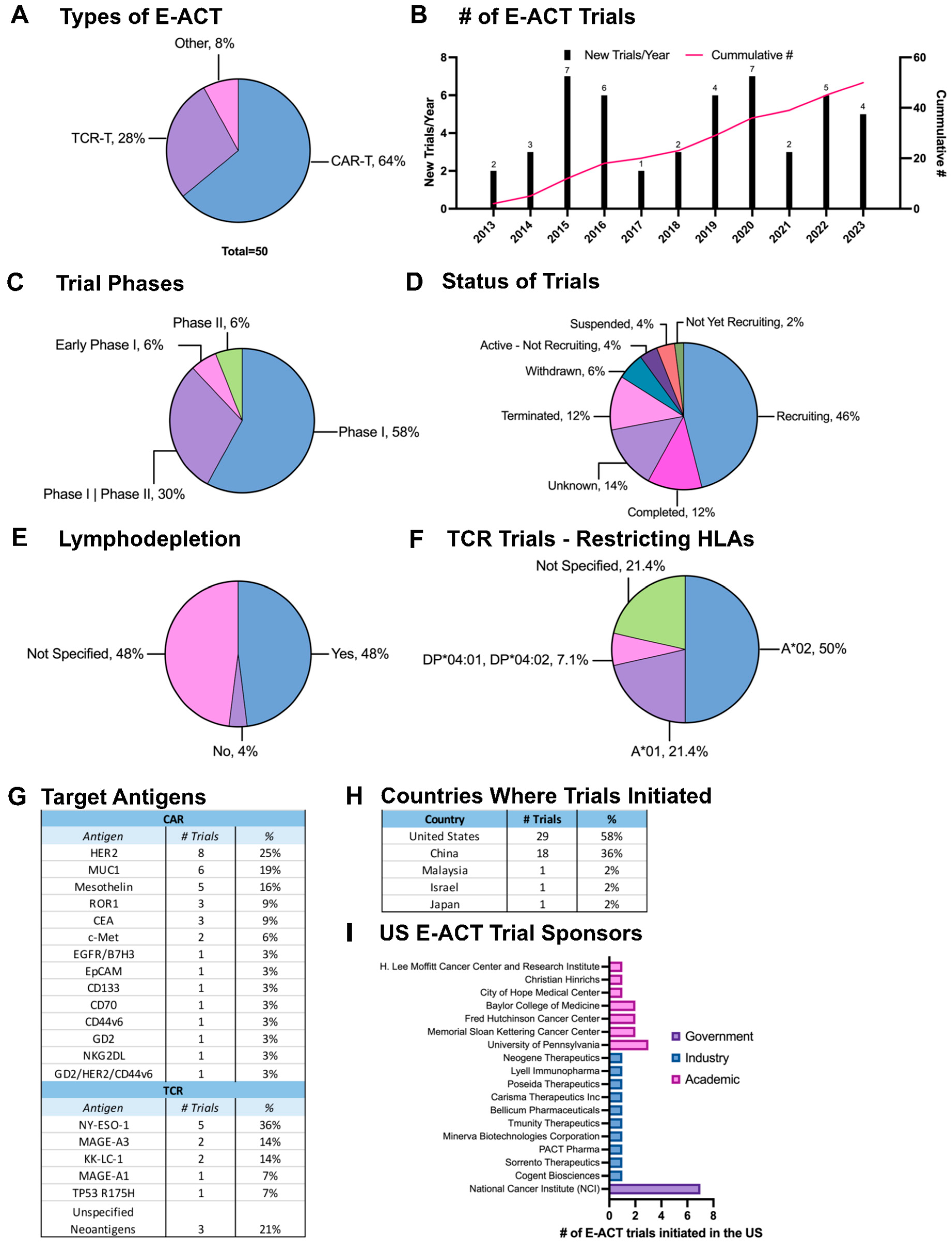

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TCR T | CAR T | |
|---|---|---|
| Constructs | Minimally engineered TCR | Fully synthetic receptor |
| MHC Restriction | Dependent | Independent |
| Affinity and Sensitivity | Lower affinity, higher sensitivity | Higher affinity, lower sensitivity |
| Antigens Recognized | Peptides presented within the MHC molecule (proteins) | Cell surface proteins/molecules |
| Origin of Antigens | Intra-/Extracellular | Cell surface |
| Co-stimulatory Molecules | Endogenous CD28, 4-1BB | Linked to scFv (CD28, 4-1BB in combination with CD3ζ |
| Probability of CRS | Lower | Higher |
| References | [8,9,10] |
| Type of Antigen | Target | Prognostic/Clinical Association | Expression in Breast Cancer | Translational Status | Ref. |
|---|---|---|---|---|---|
| TAA | HER2 | Overexpression promotes tumor proliferation, migration, and survival | HER2+ (overexpression): ~20%|HER2-low: ~45–55% | Preclinical studies Clinical trials | [3,29,36,37,38,39,40,41,42,43,44] |
| c-Met (HGFR) | Chemotherapy resistance|Poor survival|Increased tumor migration, invasion, and proliferation | ~50% of breast cancer | Preclinical studies Clinical trials | [45,46,47,48] | |
| MUC1 | Hypo-glycosylated in tumor cells|Associated with tumor invasion, metastasis, and angiogenesis | >90% of breast cancer | Preclinical studies Clinical trials | [49,50,51,52,53] | |
| Mesothelin | Metastasis|Decreased survival | 67% of TNBC | Preclinical studies Clinical trials | [54,55,56,57,58,59] | |
| EpCAM | Worse overall survival (all cases)|Unfavorable prognosis (basal-like/luminal B HER2+)|Favorable prognosis (HER2+) | 65% ER−|43% ER+|54% HER2+|47% HER2- | Preclinical studies Clinical trials | [60,61,62] | |
| ROR1 | Aggressive disease|Tumor cell growth and survival | ~40% of breast cancer 22–57% of TNBC | Preclinical studies Clinical trials | [63,64,65,66,67,68,69] | |
| CEA | Higher tumor burden|Poor overall survival | Elevated serum levels in 10.9–16.7% of patients | Clinical trials | [70,71] | |
| NKG2DL | Induced by malignant transformation of cells|May result in favorable outcomes | MIC-AB: 50%|ULBP-1: 90%|ULBP-2: 99%|ULBP-3: 100%|ULBP-4: 26%|ULBP-5: 90% | Preclinical studies Clinical trials | [72,73] | |
| CSPG4 | Disease recurrence|Poor overall survival|Tumor migration, invasion, angiogenesis, and metastasis | 77% of breast cancer | Preclinical studies | [74,75,76] | |
