

Inflammation-Associated Cytotoxic Agents in Tumorigenesis
Abstract
:Simple Summary
Abstract
1. Introduction
2. Chronic Inflammatory States and Altered Conditions in Tumors
2.1. Immunosuppression during Acute and Chronic Inflammation
2.2. Key Elements of Immunosuppression in Tumors
2.3. Poor Quality of the Tumor Vasculature
2.4. Direct Effects of Hypoxia in Tumor Cells
2.5. Stress-Related Responses in Tumor Cells
2.6. Intratumoral Hemorrhages
2.7. Reversed pH Gradient in Cancer Cells
2.8. Matrix Remodeling in the TME
3. Cytotoxic Agents in Cancer Cells
3.1. Sources for Cytotoxic Agents in Cancer Cells
3.2. Mitochondria-Derived Reactive Species in Cancer Cells
3.3. Antioxidant Defense Mechanisms and Stabilization of Redox Homeostasis in Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antioxidative System | Functions | Cancer Types | References |
|---|---|---|---|
| SOD1 | Removal of O2•−, formation of H2O2 | Lung and breast cancer | [248,249] |
| SOD2 | Removal of O2•−, formation of H2O2 | Esophageal cancer and others | [250,251,252] |
| Catalase | Removal of H2O2 | Glioblastoma | [253] |
| Peroxiredoxins (six subtypes) | Removal of H2O2 | Many types of tumors | [226,254] |
| Thioredoxin system (thioredoxin, thioredoxin reductase; NADPH) | Removal of H2O2, redox homeostasis | Lung, breast, prostate, colorectal, pancreatic, hepatocellular, gastric, and other carcinomas | [255,256] |
| Cystine/glutamate anti-porter solute carrier family 7 member 11 | Cystine/cysteine uptake in cells for GSH production | Multiple types of cancer | [117,118] |
| Glutathione (GSH) | Redox homeostasis, cofactor of glutathione peroxidases | Ovarian, breast, and lung cancers | [257,258,259] |
| Glutathione reductase | Recovery of GSH | Glioblastoma | [260] |
| Glutathione peroxidase 4 | Removal of H2O2 and lipid hydroperoxides, inhibition of ferroptosis | Thyroid, colorectal, and esophageal carcinomas | [261,262] |
| Glutathione peroxidase 1 | Removal of H2O2, peroxynitrite, and lipid hydroperoxides | Breast, hepatocellular, colorectal, esophageal, and lung carcinomas | [243] |
| Glutathione peroxidase 2 | Removal of H2O2 | Liver, colorectal, breast, lung, and prostate carcinomas | [263,264] |
| Glutaredoxins 3 and 5 | Redox homeostasis | Liver and colorectal cancers | [265] |
| sulfiredoxin | Redox homeostasis | Skin, liver, and colorectal cancers | [266] |
| Ribonucleotide reductase | Redox homeostasis | Gastric, ovarian, bladder, colorectal cancers | [267,268] |
| Methionine sulfoxide reductase B1 and B3 | Redox homeostasis | Hepatocellular carcinoma and others | [245,246,247] |
| Glucose-6-phosphate dehydrogenase | NADPH production, redox homeostasis | Multiple types of cancer | [269,270] |
| 6-phosphogluconate dehydrogenase | NADPH production, redox homeostasis | Multiple types of cancer | [271,272] |
3.4. Disturbed Iron Homeostasis in Cancer Cells
3.5. Intratumoral Hemorrhages and Free Heme
3.6. Protection against Free Heme by Heme Oxygenase 1 (HO-1)
4. Cytotoxic Agents in the TME
4.1. Special Conditions in the TME
4.2. Peculiarities of Cytotoxic Agents in the TME
4.3. Neutrophil Subpopulations with the Tumor-Promoting Phenotype
4.4. Involvement of Myeloperoxidase (MPO) in the Immunosuppression by G-MDSCs
4.5. Elastase-Mediated Effects in the TME
4.6. Role of Angiotensin II
4.7. NO Metabolism in the TME
4.8. Role of the Extracellular Matrix Components in the TME
5. Conclusions
Funding
Conflicts of Interest
References
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Smyth, M.J. Targeting cancer-related inflammation in the era of immunotherapy. Immunol. Cell Biol. 2017, 95, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Arnhold, J. Immune response and tissue damage. In Cell and Tissue Destruction. Mechanisms, Protection, Disorders; Academic Press: London, UK; San Diego, CA, USA; Cambridge, MA, USA; Oxford, UK, 2020; pp. 155–204. [Google Scholar] [CrossRef]
- Arnhold, J. Acute-phase proteins and additional protective systems. In Cell and Tissue Destruction. Mechanisms, Protection, Disorders; Academic Press: London, UK; San Diego, CA, USA; Cambridge, MA, USA; Oxford, UK, 2020; pp. 205–228. [Google Scholar] [CrossRef]
- Wang, D.; DuBois, R.N. Immunosuppression associated with chronic inflammation in the tumor microenvironment. Carcinogenesis 2015, 36, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Burnet, M. Cancer; a biological approach. I. The process of control. II. The significance of somatic mutation. Br. Med. J. 1957, 1, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L. On immunosurveillance in human cancer. Yale J. Biol. Med. 1982, 55, 329–333. [Google Scholar] [PubMed]
- Shimasaki, M.; Jain, A.; Campana, D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 2020, 19, 200–218. [Google Scholar] [CrossRef]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gögenur, I. Cytotoxic CD8+ T cells in cancer and cancer immunotherapy. Br. J. Cancer 2021, 124, 359–367. [Google Scholar] [CrossRef]
- Nakamura, K.; Smyth, M.J. Myeloid immunosuppression and immune checkpoints in the tumor microenvironment. Cell Mol. Immunol. 2020, 17, 1–12. [Google Scholar] [CrossRef]
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The tumor microenvironment: A milieu hindering and obstructing antitumor immune response. Front. Immunol. 2020, 11, 940. [Google Scholar] [CrossRef]
- Barnestein, R.; Galland, L.; Kalfeist, L.; Ghiringhelli, F.; Ladoire, S.; Limagne, E. Immunosuppressive tumor microenvironment modulation by chemotherapies and targeted therapies to enhance immunotherapy effectiveness. OncoImmunology 2022, 11, 2120676. [Google Scholar] [CrossRef]
- Tie, Y.; Tang, F.; Wei, Y.-Q.; Wie, X.-W. Immunosuppressive cells in cancer: Mechanisms and potential therapeutic targets. J. Hematol. Oncol. 2022, 15, 61. [Google Scholar] [CrossRef]
- Arnhold, J. Host-derived cytotoxic agents in chronic inflammation and disease progression. Int. J. Mol. Sci. 2023, 24, 3016. [Google Scholar] [CrossRef]
- Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef]
- Janeway, C.A.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef]
- Suresh, R.; Moser, D.M. Pattern recognition in innate immunity, host defense, and immunopathology. Adv. Physiol. Educ. 2013, 37, 284–291. [Google Scholar] [CrossRef]
- Pepys, M.B.; Baltz, M.I. Acute phase proteins with special reference to C-reactive protein and related proteins (pentraxins) and serum amyloid A protein. Adv. Immunol. 1983, 34, 141–212. [Google Scholar] [CrossRef]
- Vandivier, R.W.; Henson, P.M.; Douglas, I.S. Burying the death: The impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest 2006, 129, 1673–1682. [Google Scholar] [CrossRef]
- Pober, J.S.; Sessa, W.C. Inflammation and the blood microvascular system. Cold Spring Harbor Perspect. Biol. 2015, 7, 016345. [Google Scholar] [CrossRef]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.K.; Flavell, R.A. Transforming growth factor-beta regulation of immune response. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef]
- Li, M.O.; Flavell, R.A. Contextual regulation of inflammation: A duet of transforming growth factor-beta and interleukin-10. Immunity 2008, 28, 468–476. [Google Scholar] [CrossRef]
- Couper, K.N.; Blount, D.G.; Riley, E.M. IL-10: The master regulator of immunity to infection. J. Immunol. 2008, 180, 5771–5777. [Google Scholar] [CrossRef]
- Landén, N.X.; Li, D.; Ståhle, M. Transition from inflammation to proliferation: A critical step during wound healing. Cell Mol. Life Sci. 2016, 73, 3861–3885. [Google Scholar] [CrossRef]
- Marega, M.; Chen, C.; Bellusci, S. Cross-talk between inflammation and fibroblast growth factor 10 during organogenesis and pathogenesis: Lessons learnt from the lung and other organs. Front. Cell Dev. Biol. 2021, 9, 656883. [Google Scholar] [CrossRef]
- Serhan, C.N.; Savill, J. Resolution of inflammation: The beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef]
- Chandrasekharan, J.A.; Sharma-Walia, N. Lipoxins: Nature’s way to resolve inflammation. J. Inflamm. Res. 2015, 8, 181–192. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S.; Locati, M.; Montavani, A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: New molecules and patterns of gene expression. J. Immunol. 2006, 177, 7303–7311. [Google Scholar] [CrossRef]
