
Evolving CAR-T-Cell Therapy for Cancer Treatment: From Scientific Discovery to Cures


Department of Medicine, University of California San Francisco, San Francisco, CA 94158, USA
Cancers 2024, 16(1), 39; https://doi.org/10.3390/cancers16010039
Submission received: 21 November 2023
/
Revised: 18 December 2023
/
Accepted: 19 December 2023
/
Published: 20 December 2023
(This article belongs to the Collection Mechanism of Immunotherapy in Cancers)
Abstract
:Simple Summary
It is well recognized now that the development of drug resistance is one of the leading causes of treatment failure in conventional therapies. In comparison, recent improvements in immunotherapy showed promising results in eradicating cancer. Chimeric antigen receptor (CAR)-T-cell therapy is one of the cancer immunotherapies that uses patient’s T cells and genetically modifies them to target cancer cells. Although CAR-T-cell therapy has shown remarkable success in treating blood cancer, it has proven far more limited in the treatment of solid tumors in different organs. This review article discussed the chronological development of CAR-T-cell therapies and their treatment options in different types of cancers. This article also addresses the current clinical challenges in CAR-T-cell therapy and recent advancements in developing novel therapeutic strategies with fewer side effects.
Abstract
In recent years, chimeric antigen receptor (CAR)-T-cell therapy has emerged as the most promising immunotherapy for cancer that typically uses patients’ T cells and genetically engineered them to target cancer cells. Although recent improvements in CAR-T-cell therapy have shown remarkable success for treating hematological malignancies, the heterogeneity in tumor antigens and the immunosuppressive nature of the tumor microenvironment (TME) limits its efficacy in solid tumors. Despite the enormous efforts that have been made to make CAR-T-cell therapy more effective and have minimal side effects for treating hematological malignancies, more research needs to be conducted regarding its use in the clinic for treating various other types of cancer. The main concern for CAR-T-cell therapy is severe toxicities due to the cytokine release syndrome, whereas the other challenges are associated with complexity and immune-suppressing TME, tumor antigen heterogeneity, the difficulty of cell trafficking, CAR-T-cell exhaustion, and reduced cytotoxicity in the tumor site. This review discussed the latest discoveries in CAR-T-cell therapy strategies and combination therapies, as well as their effectiveness in different cancers. It also encompasses ongoing clinical trials; current challenges regarding the therapeutic use of CAR-T-cell therapy, especially for solid tumors; and evolving treatment strategies to improve the therapeutic application of CAR-T-cell therapy.
Keywords:
immunotherapy; immuno-oncology; combination therapy; cancer treatment; tumor microenvironment (TME); chimeric antigen receptor (CAR); checkpoint blockade; immunomodulation; solid tumor; hematological malignancies; blood cancer; B-cell lymphoma; cytokine release syndrome; TCR-T-cell receptor1. Introduction
Cancer is the primary health concern and leading cause of death worldwide. The American Cancer Society estimated that 1,958,310 new cancer cases and 609,820 cancer deaths occurred in the year 2022 in the United States [1]. Although there has been enormous progress in different types of conventional therapies for treating cancer, the development of drug resistance and drug-related toxicity makes these therapies much less effective. On the other hand, immunotherapy relies on using patients’ immune systems to identify cancer cells as foreign bodies and eradicate them via various mechanisms. Various immunotherapies have been developed for cancer, primarily by potentiating the body’s immune cells by releasing their immune suppression or empowering them to more effectively perform their immune functions. Chimeric antigen receptor (CAR)-T-cell therapy is one of the approaches of adoptive T-cell transfer (ACT) used in immunotherapy, which recently showed enormous success in terms of effectiveness and durable clinical response [2]. In this approach, T cells have been genetically altered by expressing the chimeric antigen receptor (CAR) to recognize tumor-specific antigens without the involvement of major histocompatibility complex (MHC), resulting in vigorous T-cell activation and robust anti-tumor responses [3]. CAR-T cells mediate anti-tumor effects through granzyme release, cytokine release, and other immune effectors (Figure 1).
Growing and evolving research has utilized sophisticated ex vivo culture and cellular engineering approaches to improve CAR-T-cell therapy, with long-life therapeutic responses. As a consequence of unparalleled clinical efficacy in particular B-cell malignancies, the US Food and Drug Administration (FDA) approved many CAR-T-cell therapies for treating hematological malignancy. CAR-T-cell therapy showed remarkable efficacy and profound therapeutic potencies in treating a subset of hematological malignancies by targeting lineage-restricted surface molecules [4,5,6,7,8,9]. In contrast, CAR-T-cell therapy hits roadblocks in treating solid tumors, mainly due to lack of high-quality antigen targets and poor tumor infiltration [10]. So far, most antigens targeted in solid tumors via CAR-T cells are mainly tumor-associated antigens rather than tumor-specific ones, which means that these antigens express higher levels in tumors and lower levels in some of the normal healthy tissues. These low antigen expression in normal tissue often leads to severe on-target/off-tumor toxicities. A case report showed severe side effects on a patient’s lungs after the infusion of CAR-T cells targeting HER2 antigen, which finally leads to death after a few days [11]. CAR-T cells’ clinical success also depends on CAR-T cells’ persistence; in this regard, CAR-T cells that exhibit a long-lived memory phenotype have shown much more antitumor responses than terminal effector-like CAR-T cells [12]. Antigen-independent tonic signaling is one of the main factors affecting CAR-T cells’ phenotype and longevity, there by its therapeutic response [13]. Many studies have tested innovative techniques to overcome these limitations, including targeting dual antigens, post-translational modified tumor-associated antigens, combination therapies to reduce immunosuppressive TME, and improved penetrations to the tumor site.
This article summarizes bench-to-bedside CAR-T-cell research, different clinical trials that have either been completed or are currently undergoing, limitations of this therapy, and different combinations of strategies to overcome the limitations of CAR-T-cell therapy. This review article will help future researchers and clinicians to understand the growing body of research and make newer therapeutic interventions that are more effective and have fewer side effects than current regimens of these patients.
2. Biology of CAR-T Cells in Cancer
Chimeric antigen receptors (CARs) are engineered to repurpose them to recognize specific antigens on the cancer cell’s surface. CAR contains a binding moiety in the extracellular part that recognizes and binds to target antigens in cancer cells and signaling domain(s) in the intracellular part that activates the T cells to proliferate and attack cancer cells (Figure 2). The binding moiety has a specific region called a single-chain variable fragment (scFv) that identifies specific antigens. This scFv region is derived from variable heavy (VH) and light (VL) chains of monoclonal antibodies connected via a flexible linker, as shown in Figure 2A. In comparison, the intracellular domain of CAR consists of several parts, which include a CD3ζ signaling domain (that activates the T cell) and co-stimulatory domains (that provide additional signals) [14,15,16,17,18]. Between the extra- and intra-cellular domains, there is a hinge region that provides flexibility to the extracellular domains (ECD) to help them to better recognize antigens and the transmembrane domain that anchors the CAR in the T-cell membrane.
The first generation of CAR mainly consists of scFv in the ECD and the CD3ζ chain of the T-cell receptor in the intracellular domain (Figure 2B). Although the first generation of CARs has been shown to activate T cells and induce cytokine release, they have less potency and persistency [14]. After that, the second generation of CAR was developed, with additional co-stimulatory domains to overcome these limitations of the first generations of CARs. One more co-stimulatory domain was added to the intracellular domain to develop the third generation of CARs [19,20]. This fourth generation of CARs is based on second-generation CARs, which contain an additional transgene for cytokine release. This fourth generation of CARs can activate T cells and release specific cytokines like IL-2 or IL-18, which is required to activate other immune cells and neutralize the immunosuppressive cytokines released by tumors [21,22,23]. After that, the fifth generation of CARs emerged to perform tasks such as targeting cancer cells, reducing inflammation, and recruiting other immune cells to fight cancer. In the fifth generation of CAR, a truncated intracellular domain of interleukin-2 receptor (IL-2R) was added, with a binding motif for transcription factors like STAT-3/5. In addition to activating CAR-T cells and promoting the generation of memory T-cells, the fifth generation of CARs reactivates and stimulates the immune system [24]. In addition to intracellular domains, other parts of this CAR (including the transmembrane domain and hinge region) have been modified for better recognition and binding with target antigen (Figure 2C) [25]. Due to the high antitumoral activity, potency, and persistence of the fifth generation of CARs, they have become a promising area of research in cancer immunology.
3. Generation of CAR-T Cells
The first step in CAR-T-cell therapy is to collect T cells from the patient (autologous) or a donor (allogenic). Then, these T cells need to be purified and genetically engineered to express artificially generated CARs, as shown in Figure 3 [26]. An engineered CAR gene is transduced in T cells via different methods, which include viral (lenti/retrovirus), non-viral (transposon or CRISPR/Cas9), and electroporation methods [27,28,29,30]. Next, to produce a large quantity of engineered CAR-T cells, these cells were expanded in vitro. After achieving the desired quantity and quality, they were infused back into the patient’s bloodstream. Generally, patients undergo chemotherapy before the infusion of CAR-T cells into the blood stream, which allow engineered T-cells to grow and kill cancer cells, as discussed in the Section 6.1 of this article. When CAR-T cells enter the patient’s bloodstream, they reach the cancer site, recognize specific antigens via CAR, and start performing their function, as detailed in the subsequent paragraphs and illustrated in Figure 1 [29].
4. CAR-T-Cell Therapy: From Scientific Discovery to Cures
Although CAR-T-cell therapy has recently gained much attention due to its revolutionary success in treating blood cancer, the idea has existed for several decades. It was repurposing the T-cell via genetically engineered T-cell receptors (TCRs) to kill cancer cells for the first time that was proposed in the 1980s. This approach had limitations because, at that time, it was challenging to identify specific antigens specific to cancer and make TCR specific to that antigen. This approach was even more challenging since TCR recognizes antigens in the major histocompatibility complex (MHC) setting, which may vary from patient to patient. To overcome these challenges, in 1989, it was proposed to combine the antigen-binding domain (derived from a monoclonal antibody) with the signaling domains of TCRs. This was the first generation of CAR-T cells; this approach allows the T cells to recognize the specific antigen present in cancer cells independently of MHC [31]. Although this first-generation CAR-T-cell therapy approach is more versatile than TCR, these cells have limited tumor-killing capacity because they do not grow and persist near tumors [32]. These limitations make the first generation of CAR-T cells less out of reach to the expectations of treating cancer. In the mid-1990s, the second generation of CAR-T cells was developed by adding the co-stimulatory domain to the CAR, which helps these cells to expand and survive. This approach has shown to be more efficient in preclinical models and paved the way for clinical trials. In 2002, the first effective CAR-T cells were developed by adding CD28 as a co-stimulatory domain [33]. In 2003, a study showed the complete abolition of B-cell lymphoma in a mouse model via CAR-T cells targeting the B-cell antigen CD19 [34]. In 2006, the first clinical trial was conducted with CAR targeting the tumor antigen carbonic anhydrase IX in patients with metastatic renal cell carcinoma [35]. Although this therapeutic strategy was reported to be safe for the patients, it did not yield any clinical benefits for the patients. After this step, in 2011, another clinical trial was conducted at the University of Pennsylvania, where they found clinical success in treating advanced chronic lymphocytic leukemia (CLL) using CAR-T-cell therapy [36]. This group used autologous T-cells engineered to express a CAR targeting the B-cell antigen CD19, and all patients were found to achieve complete remission after this therapy. This success led to many clinical trials with CAR-T-cell therapies, and they found various degrees of success. In 2013, a clinical trial with CAR-T-cell therapy found profound antitumor effects in patients with relapsed or refractory acute lymphoblastic leukemia (ALL) [37]. After that, in 2014, a group developed CAR-T-cell therapy for solid tumors to recognize antigen mesothelin [38]. Shortly after that, in 2015, the fourth generation of CAR-T cells was developed by adding a transgene with the second generation of CAR.
After the CRISPR system was developed, this gene-editing tool was used to make CAR-T cells in 2017 [39]. Seeing the unprecedented success of CAR-T-cell therapy, in the year 2017, the US Food and Drug Administration (FDA) approved the CAR-T-cell therapy tisagenlecleucel (Kymriah) for the treatment of relapsed or refractory ALL [40]. Consequently, the FDA approved axicabtagene ciloleucel (Yescarta), another CAR-T-cell therapy for treating relapsed or refractory diffuse large B-cell lymphoma (DLBCL) in adults [40]. After that, one by one, several CAR-T-cell therapies were approved for treating different types of cancer. Some important CAR-T-cell therapies include Abecma (idecabtagene vicleucel) for the treatment of relapsed or refractory multiple myeloma [41], Lisocabtagene maraleucel (Breyanzi) for the treatment of relapsed or refractory large B-cell lymphoma [42], Brexucabtagene autoleucel (Tecartus) for the treatment of relapsed or refractory mantle cell lymphoma [43], and Liso-cel (liso-cel) for the treatment of relapsed or refractory large B-cell lymphoma [44].
The FDA approval of CAR-T-cell therapy also expanded to other types of cancer beyond leukemia and lymphoma, and there have been many ongoing clinical trials, with some being completed, which are summarized in Table 1.
5. Current Limitations and Potential Strategies
Despite CAR-T-cell therapy’s promising success in cancer treatment, several limitations still need to be addressed. These limitations make CAR-T-cell therapy less effective for treating certain types of cancer. Several factors cause cancer cells to become resistant to this therapy, such as antigen escape, on-target/off-tumor effects, poor CAR-T-cell trafficking and tumor infiltration, immunosuppressive TME, and CAR-T-cell therapy-associated toxicities. Also, to treat cancer and prevent relapse, CAR-T cells have to persist and remain active for a longer period of time in our body. The main limiting factors for CAR-T-cell persistence include the stability of transgene expression, immune responses against CAR, and the adaptation of CAR-T cells inside the body [45]. There are different factors that contribute to severe toxicities associated with CAR-T-cell therapy, which include disease burden [46], high-dose chemotherapy regimen [47], high-dose CAR-T-cell infusion [48], and peak levels of serum cytokines and C-reactive protein [49].
Most solid tumors create immunosuppressing TMEs that obstruct tumors’ infiltration, activation, or expansion of cytotoxic T cells. Many strategies have been proposed to overcome this limitation, including the local/systemic administration of high-dose inflammatory cytokines engineering T cells to express the synthetic Notch (synNotch) receptor-mediated local production of IL-2 [50,51,52,53]. Another limitation of CAR-T-cell therapy in solid tumor is the unavailability of tumor-specific antigens. Most of the antigens that have been targeted via CAR-T-cell therapy are tumor-associated, not tumor-specific, in nature, which means these antigens also express in low levels in normal tissues, leading to severe cytotoxicity. Some strategies showed effectiveness at overcoming this issue, which include targeting two antigens rather than one antigen using the synthetic Notch receptor and CAR, as shown in Figure 4 [54], engineered CAR-T cells with synthetic Notch receptors to sense antigen density [55], and targeting tumor-associated antigens that are post-translationally modified in tumors [56,57,58,59,60,61].
The response to different doses of CAR-T-cell therapy varies from patient to patient, so another challenge of this therapy is determining the correct dose for patients. Similarly, knowing whether a single dose or an infusion of multiple small doses has the optimum effect on patients is essential [36,62]. Likewise, ex vivo T-cell expansion duration is another confounding factor. In this regard, some studies showed that less differentiated and more proliferative T cells have better anti-tumor responses in preclinical trials [63,64]. There has been a massive amount of effort made to design innovative CAR-T-cell therapy strategies to overcome these limitations, which are elaborated in the subsequent paragraph and summarized in Table 2.
6. Emerging Combination Strategies with CAR-T-Cell Therapy
CAR-T-cell therapy, in combination with other therapies, revolutionized cancer treatment, especially for solid tumors. Many studies have shown that combination therapy with CAR-T-cell therapy dramatically improved the effectiveness and reduced the side effects of treatment [51,54,95,96]. This combination of approaches was found to modulate the tumor microenvironment, enhance the CAR-T structure, connect the CAR-T cells with cancer cells, increase precision by targeting multiple antigens at the same time, bypass the tumor immune escape, and reduce the side effects. Some important combination therapies are discussed in the sections below.