| FRα | Poor outcomes (early recurrence) | 30% of breast cancer|70–80% of stage IV metastatic TNBC | Preclinical studies | [77,78,79,80] | |
| Ganglioside GD2 | Stem cell marker|Tumorigenesis and migration | 35% of breast cancer | Preclinical studies Clinical trials | [81,82,83,84] | |
| EGFR | Poor prognosis|Poor disease-free survival (high EGFR copy number) | 61.2–64% of TNBC | Preclinical studies Clinical trials | [85,86,87,88] | |
| ICAM-1 | Promotes bone metastasis|Aggressive phenotype|Metastasis|Poor prognosis | Overexpressed in TNBC (% not specified) | Preclinical studies | [62,89,90,91] | |
| CD24 | Advanced stage|Shorter survival|Resistance to chemotherapy | Highest expression seen in HER2+ and TNBC samples (% not specified) | Preclinical studies | [92] | |
| AXL | Tolerance of chromosomal instability|Therapy resistance|Reduced survival|Supports EMT and metastasis | Overexpressed (% not specified) | Preclinical studies | [93,94,95,96,97,98] | |
| CGA | NY-ESO-1 | High humoral immune response|No association with overall survival or progression-free survival | 17–28.6% TNBC|12.5% HER2+ | Preclinical studies Clinical trials | [99,100,101,102] |
| MAGE-A3 | Worse prognosis|Reduced overall survival | ~10–15% of breast cancer | Preclinical studies Clinical trials | [103,104] | |
| MAGE-A1 | Lower overall survival | ~6% of breast cancer | Clinical trials | [105] | |
| KK-LC-1 | TNBC cell stemness|Poor survival|Malignant cell behavior | 75% of TNBC | Clinical trials | [106,107,108] | |
| TSA | PIK3CA H1047L | Cell transformation|Tumor proliferation|Resistance to apoptosis|Detected in tumors with favorable characteristics | PIK3CA mutations: 30–40% of breast cancer|~4% of PIK3CA mutations are H1047L | Preclinical studies | [34,109,110,111,112] |
| TP53 R175H | Cell migration/invasion through enhanced EGFR activation|Supports tumor microenvironment|Poor survival | TP53 mutations: ~30% of breast cancer, 50% of inflammatory breast cancer|TP53 R175H: 7% of breast cancer | Preclinical studies Clinical trials | [113,114,115,116,117] |
| Year | Trial ID | Target | Total # Pts | Comments | Phase | Responses | Adverse Effects | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| CAR | 2017 | NCT01837602 | c-Met | 6 | Intratumoral administration; mRNA electroporated CAR T-cells; Tumors resected two days later | 0/I | Clinical response was not measured | All grade 3 SAEs were deemed unrelated to the study drug | [47] |