- Sanchez-Pino, M.D.; Dean, M.J.; Ochoa, A.C. Myeloid-derived suppressor cells (MDSC): When good intentions go awry. Cell Immunol. 2021, 362, 104302. [Google Scholar] [CrossRef]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumor progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Sepkowitz, K.A. Opportunistic infections in patients with and patients without acquired immunodeficiency syndrome. Clin. Infect. Dis. 2002, 34, 1098–1107. [Google Scholar] [CrossRef]
- Kampitak, T.; Suwanpimolkul, G.; Browne, S.; Suankratay, C. Anti-interferon-autoantibody and opportunistic infections: Case series and review of the literature. Infection 2011, 39, 65–71. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Beneticits, G. Inflammaging—An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Newman, A.B.; Sanders, J.L.; Kizer, J.R.; Boudrou, R.M.; Odden, M.C.; Zeki Al Hazzouri, A.; Arnold, A.M. Trajectories of function and biomarkers with age: The CHS All stars study. Int. J. Epidemiol. 2016, 45, 1135–1145. [Google Scholar] [CrossRef]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 1135–1145. [Google Scholar] [CrossRef]
- Othman, N.; Jamal, R.; Abu, N. Cancer-derived exosomes as effectors of key inflammation-related players. Front. Immunol. 2019, 10, 2103. [Google Scholar] [CrossRef]
- Wrzesinski, S.H.; Wan, Y.Y.; Flavell, R.A. Transforming growth factor-β and immune response: Implications for anticancer therapy. Clin. Cancer Res. 2007, 13, 5262–5270. [Google Scholar] [CrossRef]
- Bierie, B.; Moses, H.L. Transforming growth factor β (TGF-β) and inflammation in cancer. Cytokine Growth Factor Rev. 2010, 21, 49–59. [Google Scholar] [CrossRef]
- Szczepanski, M.J.; Szajnik, M.; Welsh, A.; Whiteside, T.L.; Boyiadzis, M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-beta 1. Haematologia 2011, 96, 1302–1309. [Google Scholar] [CrossRef]
- He, Z.; Zhang, S. Tumor-associated macrophages and their functional transformation in the hypoxic tumor microenvironment. Front. Immunol. 2021, 12, 741305. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef]
- Umansky, V.; Blattner, C.; Gebhardt, C.; Utikal, J. The role of myeloid-derived suppressor cells (MDSC) in cancer progression. Vaccines 2016, 4, 36. [Google Scholar] [CrossRef]
- Hedge, S.; Leader, A.M.; Merad, M. MDSC: Marker, development, states, and unaddressed complexity. Immunity 2021, 54, 875–884. [Google Scholar] [CrossRef]
- Cassetta, L.; Bruderek, K.; Skrzeczynska-Moncznik, J.; Osiecka, O.; Hu, X.; Rundgren, I.M.; Lin, A.; Santegoets, K.; Horzum, U.; Godinhi-Santos, A.; et al. Differential expansion of circulating human MDSC subsets in patients with cancer, infection and inflammation. J. Immunother. Cancer 2020, 8, e001223. [Google Scholar] [CrossRef]
- Casetta, L.; Baekkevold, E.S.; Brandau, S.; Bujko, A.; Casatella, M.A.; Dorhoi, A.; Krieg, C.; Lin, A.; Loré, K.; Marini, O.; et al. Deciphering myeloid-derived suppressor cells: Isolation and markers in humans, mice and non-human primates. Cancer Immunol. Immunother. 2019, 68, 687–697. [Google Scholar] [CrossRef]
- Bronte, V.; Brandau, S.; Chen, S.-H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.M.C.S.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef]
- Molinier-Frenkel, V.; Castellano, F. Immunosuppressive enzymes in the tumor microenvironment. FEBS Lett. 2017, 591, 3135–3157. [Google Scholar] [CrossRef]
- Bader, J.E.; Voss, K.; Rathmell, J.C. Targeting metabolism to improve the tumor microenvironment for cancer immunotherapy. Mol. Cell 2020, 78, 1019–1033. [Google Scholar] [CrossRef]
- Allard, B.; Longhi, M.S.; Robson, S.C.; Stagg, J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol. Rev. 2017, 276, 121–144. [Google Scholar] [CrossRef]
- Moon, Y.W.; Hajjar, J.; Hwu, P.; Naing, A. Targeting the indoleamine 2,3-dioxygenase pathway in cancer. J. Immunother. Cancer 2015, 3, 51. [Google Scholar] [CrossRef]
- Kitamura, H.; Ohno, Y.; Toyoshima, Y.; Ohtake, J.; Homma, S.; Kawamura, H.; Takahashi, N.; Taketomi, A. Interleukin-6/STAT3 signaling as a promising target to improve the efficacy of cancer immunotherapy. Cancer Sci. 2017, 108, 1947–1952. [Google Scholar] [CrossRef]
- Park, S.-J.; Nakagawa, T.; Kitamura, H.; Atsumi, T.; Kamon, H.; Sawa, S.-I.; Kamimura, D.; Ueda, N.; Iwakura, Y.; Ishihara, K. IL-6 Regulates in vivo dendritic cell differentiation through STAT3 activation. J. Immunol. 2004, 1950, 3844–3854. [Google Scholar] [CrossRef]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.-L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef]
- Ohm, J.E.; Carbone, D.P. VEGF as a mediator of tumor-associated immunodeficiency. Immunol. Res. 2001, 23, 263–272. [Google Scholar] [CrossRef]
- Huang, H.; Langenkamp, E.; Georganaki, M.; Loskog, A.; Fuchs, P.F.; Dieterich, L.C.; Kreuger, J.; Dimberg, A. VEGF suppresses T-lymphocyte infiltration in the tumor microenvironment through Inhibition of NF-κB-induced endothelial activation. FASEB J. 2015, 29, 227–238. [Google Scholar] [CrossRef]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.P.; Kurzrock, R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1994, 1, 405–413. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, S.; Inozume, T.; Kawazu, M.; Ueno, T.; Nagasaki, J.; Tanji, E.; Honobe, A.; Ohnuma, T.; Kawamura, T.; Umeda, Y. TIGIT/CD155 axis mediates resistance to immunotherapy in patients with melanoma with the inflamed tumor microenvironment. J. Immunother. Cancer 2021, 9, e003134. [Google Scholar] [CrossRef] [PubMed]
- Konerding, M.A.; Fait, E.; Gaumann, A. 3D microvascular architecture of pre-cancerous lesion and invasive carcinomas in the colon. Br. J. Cancer 2001, 84, 1354–1362. [Google Scholar] [CrossRef]
- Siemann, D.W. The unique characteristics of tumor vasculature and preclinical evidence for its selective disruption by tumor-vascular disrupting agents. Cancer Treat. Rev. 2011, 37, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Jubb, A.M.; Pham, T.Q.; Hanby, A.M.; Frantz, G.D.; Peale, F.V.; Wu, T.D.; Koeppen, H.W.; Hilan, K.J. Expression of vascular endothelial growth factor, hypoxia inducible factor 1 alpha, and carbonic anhydrase IX in human tumours. J. Clin. Pathol. 2004, 57, 504–512. [Google Scholar] [CrossRef]
- Shweiki, D.; Itin, A.; Soffer, D.; Keshet, E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 1992, 359, 843–845. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Vascular endothelial growth factor(VEGF) signaling in tumor progression. Crit. Rev. Oncol./Hematol. 2007, 62, 179–213. [Google Scholar] [CrossRef]
- Saharinen, P.; Eklund, L.; Pulkki, K.; Bono, P.; Alitalo, K. VEGF and angiopoietin signaling in tumor angiogenesis and metastasis. Trends Mol. Med. 2011, 17, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Cao, Y. The impact of VEGF on cancer metastasis and systemic disease. Semin. Cancer Biol. 2022, 86, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Carreau, A.; El Hafny-Rahbi, B.; Matejuk, A.; Grillon, C.; Kieda, C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J. Cell Mol. Med. 2011, 15, 1239–1253. [Google Scholar] [CrossRef] [PubMed]
- McKeown, S.R. Defined normoxia, physoxia and hypoxia in tumors—Implications for treatment response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Mayer, A.; Höckel, M. Impact of hemoglobin levels on tumor oxygenation: The higher, the better? Strahlenther. Onkol. 2006, 182, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, A.M.; Lone, A.; Betts, D.H.; Cumming, R.C. Lactate preconditioning promotes a HIF-1α-mediated metabolic shift from OXPHOS to glycolysis in normal human diploid fibroblasts. Sci. Rep. 2020, 10, 8388. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Fukuda, A.; Hisatake, K. Mechanisms of the metabolic shift during somatic cell reprogramming. Int. J. Mol. Sci. 2019, 20, 2254. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Choi, J.; Gyamfi, J.; Jang, H.; Koo, J.S. The role of tumor-associated macrophage in breast cancer biology. Histol. Histopathol. 2018, 33, 133–145. [Google Scholar] [CrossRef]