6.1. Combination of CAR-T-Cell Therapy with Chemotherapy
Past studies have found that chemotherapy can induce immune function and reduce tumor burden, highlighting the possible benefits of this therapy for improving CAR-T-cell function. As expected, a study noted that this combination approach upregulated the migration of DCs and T cells into the TME. Moreover, chemotherapy has been found to elevate the secretion of damage-associated molecular patterns (DAMPs) and high-mobility group box 1 (HMGB1) from apoptotic/necrotic tumor cells [97], which, consequently, may help with the CAR-T-cell function. Similarly, tumor cells that have experienced chemotherapy can produce type I interferon (IFN- I), which can also escalate the maturation of DCs [98].
Additionally, chemotherapy can suppress the function of different immunosuppressive cells, including regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs). Chemotherapy has been found to work synergistically with CAR-T cells for targeting extended ErbB family proteins in epithelial ovarian cancer (EOC) [99]. Similarly, a clinical trial identified the improved persistence and response of CD19 CAR-T cells in pediatric/young adult relapsed/refractory B-ALL after chemotherapy with cyclophosphamide (CTX) [100].
Moreover, chemotherapy reduces autoimmunity and the levels of immunosuppressive cells, and as a result, it induces the persistence of CAR-T cells [45,101]. Many chemotherapeutic agents, like cyclophosphamide, doxorubicin, and fluorouracil, have been shown to abrogate CAR-T-cell therapy’s function through various mechanisms [102,103]. Chemotherapy is found to sensitize the tumor cells by releasing granzyme B, which helps CAR-T cells to gain easy access to the antigens of cancer cells [97]. In comparison, CAR-T-cell therapy was also found to induce the efficiency of chemotherapy. A study reported that adaptive T-cells can reduce the resistance to chemotherapy by releasing the interferon-γ (IFN-γ) in ovarian cancer [104].
Chemotherapy-stimulated cancer-related macrophages are found to help with the attachment of CAR-T cells to the tumors. A study showed that the continuous synthesis of CCL5 by cancer cells attracts T-cells that release IFN-γ upon antigen identification [105]. Studies found that chemotherapeutic agents like taxanes and vinca alkaloids can induce calreticulin and create more tumor antigens, which subsequently help CAR-T cells in their activity [106]. Some clinical trials found an improvement in the event-free survival (EFS) time when patients received extensive lymphodepleting chemotherapy regimens [107,108].
6.2. Combination of CAR-T-Cell Therapy with Radiotherapy
Similar to chemotherapy, radiotherapy has been reported to modulate TME to promote CAR-T-cell infiltration and trafficking into tumor sites [109,110].
Radiotherapy can help cytotoxic lymphocytes to access tumor sites and kill cancer cells [111,112]. Additionally, it was noted that local radiotherapy might also help CAR-T cells to enter the TME by releasing different chemokines, including (CXCL) 1, 2, 9, 10, and 16, to help T-cells prepare for their entry into the TME [113].
Radiotherapy stimulates CTLs on the local site and provides an inhibitory effect against distant tumors [114]. Hence, radiotherapy can provide additional benefits via the suppression of metastasis. A past study showed a synergic effect of radiotherapy with natural killer group 2-member D (NKG2D)-based chimeric antigen receptor construct (chNKG2D) in fully immunocompetent orthotopic glioblastoma mouse models [115].
As for the monotherapy method, antigen escape can lead to the failure to treat solid tumors. A low dose of sensitizing radiation was shown to have a better treatment response for CAR-T-cell therapy via the mitigation of antigen escape [116].
6.3. Combination of CAR-T-Cell Therapy with Oncolytic Virus
Oncolytic virotherapy is a new, growing field in cancer immunotherapy, where killer viruses are selectively used to kill cancer cells. The most critical features of oncolytic viruses include a capacity to modify the genome of cancer cells, selective and direct treatment for cancer cells, and the recruitment of the innate and adaptive immune system to induce a tumor-specific immune response [117]. Oncolytic virotherapy can be used in various aspects, such as presenting tumor-associated antigens (TAA) to the immune system as a vaccine and delivering exogenous therapeutic genes to express inside the tumor; they can also be combined with other immunotherapy, including CAR-T-cell therapy [118]. Oncolytic viruses can synergize CAR-T-cell therapy to treat solid tumors for many reasons, including the diffusion of tumor antigens via the lytic effect on tumor cells, and carry potent therapeutic chemokines [119].
One of the main limitations of CAR-T-cell therapy in treating solid tumors is the low migration capacity of CAR-T cells to the TME; different studies have reported that combining CAR-T-cell therapy with an oncolytic virus improved the migration of CAR-T cells due to the enhanced M1 polarization of macrophages and the maturation of DCs [120,121,122]. Similarly, a study found improved CAR-T-cell migration when mesothelin targeted CAR-T-cell therapy combined with an oncolytic virus expressing TGF-β in a triple-negative breast cancer model [123]. Another study noted improved performance in HER2-targeted CAR-T cells by combining an oncolytic adenovirus expressing a programmed death-ligand 1 (PD-L1) blocking antibody [124]. Moreover, a report showed that when an oncolytic virus secreted IL-12p70, in addition to a PD-L1-blocking antibody combined with CAR-T-cell therapy in head and neck squamous cell carcinoma (HNSCC) xenograft models, it showed reduced growth primary and metastasized tumors compared to individual monotherapy [125]. In addition to improving the migration of CAR-T cells, this combination strategy was found to overcome antigen escape and the off-target effects of CAR-T cells [126]. In addition to the anti-tumor effects of CAR-T cells, this combination treatment was shown to induce tumor-specific immune memory.
6.4. Combination of CAR-T-Cell Therapy with Cancer Vaccines
Cancer vaccines are generally used to expose tumor-associated epitopes to activate the body’s adaptive immune system for eradicating cancer cells. Indeed, cancer vaccines have been found to enhance the function of CAR-T cells by inducing antigen-presenting cells (APCs) or human leukocyte antigen (HLA) expression and directly stimulating dual- or bispecific CAR-T-cells into the tumor site [127]. Based on the structural differences, cancer vaccines are divided into three types, as discussed in the sections below.
6.4.1. Cellular Vaccines
In this type of cancer vaccine, whole cells/cellular components were used as sources of antigens for antigen-presenting cells. Cellular vaccines can be prepared from tumor cells, irradiated immortalized cell lines, or dendritic cells (DCs) [128]. This combination approach was first used in a whole-cell vaccine by engineering the K562 cell line to express the CMV-pp65 protein and the immune stimulatory molecules CD40 ligand CD40L and OX40 Ligand. Similarly, a clinical trial used an irradiated EBV-transformed lymphoblastoid cell line (LCL) as a cellular vaccine and identified better the persistence and expansion of CD19-CAR-CTL therapy [129].
DCs regulate innate and adaptive immune responses and present tumor antigens to T cells. To induce anti-tumor immune responses, DC-based vaccines were developed [128,130]. In CAR-T-cell therapy in blood cancer, tumor relapse is one of the limitations, so combining CAR-T-cell therapy with DC-based vaccines would be a good strategy. A group showed the better expansion and persistence of CAR-T cells via the Eps8-DC-mediated cellular vaccine [131].
6.4.2. Molecular Vaccines
In molecular vaccines, different molecules have been used (including peptides, DNA, and RNA) to load specific antigens to the APCs and induce T cells. Thus, combining molecular vaccines with CAR-T-cell therapy would be beneficial for targeting specific antigens in tumor cells and inducing CAR-T-cell-medicated killing of cancer cells. Designing and using a specific vehicle is most important as this strategy requires delivering this molecular vaccine to the APC cells. A study used a nanoparticulate vaccination platform by encapsulating ovalbumin (OVA) peptides and found a durable remission in mice [132]. Similarly, another study used liposomal antigen-encoding RNA (RNA-LPX) and noted the significant expansion and cytolysis activity of claudin 6 (CLDN6) targeting CAR-T cells [133].
6.4.3. Viral Vaccines
Many studies have investigated the use of complete viruses or virus antigens as immunogens to promote T-cell function [134,135,136]. A study restimulated CAR (+)-T cells through an endogenous cytomegalovirus (CMV)-specific T-cell receptor to enhance persistence and augment the anti-tumor activity of CD19-CAR-T cells [137]. In these bispecific T cells, both TCR and CAR are located on the same T cells, and they showed that vaccination with CMVpp65 abrogated the tumor eradication effects of CAR-T cells. Likewise, viral vaccine encoding human gp100 (VV-gp100) and showed enhanced expansion of Her2 CAR-T cells.
6.5. Combination of CAR-T-Cell Therapy with Cytokines
The TME is very immunosuppressive due to the production of various types of cytokines. To protect CAR-T cells from the immunosuppressive cytokine IL-4, a group created CAR-T cells with inverted cytokine receptors (ICRs) encoding the cytokine-binding portion of the IL-4 receptor (an immunosuppressive cytokine releases from TME) exodomain linked to the IL-7 receptor (an immunostimulatory cytokine) signaling endodomain. These CAR-T cells were directed against prostate stem cell antigen (PSCA) to target pancreatic cancer, and the authors found that these CAR-T cells persisted in an IL-4-rich TME, which resulted in enhanced anti-tumor activity [83]. Likewise, a group developed CAR-T cells with a novel 4/21 ICR, a fusion protein of the IL-4 receptor ectodomain and IL-21 receptor endodomain [138]. The whole purpose of this 4/21 ICR is to inhibit the immunosuppressive effects of IL-4 and activate STAT3 phosphorylation to ensure a better immune response [139]. Similarly, another group experimented with CAR-T cells expressing the IL-7 receptor (C7R) that targets AXL in AXL-positive triple-negative breast cancer (TNBC) [140]. This study noted that IL-7 expression augmented the cytotoxicity and survival of CAR-T cells in TNBC.
IL-15 is another cytokine that stimulates the immune system by inducing T-cell persistence and trafficking [141]. In lymphoma and leukemia research, a study noted improved performance in CAR-T cells targeting CD19 via the expression of the IL-15 gene and an inducible caspase-9-based suicide gene [142]. In addition, this study noted that iC9/CAR.19/IL-15 cells reduced the expression of PD-1 receptors and blocked CAR-T-cell exhaustion. Similarly, another study investigated CAR-T/IL-15 cells in lymphoma and glioblastoma models, and they reported the downregulation of exhaustion markers and upregulation of antiapoptotic markers in CAR-T cells [143]. Likewise, combining murine IL-15 (mIL-5) with CAR-T cells was noted to upregulate Bcl-2 (an antiapoptotic marker) and decreased PD-1 expression [144]. For the multiple myeloma (MM) model, a group found IL15 to be superior when they compared different combinations of IL-15 and IL-2 cytokines in B-cell maturation antigen (BCMA)-targeted CAR-T cells [145]. For improving the persistence of CAR-T cells, T-memory stem cells (TSCM) play a significant role, so a group showed that membrane-bound chimeric IL-15 (mbIL-15) expression improved the persistence of the TSCM phenotype [59].
6.6. Combination of CAR-T-Cell Therapy with Checkpoint Inhibition
As tumor cells invade the immune system by expressing different immune checkpoint ligands, checkpoint blockade (CPB) via immune checkpoint inhibitors (CPIs) has become one of the most promising strategies for cancer treatment. Many studies investigated the combined effects of CAR-T cells with PD-1/PD-L1 CPB, which are summarized in the sections below.
6.6.1. Antibodies-Mediated Checkpoint Blockade
Several studies have already reported that combining anti-PD-1 antibodies with CAR-T-cell therapy improved the persistence of CAR-T cells in different types of cancer [146]. A study showed enhanced therapeutic outcomes when they combined anti-Her-2 CD8 + T cells with anti-PD-1 antibodies in murine breast cancer models [147]. This study found that treatment with anti-PD-1 antibodies induced T-cell immunity by improving CAR-T cells’ activity and enhancing the expression of IFN-γ and granzyme B in both in vitro and in vivo conditions. Although this study was conducted in HER2-amplified cancer, it does not consider the possibility of an autoimmune effect on normal tissue expressing normal physiological levels of HER2 [147,148,149,150]. Despite a growing body of research showing that improved outcomes by combining the anti-PD-1 antibody therapy with CAR-T-cell therapy, there are still multiple concerns, including anti-PD-1 antibody barely reaching optimum persistence in TME when systemically infused. Also, this immune-augmentation strategy may cause uncontrolled T-cell activation; as a result, it has been found to affect several organs [151,152]. CPB antibodies can be delivered via CAR-T cells to overcome these cytotoxic effects. A study showed that preconditioning the CAR-T cells in the IL-7/IL-15 cytokines’ milieu could induce therapeutic outcomes and decrease treatment-related adverse events [153].
6.6.2. DNR and shRNA-Mediated Checkpoint Blockade
PD-1 dominant negative receptor (DNR) can be expressed in CAR-T cells to reduce PD-1-mediated immunosuppression in TME [154]. In this approach, only the ECD of PD-L1 is expressed in CAR-T cells. PD-1 DNR reduces the engagement of endogenous PD 1 receptor with PD-L1 via competitive binding and reduces the immune suppression of cancer cells [154]. In this study, the authors also made CAR-T cells with PD-1–targeting shRNAs and showed the improved performance of CAR-T cells [154]. Retrovirus-mediated shRNA targeting adenosine 2A receptors (A2ARs) was examined in HER2-targeted CAR-T cells, and we found improved function in CAR-T cells [155]. Likewise, the knockdown of cytotoxic T-lymphocyte associated protein 4 (CTLA-4) in CAR-T cells via shRNA also showed beneficial effects for the performance of CAR-T cells [156].
6.6.3. Checkpoint Blockade via Gene Editing
Single-Gene Editing
In 2019, a group used the CRISPR/Cas9 tool to delete the programmed cell death protein 1 (PDCD1) gene in CAR-T cells targeting mesothelin. This study found that knocking out the PDCD1 gene improved the CAR-T performance [157]. Likewise, another study showed that the CRISPR/Cas9-mediated deletion of the PDCD1 gene increased the cytotoxic effect of CD19 targeting CAR-T cells and reduced tumor growth in a xenograft tumor model [158]. Similarly, CRISPR/Cas9 mediated the deletion of lymphocyte activation gene-3 (LAG-3) (a negative regulator of T-cell activity) was found to improve the anti-tumor effect of CAR-T cells in the murine xenograft model [159].
Multiplex Gene Editing
Similar to single-gene editing, altering multiple genes via CRISPR/Cas9 seems to be a good strategy for making universal allogeneic T cells, improving the treatment efficacy, and reducing both time and expense of CAR-T-cell therapy. Graft-versus-host disease (GVHD) is a primary concern in using allogeneic T cells, so silencing endogenous TCR and β-2 microglobulin (an essential subunit of the HLA-I) may eliminate the limitation associated with GVHD. As a result, CRISPR/Cas9-mediated CBP therapy become a novel area of interest in cancer immunotherapy. Similarly, to improve the persistence of CAR-T cells, the deletion of CTLA-4 and PD1 was achieved, and the findings suggest that it also reduces the resistance to apoptosis and the immune inhibition of CAR-T cells [160].
Co-Stimulatory Molecule
Previous studies reported that adding co-stimulatory domains like 4-1BB and CD28 in CAR-T cells improved their performance. It was also noted that the selection and positioning of the co-stimulatory molecules in the CAR construct affect CAR-T cells’ function, kinetics, and potential safety profile. Studies found that compared to CD28, the incorporation of 4-1BB in CAR-T cells improved CAR-T cells performance via the significant downregulation of T cells’ immunoglobulin and mucin domain-containing protein 3 (TIM3), LAG-3, and PD-1 [154,161]. Different types of cancer may benefit from different CAR-T-cell co-stimulatory domains; for example, using the 4-1BB co-stimulatory domain for longer persistence may be necessary for the long-term remission of the precursor B-cell malignancy B-ALL, whereas it is less critical than early antitumor activity when treating mature B-NHL malignancies [162].
Pharmacological Antagonists
Checkpoint blockade via pharmacological means has also been tested via CAR-T-cell therapy. A study showed that combining A2AR antagonist SCH58261 with anti-PD-1 antibody improved CAR-T-cell function and upregulated granzyme B expression in tumor-infiltrating CD8 + T cells [155].
6.7. Combination of CAR-T-Cell Therapy with BiTEs
Bispecific T-cell engagers (BiTEs) have a dual specificity against two different antigens [163]. The benefits of this strategy are that it can activate T cells and link T cells with tumor cells without engaging MHC [163]. A group examined the efficacy of CD3/EGFR BiTEs together with EGFRvIII (oncogenic mutation in the ECD of the EGFR)-targeted CAR-T cells in a glioblastoma mouse model and showed that this strategy induced the efficiency of CAR-T cells function by removing heterogeneous tumor cells without significant toxicity [164,165].