| 2023 | NCT03060356 | c-Met | 7 | mRNA electroporated CAR T-cells; Up to six infusions of CAR T-cells without LD; CAR T not found in tumor biopsy; 4 TNBC patients. | I | 4/7 = 57.1% SD | No grade 3 or higher toxicity | [272] | |
| 2021 | NCT02706392 | ROR1 | 21 | CAR T-cells were seen in tumor biopsy; CAR T-cells upregulated inhibitory receptors and lost the ability to produce effector cytokines; 3 TNBC patients | I | 2/21 = 9.5% SD | All patients noted as experiencing adverse events | [273] | |
| 2021 | NCT02414269 | Mesothelin | 27 | LD, Intrapleural administration, +Pembrolizumab; 1 BC patient | I/II | 56% SD; ORR: 12.5% (PR); BC patient did not respond | No adverse events were noted | [274] | |
| TCR | 2017 | NCT02111850 | MAGE-A3 | 17 | LD and high-dose IL-2 were given; 2 BC patients | I/II | ORR: 23.5% 5.9% (CR), 17.6% (PR) BC patients did not respond | Transient G3 transaminitis (2 pts) | [275] |
| 2022 | NCT01967823 | NY-ESO-1 | 9 | LD; 1 BC patient | I | ORR: 33% (PR) BC patient did not respond | 1 Grade 3 Lung injury 3 CRS | [276] | |
| 2022 | NCT03412877 | p53 R175H | 1 | LD; Pembrolizumab given on day 16 after TCR T-cells | II | 55% decrease in tumor burden. Progressed six months post-treatment due to loss of HLA expression | Grade 3 acute CRS, resolved | [116] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chamorro, D.F.; Somes, L.K.; Hoyos, V. Engineered Adoptive T-Cell Therapies for Breast Cancer: Current Progress, Challenges, and Potential. Cancers 2024, 16, 124. https://doi.org/10.3390/cancers16010124
Chamorro DF, Somes LK, Hoyos V. Engineered Adoptive T-Cell Therapies for Breast Cancer: Current Progress, Challenges, and Potential. Cancers. 2024; 16(1):124. https://doi.org/10.3390/cancers16010124
Chicago/Turabian StyleChamorro, Diego F., Lauren K. Somes, and Valentina Hoyos. 2024. "Engineered Adoptive T-Cell Therapies for Breast Cancer: Current Progress, Challenges, and Potential" Cancers 16, no. 1: 124. https://doi.org/10.3390/cancers16010124
APA StyleChamorro, D. F., Somes, L. K., & Hoyos, V. (2024). Engineered Adoptive T-Cell Therapies for Breast Cancer: Current Progress, Challenges, and Potential. Cancers, 16(1), 124. https://doi.org/10.3390/cancers16010124