- Mu, X.; Shi, W.; Xu, Y.; Xu, C.; Zhao, T.; Geng, B.; Yang, J.; Pan, J.; Hu, S.; Zhang, C.; et al. Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle 2018, 17, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Ratter, J.M.; Rooljackers, H.M.M.; Hoolveld, G.J.; Hijmans, A.G.M.; de Galan, B.E.; Tack, C.J.; Stienstra, R. In vitro and in vivo effects of lactate on metabolism and cytokine production of human primary PBMCs and monocytes. Front. Immunol. 2018, 9, 2564. [Google Scholar] [CrossRef] [PubMed]
- De Saedeleer, C.J.; Copetti, T.; Porporato, P.E.; Verrax, J.; Feron, O.; Sonveaux, P. Lactate activates HIF-1 in oxidative but not in Warburg-phenotype human tumor cells. PLoS ONE 2012, 7, e46571. [Google Scholar] [CrossRef] [PubMed]
- Pires, B.R.B.; Mencalha, A.L.; Ferreira, G.M.; Panis, C.; Silva, R.C.M.C.; Abdelhay, E. The hypoxia-inducible factor-1 alpha signaling pathway and its relation to cancer and immunology. Am. J. Immunol. 2014, 10, 215–224. [Google Scholar] [CrossRef]
- Davis, L.; Recktenwald, M.; Hutt, E.; Fuller, S.; Briggs, M.; Goel, A.; Daringer, N. Targeting HIF-2α in the tumor microenvironment: Redefining the role of HIF-2α for solid cancer therapy. Cancers 2022, 14, 1259. [Google Scholar] [CrossRef] [PubMed]
- Löfstedt, T.; Fredlund, E.; Holmquist-Mengelbier, L.; Pietras, A.; Overnberger, M.; Pollinger, L.; Påhlman, S. Hypoxia inducible factor-2α in cancer. Cell Cycle 2007, 6, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Schödel, J.; Oikonomopoulos, S.; Ragoussis, J.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. High-resolution genome-wide mapping of HIF-binding sites by CHIP-seq. Blood 2011, 117, e207. [Google Scholar] [CrossRef] [PubMed]
- Stalnecker, C.A.; Cluntun, A.A.; Cerione, R.A. Balancing redox stress: Anchorage-independent growth requires reductive carboxylation. Transl. Cancer Res. 2016, 5, S433–S437. [Google Scholar] [CrossRef]
- Jin, J.; Byun, J.-K.; Choi, Y.-K.; Park, K.-G. Targeting glutamine metabolism as a therapeutic strategy for cancer. Exp. Mol. Med. 2023, 55, 706–715. [Google Scholar] [CrossRef]
- Végran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports on NFκB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011, 71, 2550–2560. [Google Scholar] [CrossRef]
- Chamnee, T.; Ontong, P.; Itano, N. Hyaluronan: A modulator of the tumor microenvironment. Cancer Lett. 2016, 375, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Lennicke, C.; Rahn, J.; Lichtenfels, R.; Wessjohann, L.A.; Seliger, B. Hydrogen peroxide—Production, fate and role in redox signaling of tumor cells. Cell Commun. Signal. 2015, 13, 39. [Google Scholar] [CrossRef] [PubMed]
- Cyran, A.M.; Zhitkovich, A. HIF1, HSF1, and NRF2: Oxidant-responsive trio raising cellular defenses and engaging immune system. Chem. Res. Toxicol. 2022, 35, 1690–1700. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Song, P.; Zou, M.-H. AMP-activated protein kinase, stress responses and cardiovascular diseases. Clin. Sci. 2012, 122, 555–573. [Google Scholar] [CrossRef] [PubMed]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef]
- Barnard, R.A.; Regan, D.P.; Hansen, R.J.; Maycotte, P.; Thorborn, A.; Gustafson, D.L. Autophagy inhibition delays early but not late-stage metastatic disease. J. Pharmacol. Exp. Ther. 2016, 358, 282–293. [Google Scholar] [CrossRef]
- Guo, J.Y.; Xia, B.; White, E. Autophagy-mediated tumor promotion. Cell 2013, 155, 1216–1219. [Google Scholar] [CrossRef]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef]
- Jiang, X.; Overholtzer, M.; Thompson, C.B. Autophagy in cellular metabolism and cancer. J. Clin. Investig. 2015, 125, 47–54. [Google Scholar] [CrossRef]
- Jin, S.; DiPaola, R.S.; Mathew, R.; White, E. Metabolic catastrophe as a means to cancer cell death. J. Cell Sci. 2007, 120, 379–383. [Google Scholar] [CrossRef]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Lum, J.J.; Bauer, D.E.; Kong, M.; Harris, M.H.; Li, C.; Lindsten, Z.; Thompson, C.B. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 2005, 120, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Fung, C.; Lock, R.; Gao, S.; Salas, E.; Debnath, J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol. Biol. Cell 2008, 19, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Macintosh, R.L.; Timpson, P.; Thorborn, J.; Anderson, K.I.; Thorborn, A.; Ryan, K.M. Inhibition of autophagy impairs tumor cell invasion in an organotypic model. Cell Cycle 2012, 11, 2022–2029. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.-F.; Shi, Y.-H.; Ding, Z.-B.; Ke, A.-W.; Gu, C.-Y.; Zhou, J.; Qiu, S.-J.; Dai, Z.; Fan, J. Autophagy inhibition suppresses pulmonary metastasis of HCC in mice via impairing anoikis resistance and colonization of HCC cells. Autophagy 2013, 9, 2056–2068. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef] [PubMed]
- Moenner, M.; Pluquet, O.; Bouchecareilh, M.; Chevet, E. Integrated endoplasmic reticulum stress responses in cancer. Cancer Res. 2007, 67, 10361–10364. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, P.M.; Tabbara, S.O.; Jacobs, L.K.; Manning, F.C.; Tsangaris, T.N.; Schwartz, A.M.; Kennedy, K.A.; Patierno, S.R. Overexpression of the glucose-regulated stress gene GRP78 in malignant but not benign human breast lesions. Breast Cancer Res. Treat. 2000, 59, 15–26. [Google Scholar] [CrossRef]
- Xue, Z.; He, Y.; Ye, K.; Gu, Z.; Mao, Y.; Qi, L. A conserved structural determinant located at the interdomain region of mammalian inositol-requiring enzyme 1alpha. J. Biol. Chem. 2011, 286, 30859–30866. [Google Scholar] [CrossRef]
- Hong, S.Y.; Hagen, T. Multiple myeloma Leu167Ile (c.499C>A) mutation prevents XBP1 mRNA splicing. Br. J. Haematol. 2013, 161, 898–901. [Google Scholar] [CrossRef]
- Ghosh, R.; Wang, L.; Wang, E.S.; Perera, B.G.; Igbaria, A.; Morita, S.; Prado, K.; Thamsen, M.; Caswell, D.; Macias, H.; et al. Allosteric inhibition of the IRE1α RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 2014, 158, 534–548. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A. Endoplasmic reticulum stress signaling in cancer cells. Am. J. Pathol. 2020, 190, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Min, J.-Y.; Chun, K.-S.; Kim, D.-H. The versatile utility of cysteine as a target for cancer treatment. Front. Oncol. 2023, 12, 997919. [Google Scholar] [CrossRef] [PubMed]
- Banjac, A.; Perisic, T.; Sato, H.; Seiler, A.; Bannai, S.; Weiss, N.; Kölle, P.; Tschoep, K.; Issels, R.D.; Daniel, P.T.; et al. The cystine/cysteine cycle: A redox cycle regulating susceptibility versus resistance to cell death. Oncogene 2008, 27, 1618–1628. [Google Scholar] [CrossRef] [PubMed]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system xc− in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Wang, C.; Liu, G.; Bi, C.; Wang, X.; Zhou, Q.; Jin, H. SLC7A11/xCT in cancer: Biological functions and therapeutic implications. Am. J. Cancer Res. 2020, 10, 3106–3126. [Google Scholar]
- Sen, N.; Paul, B.D.; Gadalla, M.M.; Mustafa, A.K.; Sen, T.; Xu, R.; Kim, S.; Snyder, S.H. Hydrogen sulfide-linked sulfhydration of NF-κB mediates its antiapoptotic actions. Mol. Cell 2012, 45, 13–24. [Google Scholar] [CrossRef]
- Yang, G.; Zhao, K.; Ju, Y.; Mani, S.; Cao, Q.; Puukila, S.; Khaper, N.; Wu, L.; Wang, R. Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxid. Redox Signal. 2013, 18, 1906–1919. [Google Scholar] [CrossRef]
- Ma, Z.; Bi, Q.; Wang, Y. Hydrogen sulfide accelerates cell cycle progression in oral squamous cell carcinoma cell lines. Oral Dis. 2015, 21, 156–162. [Google Scholar] [CrossRef]
- Jiang, L.; Shestov, A.A.; Swain, P.; Yang, C.; Parker, S.J.; Wang, Q.A.; Terada, L.S.; Adams, N.D.; McCabe, M.T.; Pietrak, B.; et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 2016, 532, 255–258. [Google Scholar] [CrossRef]