As the loss of CD19 was the main limitation in using anti-CD19-CAR-T-cell therapy, to overcome this limitation, blinatumomab (a BiTE composed of a CD3 and a CD19 site) was combined with anti-CD19 CAR-T cells and tested in relapsed/refractory B-ALL cases, and the findings showed that blinatumomab augmented CAR-T performance [166]. In another study, when anti-biotin CAR-T cells were combined with biotinylated bispecific antibody (anti-CD19 or anti-CD20) coated on tumor cells, the function of CAR-T cells improved [167]. As the folate receptor (FR) is found to be overexpressed in tumors (including lung, ovarian, uterus, and breast tumors) compared to normal tissue, folate fluorescein isothiocyanate (FITC) conjugate (BiTE) was developed to control CAR-T cells’ function, as well as target FR [168]. Combining anti-FITC-CAR-T cells with BiTE (folate-FITC conjugate) was found to recruit the CAR-T cells in the tumor site and induce their function in tumors with high FR expression [168]. Using this strategy, folate-FITC can be conjugated with diverse anti-tumor antibodies and directed to specific cancers [169].
6.8. Combination of CAR-T-Cell Therapy with Immunomodulatory Agents
Different immunomodulatory drugs have been tested with CAR-T-cell therapy and showed that this combination strategy significantly improved CAR-T cells’ proliferation, persistence, and cytokine production in the TME. Some of the immune modulatory drugs combined with CAR-T-cell therapy have been described in the following sections.
6.8.1. Lenalidomide
Lenalidomide has been shown to induce the proliferation and cytokine production of T cells by inhibiting the immunosuppressive effect of CTLA4-Ig (a blocker of the B7-CD28 pathway) [170,171]. In these studies, lenalidomide was found to induce the expression of NF-κB and enhance the phosphorylation of the CD28 co-stimulatory molecule in T cells. Hence, combination treatment with lenalidomide and CAR-T-cell therapy will be an excellent strategy to overcome the limitations associated with the immunosuppressive environment of TME. Likewise, another study found that lenalidomide, in combination with CD19-CAR-T-cell therapy, induced IFN-γ production; as a result, it caused elevated activity and infiltration of T cells in TME and reduced tumor burden in mice [172]. Similarly, a study reported the lenalidomide-mediated increased persistence and cytotoxicity of CAR-T cells in multiple myeloma models [173]. This study noted that this combination strategy not only reinforced the immunological synapse formation between CAR-T cells and myeloma cells but also induced the proliferation of CTLs, enhanced the production of immunogenic cytokines like IFN-γ and TNF-α, and inhibited the production of immunosuppressive cytokines, like IL-5 and IL-10.
6.8.2. miRNAs (miR-153)
Similar to small-molecule drugs, non-coding RNAs also emerge as epigenetic immunomodulatory molecules [174,175,176,177,178], which are reported to affect naive, effector, and memory T-cell function. miR-153 was found to inhibit the expression of Indol amine 2,3-dioxygenase 1 (IDO1), which catalyzes the production of tryptophan kynurenine and 3-hydroxy anthranilic acid (immunosuppressive metabolites). A study reported that the overexpression of miR-153 with anti-EGFRvIII-CAR-T cells therapy reduced tumor burden in the xenografts model of colon cancer [179]. As IDO catalyzes the conversion of tryptophan into inhibitory metabolites that inhibit T-cell activity, IDO inhibitors were noted to enhance the efficacy of CD19-CAR-T-cell therapy in a xenograft lymphoma model [180].
6.8.3. Decitabine
Antigen heterogeneity and the loss of antigen is one of the main limitations of CAR-T-cell therapy [181,182]. Decitabine, a hypomethylating compound, was found to augment the effect of CAR-T cells. A study showed that decitabine increased the expression of mucin 1 (MUC1) by demethylating DNA in pancreatic tumor cells, and decitabine was found to increase the potency of MUC1-CAR-T cells. Another study reported that decitabine improved the efficacy of CD19-CAR-T-cell therapy by upregulating the expression level of CD19 in lymphoma cell lines [183]. Similarly, on acute myeloid leukemia (AML) cell lines, decitabine was found to improve the effectiveness of CD33-CAR-T cells by enhancing the expression of CD33 [184].
6.8.4. HDAC Inhibitors (HDACis)
The acetylation of histone in chromatin was found to open a compact chromatin structure, and so it helps with transcription factor binding for gene expression process (transcription). Similarly, histone deacetylase (HDAC) can deacetylate histones and suppress the transcription of related genes [185]; hence, utilizing HDAC inhibitors (HDACis) seems to be an excellent strategy for inducing specific gene expression [186]. Studies reported that when Romidepsin (HDAC inhibitors) is combined with CD20-CAR-T-cell therapy, it induces the expression of CD20, elevates the potency of CD20-CAR-T cells, and prolongs the overall survival of the mice in the lymphoma model [187]. Similarly, another study showed that administration of HDACi valproic acid before CAR-T-cell therapy induced the expression levels of NKG2DL on low-level expressing AML cell lines, and as a result, it enhanced the anti-tumor effects of CAR-T cells [188].
6.8.5. SMAC Mimetics
The second mitochondria-derived activator of caspase (SMAC) mimetics binds to inhibitor of apoptosis proteins (IAPs), thus freeing caspases to activate apoptosis [189]. Inhibition of IAPs via SMAC mimetics (SMs) seems to be a good strategy for improving the CAR-T cell’s function. A study found three SMs to be effective boosters of CD19-CAR-T cells in B-ALL [190]. Similarly, another study showed that administering birinapant (a potent SM) with HER2-CAR-T cells improved its efficacy by engaging the TNF receptor pathway; as a result, it reduced tumor growth in the murine model [191].
6.9. Combination of CAR-T-Cell Therapy with Allo-HSCT
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is a treatment strategy of adoptive cell therapy (ACT), and it can be combined with CAR-T-cell therapy for cancer treatment [192]. Studies have shown that using allo-HSCT before CAR-T-cell therapy improved the effectiveness of CAR-T cells in relapsed/refractory (R/R) B-ALL patients with minimal residual disease (MRD) [193,194]. This combination therapy with CD19-CAR-T cells and allo-HSCT was shown to prolong leukemia-free survival (LFS) with acceptable safety and efficacy compared to CAR-T-cell monotherapy [195,196]. Similarly, augmented effects in tumor eradication with prolonged EFS (event free survival) and OS (overall survival) were shown in combination therapy with CD22 CAR-T-cell therapy and allo-HSCT [197,198]. Likewise, allo-HSCT after CD19-targeted CAR-T-cell therapy was found to be safe and effective at treating B-cell chronic lymphocytic leukemia (B-CLL) and non-Hodgkin lymphoma (NHL) [199].
6.10. Combination of CAR-T-Cell Therapy with Metabolic Inhibitors
In particular, for solid tumors, manipulating the metabolic profile of TME to induce the effectiveness and persistence of CAR-T-cell therapy is a promising strategy [200]. Different genetic modifications have been made in T cells to modify the metabolic profile of TME, such as upregulating mitochondrial function via the ectopic expression of PCG1α (PPAR-γ co-activator 1α), inducing effector T-cell functions via the knocking out of ACAT1 (acyl-CoA cholesterol acyltransferase 1), reducing hypoxia by upregulating the expression of catalase, etc. [201,202,203,204,205,206,207]. In addition to these, different co-stimulatory molecules have been added to CAR-T cells to enhance the survival, proliferation, and effectiveness of CAR-T cells in different TMEs, which include adding CD28 for aerobic glycolysis, adding 4-1BB for fatty acid oxidative breakdown and mitochondrial biogenesis, etc. [161,208,209,210,211]. The presence of immunosuppressive cytokines like PGE2, IDO, CTLA4, PD-L1, IL-10, and TGF-β released in TME can affect the efficacy of CAR-T-cell therapy [212]. A study showed that the treatment with all-trans retinoic acid (ATRA) improved the efficacy of CAR-T cells by reducing the suppressive effects of myeloid-derived suppressor cells (MDSCs) in pediatric sarcomas [213]. Similarly, another group found that the expression of NKG2D ligands via NK cells significantly abolished MDSCs and augmented the anti-tumor tumor effects of CAR-T cells [214]. In the same way, to inhibit the immunosuppressive effects of IDO, IDO inhibitory drugs such as cyclophosphamide and fludarabine have been used before CAR-T-cell therapy, reporting significant tumor regression and increased survival [180].
7. Conclusions
So far, we have outlined the considerable efforts of researchers in the field of CAR-T cells’ evolution, and it is an integral part of the armamentarium in the field of hematological malignancies. Although this treatment can be indicated for the treatment of both hematological malignancies and solid tumors, a game-changing role for these agents has clearly emerged in treating hematological malignancies. In contrast, the promises in the field of solid tumors have yet to be fulfilled due to different limitations in the treatment of solid tumors. Lack of specific TAA is a major limitation for solid tumors, contributing to antigen escape, on-target/off-tumor effects, and life-threatening adverse effects such as cytokine storm syndrome and tumor lysis syndrome. It has been noted that major regulatory agencies like the FDA in the US and the European Medicines Agency (EMA) in Europe have approved a large number of CAR-T therapies for clinical use in hematological malignancies. After many years of research into CAR-T therapies, the technical modifications of this therapy have already been achieved in treating solid tumors. These modifications include targeting multiple targets, immune checkpoint blockade, etc. Many combination strategies with existing treatments were also applied, which showed promising results for improving the efficacy of CAR-T therapy in both solid and hematological malignancies [215,216]. Indeed, more research and advanced knowledge in CAR-T-cell therapy are necessary to treat solid tumors in the near future.
Funding
This research received no external funding.
Data Availability Statement
No new data were created for this study.
Conflicts of Interest
The author declares no conflict of interest.
Abbreviations
| CAR | Chimeric antigen receptor |
| TME | Tumor microenvironment |
| ACT | Adoptive T-cell transfer |
| MHC | Major histocompatibility complex |
| TAA | Tumor-associated antigen |
| FasL | Fas ligand |
| TRAIL | Tumor necrosis factor-related apoptosis-inducing ligand |
| FDA | The Food and Drug Administration |
| EMA | The European Medicines Agency |
| EGFR | Epidermal growth factor receptor |
| HER2 | Human epidermal growth factor receptor 2 |
| HNSCC | Head and neck squamous cell carcinoma |
| CEA | Carcinoembryonic antigen |
| PSMA | Prostate-specific membrane antigen |
| scFv | Single-chain variable fragment |
| CD | Cluster of differentiation |
| NSCLC | Non-small-cell lung cancers |
| OS | Overall survival |
| PD-1 | Programmed cell death-1 |
| PFS | Progression-free survival |
| CSD | Co-stimulatory domain |
| IL | Interleukin |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| TCR | T-cell receptor |
| CLL | Chronic lymphocytic leukemia |
| ALL | Acute lymphoblastic leukemia |
| DLBCL | Diffuse large B-cell lymphoma |
| TNBC | Triple-negative breast cancer |
| TLR4 | Toll-like receptor 4 |
| DCs | Dendritic cells |
| DAMPs | Damage-associated molecular patterns |
| HMGB1 | High-mobility group box 1 |
| IFN-I | Type I interferon |
| Tregs | Regulatory T cells |
| MDSC | Myeloid-derived suppressor cells |
| TGF-β | Transforming growth factor-Β |
| EOC | Epithelial ovarian cancer |
| CTX | Cyclophosphamide |
| M6P | Mannose-6-phosphate |
| HLA | Human leukocyte antigen |
| APCs | Antigen-presenting cells |
| BCMA | B-Cell maturation antigen |
| CPIs | Checkpoint inhibitors |
| DNR | PD-1 dominant negative receptor |
| shRNA | Short hairpin RNA |
| A2ARs | Adenosine 2A receptors |
| CTLA-4 | Cytotoxic T-lymphocyte associated protein 4 |
| PDCD1 | Programmed cell death protein 1 |
| TRAC | T-Cell receptor alpha constant |
| BiTEs | Bispecific T-Cell engager |
| HDAC | Histone deacetylase |
References
- Kratzer, T.B.; Jemal, A.; Miller, K.D.; Nash, S.; Wiggins, C.; Redwood, D.; Smith, R.; Siegel, R.L. Cancer statistics for American Indian and Alaska Native individuals, 2022: Including increasing disparities in early onset colorectal cancer. CA Cancer J. Clin. 2023, 73, 120–146. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Sadelain, M.; Brentjens, R.; Rivière, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Zhao, J.; Song, Y.; Liu, D. Clinical trials of dual-target CAR T cells, donor-derived CAR T cells, and universal CAR T cells for acute lymphoid leukemia. J. Hematol. Oncol. 2019, 12, 17. [Google Scholar] [CrossRef] [PubMed]
- Mancikova, V.; Smida, M. Current State of CAR T-Cell Therapy in Chronic Lymphocytic Leukemia. Int. J. Mol. Sci. 2021, 22, 5536. [Google Scholar] [CrossRef]
- Wang, D.; Wang, J.; Hu, G.; Wang, W.; Xiao, Y.; Cai, H.; Jiang, L.; Meng, L.; Yang, Y.; Zhou, X.; et al. A phase 1 study of a novel fully human BCMA-targeting CAR (CT103A) in patients with relapsed/refractory multiple myeloma. Blood 2021, 137, 2890–2901. [Google Scholar] [CrossRef]
- Mei, H.; Li, C.; Jiang, H.; Zhao, X.; Huang, Z.; Jin, D.; Guo, T.; Kou, H.; Liu, L.; Tang, L.; et al. A bispecific CAR-T cell therapy targeting BCMA and CD38 in relapsed or refractory multiple myeloma. J. Hematol. Oncol. 2021, 14, 161. [Google Scholar] [CrossRef]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- McLellan, A.D.; Ali Hosseini Rad, S.M. Chimeric antigen receptor T cell persistence and memory cell formation. Immunol. Cell Biol. 2019, 97, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Ajina, A.; Maher, J. Strategies to Address Chimeric Antigen Receptor Tonic Signaling. Mol. Cancer Ther. 2018, 17, 1795–1815. [Google Scholar] [CrossRef] [PubMed]
- James, S.E.; Greenberg, P.D.; Jensen, M.C.; Lin, Y.; Wang, J.; Till, B.G.; Raubitschek, A.A.; Forman, S.J.; Press, O.W. Antigen sensitivity of CD22-specific chimeric TCR is modulated by target epitope distance from the cell membrane. J. Immunol. 2008, 180, 7028–7038. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Santos, E.; Nikhamin, Y.; Yeh, R.; Matsushita, M.; La Perle, K.; Quintás-Cardama, A.; Larson, S.M.; Sadelain, M. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin. Cancer Res. 2007, 13, 5426–5435. [Google Scholar] [CrossRef] [PubMed]
- Finney, H.M.; Akbar, A.N.; Lawson, A.D. Activation of resting human primary T cells with chimeric receptors: Costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J. Immunol. 2004, 172, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.H.; Geiger, T.L.; Campana, D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004, 18, 676–684. [Google Scholar] [CrossRef]
- Teng, M.W.; Kershaw, M.H.; Moeller, M.; Smyth, M.J.; Darcy, P.K. Immunotherapy of cancer using systemically delivered gene-modified human T lymphocytes. Hum. Gene Ther. 2004, 15, 699–708. [Google Scholar] [CrossRef]
- Tang, X.Y.; Sun, Y.; Zhang, A.; Hu, G.L.; Cao, W.; Wang, D.H.; Zhang, B.; Chen, H. Third-generation CD28/4-1BB chimeric antigen receptor T cells for chemotherapy relapsed or refractory acute lymphoblastic leukaemia: A non-randomised, open-label phase I trial protocol. BMJ Open 2016, 6, e013904. [Google Scholar] [CrossRef]
- Zhong, X.S.; Matsushita, M.; Plotkin, J.; Riviere, I.; Sadelain, M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol. Ther. 2010, 18, 413–420. [Google Scholar] [CrossRef]
- Maus, M.V.; June, C.H. Making Better Chimeric Antigen Receptors for Adoptive T-cell Therapy. Clin. Cancer Res. 2016, 22, 1875–1884. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Li, A.; Liu, Q.; Li, T.; Yuan, X.; Han, X.; Wu, K. Chimeric antigen receptor T cells: A novel therapy for solid tumors. J. Hematol. Oncol. 2017, 10, 78. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, J.; Mellody, M.P.; Hou, A.J.; Desai, R.P.; Fung, A.W.; Pham, A.H.T.; Chen, Y.Y.; Zhao, W. CAR-T design: Elements and their synergistic function. eBioMedicine 2020, 58, 102931. [Google Scholar] [CrossRef]
- Ellis, G.I.; Sheppard, N.C.; Riley, J.L. Genetic engineering of T cells for immunotherapy. Nat. Rev. Genet. 2021, 22, 427–447. [Google Scholar] [CrossRef]
- McGuirk, J.; Waller, E.K.; Qayed, M.; Abhyankar, S.; Ericson, S.; Holman, P.; Keir, C.; Myers, G.D. Building blocks for institutional preparation of CTL019 delivery. Cytotherapy 2017, 19, 1015–1024. [Google Scholar] [CrossRef]
- Milone, M.C.; Fish, J.D.; Carpenito, C.; Carroll, R.G.; Binder, G.K.; Teachey, D.; Samanta, M.; Lakhal, M.; Gloss, B.; Danet-Desnoyers, G.; et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. 2009, 17, 1453–1464. [Google Scholar] [CrossRef]
- Pang, Y.; Hou, X.; Yang, C.; Liu, Y.; Jiang, G. Advances on chimeric antigen receptor-modified T-cell therapy for oncotherapy. Mol. Cancer 2018, 17, 91. [Google Scholar] [CrossRef]
- Riet, T.; Holzinger, A.; Dörrie, J.; Schaft, N.; Schuler, G.; Abken, H. Nonviral RNA transfection to transiently modify T cells with chimeric antigen receptors for adoptive therapy. Methods Mol. Biol. 2013, 969, 187–201. [Google Scholar] [CrossRef]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [PubMed]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Rivière, I.; Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.J.; Latouche, J.B.; Santos, E.; Marti, F.; Gong, M.C.; Lyddane, C.; King, P.D.; Larson, S.; Weiss, M.; Rivière, I.; et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat. Med. 2003, 9, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C.H.; Sleijfer, S.; van Steenbergen, S.; van Elzakker, P.; van Krimpen, B.; Groot, C.; Vulto, A.; den Bakker, M.; Oosterwijk, E.; Debets, R.; et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: Clinical evaluation and management of on-target toxicity. Mol. Ther. 2013, 21, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013, 5, 177ra138. [Google Scholar] [CrossRef] [PubMed]
- Adusumilli, P.S.; Cherkassky, L.; Villena-Vargas, J.; Colovos, C.; Servais, E.; Plotkin, J.; Jones, D.R.; Sadelain, M. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci. Transl. Med. 2014, 6, 261ra151. [Google Scholar] [CrossRef]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef]
- Fala, L. Yescarta (Axicabtagene Ciloleucel) Second CAR T-cell Therapy Approved for Patients with Certain Types of Large B-Cell Lymphoma. 2018. Available online: https://jons-online.com/browse-by-topic/fda-approvals-news-updates/1829-yescarta-axicabtagene-ciloleucel-second-car-t-cell-therapy-approved-for-patients-with-certain-types-of-large-b-cell-lymphoma (accessed on 20 November 2023).