- Kaya, B.; Çiçek, O.; Erdi, F.; Findig, S.; Karatas, Y.; Esen, H.; Keskin, F.; Kalkan, E. Intratumoral hemorrhage-related differences in the expression of vascular endothelial growth factor, basic fibroblast growth factor and thioredoxin reductase 1 in human glioblastoma. Mol. Clin. Oncol. 2006, 5, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Yin, T.; He, S.; Liu, X.; Jiang, W.; Ye, T.; Lin, Z.; Sang, Y.; Su, C.; Wan, Y.; Shen, G.; et al. Extravascular red blood cells and hemoglobin promote tumor growth and therapeutic resistance as endogenous danger signals. J. Immunol. 2015, 194, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Poon, L.C.; Methot, S.P.; Morabi-Pazocki, W.; Pio, F.; Bennet, A.J.; Sen, D. Guanine-rich RNAs and DNAs that bind heme robustly catalyze oxygen transfer reactions. J. Am. Chem. Soc. 2011, 133, 1877–1884. [Google Scholar] [CrossRef] [PubMed]
- Gray, L.T.; Lombardi, E.P.; Verga, D.; Nicolas, A.; Teulade-Fichou, M.-P.; Londoño-Vallejo, A.; Maizels, N. G-Quadruplexes sequester free heme in living cells. Cell Chem. Biol. 2019, 26, 1681–1689. [Google Scholar] [CrossRef] [PubMed]
- Brooks, T.A.; Hurley, L.H. Targeting MYC expression through G-quadruplexes. Genes Cancer 2010, 1, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Brooks, T.A.; Kendrick, S.; Hurley, L.H. Making sense of G-quadruplex and i-motif functions in oncogene promoters. FEBS J. 2010, 277, 3459–3469. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; White, K.A.; Barber, D.L. Intracellular pH regulates cancer and stem cell behaviors: A protein dynamics perspective. Front. Oncol. 2020, 10, 1401. [Google Scholar] [CrossRef]
- Hao, G.; Xu, Z.P.; Li, L. Manipulating extracellular tumour pH: An effective target for cancer therapy. RSC Adv. 2018, 8, 22182–22192. [Google Scholar] [CrossRef]
- Casey, J.R.; Grinstein, S.; Orlowski, J. Sensors and regulators of intracellular pH. Nat. Rev. Mol. Cell Biol. 2010, 11, 50–61. [Google Scholar] [CrossRef]
- Flinck, M.; Kramer, S.H.; Schnipper, J.; Andersen, A.P.; Pedersen, S.F. The acid-base transport proteins NHE1 and NBCn1 regulate cell cycle progression in human breast cancer cells. Cell Cycle 2018, 17, 1056–1067. [Google Scholar] [CrossRef] [PubMed]
- Putney, L.K.; Barber, D.L. Na-H exchange-dependent increase in intracellular pH times G2/M entry and transition. J. Biol. Chem. 2003, 278, 44645–44649. [Google Scholar] [CrossRef] [PubMed]
- Parks, S.K.; Pouyssegur, J. The Na(+)/HCO3(-) co-transporter SLC4A4 plays a role in growth and migration of colon and breast cancer cells. J. Cell Physiol. 2015, 230, 1954–1963. [Google Scholar] [CrossRef] [PubMed]
- Schwab, A.; Stock, C. Ion channels and transporters in tumour cell migration and invasion. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130102. [Google Scholar] [CrossRef]
- Schulze, A.; Harris, A.L. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 2012, 491, 364–373. [Google Scholar] [CrossRef]
- Lee, S.; Shanti, A. Effect of exogenous pH on cell growth of breast cancer cells. Int. J. Mol. Sci. 2021, 22, 9910. [Google Scholar] [CrossRef]
- Makris, E.A.; Responte, D.J.; Paschos, N.K.; Hu, J.C.; Athanasiou, K.A. Developing functional musculoskeletal tissues through hypoxia and lysyl oxidase-induced collagen cross-linking. Proc. Natl. Acad. Sci. USA 2014, 111, E4832–E4841. [Google Scholar] [CrossRef]
- Makris, E.A.; Hu, J.C.; Athanasiou, K.A. Hypoxia-induced collagen crosslinking as a mechanism for enhancing mechanical properties of engineered articular cartilage. Osteoarthr. Cartil. 2013, 21, 634–641. [Google Scholar] [CrossRef]
- Najafi, M.; Farhood, B.; Mortezaee, K. Extracellular matrix (ECM) stiffness and degradation as cancer drivers. J. Cell Biochem. 2019, 120, 2782–2790. [Google Scholar] [CrossRef]
- Aro, E.; Khatri, R.; Gerard-O’Riley, R.; Mangiavini, L.; Myllyharju, J.; Schipani, E. Hypoxia-inducible factor-1 (HIF-1) but not HIF-2 is essential for hypoxic induction of collagen prolyl 4-hydroxylases in primary newborn mouse epiphyseal growth plate chondrocytes. J. Biol. Chem. 2012, 287, 37134–37144. [Google Scholar] [CrossRef]
- Xiong, G.; Stewart, R.L.; Chen, J.; Gao, T.; Scott, T.L.; Samayoa, L.M.; O’Connor, K.; Lane, A.N.; Xu, R. Collagen prolyl 4-hydroxylase 1 is essential for HIF-1alpha stabilization and TNBC chemoresistance. Nat. Commun. 2018, 9, 4456. [Google Scholar] [CrossRef] [PubMed]
- Saatci, O.; Kaymak, A.; Raza, U.; Ersan, P.G.; Akbulut, O.; Banister, C.E.; Sikirzhytski, V.; Tokat, U.M.; Aykut, G.; Ansari, S.A.; et al. Targeting lysyl oxidase (LOX) overcomes chemotherapy resistance in triple negative breast cancer. Nat. Commun. 2020, 11, 2416. [Google Scholar] [CrossRef] [PubMed]
- Janjanam, J.; Pano, G.; Wang, R.; Minden-Birkenmaier, B.A.; Breeze-Jones, H.; Baker, E.; Garcin, C.; Clayton, G.; Shirinifard, A.; Zaske, A.M.; et al. Matricellular protein WISP2 is an endogenous inhibitor of collagen linearization and cancer metastasis. Cancer Res. 2021, 81, 5666–5677. [Google Scholar] [CrossRef] [PubMed]
- Murdocca, M.; De Masi, C.; Pucci, S.; Mango, R.; Novelli, G.; Di Natale, C.; Sangiuolo, F. LOX-1 and cancer: An indissoluble liaison. Cancer Gene Ther. 2021, 28, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
- Bodelon, C.; Mullooly, M.; Pfeiffer, R.M.; Fan, S.; Abubakar, M.; Lenz, P.; Vacek, P.M.; Weaver, D.L.; Herschorn, S.D.; Johnson, J.M.; et al. Mammary collagen architecture and its association with mammographic density and lesion severity among women undergoing image-guided breast biopsy. Breast Cancer Res. 2021, 23, 105. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Liu, Z.; Liu, W.; Fu, M.; Jiang, W.; Xu, S.; Wang, G.; Chen, F.; Lu, J.; Chen, H.; et al. Predicting postoperative peritoneal metastasis in gastric cancer with serosal invasion using a collagen nomogram. Nat. Commun. 2021, 12, 179. [Google Scholar] [CrossRef]
- Angenendt, L.; Mikesch, J.H.; Gorlich, D.; Busch, A.; Arnhold, I.; Rudack, C.; Hartmann, W.; Wardelmann, E.; Berdel, W.E.; Stenner, M.; et al. Stromal collagen type VI associates with features of malignancy and predicts poor prognosis in salivary gland cancer. Cell. Oncol. Dordr. 2018, 41, 517–525. [Google Scholar] [CrossRef]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910. [Google Scholar] [CrossRef]
- Piersma, B.; Hayward, M.K.; Weaver, V.M. Fibrosis and cancer: A strained relationship. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188356. [Google Scholar] [CrossRef]
- Landoldt, L.; Spagnoli, G.C.; Hertig, A.; Brocheriou, I.; Marti, H.-P. Fibrosis and cancer: Shared features and mechanisms suggest common targeted therapeutic approaches. Nephrol. Dial. Transplant. 2022, 37, 1024–1032. [Google Scholar] [CrossRef]
- Wu, B.; Sodji, Q.H.; Oyelere, A.K. inflammation, fibrosis and cancer: Mechanisms, therapeutic options and challenges. Cancers 2022, 14, 552. [Google Scholar] [CrossRef]
- Luo, L.; Yang, J.-X.; Luo, T.; Liu, D.; Wu, G.-H.; He, J.-M. A study on the mechanism of PP2A in the recovery of SCI in rats through downregulation of MMP-9 via MAPK signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 7195–7203. [Google Scholar] [CrossRef] [PubMed]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta 2012, 1824, 68–88. [Google Scholar] [CrossRef] [PubMed]
- Piperigkou, Z.; Kyriakopoulou, K.; Koutsakis, C.; Mastronikolis, S.; Karamanos, N.K. Key Matrix Remodeling Enzymes: Functions and Targeting in Cancer. Cancers 2021, 13, 1441. [Google Scholar] [CrossRef] [PubMed]
- Barash, U.; Cohen-Kaplan, V.; Dowek, I.; Sanderson, R.D.; Ilan, N.; Vlodavsky, I. Proteoglycans in health and disease: New concepts for heparanase function in tumor progression and metastasis. FEBS J. 2010, 277, 3890–3903. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.-R.; Sun, D.-N.; Yang, H.; Yan, J.; Zhang, X.; Zheng, X.-L.; Fu, X.-H.; Geng, M.-Y.; Huang, X.; Ding, J. CTC clusters induced by heparanase enhance breast cancer metastasis. Acta Pharmacol. Sin. 2018, 39, 1326–1337. [Google Scholar] [CrossRef] [PubMed]
- Song, J.M.; Im, J.; Nho, R.S.; Han, Y.H.; Upadhyaya, P.; Kassie, F. Hyaluronan-CD44/RHAMM interaction-dependent cell proliferation and survival in lung cancer cells. Mol. Carcinog. 2019, 58, 321–333. [Google Scholar] [CrossRef]
- Makkar, S.; Riehl, T.E.; Chen, B.; Yan, Y.; Alvarado, D.M.; Ciorba, M.A.; Stenson, W.F. Hyaluronic acid binding to TLR4 promotes proliferation and blocks apoptosis in colon cancer. Mol. Cancer Ther. 2019, 18, 2446–2456. [Google Scholar] [CrossRef]
- Yang, Y.M.; Noureddin, M.; Liu, C.; Ohashi, K.; Kim, S.Y.; Ramnath, D.; Powell, E.E.; Sweet, M.J.; Roh, Y.S.; Hsin, I.-F.; et al. Hyaluronan synthase 2-mediated hyaluronan production mediates Notch1 activation and liver fibrosis. Sci. Transl. Med. 2019, 11, eaat9284. [Google Scholar] [CrossRef]
- Chen, J.E.; Lumibao, J.; Blazek, A.; Gaskins, H.R.; Harley, B. Hypoxia activates enhanced invasive potential and endogenous hyaluronic acid production by glioblastoma cells. Biomater. Sci. 2018, 6, 854–862. [Google Scholar] [CrossRef]
- Kobayashi, N.; Miyoshi, S.; Mikami, T.; Koyama, H.; Kitazawa, M.; Takeoka, M.; Sano, K.; Amano, J.; Isogai, Z.; Niida, S.; et al. Hyaluronan deficiency in tumor stroma impairs macrophage trafficking and tumor neovascularization. Cancer Res. 2010, 70, 7073–7083. [Google Scholar] [CrossRef] [PubMed]
- Kuang, D.M.; Wu, Y.; Chen, N.; Cheng, J.; Zhuang, S.M.; Zheng, L. Tumor-derived hyaluronan induces formation of immunosuppressive macrophages through transient early activation of monocytes. Blood 2007, 110, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Ma, J.; Lu, W. The significance of mitochondrial dysfunction in cancer. Int. J. Mol. Sci. 2020, 21, 5598. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by lipid peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 360–379. [Google Scholar] [CrossRef] [PubMed]
- Arnhold, J. Role of reactive species in destructions. In Cell and Tissue Destruction. Mechanisms, Protection, Disorders; Academic Press: London, UK; San Diego, CA, USA; Cambridge, MA, USA; Oxford, UK, 2020; pp. 23–54. [Google Scholar] [CrossRef]
- Arnhold, J.; Flemmig, J. Human myeloperoxidase in innate and acquired immunity. Arch. Biochem. Biophys. 2010, 500, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, S.J. Myeloperoxidase: Friend and foe. J. Leukoc. Biol. 2005, 77, 598–625. [Google Scholar] [CrossRef]
- Rothenberg, M.E.; Hogan, S.P. The eosinophil. Annu. Rev. Immunol. 2006, 24, 147–174. [Google Scholar] [CrossRef]
- Cadet, J.; Teoule, R. Comparative study of oxidation of nucleic acid components by hydroxyl radicals, singlet oxygen and superoxide anion radicals. Photochem. Photobiol. 1978, 28, 661–667. [Google Scholar] [CrossRef]
- Agnez-Lima, L.F.; Melo, J.T.A.; Silva, A.E.; Oliviera, A.H.S.; Timoteo, A.R.S.; Lima-Bessa, K.M.; Martinez, G.R.; Medeiros, M.H.G.; Di Mascio, P.; Galhardo, R.S.; et al. DNA damage by singlet oxygen and cellular protective mechanisms. Mutat. Res. 2012, 751, 15–28. [Google Scholar] [CrossRef]
- Brand, M.D. mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef] [PubMed]
- Robb, E.L.; Hall, A.R.; Prime, T.A.; Eaton, S.; Szibor, M.; Viscomi, C.; James, A.M.; Murphy, M.P. Control of mitochondrial superoxide production by reverse electron transport at complex I. J. Biol. Chem. 2011, 286, 9869–9879. [Google Scholar] [CrossRef]
- Demin, O.V.; Khodolenko, B.N.; Skulachev, V.P. A model of O2•− generation in the complex III of the electron transport chain. Mol. Cell Biochem. 1998, 184, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Gardner, P.R. Superoxide-driven aconitase Fe-S cycling. Biosci. Rep. 1997, 17, 33–42. [Google Scholar] [CrossRef]
- Gardner, P.R. Aconitase: Sensitive target and measure of superoxide. Meth. Enzymol. 2002, 349, 9–23. [Google Scholar] [CrossRef]
- Tengan, C.H.; Moraes, C.T. NO control of mitochondrial function in normal and transformed cells. Biochim. Biophys. Acta 2017, 1858, 573–581. [Google Scholar] [CrossRef]
- Tengan, C.H.; Rodrigues, G.S.; Godinho, R.O. Nitric oxide in skeletal muscle: Role on mitochondrial biogenesis and function. Int. J. Mol. Sci. 2012, 13, 17160–17184. [Google Scholar] [CrossRef]
- Figueira, T.R.; Barros, M.H.; Camargo, A.A.; Castilho, R.F.; Ferreira, J.C.; Kowaltowski, A.J.; Sluse, F.E.; Souza-Pinto, N.C.; Vercesi, A.E. Mitochondria as a source of reactive oxygen and nitrogen species: From molecular mechanisms to human health. Antioxid. Redox Signal. 2013, 18, 2029–2074. [Google Scholar] [CrossRef]
- Aaltoma, S.H.; Lipponen, P.K.; Kosma, V.M. Inducible nitric oxide synthase (iNOS) expression and its prognostic value in prostate cancer. Anticancer Res. 2001, 21, 3101–3106. [Google Scholar]
- Jabłońska, E.; Puzewska, W.; Marcińczyk, M.; Grabowska, Z.; Jabłoński, J. iNOS expression and NO production by neutrophils in cancer patients. Arch. Immunol. Ther. Exp. 2005, 53, 175–179. [Google Scholar]
- Barani, R.; Motalleb, G.; Maghsoudi, H. Evaluation of iNOS expression in esophageal eancer eatients. Gastrointest. Tumors 2016, 3, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Huie, R.E.; Padmaja, S. Reactions of NO and O2•−. Free Radic. Res. Commun. 1993, 18, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Kissner, R.; Nauser, T.; Bugnon, P.; Lye, P.G.; Koppenol, W.H. Formation and properties of peroxynitrite as studied by laser flash photolysis, high-pressure stopped-flow technique, and pulse radiolysis. Chem. Res. Toxicol. 1997, 10, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, U.; Kuntz, S.; De Sousa, U.J.; Daniel, H. Nitric oxide suppresses apoptosis in human colon cancer cells by scavenging mitochondrial superoxide anions. Int. J. Cancer 2003, 106, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ye, S.; Chen, X.; Xu, P.; Li, K.; Zeng, S.; Huang, M.; Gao, W.; Chen, J.; Zhang, Q.; et al. Mitochondrial NOS1 suppresses apoptosis in colon cancer cells through increasing SIRT3 activity. Biochem. Biophys. Res. Commun. 2019, 515, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Radi, R.; Beckman, J.S.; Bush, K.M.; Freeman, B.A. Peroxynitrite-induced membrane lipid peroxidation: The cytotoxic potential of superoxide and nitric oxide. Arch. Biochem. Biophys. 1991, 288, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, S.; Czapski, G. Mechanism of the nitrosation of thiols and amines by oxygenated •NO solutions: The nature of the nitrosating intermediates. J. Am. Chem. Soc. 1996, 118, 3419–3425. [Google Scholar] [CrossRef]
- Schopfer, F.J.; Baker, P.R.S.; Freeman, B.A. NO-dependent protein nitration: A cell signaling event or an oxidative inflammatory response? Trends Biochem. Sci. 2003, 28, 646–654. [Google Scholar] [CrossRef]
- Ferrer-Sueta, G.; Radi, R. Chemical biology of peroxynitrite: Kinetics, diffusion, and radicals. ACS Chem. Biol. 2009, 4, 161–177. [Google Scholar] [CrossRef]
- Furtmüller, P.G.; Jantschko, W.; Zederbauer, M.; Schwanninger, M.; Jakoptisch, C.; Herold, S.; Koppenol, W.H.; Obinger, C. Peroxynitrite efficiently mediates the interconversion of redox intermediates of myeloperoxidase. Biochem. Biophys. Res. Commun. 2005, 337, 944–954. [Google Scholar] [CrossRef]
- Koyani, C.N.; Flemmig, J.; Malle, E.; Arnhold, J. Myeloperoxidase scavenges peroxynitrite: A novel anti-inflammatory action of the heme enzyme. Arch. Biochem. Biophys. 2015, 571, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Groves, J.T. Mechanisms of peroxynitrite interactions with heme proteins. Inorg. Chem. 2010, 49, 6317–6329. [Google Scholar] [CrossRef] [PubMed]
- Gebicka, L.; Gebicki, J.L. Reaction of heme peroxidases with peroxynitrite. IUBMB Life 2000, 49, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M.C. Iron as a biological pro-oxidant. ISI Atlas Sci. Biochem. 1988, 1, 48–52. [Google Scholar]
- Koppenol, W.H. The centennial of the Fenton reaction. Free Radic. Biol. Chem. 1993, 15, 645–651. [Google Scholar] [CrossRef]
- Gunther, M.R.; Hanna, P.M.; Mason, R.P.; Cohen, M.S. Hydroxyl radical formation from cuprous ion and hydrogen peroxide: A spin-trapping study. Arch. Biochem. Biophys. 1995, 316, 515–522. [Google Scholar] [CrossRef]
- Bors, W.; Erben-Russ, M.; Saran, M. Fatty acid peroxyl radicals: Their generation and reactivities. Bioelectrochem. Bioenergy 1987, 18, 37–49. [Google Scholar] [CrossRef]
- Zhao, N.; Zhang, A.-S.; Enns, C.A. Iron regulation by hepcidin. J. Clin. Investig. 2013, 123, 2337–2343. [Google Scholar] [CrossRef]
- Gkouvatsos, K.; Papanikolaou, G.; Pantopoulos, K. Regulation of iron transport and the role of transferrin. Biochim. Biophys. Acta 2011, 1820, 188–202. [Google Scholar] [CrossRef]