- Warnings, B. US Food and Drug Administration Approves Bristol Myers Squibb’s and Bluebird bio’s Abecma (Idecabtagene Vicleucel), the First Anti-BCMA CAR T Cell Therapy for Relapsed or Refractory Multiple Myeloma. 2021. Available online: https://www.businesswire.com/news/home/20210326005507/en/U.S.-Food-and-Drug-Administration-Approves-Bristol-Myers-Squibb%E2%80%99s-and-bluebird-bio%E2%80%99s-Abecma-idecabtagene-vicleucel-the-First-Anti-BCMA-CAR-T-Cell-Therapy-for-Relapsed-or-Refractory- (accessed on 20 November 2023).
- Cartron, G.; Fox, C.P.; Liu, F.F.; Kostic, A.; Hasskarl, J.; Li, D.; Bonner, A.; Zhang, Y.; Maloney, D.G.; Kuruvilla, J. Matching-adjusted indirect treatment comparison of chimeric antigen receptor T-cell therapies for third-line or later treatment of relapsed or refractory large B-cell lymphoma: Lisocabtagene maraleucel versus tisagenlecleucel. Exp. Hematol. Oncol. 2022, 11, 17. [Google Scholar] [CrossRef]
- Mian, A.; Hill, B.T. Brexucabtagene autoleucel for the treatment of relapsed/refractory mantle cell lymphoma. Expert Opin. Biol. Ther. 2021, 21, 435–441. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, F.; Gordon, L.I. Lisocabtagene maraleucel in the treatment of relapsed/refractory large B-cell lymphoma. Future Oncol. 2023, 19, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C.H.; Willemsen, R.; van Elzakker, P.; van Steenbergen-Langeveld, S.; Broertjes, M.; Oosterwijk-Wakka, J.; Oosterwijk, E.; Sleijfer, S.; Debets, R.; Gratama, J.W. Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood 2011, 117, 72–82. [Google Scholar] [CrossRef]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Somerville, R.P.T.; Lu, T.; Shi, V.; Bot, A.; Rossi, J.; Xue, A.; Goff, S.L.; Yang, J.C.; Sherry, R.M.; et al. Lymphoma Remissions Caused by Anti-CD19 Chimeric Antigen Receptor T Cells Are Associated With High Serum Interleukin-15 Levels. J. Clin. Oncol. 2017, 35, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Turtle, C.J.; Hanafi, L.A.; Berger, C.; Hudecek, M.; Pender, B.; Robinson, E.; Hawkins, R.; Chaney, C.; Cherian, S.; Chen, X.; et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci. Transl. Med. 2016, 8, 355ra116. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Dudley, M.E.; Kassim, S.H.; Somerville, R.P.; Carpenter, R.O.; Stetler-Stevenson, M.; Yang, J.C.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J. Clin. Oncol. 2015, 33, 540–549. [Google Scholar] [CrossRef]
- Tang, L.; Zheng, Y.; Melo, M.B.; Mabardi, L.; Castaño, A.P.; Xie, Y.Q.; Li, N.; Kudchodkar, S.B.; Wong, H.C.; Jeng, E.K.; et al. Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat. Biotechnol. 2018, 36, 707–716. [Google Scholar] [CrossRef]
- Allen, G.M.; Frankel, N.W.; Reddy, N.R.; Bhargava, H.K.; Yoshida, M.A.; Stark, S.R.; Purl, M.; Lee, J.; Yee, J.L.; Yu, W.; et al. Synthetic cytokine circuits that drive T cells into immune-excluded tumors. Science 2022, 378, eaba1624. [Google Scholar] [CrossRef]
- George, A.K.; Singh, M.; Homme, R.P.; Majumder, A.; Sandhu, H.S.; Tyagi, S.C. A hypothesis for treating inflammation and oxidative stress with hydrogen sulfide during age-related macular degeneration. Int. J. Ophthalmol. 2018, 11, 881–887. [Google Scholar] [CrossRef]
- Homme, R.P.; Singh, M.; Majumder, A.; George, A.K.; Nair, K.; Sandhu, H.S.; Tyagi, N.; Lominadze, D.; Tyagi, S.C. Remodeling of Retinal Architecture in Diabetic Retinopathy: Disruption of Ocular Physiology and Visual Functions by Inflammatory Gene Products and Pyroptosis. Front. Physiol. 2018, 9, 1268. [Google Scholar] [CrossRef] [PubMed]
- Hyrenius-Wittsten, A.; Su, Y.; Park, M.; Garcia, J.M.; Alavi, J.; Perry, N.; Montgomery, G.; Liu, B.; Roybal, K.T. SynNotch CAR circuits enhance solid tumor recognition and promote persistent antitumor activity in mouse models. Sci. Transl. Med. 2021, 13, eabd8836. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Lopez, R.A.; Yu, W.; Cabral, K.A.; Creasey, O.A.; Lopez Pazmino, M.D.P.; Tonai, Y.; De Guzman, A.; Mäkelä, A.; Saksela, K.; Gartner, Z.J.; et al. T cell circuits that sense antigen density with an ultrasensitive threshold. Science 2021, 371, 1166–1171. [Google Scholar] [CrossRef] [PubMed]
- Hege, K.M.; Bergsland, E.K.; Fisher, G.A.; Nemunaitis, J.J.; Warren, R.S.; McArthur, J.G.; Lin, A.A.; Schlom, J.; June, C.H.; Sherwin, S.A. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J. Immunother. Cancer 2017, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Hirabayashi, K.; Ahn, S.; Kren, N.P.; Montgomery, S.A.; Wang, X.; Tiruthani, K.; Mirlekar, B.; Michaud, D.; Greene, K.; et al. Antitumor Responses in the Absence of Toxicity in Solid Tumors by Targeting B7-H3 via Chimeric Antigen Receptor T Cells. Cancer Cell 2019, 35, 221–237. [Google Scholar] [CrossRef]
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C.; et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin. Cancer Res. 2019, 25, 2560–2574. [Google Scholar] [CrossRef] [PubMed]
- Hurton, L.V.; Singh, H.; Najjar, A.M.; Switzer, K.C.; Mi, T.; Maiti, S.; Olivares, S.; Rabinovich, B.; Huls, H.; Forget, M.A.; et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E7788–E7797. [Google Scholar] [CrossRef]
- Koneru, M.; O’Cearbhaill, R.; Pendharkar, S.; Spriggs, D.R.; Brentjens, R.J. A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16(ecto) directed chimeric antigen receptors for recurrent ovarian cancer. J. Transl. Med. 2015, 13, 102. [Google Scholar] [CrossRef]
- Chekmasova, A.A.; Rao, T.D.; Nikhamin, Y.; Park, K.J.; Levine, D.A.; Spriggs, D.R.; Brentjens, R.J. Successful eradication of established peritoneal ovarian tumors in SCID-Beige mice following adoptive transfer of T cells genetically targeted to the MUC16 antigen. Clin. Cancer Res. 2010, 16, 3594–3606. [Google Scholar] [CrossRef]
- Maus, M.V.; Haas, A.R.; Beatty, G.L.; Albelda, S.M.; Levine, B.L.; Liu, X.; Zhao, Y.; Kalos, M.; June, C.H. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol. Res. 2013, 1, 26–31. [Google Scholar] [CrossRef]
- Gattinoni, L.; Klebanoff, C.A.; Palmer, D.C.; Wrzesinski, C.; Kerstann, K.; Yu, Z.; Finkelstein, S.E.; Theoret, M.R.; Rosenberg, S.A.; Restifo, N.P. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J. Clin. Investig. 2005, 115, 1616–1626. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, M.; Hu, J.; Sommariva, M.; Gautam, S.; Fellowes, V.; Hocker, J.D.; Dougherty, S.; Qin, H.; Klebanoff, C.A.; Fry, T.J.; et al. Generation of clinical-grade CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human B-cell malignancies. Blood 2016, 128, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gao, L.; Liu, L.; Wang, J.; Wang, S.; Gao, L.; Zhang, C.; Liu, Y.; Kong, P.; Liu, J. A Bcma and CD19 bispecific CAR-T for relapsed and refractory multiple myeloma. Blood 2019, 134, 3147. [Google Scholar] [CrossRef]
- Lin, Q.; Zhao, J.; Song, Y.; Liu, D. Recent updates on CAR T clinical trials for multiple myeloma. Mol. Cancer 2019, 18, 154. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Wu, Z.; Jia, H.; Tong, C.; Guo, Y.; Ti, D.; Han, X.; Liu, Y.; Zhang, W.; Wang, C.; et al. Bispecific CAR-T cells targeting both CD19 and CD22 for therapy of adults with relapsed or refractory B cell acute lymphoblastic leukemia. J. Hematol. Oncol. 2020, 13, 30. [Google Scholar] [CrossRef] [PubMed]
- Hossain, N.; Sahaf, B.; Abramian, M.; Spiegel, J.Y.; Kong, K.; Kim, S.; Mavroukakis, S.; Oak, J.; Natkunam, Y.; Meyer, E.H. Phase I experience with a bi-specific CAR targeting CD19 and CD22 in adults with B-cell malignancies. Blood 2018, 132, 490. [Google Scholar] [CrossRef]
- Wilkie, S.; van Schalkwyk, M.C.; Hobbs, S.; Davies, D.M.; van der Stegen, S.J.; Pereira, A.C.; Burbridge, S.E.; Box, C.; Eccles, S.A.; Maher, J. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J. Clin. Immunol. 2012, 32, 1059–1070. [Google Scholar] [CrossRef]
- Han, X.; Wang, Y.; Wei, J.; Han, W. Multi-antigen-targeted chimeric antigen receptor T cells for cancer therapy. J. Hematol. Oncol. 2019, 12, 128. [Google Scholar] [CrossRef]
- Priceman, S.J.; Tilakawardane, D.; Jeang, B.; Aguilar, B.; Murad, J.P.; Park, A.K.; Chang, W.C.; Ostberg, J.R.; Neman, J.; Jandial, R.; et al. Regional Delivery of Chimeric Antigen Receptor-Engineered T Cells Effectively Targets HER2(+) Breast Cancer Metastasis to the Brain. Clin. Cancer Res. 2018, 24, 95–105. [Google Scholar] [CrossRef]
- Brown, C.E.; Aguilar, B.; Starr, R.; Yang, X.; Chang, W.C.; Weng, L.; Chang, B.; Sarkissian, A.; Brito, A.; Sanchez, J.F.; et al. Optimization of IL13Rα2-Targeted Chimeric Antigen Receptor T Cells for Improved Anti-tumor Efficacy against Glioblastoma. Mol. Ther. 2018, 26, 31–44. [Google Scholar] [CrossRef]
- Whilding, L.M.; Halim, L.; Draper, B.; Parente-Pereira, A.C.; Zabinski, T.; Davies, D.M.; Maher, J. CAR T-Cells Targeting the Integrin αvβ6 and Co-Expressing the Chemokine Receptor CXCR2 Demonstrate Enhanced Homing and Efficacy against Several Solid Malignancies. Cancers 2019, 11, 674. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Rui, W.; Zheng, H.; Huang, D.; Yu, F.; Zhang, Y.; Dong, J.; Zhao, X.; Lin, X. CXCR2-modified CAR-T cells have enhanced trafficking ability that improves treatment of hepatocellular carcinoma. Eur. J. Immunol. 2020, 50, 712–724. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Tao, H.; Karachi, A.; Long, Y.; Hou, A.Y.; Na, M.; Dyson, K.A.; Grippin, A.J.; Deleyrolle, L.P.; Zhang, W.; et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat. Commun. 2019, 10, 4016. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.C.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Li, A.M.; Hucks, G.E.; Dinofia, A.M.; Seif, A.E.; Teachey, D.T.; Baniewicz, D.; Callahan, C.; Fasano, C.; McBride, B.; Gonzalez, V. Checkpoint inhibitors augment CD19-directed chimeric antigen receptor (CAR) T cell therapy in relapsed B-cell acute lymphoblastic leukemia. Blood 2018, 132, 556. [Google Scholar] [CrossRef]
- Yin, Y.; Boesteanu, A.C.; Binder, Z.A.; Xu, C.; Reid, R.A.; Rodriguez, J.L.; Cook, D.R.; Thokala, R.; Blouch, K.; McGettigan-Croce, B.; et al. Checkpoint Blockade Reverses Anergy in IL-13Rα2 Humanized scFv-Based CAR T Cells to Treat Murine and Canine Gliomas. Mol. Ther. Oncolytics 2018, 11, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Chong, E.A.; Melenhorst, J.J.; Lacey, S.F.; Ambrose, D.E.; Gonzalez, V.; Levine, B.L.; June, C.H.; Schuster, S.J. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: Refueling the CAR. Blood 2017, 129, 1039–1041. [Google Scholar] [CrossRef]
- Koneru, M.; Purdon, T.J.; Spriggs, D.; Koneru, S.; Brentjens, R.J. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology 2015, 4, e994446. [Google Scholar] [CrossRef]
- Krenciute, G.; Prinzing, B.L.; Yi, Z.; Wu, M.F.; Liu, H.; Dotti, G.; Balyasnikova, I.V.; Gottschalk, S. Transgenic Expression of IL15 Improves Antiglioma Activity of IL13Rα2-CAR T Cells but Results in Antigen Loss Variants. Cancer Immunol. Res. 2017, 5, 571–581. [Google Scholar] [CrossRef]
- Mohammed, S.; Sukumaran, S.; Bajgain, P.; Watanabe, N.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Fisher, W.E.; Leen, A.M.; Vera, J.F. Improving Chimeric Antigen Receptor-Modified T Cell Function by Reversing the Immunosuppressive Tumor Microenvironment of Pancreatic Cancer. Mol. Ther. 2017, 25, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jiang, S.; Fang, C.; Yang, S.; Olalere, D.; Pequignot, E.C.; Cogdill, A.P.; Li, N.; Ramones, M.; Granda, B.; et al. Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res. 2015, 75, 3596–3607. [Google Scholar] [CrossRef] [PubMed]