- Gamella, E.; Buratti, P.; Cairo, G.; Recalcati, S. The transferrin receptor: The cellular iron gate. Metallomics 2017, 9, 1367–1375. [Google Scholar] [CrossRef]
- Massover, W.H. Ultrastructure of ferritin and apoferritin: A review. Micron 1993, 24, 389–437. [Google Scholar] [CrossRef]
- Prohaska, J.R. Role of copper transporters in copper homeostasis. Am. J. Clin. Nutr. 2008, 88, 826S–829S. [Google Scholar] [CrossRef] [PubMed]
- Stoj, C.; Kosman, D.J. Cuprous oxidase activity of yeast Fet3p and human ceruloplasmin: Implication for function. FEBS Lett. 2003, 554, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Sokolov, A.V.; Pulina, M.O.; Ageeva, K.V.; Runova, O.I.; Zakharova, E.T.; Vasilyev, V.B. Identification of leukocyte cationic proteins that interact with ceruloplasmin. Biochemistry 2007, 72, 872–877. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.; Fridovich, I. Superoxide dismutase: An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1960, 224, 6049–6055. [Google Scholar]
- Chang, L.Y.; Slot, J.W.; Geuze, H.J.; Crapo, J.D. Molecular immunocytochemistry of the CuZn superoxide dismutase in rat hepatocytes. J. Cell Biol. 1988, 107, 2169–2179. [Google Scholar] [CrossRef]
- Weisiger, R.A.; Fridovich, I. Mitochondrial superoxide dismutase. Site of synthesis and intramolecular localization. J. Biol. Chem. 1973, 248, 4793–4796. [Google Scholar] [CrossRef]
- Antonyuk, S.V.; Strange, R.W.; Marklund, S.L.; Hasnain, S.S. The structure of human extracellular copper-zinc superoxide dismutase at 1.7 Å resolution: Insights into heparin and collagen binding. J. Mol. Biol. 2009, 388, 310–326. [Google Scholar] [CrossRef]
- Starkov, A.A. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52. [Google Scholar] [CrossRef]
- Pasdois, P.; Parker, J.E.; Griffiths, E.J.; Halestrap, A.P. The role of oxidized cytochrome c in regulating mitochondrial reactive species production and its perturbation in ischemia. Biochem. J. 2011, 436, 493–505. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M.; Roveri, A. Phospholipid hydroperoxide glutathione peroxidase (PHGPx): More than an antioxidant enzyme? Biomed. Environm. Sci. 1997, 10, 327–332. [Google Scholar]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione peroxidase-1 in health and disease: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef] [PubMed]
- Low, F.M.; Hampton, M.P.; Winterbourn, C.C. Prx2 and peroxide metabolism in the erythrocyte. Antioxid. Redox Signal. 2008, 10, 1621–1630. [Google Scholar] [CrossRef] [PubMed]
- Goyal, M.M.; Basak, A. Human catalase: Looking for complete identity. Protect. Cell 2010, 1, 888–897. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, S.S.; Waypa, G.B.; Marks, J.D.; Schumacker, P.T. Peroxiredoxin-5 targeted to the mitochondrial intermembrane space attenuates hypoxia-induced reactive oxygen species signalling. Biochem. J. 2013, 456, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar]
- Kamiguti, A.S.; Serrander, L.; Lin, K.; Harris, R.J.; Cawley, J.C.; Allsup, D.J.; Slupsky, J.R.; Krause, K.H.; Zuzel, M. Expression and activity of NOX5 in the circulating malignant B cells of hairy cell leukemia. J. Immunol. 2005, 175, 8424–8430. [Google Scholar] [CrossRef]
- Tsao, S.M.; Yin, M.C.; Liu, W.H. Oxidant stress and B vitamins status in patients with non-small cell lung cancer. Nutr. Cancer 2007, 59, 8–13. [Google Scholar] [CrossRef]
- Khandrika, L.; Kumar, B.; Koul, S.; Maroni, P.; Koul, H.K. Oxidative stress in prostate cancer. Cancer Lett. 2009, 282, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.P.; Rawal, U.M.; Dave, T.K.; Rawal, R.M.; Shukla, S.N.; Shah, P.M.; Patel, P.S. Lipid peroxidation, total antioxidant status, and total thiol levels predict overall survival in patients with oral squamous cell carcinoma. Integr. Cancer Ther. 2007, 6, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Nicolussi, A.; D’Inezo, S.; Capalbo, C.; Giannini, G.; Coppa, A. the role of peroxiredoxins in cancer. Mol. Clin. Oncol. 2017, 6, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Jo, M.; Kim, Y.R.; Lee, C.K.; Hong, J.T. Roles of peroxiredoxins in cancer, neurodegenerative diseases and inflammatory diseases. Pharmacol. Ther. 2016, 163, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef]
- Jin, L.; Zhou, Y. Crucial role of the pentose phosphate pathway in malignant tumors. Oncol. Lett. 2019, 17, 4213–4221. [Google Scholar] [CrossRef]
- Menegon, S.; Columbano, A.; Giordano, S. The dual role of NRF2 in cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef]
- Schmidlin, C.J.; Shakya, A.; Dodson, M.; Chapman, E.; Zhang, D.D. The intricacies of NFR2 regulation in cancer. Semin. Cancer Biol. 2021, 76, 110–119. [Google Scholar] [CrossRef]
- Singh, A.; Misra, V.; Thimmulappa, R.K.; Lee, H.; Ames, S.; Hoque, M.O.; Herman, J.G.; Baylin, S.B.; Sidransky, D.; Gabrielson, E.; et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006, 3, e420. [Google Scholar] [CrossRef] [PubMed]
- Shibata, T.; Kokubu, A.; Gotoh, M.; Ojima, H.; Ohta, T.; Yamamoto, M.; Hirohashi, S. Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology 2008, 135, 1358–1368.e4. [Google Scholar] [CrossRef] [PubMed]
- Solis, L.M.; Behrens, C.; Dong, W.; Suraokar, M.; Ozburn, N.C.; Moran, C.A.; Corvalan, A.H.; Biswal, S.; Swisher, S.G.; Bekele, B.N.; et al. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin. Cancer Res. 2010, 16, 3743–3753. [Google Scholar] [CrossRef] [PubMed]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef] [PubMed]
- Mullarky, E.; Cantley, L.C. Diverting glycolysis to combat oxidative stress. In Innovative Medicine. Basic Research and Development; Nakao, K., Minato, N., Uemoto, S., Eds.; Springer Open: Tokyo, Japan, 2015; pp. 3–24. [Google Scholar] [CrossRef]
- Arnhold, J. Oxidation and reduction of biological material. In Cell and Tissue Destruction. Mechanisms, Protection, Disorders; Academic Press: London, UK; San Diego, CA, USA; Cambridge, MA, USA; Oxford, UK, 2020; pp. 55–97. [Google Scholar] [CrossRef]
- Meister, A. Glutathione metabolism and its selective modification. J. Biol. Chem. 1988, 263, 17205–17208. [Google Scholar] [CrossRef] [PubMed]
- Arnér, E.S.; Holmgren, A. The thioredoxin system in cancer. Semin. Cancer Biol. 2006, 16, 420–426. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, H.; Zhou, J.; Shao, Q. Glutathione peroxidase GPX1 and its dichotomous roles in cancer. Cancers 2022, 14, 2560. [Google Scholar] [CrossRef]
- De Luca, A.; Sanna, F.; Sallese, M.; Ruggiero, C.; Grossi, M.; Sacchetta, P.; Rossi, C.; de Laurenzi, V.; de Ilio, C.; Favaloro, B. Methionine sulfoxide reductase A down-regulation in human breast cancer cells results in a more aggressive phenotype. Proc. Natl. Acad. Sci. USA 2010, 107, 18628–18633. [Google Scholar] [CrossRef]
- Kwak, G.-H.; Kim, T.-H.; Kim, H.-Y. Down-regulation of MsrB3 induces cancer cell apoptosis through reactive oxygen species production and intrinsic mitochondrial pathway activation. Biochem. Biophys. Res. Commun. 2017, 483, 468–474. [Google Scholar] [CrossRef]
- Jiang, B.; Moskovitz, J. The functions of the mammalian sulfoxide reductase system and related diseases. Antioxidants 2018, 7, 122. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Feng, H.; Sundberg, B.; Yang, J.; Powers, J.; Christian, A.H.; Wilkinson, J.E.; Monnin, C.; Avizonis, D.; Thomas, C.J.; et al. Methionine oxidation activates pyruvate kinase M2 to promote pancreatic cancer metastasis. Mol. Cell 2022, 82, 3045–3060. [Google Scholar] [CrossRef]
- Glasauer, A.; Sena, L.A.; Diebold, L.P.; Mazar, A.P.; Chandel, N.S. Targeting SOD1 reduces experimental non-small-cell lung cancer. J. Clin. Investig. 2014, 124, 117–128. [Google Scholar] [CrossRef]
- Papa, L.; Hahn, M.; Marsh, E.L.; Evans, B.S.; Germain, D. SOD2 to SOD1 switch in breast cancer. J. Biol. Chem. 2014, 289, 5412–5416. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.K.; St. Clair, D.K. Manganese superoxide dismutase regulation and cancer. Free Radic. Biol. Med. 2012, 52, 2209–2222. [Google Scholar] [CrossRef] [PubMed]
- Che, M.; Wang, R.; Wang, H.-Y.; Zheng, X.F.S. Expanding roles of superoxide dismutases in cell regulation and cancer. Drug Discov. Today 2016, 21, 143–149. [Google Scholar] [CrossRef]
- Kinnula, V.L.; Crapo, J.D. Superoxide dismutases in malignant cells and human tumors. Free Radic. Biol. Med. 2004, 36, 718–744. [Google Scholar] [CrossRef]
- Flor, S.; Oliva, C.R.; Ali, M.Y.; Coleman, K.L.; Greenlee, J.D.; Jones, K.A.; Monga, V.; Griguer, C.E. Catalase overexpression drives an aggressive phenotype in glioblastoma. Antioxidants 2021, 10, 1988. [Google Scholar] [CrossRef]
- Forshaw, T.E.; Holmila, R.; Nelson, K.J.; Lewis, J.E.; Kemp, M.L.; Tsang, A.W.; Poole, L.B.; Lowther, W.T.; Furdui, C.M. Peroxiredoxins in cancer and response to radiation therapies. Antioxidants 2019, 8, 11. [Google Scholar] [CrossRef]
- Jia, J.-J.; Geng, W.-S.; Wang, Z.-Q.; Chen, L.; Zeng, X.-S. The role of thioredoxin system in cancer: Strategy for cancer therapy. Cancer Chemother. Pharmacol. 2019, 84, 453–470. [Google Scholar] [CrossRef]
- Ghareeb, H.; Metanis, N. The thioredoxin system: A promising target for cancer drug development. Chem. Eur. J. 2020, 26, 10175–10184. [Google Scholar] [CrossRef] [PubMed]
- Gamcsik, M.P.; Kasibhatla, M.S.; Teeter, S.D.; Colvin, O.M. Glutathione levels in human tumors. Biomarkers 2012, 17, 671–691. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, L.; Sandhu, J.K.; Harper, M.-E.; Cuperlovic-Culf, M. Role of glutathione in cancer: From mechanisms to therapies. Biomolecules 2020, 10, 1429. [Google Scholar] [CrossRef] [PubMed]
- Valenti, G.E.; Tasso, B.; Traverso, N.; Domenicotti, C.; Marengo, B. Glutathione in cancer progression and chemoresistance: An update. Redox Exp. Med. 2023, 2023, e220023. [Google Scholar] [CrossRef]
- Zhu, Z.; Du, S.; Du, Y.; Ren, J.; Ying, G.; Yan, Z. Glutathione reductase mediates drug resistance in glioblastoma cells by regulating redox homeostasis. J. Neurochem. 2018, 144, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, K.R.; Hanna, D.N.; Cyr, S.; Baechle, J.J.; Kuravi, S.; Balusu, R.; Rathmell, K.; Baregamian, N. Glutathione peroxidase 4 inhibition induces ferroptosis and mTOR pathway suppression in thyroid cancer. Sci. Rep. 2022, 12, 19396. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.-Z.; Tao, H.; Fan, Z.-W.; Song, S.-J.; Bal, J. Prognostic and immunological role of key genes of ferroptosis in pan-cancer. Front. Cell Dev. Biol. 2021, 9, 748925. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.M.; Veeramachaneni, R.; Deng, D.; Putluri, N.; Putluri, V.; Cardenas, M.F.; Wheeler, D.A.; Decker, W.K.; Frederick, A.I.; Kazi, S.; et al. Glutathione peroxidase 2 is a metabolic driver of the tumor immune microenvironment and immune checkpoint inhibitor response. J. Immunother. Cancer 2022, 10, e004752. [Google Scholar] [CrossRef]
- Wang, M.; Chen, X.; Fu, G.; Ge, M. Glutathione peroxidase 2 overexpression promotes malignant progression and cisplatin resistance of KRAS-mutated lung cancer cells. Oncol. Rep. 2022, 48, 207. [Google Scholar] [CrossRef]
- Mollbrink, A.; Jawad, R.; Vlamis-Gardikas, A.; Edenvik, P.; Isaksson, B.; Danielsson, O.; Stål, F.A.P. Expression of thioredoxins and glutaredoxins in human hepatocellular carcinoma: Correlation to cell proliferation, tumor size and metabolic syndrome. Int. J. Immunopathol. Pharmacol. 2014, 27, 169–183. [Google Scholar] [CrossRef]
- Kim, H.; Lee, G.-R.; Kim, J.; Baek, J.Y.; Jo, Y.-J.; Hong, S.-E.; Kim, S.H.; Lee, J.; Lee, H.I.; Park, S.-K.; et al. Sulfiredoxin inhibitor induces preferential death of cancer cells through reactive species-mediated mitochondrial damage. Free Radic. Biol. Med. 2016, 91, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Aye, Y.; Li, M.; Long, M.J.C.; Weiss, R.S. Ribonucleotide reductase and cancer: Biological mechanisms and targeted therapies. Oncogene 2015, 34, 2011–2021. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Jiang, L.; Jin, X.; Ying, S.; Wu, Z.; Wang, L.; Yu, W.; Tong, J.; Zhang, L.; Lou, Y.; et al. Inhibiting RRM2 to enhance the anticancer activity of chemotherapy. Biomed. Pharmacother. 2021, 133, 110996. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Sun, H.; Zhang, S.; Shan, C. The multiple roles of glucose-6-phosphate dehydrogenase in tumorigenesis and cancer chemoresistance. Life 2022, 12, 271. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wang, W.; Yang, Y.; Gu, C. Exploring the role of glucose-6-phospahte dehydrogenase in cancer. Oncol. Rep. 2020, 44, 2325–2336. [Google Scholar] [CrossRef] [PubMed]
- Sarfraz, I.; Rasul, A.; Hussain, G.; Shah, M.A.; Zahoor, A.F.; Asrar, M.; Selamoglu, Z.; Ji, X.-Y.; Adem, S.; Sarker, S.D. 6-Phosphogluconate dehydrogenase fuels multiple aspects of cancer cells: From cancer initiation to metastasis and chemoresistance. BioFactors 2020, 46, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Du, P.; Zhang, Z.; Bao, D. 6-Phosphoglucoronate dehydrogenase inhibition arrests growth and induces apoptosis in gastric cancer via AMPK activation and oxidative stress. Open Life Sci. 2023, 18, 20220514. [Google Scholar] [CrossRef] [PubMed]
- Chrichton, R.R.; Ward, R.J. Iron homeostasis. Met. Ions Biol. Syst. 1998, 35, 633–665. [Google Scholar]
- Ponka, P. Cellular iron metabolism. Kidney Int. 1999, 55, S2–S11. [Google Scholar] [CrossRef]
- Adachi, M.; Kai, K.; Yamaji, K.; Ide, T.; Noshiro, H.; Kawaguchi, A.; Aishima, S. Transferrin receptor 1 overexpression is associated with tumour de-differentiation and acts as a potential prognostic indicator of hepatocellular carcinoma. Histopathology 2019, 75, 63–73. [Google Scholar] [CrossRef]
- Brown, R.A.; Richardson, K.L.; Kabir, T.D.; Trinder, D.; Ganss, R.; Leedman, P.J. Altered iron metabolism and impact in cancer biology, metastasis, and immunology. Front. Oncol. 2020, 10, 476. [Google Scholar] [CrossRef] [PubMed]
- Xue, D.; Zhou, C.X.; Shi, Y.B.; Lu, H.; He, X.Z. Decreased expression of ferroportin in prostate cancer. Oncol. Lett. 2015, 10, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Pinnix, Z.K.; Miller, L.D.; Wang, W.; D’Agostino, R.; Kute, T.; Willingham, M.C.; Hatcher, H.; Tesfay, L.; Sui, G.; Di, X.; et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci. Transl. Med. 2010, 2, 43ra56. [Google Scholar] [CrossRef] [PubMed]
- Toshiyama, R.; Konno, M.; Eguchi, H.; Asai, A.; Noda, T.; Koseki, J.; Asukai, K.; Ohashi, T.; Matsushita, K.; Iwagami, Y.; et al. Association of iron metabolic enzyme hepcidin expression levels with the prognosis of patients with pancreatic cancer. Oncol. Lett. 2018, 15, 8125–8133. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Li, X.; Dong, D.; Zhang, B.; Xue, Y.; Shang, P. Transferrin receptor 1 in cancer: A new sight for cancer therapy. Am. J. Cancer Res. 2018, 8, 916–931. [Google Scholar] [PubMed]
- Forciniti, S.; Greco, L.; Grizzi, F.; Malesci, A.; Laghi, L. Iron metabolism in cancer progression. Int. J. Mol. Sci. 2020, 21, 2257. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cheng, Q.; Bao, L.; Li, M.; Chang, K.; Yi, X. Cytoprotective role of heme oxygenase-1 in cancer chemoresistance: Focus on antioxidant, antiapoptotic, and pro-autophagy properties. Antioxidants 2023, 12, 1217. [Google Scholar] [CrossRef]
- Reiter, C.D.; Wang, X.; Tanus-Santos, J.E.; Hogg, N.; Cannon, R.O., III; Schechter, A.N.; Gladwin, M.T. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat. Med. 2002, 8, 1383–1389. [Google Scholar] [CrossRef]
- Rother, R.P.; Bell, L.; Hillman, P.; Gladwin, M.T. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin. J. Am. Med. Assoc. 2005, 293, 1653–1662. [Google Scholar] [CrossRef]
- Kristiansen, M.; Graversen, J.H.; Jacobsen, C.; Sonne, O.; Hoffmann, H.J.; Law, S.K.; Moestrup, S.K. Identification of the haemoglobin scavenger receptor. Nature 2001, 409, 198–201. [Google Scholar] [CrossRef]
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Tolosano, E. Haptoglobin and hemopexin in heme detoxification and iron recycling. In Acute Phase Proteins-Regulation and Functions of Acute Phase Proteins; Veas, F., Ed.; Intech: Rijeka, Croatia, 2011; pp. 261–288. [Google Scholar] [CrossRef]
- Sauret, J.M.; Marinides, G.; Wang, G.K. Rhabdomyolysis. Am. Fam. Phys. 2002, 65, 907–912. [Google Scholar]