- Ying, Z.; Huang, X.F.; Xiang, X.; Liu, Y.; Kang, X.; Song, Y.; Guo, X.; Liu, H.; Ding, N.; Zhang, T.; et al. A safe and potent anti-CD19 CAR T cell therapy. Nat. Med. 2019, 25, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Salter, A.I.; Ivey, R.G.; Kennedy, J.J.; Voillet, V.; Rajan, A.; Alderman, E.J.; Voytovich, U.J.; Lin, C.; Sommermeyer, D.; Liu, L.; et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal. 2018, 11, eaat6753. [Google Scholar] [CrossRef] [PubMed]
- Sommermeyer, D.; Hill, T.; Shamah, S.M.; Salter, A.I.; Chen, Y.; Mohler, K.M.; Riddell, S.R. Fully human CD19-specific chimeric antigen receptors for T-cell therapy. Leukemia 2017, 31, 2191–2199. [Google Scholar] [CrossRef]
- Jonnalagadda, M.; Mardiros, A.; Urak, R.; Wang, X.; Hoffman, L.J.; Bernanke, A.; Chang, W.C.; Bretzlaff, W.; Starr, R.; Priceman, S.; et al. Chimeric antigen receptors with mutated IgG4 Fc spacer avoid fc receptor binding and improve T cell persistence and antitumor efficacy. Mol. Ther. 2015, 23, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.M.; Sakemura, R.; Cox, M.J.; Yang, N.; Khadka, R.H.; Forsman, C.L.; Hansen, M.J.; Jin, F.; Ayasoufi, K.; Hefazi, M.; et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood 2019, 133, 697–709. [Google Scholar] [CrossRef]
- Sterner, R.M.; Cox, M.J.; Sakemura, R.; Kenderian, S.S. Using CRISPR/Cas9 to Knock Out GM-CSF in CAR-T Cells. J. Vis. Exp. 2019. [Google Scholar] [CrossRef]
- Giavridis, T.; van der Stegen, S.J.C.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018, 24, 731–738. [Google Scholar] [CrossRef]
- Philip, B.; Kokalaki, E.; Mekkaoui, L.; Thomas, S.; Straathof, K.; Flutter, B.; Marin, V.; Marafioti, T.; Chakraverty, R.; Linch, D.; et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood 2014, 124, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Mestermann, K.; Giavridis, T.; Weber, J.; Rydzek, J.; Frenz, S.; Nerreter, T.; Mades, A.; Sadelain, M.; Einsele, H.; Hudecek, M. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci. Transl. Med. 2019, 11, eaau5907. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Deng, J.; Gao, L.; Zhou, J. Innovative strategies to advance CAR T cell therapy for solid tumors. Am. J. Cancer Res. 2020, 10, 1979–1992. [Google Scholar] [PubMed]
- Ramello, M.C.; Haura, E.B.; Abate-Daga, D. CAR-T cells and combination therapies: What’s next in the immunotherapy revolution? Pharmacol. Res. 2018, 129, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Adjemian, S.; Mattarollo, S.R.; Yamazaki, T.; Aymeric, L.; Yang, H.; Portela Catani, J.P.; Hannani, D.; Duret, H.; Steegh, K.; et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity 2013, 38, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remédios, C.; et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Parente-Pereira, A.C.; Whilding, L.M.; Brewig, N.; van der Stegen, S.J.; Davies, D.M.; Wilkie, S.; van Schalkwyk, M.C.; Ghaem-Maghami, S.; Maher, J. Synergistic Chemoimmunotherapy of Epithelial Ovarian Cancer Using ErbB-Retargeted T Cells Combined with Carboplatin. J. Immunol. 2013, 191, 2437–2445. [Google Scholar] [CrossRef] [PubMed]
- Curran, K.J.; Margossian, S.P.; Kernan, N.A.; Silverman, L.B.; Williams, D.A.; Shukla, N.; Kobos, R.; Forlenza, C.J.; Steinherz, P.; Prockop, S.; et al. Toxicity and response after CD19-specific CAR T-cell therapy in pediatric/young adult relapsed/refractory B-ALL. Blood 2019, 134, 2361–2368. [Google Scholar] [CrossRef]
- Muranski, P.; Boni, A.; Wrzesinski, C.; Citrin, D.E.; Rosenberg, S.A.; Childs, R.; Restifo, N.P. Increased intensity lymphodepletion and adoptive immunotherapy—How far can we go? Nat. Clin. Pract. Oncol. 2006, 3, 668–681. [Google Scholar] [CrossRef]
- Xu, J.; Wang, Y.; Shi, J.; Liu, J.; Li, Q.; Chen, L. Combination therapy: A feasibility strategy for CAR-T cell therapy in the treatment of solid tumors. Oncol. Lett. 2018, 16, 2063–2070. [Google Scholar] [CrossRef]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Kryczek, I.; Dostál, L.; Lin, H.; Tan, L.; Zhao, L.; Lu, F.; Wei, S.; Maj, T.; Peng, D.; et al. Effector T Cells Abrogate Stroma-Mediated Chemoresistance in Ovarian Cancer. Cell 2016, 165, 1092–1105. [Google Scholar] [CrossRef] [PubMed]
- Dangaj, D.; Bruand, M.; Grimm, A.J.; Ronet, C.; Barras, D.; Duttagupta, P.A.; Lanitis, E.; Duraiswamy, J.; Tanyi, J.L.; Benencia, F.; et al. Cooperation between Constitutive and Inducible Chemokines Enables T Cell Engraftment and Immune Attack in Solid Tumors. Cancer Cell 2019, 35, 885–900. [Google Scholar] [CrossRef] [PubMed]
- Paulsson, J.; Micke, P. Prognostic relevance of cancer-associated fibroblasts in human cancer. Semin. Cancer Biol. 2014, 25, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.A.; Finney, O.; Annesley, C.; Brakke, H.; Summers, C.; Leger, K.; Bleakley, M.; Brown, C.; Mgebroff, S.; Kelly-Spratt, K.S.; et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 2017, 129, 3322–3331. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Hwang, W.T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef]
- Lee, Y.; Auh, S.L.; Wang, Y.; Burnette, B.; Wang, Y.; Meng, Y.; Beckett, M.; Sharma, R.; Chin, R.; Tu, T.; et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: Changing strategies for cancer treatment. Blood 2009, 114, 589–595. [Google Scholar] [CrossRef]
- Minn, I.; Rowe, S.P.; Pomper, M.G. Enhancing CAR T-cell therapy through cellular imaging and radiotherapy. Lancet Oncol. 2019, 20, e443–e451. [Google Scholar] [CrossRef]
- Reits, E.A.; Hodge, J.W.; Herberts, C.A.; Groothuis, T.A.; Chakraborty, M.; Wansley, E.K.; Camphausen, K.; Luiten, R.M.; de Ru, A.H.; Neijssen, J.; et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J. Exp. Med. 2006, 203, 1259–1271. [Google Scholar] [CrossRef]
- Higgins, J.P.; Bernstein, M.B.; Hodge, J.W. Enhancing immune responses to tumor-associated antigens. Cancer Biol. Ther. 2009, 8, 1440–1449. [Google Scholar] [CrossRef]
- Zhang, B.; Bowerman, N.A.; Salama, J.K.; Schmidt, H.; Spiotto, M.T.; Schietinger, A.; Yu, P.; Fu, Y.X.; Weichselbaum, R.R.; Rowley, D.A.; et al. Induced sensitization of tumor stroma leads to eradication of established cancer by T cells. J. Exp. Med. 2007, 204, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Demaria, S.; Ng, B.; Devitt, M.L.; Babb, J.S.; Kawashima, N.; Liebes, L.; Formenti, S.C. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Weiss, T.; Weller, M.; Guckenberger, M.; Sentman, C.L.; Roth, P. NKG2D-Based CAR T Cells and Radiotherapy Exert Synergistic Efficacy in Glioblastoma. Cancer Res. 2018, 78, 1031–1043. [Google Scholar] [CrossRef] [PubMed]
- DeSelm, C.; Palomba, M.L.; Yahalom, J.; Hamieh, M.; Eyquem, J.; Rajasekhar, V.K.; Sadelain, M. Low-Dose Radiation Conditioning Enables CAR T Cells to Mitigate Antigen Escape. Mol. Ther. 2018, 26, 2542–2552. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, Y.; Chen, K.; Qian, L.; Wang, P. Oncolytic virotherapy reverses the immunosuppressive tumor microenvironment and its potential in combination with immunotherapy. Cancer Cell Int. 2021, 21, 262. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Alemany, R. CAR-T Cells and Oncolytic Viruses: Joining Forces to Overcome the Solid Tumor Challenge. Front. Immunol. 2018, 9, 2460. [Google Scholar] [CrossRef]
- Nishio, N.; Diaconu, I.; Liu, H.; Cerullo, V.; Caruana, I.; Hoyos, V.; Bouchier-Hayes, L.; Savoldo, B.; Dotti, G. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. 2014, 74, 5195–5205. [Google Scholar] [CrossRef]
- Moon, E.K.; Wang, L.S.; Bekdache, K.; Lynn, R.C.; Lo, A.; Thorne, S.H.; Albelda, S.M. Intra-tumoral delivery of CXCL11 via a vaccinia virus, but not by modified T cells, enhances the efficacy of adoptive T cell therapy and vaccines. Oncoimmunology 2018, 7, e1395997. [Google Scholar] [CrossRef]
- Watanabe, K.; Luo, Y.; Da, T.; Guedan, S.; Ruella, M.; Scholler, J.; Keith, B.; Young, R.M.; Engels, B.; Sorsa, S.; et al. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight 2018, 3, e99573. [Google Scholar] [CrossRef]
- Li, Y.; Xiao, F.; Zhang, A.; Zhang, D.; Nie, W.; Xu, T.; Han, B.; Seth, P.; Wang, H.; Yang, Y.; et al. Oncolytic adenovirus targeting TGF-β enhances anti-tumor responses of mesothelin-targeted chimeric antigen receptor T cell therapy against breast cancer. Cell Immunol. 2020, 348, 104041. [Google Scholar] [CrossRef] [PubMed]
- Tanoue, K.; Rosewell Shaw, A.; Watanabe, N.; Porter, C.; Rana, B.; Gottschalk, S.; Brenner, M.; Suzuki, M. Armed Oncolytic Adenovirus-Expressing PD-L1 Mini-Body Enhances Antitumor Effects of Chimeric Antigen Receptor T Cells in Solid Tumors. Cancer Res. 2017, 77, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Rosewell Shaw, A.; Porter, C.E.; Watanabe, N.; Tanoue, K.; Sikora, A.; Gottschalk, S.; Brenner, M.K.; Suzuki, M. Adenovirotherapy Delivering Cytokine and Checkpoint Inhibitor Augments CAR T Cells against Metastatic Head and Neck Cancer. Mol. Ther. 2017, 25, 2440–2451. [Google Scholar] [CrossRef] [PubMed]
- Park, A.K.; Fong, Y.; Kim, S.I.; Yang, J.; Murad, J.P.; Lu, J.; Jeang, B.; Chang, W.C.; Chen, N.G.; Thomas, S.H.; et al. Effective combination immunotherapy using oncolytic viruses to deliver CAR targets to solid tumors. Sci. Transl. Med. 2020, 12, eaaz1863. [Google Scholar] [CrossRef] [PubMed]
- Avigan, D.; Rosenblatt, J. Vaccine therapy in hematologic malignancies. Blood 2018, 131, 2640–2650. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Pardoll, D.M.; Jaffee, E.M. Cellular vaccine approaches. Cancer J. 2010, 16, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Rossig, C.; Pule, M.; Altvater, B.; Saiagh, S.; Wright, G.; Ghorashian, S.; Clifton-Hadley, L.; Champion, K.; Sattar, Z.; Popova, B.; et al. Vaccination to improve the persistence of CD19CAR gene-modified T cells in relapsed pediatric acute lymphoblastic leukemia. Leukemia 2017, 31, 1087–1095. [Google Scholar] [CrossRef]
- Weinstock, M.; Rosenblatt, J.; Avigan, D. Dendritic Cell Therapies for Hematologic Malignancies. Mol. Ther. Methods Clin. Dev. 2017, 5, 66–75. [Google Scholar] [CrossRef]
- Wu, M.; Zhang, L.; Zhang, H.; Ning, J.; Tu, S.; He, Y.; Li, Y. CD19 chimeric antigen receptor-redirected T cells combined with epidermal growth factor receptor pathway substrate 8 peptide-derived dendritic cell vaccine in leukemia. Cytotherapy 2019, 21, 659–670. [Google Scholar] [CrossRef]
- Chan, J.D.; von Scheidt, B.; Zeng, B.; Oliver, A.J.; Davey, A.S.; Ali, A.I.; Thomas, R.; Trapani, J.A.; Darcy, P.K.; Kershaw, M.H.; et al. Enhancing chimeric antigen receptor T-cell immunotherapy against cancer using a nanoemulsion-based vaccine targeting cross-presenting dendritic cells. Clin. Transl. Immunol. 2020, 9, e1157. [Google Scholar] [CrossRef]
- Reinhard, K.; Rengstl, B.; Oehm, P.; Michel, K.; Billmeier, A.; Hayduk, N.; Klein, O.; Kuna, K.; Ouchan, Y.; Wöll, S.; et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science 2020, 367, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Cooper, L.J.; Al-Kadhimi, Z.; Serrano, L.M.; Pfeiffer, T.; Olivares, S.; Castro, A.; Chang, W.C.; Gonzalez, S.; Smith, D.; Forman, S.J.; et al. Enhanced antilymphoma efficacy of CD19-redirected influenza MP1-specific CTLs by cotransfer of T cells modified to present influenza MP1. Blood 2005, 105, 1622–1631. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Tashiro, H.; Omer, B.; Lapteva, N.; Ando, J.; Ngo, M.; Mehta, B.; Dotti, G.; Kinchington, P.R.; Leen, A.M.; et al. Vaccination Targeting Native Receptors to Enhance the Function and Proliferation of Chimeric Antigen Receptor (CAR)-Modified T Cells. Clin. Cancer Res. 2017, 23, 3499–3509. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.R.; Micklethwaite, K.P.; Savoldo, B.; Ramos, C.A.; Lam, S.; Ku, S.; Diouf, O.; Liu, E.; Barrett, A.J.; Ito, S.; et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: A phase 1 study. Blood 2013, 122, 2965–2973. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wong, C.W.; Urak, R.; Mardiros, A.; Budde, L.E.; Chang, W.C.; Thomas, S.H.; Brown, C.E.; La Rosa, C.; Diamond, D.J.; et al. CMVpp65 Vaccine Enhances the Antitumor Efficacy of Adoptively Transferred CD19-Redirected CMV-Specific T Cells. Clin. Cancer Res. 2015, 21, 2993–3002. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, H.; Luo, H.; Sun, Y.; Shi, B.; Sun, R.; Li, Z. An IL-4/21 Inverted Cytokine Receptor Improving CAR-T Cell Potency in Immunosuppressive Solid-Tumor Microenvironment. Front. Immunol. 2019, 10, 1691. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, Y.; Liu, W.; Li, X. Engineered IL-7 Receptor Enhances the Therapeutic Effect of AXL-CAR-T Cells on Triple-Negative Breast Cancer. BioMed Res. Int. 2020, 2020, 4795171. [Google Scholar] [CrossRef]
- Hsu, C.; Hughes, M.S.; Zheng, Z.; Bray, R.B.; Rosenberg, S.A.; Morgan, R.A. Primary human T lymphocytes engineered with a codon-optimized IL-15 gene resist cytokine withdrawal-induced apoptosis and persist long-term in the absence of exogenous cytokine. J. Immunol. 2005, 175, 7226–7234. [Google Scholar] [CrossRef]
- Hoyos, V.; Savoldo, B.; Quintarelli, C.; Mahendravada, A.; Zhang, M.; Vera, J.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Dotti, G. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia 2010, 24, 1160–1170. [Google Scholar] [CrossRef]
- Alizadeh, D.; Wong, R.A.; Yang, X.; Wang, D.; Pecoraro, J.R.; Kuo, C.F.; Aguilar, B.; Qi, Y.; Ann, D.K.; Starr, R.; et al. IL15 Enhances CAR-T Cell Antitumor Activity by Reducing mTORC1 Activity and Preserving Their Stem Cell Memory Phenotype. Cancer Immunol. Res. 2019, 7, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Lanitis, E.; Rota, G.; Kosti, P.; Ronet, C.; Spill, A.; Seijo, B.; Romero, P.; Dangaj, D.; Coukos, G.; Irving, M. Optimized gene engineering of murine CAR-T cells reveals the beneficial effects of IL-15 coexpression. J. Exp. Med. 2021, 218, e20192203. [Google Scholar] [CrossRef] [PubMed]
- Battram, A.M.; Bachiller, M.; Lopez, V.; Fernández de Larrea, C.; Urbano-Ispizua, A.; Martín-Antonio, B. IL-15 Enhances the Persistence and Function of BCMA-Targeting CAR-T Cells Compared to IL-2 or IL-15/IL-7 by Limiting CAR-T Cell Dysfunction and Differentiation. Cancers 2021, 13, 3534. [Google Scholar] [CrossRef] [PubMed]
- Gargett, T.; Yu, W.; Dotti, G.; Yvon, E.S.; Christo, S.N.; Hayball, J.D.; Lewis, I.D.; Brenner, M.K.; Brown, M.P. GD2-specific CAR T Cells Undergo Potent Activation and Deletion Following Antigen Encounter but can be Protected From Activation-induced Cell Death by PD-1 Blockade. Mol. Ther. 2016, 24, 1135–1149. [Google Scholar] [CrossRef]
- John, L.B.; Devaud, C.; Duong, C.P.; Yong, C.S.; Beavis, P.A.; Haynes, N.M.; Chow, M.T.; Smyth, M.J.; Kershaw, M.H.; Darcy, P.K. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin. Cancer Res. 2013, 19, 5636–5646. [Google Scholar] [CrossRef] [PubMed]
- Majumder, A.; Sandhu, M.; Banerji, D.; Steri, V.; Olshen, A.; Moasser, M.M. The role of HER2 and HER3 in HER2-amplified cancers beyond breast cancers. Sci. Rep. 2021, 11, 9091. [Google Scholar] [CrossRef]
- Majumder, A. HER3: Toward the Prognostic Significance, Therapeutic Potential, Current Challenges, and Future Therapeutics in Different Types of Cancer. Cells 2023, 12, 2417. [Google Scholar] [CrossRef]
- Majumder, A.; Steri, V.; Salangsang, F.; Moasser, M. Abstract LB-326: The role of HER3 in HER2-amplified cancers other than breast cancers. Cancer Res. 2020, 80, LB-326. [Google Scholar] [CrossRef]
- Hofmann, L.; Forschner, A.; Loquai, C.; Goldinger, S.M.; Zimmer, L.; Ugurel, S.; Schmidgen, M.I.; Gutzmer, R.; Utikal, J.S.; Göppner, D.; et al. Cutaneous, gastrointestinal, hepatic, endocrine, and renal side-effects of anti-PD-1 therapy. Eur. J. Cancer 2016, 60, 190–209. [Google Scholar] [CrossRef]
- Shivaji, U.N.; Jeffery, L.; Gui, X.; Smith, S.C.L.; Ahmad, O.F.; Akbar, A.; Ghosh, S.; Iacucci, M. Immune checkpoint inhibitor-associated gastrointestinal and hepatic adverse events and their management. Ther. Adv. Gastroenterol. 2019, 12, 1756284819884196. [Google Scholar] [CrossRef]
- Giuffrida, L.; Sek, K.; Henderson, M.A.; House, I.G.; Lai, J.; Chen, A.X.Y.; Todd, K.L.; Petley, E.V.; Mardiana, S.; Todorovski, I.; et al. IL-15 Preconditioning Augments CAR T Cell Responses to Checkpoint Blockade for Improved Treatment of Solid Tumors. Mol. Ther. 2020, 28, 2379–2393. [Google Scholar] [CrossRef] [PubMed]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef] [PubMed]
- Beavis, P.A.; Henderson, M.A.; Giuffrida, L.; Mills, J.K.; Sek, K.; Cross, R.S.; Davenport, A.J.; John, L.B.; Mardiana, S.; Slaney, C.Y.; et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J. Clin. Investig. 2017, 127, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Condomines, M.; Arnason, J.; Benjamin, R.; Gunset, G.; Plotkin, J.; Sadelain, M. Tumor-Targeted Human T Cells Expressing CD28-Based Chimeric Antigen Receptors Circumvent CTLA-4 Inhibition. PLoS ONE 2015, 10, e0130518. [Google Scholar] [CrossRef]
- Hu, W.; Zi, Z.; Jin, Y.; Li, G.; Shao, K.; Cai, Q.; Ma, X.; Wei, F. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol. Immunother. 2019, 68, 365–377. [Google Scholar] [CrossRef]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Cheng, C.; Mu, W.; Liu, X.; Li, N.; Wei, X.; Liu, X.; Xia, C.; Wang, H. CRISPR-Cas9 mediated LAG-3 disruption in CAR-T cells. Front. Med. 2017, 11, 554–562. [Google Scholar] [CrossRef]
- Ren, J.; Zhang, X.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget 2017, 8, 17002–17011. [Google Scholar] [CrossRef]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef]
- Salter, A.I.; Pont, M.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells: CD19 and the road beyond. Blood 2018, 131, 2621–2629. [Google Scholar] [CrossRef]
- Klinger, M.; Benjamin, J.; Kischel, R.; Stienen, S.; Zugmaier, G. Harnessing T cells to fight cancer with BiTE® antibody constructs--past developments and future directions. Immunol. Rev. 2016, 270, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 2019, 37, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [PubMed]
- Shalabi, H.; Koegel, A.; Ponduri, A.; Qin, H.; Salem, D.; Stetler-Stevenson, M.; Yuan, C.; Yates, B.; Delbrook, C.; Loh, M. Case report: Impact of BITE on CAR-T cell expansion. Adv. Cell Gene Ther. 2019, 2, e50. [Google Scholar] [CrossRef]
- Lohmueller, J.J.; Ham, J.D.; Kvorjak, M.; Finn, O.J. mSA2 affinity-enhanced biotin-binding CAR T cells for universal tumor targeting. Oncoimmunology 2017, 7, e1368604. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Ma, J.S.; Yun, H.; Cao, Y.; Kim, J.Y.; Chi, V.; Wang, D.; Woods, A.; Sherwood, L.; Caballero, D.; et al. Redirection of genetically engineered CAR-T cells using bifunctional small molecules. J. Am. Chem. Soc. 2015, 137, 2832–2835. [Google Scholar] [CrossRef]
- Tamada, K.; Geng, D.; Sakoda, Y.; Bansal, N.; Srivastava, R.; Li, Z.; Davila, E. Redirecting gene-modified T cells toward various cancer types using tagged antibodies. Clin. Cancer Res. 2012, 18, 6436–6445. [Google Scholar] [CrossRef]
- Kim, B.S.; Kim, J.Y.; Kim, E.J.; Lee, J.G.; Joo, D.J.; Huh, K.H.; Kim, M.S.; Kim, Y.S. Role of Thalidomide on the Expression of OX40, 4-1BB, and GITR in T Cell Subsets. Transplant. Proc. 2016, 48, 1270–1274. [Google Scholar] [CrossRef]
- LeBlanc, R.; Hideshima, T.; Catley, L.P.; Shringarpure, R.; Burger, R.; Mitsiades, N.; Mitsiades, C.; Cheema, P.; Chauhan, D.; Richardson, P.G.; et al. Immunomodulatory drug costimulates T cells via the B7-CD28 pathway. Blood 2004, 103, 1787–1790. [Google Scholar] [CrossRef]
- Otáhal, P.; Průková, D.; Král, V.; Fabry, M.; Vočková, P.; Latečková, L.; Trněný, M.; Klener, P. Lenalidomide enhances antitumor functions of chimeric antigen receptor modified T cells. Oncoimmunology 2016, 5, e1115940. [Google Scholar] [CrossRef]
- Wang, X.; Walter, M.; Urak, R.; Weng, L.; Huynh, C.; Lim, L.; Wong, C.W.; Chang, W.C.; Thomas, S.H.; Sanchez, J.F.; et al. Lenalidomide Enhances the Function of CS1 Chimeric Antigen Receptor-Redirected T Cells Against Multiple Myeloma. Clin. Cancer Res. 2018, 24, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; George, A.K.; Homme, R.P.; Majumder, A.; Laha, A.; Sandhu, H.S.; Tyagi, S.C. Circular RNAs profiling in the cystathionine-β-synthase mutant mouse reveals novel gene targets for hyperhomocysteinemia induced ocular disorders. Exp. Eye Res. 2018, 174, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; George, A.K.; Homme, R.P.; Majumder, A.; Laha, A.; Sandhu, H.S.; Tyagi, S.C. Expression Analysis of the Circular RNA Molecules in the Human Retinal Cells Treated with Homocysteine. Curr. Eye Res. 2019, 44, 287–293. [Google Scholar] [CrossRef] [PubMed]
- George, A.K.; Master, K.; Majumder, A.; Homme, R.P.; Laha, A.; Sandhu, H.S.; Tyagi, S.C.; Singh, M. Circular RNAs constitute an inherent gene regulatory axis in the mammalian eye and brain (1). Can. J. Physiol. Pharmacol. 2019, 97, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Majumder, A.; Behera, J.; Jeremic, N.; Tyagi, S.C. Hypermethylation: Causes and Consequences in Skeletal Muscle Myopathy. J. Cell Biochem. 2017, 118, 2108–2117. [Google Scholar] [CrossRef]
- Laha, A.; Majumder, A.; Singh, M.; Tyagi, S.C. Connecting homocysteine and obesity through pyroptosis, gut microbiome, epigenetics, peroxisome proliferator-activated receptor γ, and zinc finger protein 407. Can. J. Physiol. Pharmacol. 2018, 96, 971–976. [Google Scholar] [CrossRef]
- Huang, Q.; Xia, J.; Wang, L.; Wang, X.; Ma, X.; Deng, Q.; Lu, Y.; Kumar, M.; Zhou, Z.; Li, L.; et al. miR-153 suppresses IDO1 expression and enhances CAR T cell immunotherapy. J. Hematol. Oncol. 2018, 11, 58. [Google Scholar] [CrossRef]
- Ninomiya, S.; Narala, N.; Huye, L.; Yagyu, S.; Savoldo, B.; Dotti, G.; Heslop, H.E.; Brenner, M.K.; Rooney, C.M.; Ramos, C.A. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood 2015, 125, 3905–3916. [Google Scholar] [CrossRef]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018, 24, 20–28. [Google Scholar] [CrossRef]
- Watanabe, K.; Terakura, S.; Martens, A.C.; van Meerten, T.; Uchiyama, S.; Imai, M.; Sakemura, R.; Goto, T.; Hanajiri, R.; Imahashi, N.; et al. Target antigen density governs the efficacy of anti-CD20-CD28-CD3 ζ chimeric antigen receptor-modified effector CD8+ T cells. J. Immunol. 2015, 194, 911–920. [Google Scholar] [CrossRef]
- Li, S.; Xue, L.; Wang, M.; Qiang, P.; Xu, H.; Zhang, X.; Kang, W.; You, F.; Xu, H.; Wang, Y.; et al. Decitabine enhances cytotoxic effect of T cells with an anti-CD19 chimeric antigen receptor in treatment of lymphoma. Onco Targets Ther. 2019, 12, 5627–5638. [Google Scholar] [CrossRef] [PubMed]
- Xue, T.; Del Real, M.; Marcucci, E.; Toribio, C.; Setayesh, S.M.; Forman, S.J.; Horne, D.A.; Budde, L.E. Checkpoint blockade in combination with CD33 chimeric antigen receptor T cell therapy and hypomethylating agent against acute myeloid leukemia. Blood 2019, 134, 1383. [Google Scholar] [CrossRef]
- Bradner, J.E.; West, N.; Grachan, M.L.; Greenberg, E.F.; Haggarty, S.J.; Warnow, T.; Mazitschek, R. Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 2010, 6, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Hochberg, J.; Yahr, A.; Ayello, J.; van de Ven, C.; Barth, M.; Czuczman, M.; Cairo, M.S. Targeting CD20+ Aggressive B-cell Non-Hodgkin Lymphoma by Anti-CD20 CAR mRNA-Modified Expanded Natural Killer Cells In Vitro and in NSG Mice. Cancer Immunol. Res. 2015, 3, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, S.; Wang, Y.; Liu, J.; Mao, X.; Xing, H.; Tian, Z.; Tang, K.; Liao, X.; Rao, Q.; et al. Induced CD20 Expression on B-Cell Malignant Cells Heightened the Cytotoxic Activity of Chimeric Antigen Receptor Engineered T Cells. Hum. Gene Ther. 2019, 30, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Driouk, L.; Gicobi, J.K.; Kamihara, Y.; Rutherford, K.; Dranoff, G.; Ritz, J.; Baumeister, S.H.C. Chimeric Antigen Receptor T Cells Targeting NKG2D-Ligands Show Robust Efficacy Against Acute Myeloid Leukemia and T-Cell Acute Lymphoblastic Leukemia. Front. Immunol. 2020, 11, 580328. [Google Scholar] [CrossRef] [PubMed]
- Petersen, S.L.; Wang, L.; Yalcin-Chin, A.; Li, L.; Peyton, M.; Minna, J.; Harran, P.; Wang, X. Autocrine TNFalpha signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer Cell 2007, 12, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Dufva, O.; Koski, J.; Maliniemi, P.; Ianevski, A.; Klievink, J.; Leitner, J.; Pölönen, P.; Hohtari, H.; Saeed, K.; Hannunen, T.; et al. Integrated drug profiling and CRISPR screening identify essential pathways for CAR T-cell cytotoxicity. Blood 2020, 135, 597–609. [Google Scholar] [CrossRef]
- Michie, J.; Beavis, P.A.; Freeman, A.J.; Vervoort, S.J.; Ramsbottom, K.M.; Narasimhan, V.; Lelliott, E.J.; Lalaoui, N.; Ramsay, R.G.; Johnstone, R.W.; et al. Antagonism of IAPs Enhances CAR T-cell Efficacy. Cancer Immunol. Res. 2019, 7, 183–192. [Google Scholar] [CrossRef]
- Zhao, X.S.; Liu, Y.R.; Xu, L.P.; Wang, Y.; Zhang, X.H.; Chen, H.; Chen, Y.H.; Han, W.; Sun, Y.Q.; Yan, C.H.; et al. Minimal residual disease status determined by multiparametric flow cytometry pretransplantation predicts the outcome of patients with ALL receiving unmanipulated haploidentical allografts. Am. J. Hematol. 2019, 94, 512–521. [Google Scholar] [CrossRef]
- Khazal, S.; Kebriaei, P. Debate: Transplant Is Still Necessary in the Era of Targeted Cellular Therapy for Acute Lymphoblastic Leukemia. Clin. Lymphoma Myeloma Leuk. 2020, 20, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Taraseviciute, A.; Broglie, L.; Phelan, R.; Bhatt, N.S.; Becktell, K.; Burke, M.J. What is the Role of Hematopoietic Cell Transplantation (HCT) for Pediatric Acute Lymphoblastic Leukemia (ALL) in the Age of Chimeric Antigen Receptor T-Cell (CART) Therapy? J. Pediatr. Hematol. Oncol. 2019, 41, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Li, C.; Yin, P.; Guo, T.; Liu, L.; Xia, L.; Wu, Y.; Zhou, F.; Ai, L.; Shi, W.; et al. Anti-CD19 chimeric antigen receptor-modified T-cell therapy bridging to allogeneic hematopoietic stem cell transplantation for relapsed/refractory B-cell acute lymphoblastic leukemia: An open-label pragmatic clinical trial. Am. J. Hematol. 2019, 94, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Hay, K.A.; Gauthier, J.; Hirayama, A.V.; Voutsinas, J.M.; Wu, Q.; Li, D.; Gooley, T.A.; Cherian, S.; Chen, X.; Pender, B.S.; et al. Factors associated with durable EFS in adult B-cell ALL patients achieving MRD-negative CR after CD19 CAR T-cell therapy. Blood 2019, 133, 1652–1663. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, J.; Xu, M.; Yu, H.; Lv, C.; Cao, F.; Wang, Z.; Fu, Y.; Zhang, M.; Meng, H.; et al. Treatment response, survival, safety, and predictive factors to chimeric antigen receptor T cell therapy in Chinese relapsed or refractory B cell acute lymphoblast leukemia patients. Cell Death Dis. 2020, 11, 207. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Hu, Y.; Mei, H. Consolidative allogeneic hematopoietic stem cell transplantation after chimeric antigen receptor T-cell therapy for relapsed/refractory B-cell acute lymphoblastic leukemia: Who? When? Why? Biomark. Res. 2020, 8, 66. [Google Scholar] [CrossRef]
- Shadman, M.; Gauthier, J.; Hay, K.A.; Voutsinas, J.M.; Milano, F.; Li, A.; Hirayama, A.V.; Sorror, M.L.; Cherian, S.; Chen, X.; et al. Safety of allogeneic hematopoietic cell transplant in adults after CD19-targeted CAR T-cell therapy. Blood Adv. 2019, 3, 3062–3069. [Google Scholar] [CrossRef]
- Irving, M.; Vuillefroy de Silly, R.; Scholten, K.; Dilek, N.; Coukos, G. Engineering Chimeric Antigen Receptor T-Cells for Racing in Solid Tumors: Don’t Forget the Fuel. Front. Immunol. 2017, 8, 267. [Google Scholar] [CrossRef]
- Ligtenberg, M.A.; Mougiakakos, D.; Mukhopadhyay, M.; Witt, K.; Lladser, A.; Chmielewski, M.; Riet, T.; Abken, H.; Kiessling, R. Coexpressed Catalase Protects Chimeric Antigen Receptor-Redirected T Cells as well as Bystander Cells from Oxidative Stress-Induced Loss of Antitumor Activity. J. Immunol. 2016, 196, 759–766. [Google Scholar] [CrossRef]
- Scharping, N.E.; Menk, A.V.; Moreci, R.S.; Whetstone, R.D.; Dadey, R.E.; Watkins, S.C.; Ferris, R.L.; Delgoffe, G.M. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 2016, 45, 374–388. [Google Scholar] [CrossRef]
- Yang, W.; Bai, Y.; Xiong, Y.; Zhang, J.; Chen, S.; Zheng, X.; Meng, X.; Li, L.; Wang, J.; Xu, C.; et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature 2016, 531, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Majumder, A.; Singh, M.; Behera, J.; Theilen, N.T.; George, A.K.; Tyagi, N.; Metreveli, N.; Tyagi, S.C. Hydrogen sulfide alleviates hyperhomocysteinemia-mediated skeletal muscle atrophy via mitigation of oxidative and endoplasmic reticulum stress injury. Am. J. Physiol. Cell Physiol. 2018, 315, C609–C622. [Google Scholar] [CrossRef] [PubMed]
- Majumder, A.; Singh, M.; George, A.K.; Behera, J.; Tyagi, N.; Tyagi, S.C. Hydrogen sulfide improves postischemic neoangiogenesis in the hind limb of cystathionine-β-synthase mutant mice via PPAR-γ/VEGF axis. Physiol. Rep. 2018, 6, e13858. [Google Scholar] [CrossRef] [PubMed]
- Majumder, A.; Singh, M.; George, A.K.; Homme, R.P.; Laha, A.; Tyagi, S.C. Remote ischemic conditioning as a cytoprotective strategy in vasculopathies during hyperhomocysteinemia: An emerging research perspective. J. Cell Biochem. 2019, 120, 77–92. [Google Scholar] [CrossRef]
- Majumder, A.; Singh, M.; George, A.K.; Tyagi, S.C. Restoration of skeletal muscle homeostasis by hydrogen sulfide during hyperhomocysteinemia-mediated oxidative/ER stress condition (1). Can. J. Physiol. Pharmacol. 2019, 97, 441–456. [Google Scholar] [CrossRef]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D., Jr.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef]
- George, A.K.; Homme, R.P.; Majumder, A.; Tyagi, S.C.; Singh, M. Effect of MMP-9 gene knockout on retinal vascular form and function. Physiol. Genom. 2019, 51, 613–622. [Google Scholar] [CrossRef]
- George, A.K.; Majumder, A.; Ice, H.; Homme, R.P.; Eyob, W.; Tyagi, S.C.; Singh, M. Genes and genetics in hyperhomocysteinemia and the “1-carbon metabolism”: Implications for retinal structure and eye functions. Can. J. Physiol. Pharmacol. 2020, 98, 51–60. [Google Scholar] [CrossRef]
- George, A.K.; Homme, R.P.; Majumder, A.; Laha, A.; Metreveli, N.; Sandhu, H.S.; Tyagi, S.C.; Singh, M. Hydrogen sulfide intervention in cystathionine-β-synthase mutant mouse helps restore ocular homeostasis. Int. J. Ophthalmol. 2019, 12, 754–764. [Google Scholar] [CrossRef]
- Rabinovich, G.A.; Gabrilovich, D.; Sotomayor, E.M. Immunosuppressive strategies that are mediated by tumor cells. Annu. Rev. Immunol. 2007, 25, 267–296. [Google Scholar] [CrossRef]
- Long, A.H.; Highfill, S.L.; Cui, Y.; Smith, J.P.; Walker, A.J.; Ramakrishna, S.; El-Etriby, R.; Galli, S.; Tsokos, M.G.; Orentas, R.J.; et al. Reduction of MDSCs with All-trans Retinoic Acid Improves CAR Therapy Efficacy for Sarcomas. Cancer Immunol. Res. 2016, 4, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Parihar, R.; Rivas, C.; Huynh, M.; Omer, B.; Lapteva, N.; Metelitsa, L.S.; Gottschalk, S.M.; Rooney, C.M. NK Cells Expressing a Chimeric Activating Receptor Eliminate MDSCs and Rescue Impaired CAR-T Cell Activity against Solid Tumors. Cancer Immunol. Res. 2019, 7, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Majumder, A. Targeting Homocysteine and Hydrogen Sulfide Balance as Future Therapeutics in Cancer Treatment. Antioxidants 2023, 12, 1520. [Google Scholar] [CrossRef] [PubMed]
- Majumder, A.; Singh, M.; Tyagi, S.C. Post-menopausal breast cancer: From estrogen to androgen receptor. Oncotarget 2017, 8, 102739–102758. [Google Scholar] [CrossRef]
Figure 1.
Cartoon diagram representing a typical interaction mechanism of CAR-T cells with targeted cancer cells: CAR-T cells can recognize tumor cells by binding with specific tumor-associated antigens (TAA) independent of TCR-MHC/peptide interactions. As a result, T cells are activated. CAR-T cells can induce apoptosis via the secretion of perforin, granzymes, and different pro-inflammatory cytokines, as well as the expression of the Fas ligand (FasL) and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL).
Figure 1.
Cartoon diagram representing a typical interaction mechanism of CAR-T cells with targeted cancer cells: CAR-T cells can recognize tumor cells by binding with specific tumor-associated antigens (TAA) independent of TCR-MHC/peptide interactions. As a result, T cells are activated. CAR-T cells can induce apoptosis via the secretion of perforin, granzymes, and different pro-inflammatory cytokines, as well as the expression of the Fas ligand (FasL) and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL).
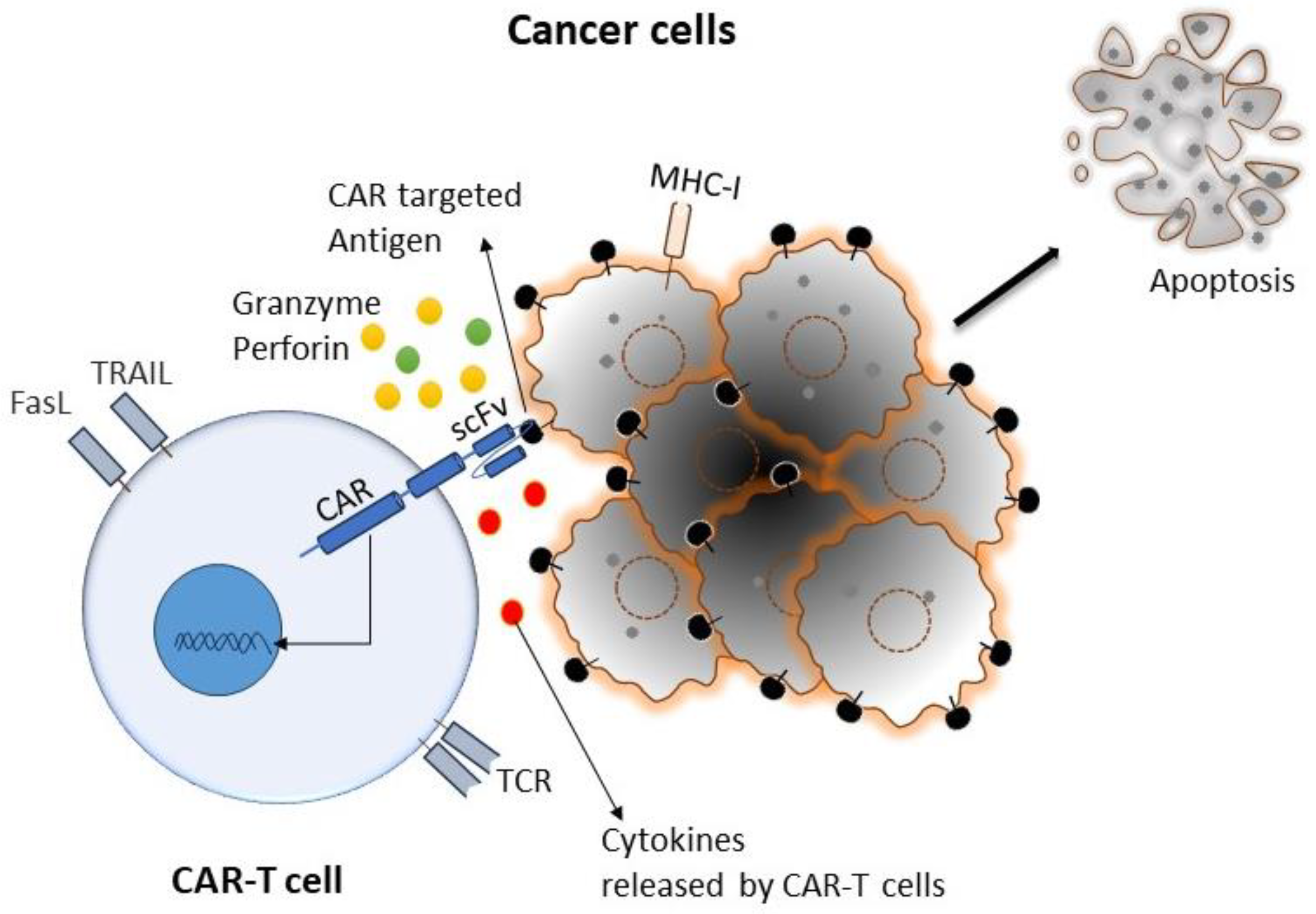
Figure 2.
Schematic representation of the basic principle of CAR architecture. (A) Cartoon diagram showing the binding moiety of CAR, where scFv comes from a TAA-specific monoclonal antibody and the signaling domain(s) comes from activating and co-stimulatory immune receptors. (B) Cartoon diagram showing the progressive evolution of CAR-T cells from first generation to fifth generation: first-generation CAR contains a single-chain variable fragment (scFv) and CD3ζ for signal transduction; an additional co-stimulatory domain (co-stimulatory domain 1: CSD 1) was added on first-generation CAR to make a second generation, whereas two additional co-stimulatory domains (CSD1 and CSD 2) were added on first-generation CAR to make third-generation CAR; one additional domain for cytokine-expression (cytokine inducer: CI) was added on second-generation CAR to make fourth-generation CAR; fifth-generation CAR contains a co-stimulatory domain (co-stimulatory domain 1) and an additional domain that activates signaling pathways such as STATE3/5. (C) Cartoon diagram showing the basic structure of a typical CAR with the functions of individual domains.
Figure 2.
Schematic representation of the basic principle of CAR architecture. (A) Cartoon diagram showing the binding moiety of CAR, where scFv comes from a TAA-specific monoclonal antibody and the signaling domain(s) comes from activating and co-stimulatory immune receptors. (B) Cartoon diagram showing the progressive evolution of CAR-T cells from first generation to fifth generation: first-generation CAR contains a single-chain variable fragment (scFv) and CD3ζ for signal transduction; an additional co-stimulatory domain (co-stimulatory domain 1: CSD 1) was added on first-generation CAR to make a second generation, whereas two additional co-stimulatory domains (CSD1 and CSD 2) were added on first-generation CAR to make third-generation CAR; one additional domain for cytokine-expression (cytokine inducer: CI) was added on second-generation CAR to make fourth-generation CAR; fifth-generation CAR contains a co-stimulatory domain (co-stimulatory domain 1) and an additional domain that activates signaling pathways such as STATE3/5. (C) Cartoon diagram showing the basic structure of a typical CAR with the functions of individual domains.
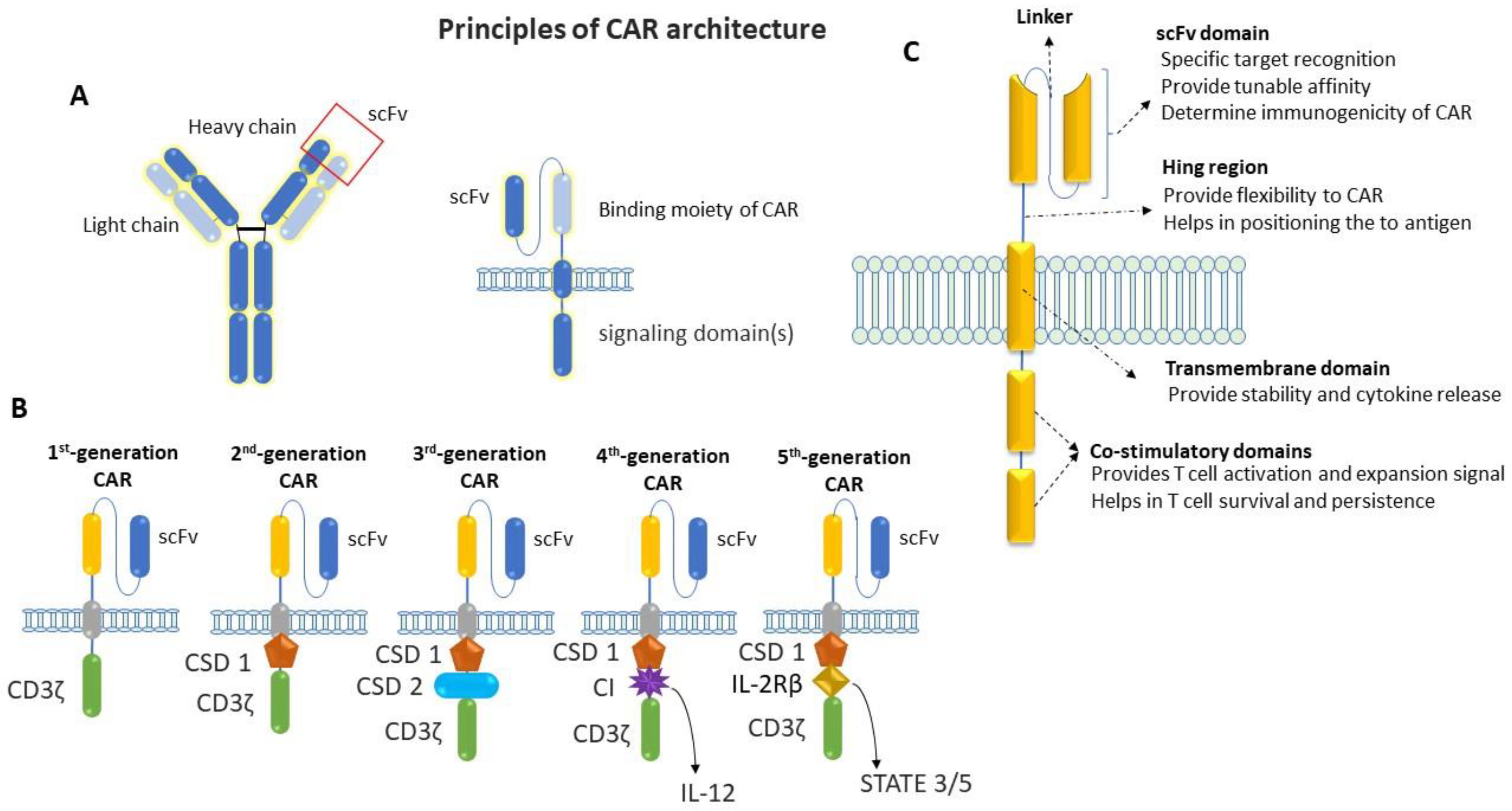
Figure 3.
Cartoon diagram showing different stages of autologous CAR-T-cell production: The generation of autologous CAR-T cells started with the leukapheresis of a patient (A), followed by the separation of T cells (B); then, T cells were transduced to permanently integrate CAR transgene (C). After that, these genetically modified CAR-T cells expanded (D) and infused back into the patient (E).
Figure 3.
Cartoon diagram showing different stages of autologous CAR-T-cell production: The generation of autologous CAR-T cells started with the leukapheresis of a patient (A), followed by the separation of T cells (B); then, T cells were transduced to permanently integrate CAR transgene (C). After that, these genetically modified CAR-T cells expanded (D) and infused back into the patient (E).
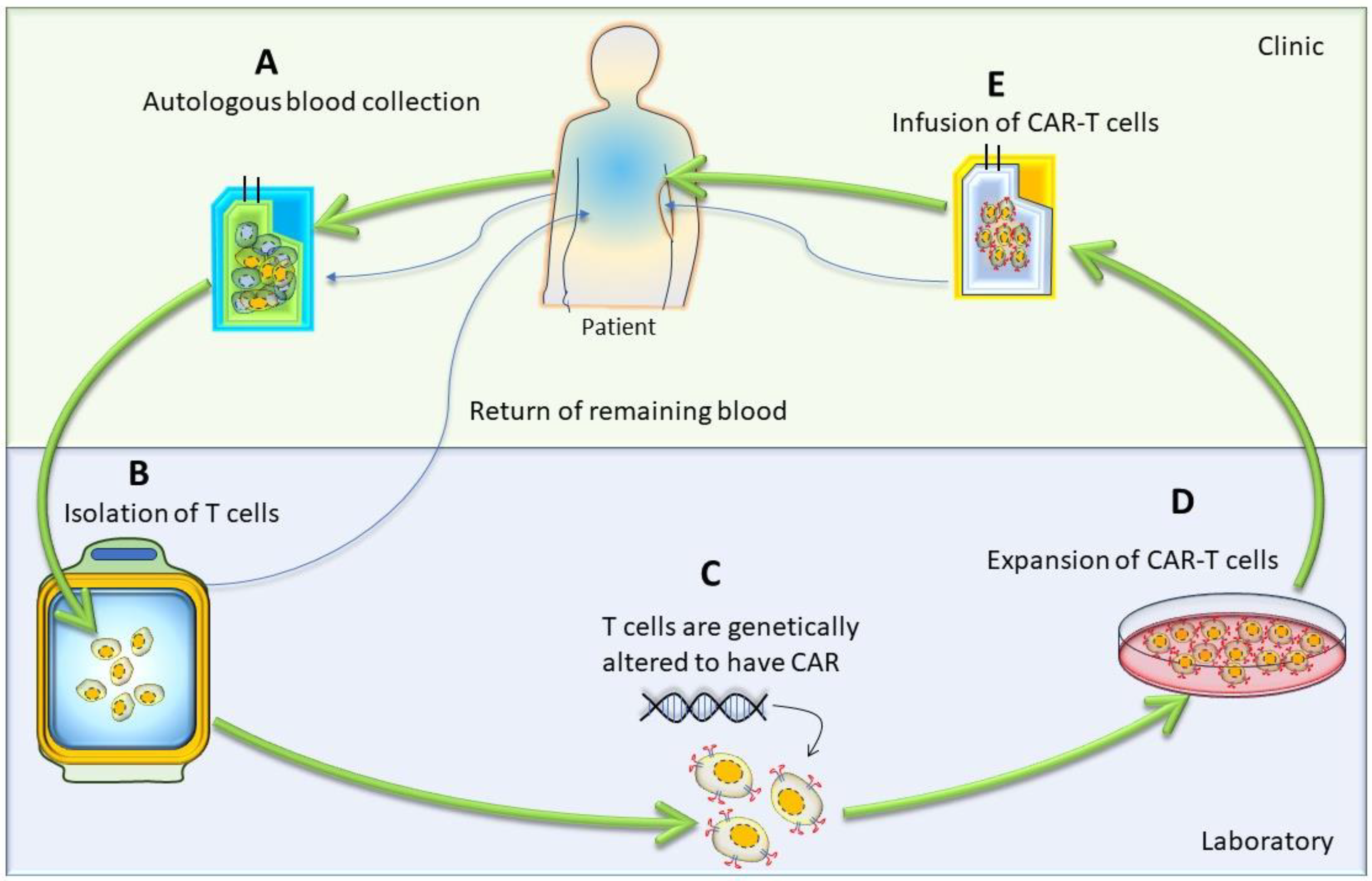
Figure 4.
Cartoon diagram showing the strategy of targeting two antigens using an engineered synthetic Notch receptor-CAR circuit that drives the expression of CAR.
Figure 4.
Cartoon diagram showing the strategy of targeting two antigens using an engineered synthetic Notch receptor-CAR circuit that drives the expression of CAR.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Important clinical trials for CAR-T-cell therapy in cancer that are currently ongoing or completed.
Table 1.
Important clinical trials for CAR-T-cell therapy in cancer that are currently ongoing or completed.
| NCT Number | CAR-T Strategy | Conditions | Type of Tumor | Phase | Current Status | Enrollment | Estimated/Actual Completion Date (DD Month YYYY) | Sponsor |
|---|---|---|---|---|---|---|---|---|
| NCT02348216 | CD19-specific CAR-T cells | Diffuse large B-cell lymphoma (DLBCL), primary mediastinal large B-cell lymphoma (PMBCL), transformation follicular lymphoma (TFL), high-grade B-cell lymphoma (HGBCL) | Hematological malignancy | Phase 1 Phase 2 | Completed | 307 | 27 July 2023 | Kite, A Gilead Company, Santa Monica, CA, USA |
| NCT02445248 | CD19-specific CAR-T cells | Relapsed or refractory DLBCL | Phase 2 | Completed | 115 | 22 December 2022 | Novartis Pharmaceuticals, Basel Switzerland | |
| NCT02601313 | CD19-specific CAR-T cells | Relapsed/refractory mantle cell lymphoma | Phase 2 | Active, not recruiting | 105 | July 2025 | Kite, A Gilead Company | |
| NCT02614066 | CD19-specific CAR-T cells | Relapsed/refractory B-precursor acute lymphoblastic leukemia | Phase 1 Phase 2 | Active, not recruiting | 125 | November 2034 | Kite, A Gilead Company | |
| NCT02631044 | CD19-specific CAR-T cells | Non-Hodgkin lymphoma, diffuse large B cell lymphoma, follicular lymphoma, mantle-cell lymphoma, primary mediastinal B-cell lymphoma | Phase 1 | Active, not recruiting | 385 | 10 May 2024 | Juno Therapeutics, a Subsidiary of Celgene, Seattle, WA, USA | |
| NCT02926833 | CD19-specific CAR-T cells in combination with PD-1 antibodies | DLBCL | Phase 1 Phase 2 | Completed | 37 | 12 January 2023 | Kite, A Gilead Company | |
| NCT03105336 | CD19-specific CAR-T cells | Refractory/relapse large B cell lymphoma | Phase 2 | Active, not recruiting | 159 | Spetember 2036 | Kite, A Gilead Company | |
| NCT03287817 | CD19- and CD22-specific CAR-T cells, followed by anti-PD1 antibody | DLBCL | Phase 1 Phase 2 | Active, not recruiting | 73 | November 2024 | Autolus Limited, London, UK | |
| NCT03310619 | CD19-specific CAR-T cells | Aggressive B-NHL | Phase 1 Phase 2 | Completed | 62 | 15 February 2023 | Celgene, Summit, NJ, USA | |
| NCT03568461 | CD19-specific CAR-T cells | Refractory follicular lymphoma | Phase 2 | Active, not recruiting | 98 | 22 May 2025 | Novartis Pharmaceuticals | |
| NCT00004178 | CEA CAR | Adenocarcinoma | Solid tumor | Phase 1 | Completed | Not given | December 2001 | Roger Williams Medical Center, Providence, RI, USA |
| NCT00019136 | Folate receptor CAR ± IL-2 | Ovarian cancer | Phase 1 | Completed | Not given | Not given | National Cancer Institute (NCI), Rockville, MD, USA | |
| NCT00085930 | GD2 CAR, EBV T cells | Neuroblastoma | Phase 1 | Active, not recruiting | 19 | December 2023 | Baylor College of Medicine, Houston, TX, USA | |
| NCT00730613 | IL-13Ra2 targeting CAR-T cells | Glioblastoma with Hy/TK suicide switch | Phase 1 | Completed | 3 | August 2011 | City of Hope Medical Center, Duarte, CA, USA | |
| NCT00889954 | HER2 CAR, EBV T cells + TGFb DNR | HER2-positive lung cancer | Phase 1 | Completed | 20 | 21 January 2018 | Baylor College of Medicine | |
| NCT00902044 | HER2 CD28 CAR | HER2-positive sarcoma | Phase 1 | Active, not recruiting | 36 | July 2032 | Baylor College of Medicine | |
| NCT01109095 | Her2 CAR, CMV T cells | HER2-positive glioblastoma | Phase 1 | Completed | 16 | 7 March 2018 | Baylor College of Medicine | |
| NCT01140373 | PSMA CAR 2nd | Castrate metastatic | Phase 1 | Active, not recruiting | 13 | June 2024 | Memorial Sloan Kettering Cancer Center, New York, NY, USA | |
| NCT01373047 | CEA CAR | CEA-positive liver metastases | Phase 1 | Completed | 8 | July 2013 | Roger Williams Medical Center | |
| NCT01454596 | EGFRvIII CAR 3rd 28 and 4-1BB ± IL-2 | Glioblastoma | Phase 1 Phase 2 | Completed | 18 | 17 January 2019 | National Cancer Institute (NCI) | |
| NCT01460901 | GD2 CAR multivirus specific | Post-allo HSCT neuroblastoma | Phase 1 | Completed | 5 | January 2015 | Children’s Mercy Hospital Kansas City, Kanzas City, MO, USA | |
| NCT01822652 | GD-2-CAR-T with iCaspase9 Suicide safety switch | Neuroblastoma | Phase 1 | Active, not recruiting | 11 | October 2030 | Baylor College of Medicine | |
| NCT02414269 | Meso-CART cells, modified with iCasp9/M284 | Malignant pleural disease | Phase 1 Phase 2 | Active, not recruiting | 113 | 30 April 2024 | Memorial Sloan Kettering Cancer Center | |
| NCT03170141 | EGFRvIlI-specific CAR-T cells producing PD-1 and PD-L1 antibodies | Glioblastoma multiforme | Phase 1 | Enrolling by invitation | 20 | 31 Decemebr 2023 | Shenzhen Geno-Immune Medical Institute, Shenzhen, China |
Table 2.
Current limitations and potential strategies of CAR-T-cell therapy.
| Limitations | Potential Strategy | Supporting Reports | References |
|---|---|---|---|
| Antigen escape | Targeting multiple antigens | In preclinical trails, CAR-T cells targeting both CD19 and CD22 antigens in ALL/DLBCL and CAR-T cells targeting both CD19 and BCMA antigens in multiple myeloma have shown promising results. | [65,66,67,68] |
| Similarly, in solid tumors, CAR-T cells targeting both HER2 and IL13Ra2 antigens in glioblastoma and CAR-T cells targeting both HER2 and MUC1 in breast cancer showed better antitumor effects compared to targeting a single antigen. | [56,69] | ||
| On-target/off-tumor effects | Targeting tumor-associated antigens that are post-translationally modified and only express in tumors | Different post-translationally modified tumor associated antigens such as TAG72, B7-H3, MUC1, and MUC16 have been targeted using CAR-T-cell therapy which showed very effective. | [56,57,58,59,60,61] |
| Targeting two antigens rather than one antigen | Engineered CAR-T cells with synthetic Notch receptors to activate CAR targeting of the second antigen in the presence of the first antigen. | [54,70] | |
| Engineered CAR-T cells to sense high antigen densities in tumors | Engineered CAR-T cells with synthetic Notch receptors to activate CAR in a high antigen threshold. | [55] | |
| Poor CAR-T-cell trafficking and tumor infiltration | Local administration vs. systemic delivery | Better antitumor effects were noted via the local delivery of CAR-T cells in glioblastoma cancer patients and the systemic delivery of CAR-T cells in mesothelioma patients. | [38,71,72] |
| Expressing chemokine receptors on CAR-T cells that respond to tumor-derived chemokines | CAR-T cells that express CXCR1/CXCR2 enhanced trafficking and significantly improved antitumor efficacy. | [73,74,75] | |
| Genetically modified CAR-T cells that express proteins that help in the penetration of tumor stoma | CAR-T cells that express heparanase or CAR-T cells that target fibroblast activation protein have shown enhanced infiltration and antitumor activity. | [76,77] | |
| Immunosuppressive TME | Combination checkpoint blockade with CAR-T-cell therapy | Administration of PD-1 inhibitor with CD19 CAR-T-cell therapy showed improved CAR-T-cell persistence in B-ALL patients. | [78] |
| Likewise, in solid tumors, checkpoint blockade has been combined with CAR-T therapy and shown improved persistence of CAR-T cells. | [79,80] | ||
| Engineering CAR-T cells to provide immunostimulatory signals to the TME | Better response was observed when CAR-T cells were genetically engineered to express immunostimulatory molecules like IL-12 and IL-15 and redirect immunosuppressive molecules like IL-4 | [81,82,83] | |
| Genetic alteration of CAR-T cells to make them resistant to immunosuppressive factors like TGF β. | [84] | ||
| CAR-T-cell therapy associated toxicities | Altering CAR structure to ameliorate toxicity | Decreasing CAR antigen-binding domain affinity to micromolar affinity. | [85] |
| Modulation of cytokine secretion via modifying the CAR hinge and transmembrane regions. | [86] | ||
| Tailoring the costimulatory domain of CAR based on tumor type, tumor burden, antigen density, etc. | [87] | ||
| CAR-mediated immune response can be decreased using human/humanized antibody fragments instead of murine-derived CARs. | [88,89] | ||
| Modifying CAR transduced T cells and neurotoxicity | Inhibition of macrophage-activating and monocyte-activating cytokine GM-CSF with lenzilumab decreases cytokine-release syndrome and neurotoxicity. | [90,91] | |
| Administration of IL-1 receptor antagonists reduced a form of neuroinflammation in leukemia/lymphoma mouse models. | [92] | ||
| CAR “off-switches” | CAR constructs engineered to express CD20 that helped with the depletion of CAR-T cells via rituximab treatment. | [93] | |
| Dasatinib treatment has exciting potential, as it provides the temporary inhibition of CAR-T-cell function. | [94] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Majumder, A. Evolving CAR-T-Cell Therapy for Cancer Treatment: From Scientific Discovery to Cures. Cancers 2024, 16, 39. https://doi.org/10.3390/cancers16010039
AMA Style
Majumder A. Evolving CAR-T-Cell Therapy for Cancer Treatment: From Scientific Discovery to Cures. Cancers. 2024; 16(1):39. https://doi.org/10.3390/cancers16010039
Chicago/Turabian StyleMajumder, Avisek. 2024. "Evolving CAR-T-Cell Therapy for Cancer Treatment: From Scientific Discovery to Cures" Cancers 16, no. 1: 39. https://doi.org/10.3390/cancers16010039
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.