- Hunter, J.D.; Gregg, K.; Damani, Z. Rhabdomyolysis. Contin. Educ. Anaesth. Crit. Care Pain 2006, 6, 141–143. [Google Scholar] [CrossRef]
- Bunn, H.F.; Jandl, J.H. Exchange of heme among hemoglobins and hemoglobin and albumin. J. Biol. Chem. 1968, 243, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Hvidberg, V.; Maniecki, M.B.; Jacobson, C.; Højrup, P.; Møller, H.J.; Moestrup, S.K. Identification of the receptor scavenging hemopexin-heme complexes. Blood 2005, 106, 2572–2579. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, R.T.; Fernandez, P.L.; Mourao-Sa, D.S.; Porto, B.N.; Dutra, F.F.; Alves, L.S.; Oliviera, M.F.; Oliviera, P.L.; Graca-Souza, A.V.; Bozza, M.T. Characterization of heme as activator of toll-like receptor 4. J. Biol. Chem. 2007, 282, 20221–20229. [Google Scholar] [CrossRef] [PubMed]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Milbauer, L.; Abdulla, F.; Alayash, A.I.; Smith, A.; Nath, K.A.; Hebbel, R.P.; Vercelotti, G.M. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 2014, 123, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Balla, G.; Jacob, H.S.; Eaton, J.W.; Belcher, J.D.; Vercelotti, G.M. Hemin: A possible physiological mediator of low density lipoprotein oxidation and endothelial injury. Arterioscler. Thromb. 1991, 11, 1700–1711. [Google Scholar] [CrossRef] [PubMed]
- Jeney, V.; Balla, J.; Yachie, A.; Varga, Z.; Vercelotti, G.M.; Eaton, J.W.; Balla, G. Pro-oxidant and cytotoxic effects of circulating heme. Blood 2002, 100, 879–887. [Google Scholar] [CrossRef]
- Kumar, S.; Bandyopadhyay, U. Free heme toxicity and its detoxification systems in human. Toxicol. Lett. 2005, 157, 175–188. [Google Scholar] [CrossRef]
- Flemmig, J.; Schlorke, D.; Kühne, F.-W.; Arnhold, J. Inhibition of the heme-induced hemolysis of red blood cells by the chlorite-based drug WF10. Free Radic. Res. 2016, 50, 1386–1395. [Google Scholar] [CrossRef]
- Lin, T.; Sammy, F.; Yang, H.; Thundivalappil, S.; Hellman, J.; Tracey, K.C.; Warren, H.S. Identification of hemopexin as an anti-inflammatory factor that inhibits synergy of hemoglobin with HMGB1 in sterile and infectious inflammation. J. Immunol. 2012, 189, 2017–2022. [Google Scholar] [CrossRef] [PubMed]
- Schaer, D.J.; Buehler, P.W.; Alayash, A.I.; Belcher, J.D.; Vercelotti, G.M. Hemolysis and free hemoglobin revisited: Exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 2013, 121, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Canesin, G.; Di Ruscio, A.; Li, M.; Ummarino, S.; Hedblom, A.; Choudhury, R.; Krzyzanowska, A.; Csizmadia, E.; Palominos, M.; Stiehm, A.; et al. Scavenging of labile heme by hemopexin is a key checkpoint in cancer growth and metastases. Cell Rep. 2020, 32, 108181. [Google Scholar] [CrossRef] [PubMed]
- Tarnawski, R.; Skladowski, K.; Maciejewski, B. Prognostic value of hemoglobin concentration in radiotherapy for cancer of supraglottic larynx. Int. J. Radiat. Oncol. Biol. Phys. 1997, 38, 1007–1011. [Google Scholar] [CrossRef] [PubMed]
- Grogan, M.; Thomas, G.M.; Malemed, J.; Wong, F.L.; Pearcey, R.G.; Joseph, P.K.; Portelance, L.; Crook, J.; Jones, K.D. The importance of hemoglobin levels during radiotherapy for carcinoma of the cervix. Cancer 1999, 86, 1528–1536. [Google Scholar] [CrossRef]
- Dejure, F.R.; Eilers, M. MYC and tumor metabolism: Chicken and egg. EMBO J. 2017, 36, 3409–3420. [Google Scholar] [CrossRef] [PubMed]
- Canesin, G.; Muralidharam, A.M.; Swanson, K.D.; Wegiel, B. HO-1 and heme: G-quadruplex interaction choreograph DNA damage response and cancer growth. Cells 2021, 10, 1801. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.M.; Lo Giudice, M.C.; D’Urso, A.; Lauceri, R.; Purello, R.; Milardi, D. Cationic porphyrins are reversible proteasome inhibitors. J. Am. Chem. Soc. 2012, 134, 10451–10457. [Google Scholar] [CrossRef]
- Vallelian, F.; Deuel, J.W.; Opitz, L.; Schaer, C.A.; Puglia, M.; Lönn, M.; Engelsberger, W.; Schauer, S.; Karnaukhova, E.; Spahn, D.R.; et al. Proteasome inhibition and oxidative reactions disrupt cellular homeostasis during heme stress. Cell Death Differ. 2015, 22, 597–611. [Google Scholar] [CrossRef]
- Nowis, D.; Legat, M.; Grzela, T.; Niderla, J.; Wilczek, E.; Wilczynski, G.M.; Głodkowska, E.; Mrówka, P.; Issat, T.; Dulak, J.; et al. Heme oxygenase-1 protects tumor cells against photodynamic therapy-mediated cytotoxicity. Oncogene 2006, 25, 3365–3374. [Google Scholar] [CrossRef]
- Pei, L.; Kong, Y.; Shao, C.; Yue, X.; Wang, Z.; Zhang, N. Heme oxygenase-1 induction mediates chemoresistance of breast cancer cells to pharmorubicin by promoting autophagy via PI3K/Akt pathway. J. Cell Mol. Med. 2018, 22, 5311–5321. [Google Scholar] [CrossRef] [PubMed]
- Slebos, D.J.; Ryter, S.W.; van der Toorn, M.; Liu, F.; Guo, F.; Baty, C.J.; Karlsson, J.M.; Watkins, S.C.; Kim, H.P.; Wang, X.; et al. Mitochondrial localization and function of heme oxygenase-1 in cigarette smoke-induced cell death. Am. J. Respir. Cell Mol. Biol. 2007, 36, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.L.; Sun, P.; Li, Y.; Liu, S.-S.; Lu, Y. Exosomes as critical mediators to cell-to-cell communication in cancer pathogenesis and their potential clinical application. Transl. Cancer Res. 2019, 8, 298–311. [Google Scholar] [CrossRef] [PubMed]
- Osaki, M.; Okada, F. Exosomes and their role in cancer progression. Yonago Acta Med. 2019, 62, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Weigel, K.J.; Zhou, H.; Wang, X.J. paradoxical roles of TGF-β signaling in suppressing and promoting squamous cell carcinoma. Acta Biochim. Biophys. Sin. 2018, 50, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.W.; Lee, S.S.; Chang, C.Y.; Lang, Y.D.; Jou, Y.S. A new switch for TGFβ in cancer. Cancer Res. 2019, 79, 3797–3805. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, Y.Y.; Chen, Y.; Wang, J.; Wang, Q.; Lu, H. TGF-β signaling and resistance to cancer therapy. Front. Cell Dev. Biol. 2021, 9, 786728. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Yang, J.; Deng, S.; Xu, H.; Wu, D.; Zeng, Q.; Wang, S.; Hu, T.; Wu, F.; Zhou, H. TGF-β signaling in the tumor metabolic microenvironment and targeted therapies. J. Hematol. Oncol. 2022, 15, 135. [Google Scholar] [CrossRef]
- Hua, W.; Ten Dijke, P.; Kostidis, S.; Giera, M.; Hornsveld, M. TGFβ-induced metabolic reprogramming during epithelial-to-mesenchymal transition in cancer. Cell Mol. Life Sci. 2020, 77, 2103–2123. [Google Scholar] [CrossRef]
- Angioni, R.; Sanchez-Rodriguez, R.; Viola, A.; Molon, B. TGF-β in cancer: Metabolic driver of the tolerogenic crosstalk in the tumor microenvironment. Cancers 2021, 13, 401. [Google Scholar] [CrossRef]
- Arnhold, J. The dual role of myeloperoxidase in immune response. Int. J. Mol. Sci. 2020, 21, 8057. [Google Scholar] [CrossRef] [PubMed]
- Arnhold, J.; Malle, E. Halogenation activity of mammalian heme peroxidases. Antioxidants 2022, 11, 890. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, B.; Horwitz, M.S.; Jenne, D.E.; Gauthier, F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol. Rev. 2010, 62, 726–759. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, R.; Shiba-Ishii, A.; Nakano, N.; Togayachi, A.; Sakashita, S.; Sato, Y.; Minami, Y.; Noguchi, M. Heterotopic production of ceruloplasmin by lung adenocarcinoma is significantly correlated with prognosis. Lung Cancer 2018, 118, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Han, B.; Meng, Y.; Han, Y.; Liu, B.; Zhang, B.; Chang, Y.; Cao, P.; Fan, Y.; Tan, K. Ceruloplasmin correlates with immune infiltration and serves as a prognostic factor in breast cancer. Aging 2021, 13, 20438–20467. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cheuk, I.W.-J.; Siu, M.-T.; Yang, W.; Chen, A.S.L.; Sin, V.Y.; Kwong, A. Human haptoglobin contributes to breast cancer oncogenesis through glycolytic activity modulation. Am. J. Cancer Res. 2020, 10, 2865–2877. [Google Scholar] [PubMed]
- Fiorito, V.; Tolosano, E. Hemopexin and cancer. Int. J. Mol. Sci. 2022, 23, 997. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Liu, G.; Jia, R.; Zhang, W.; Li, L.; Zhang, Y.; Wang, Z.; Bai, X. Targeting secretory leukocyte protease inhibitor (SLPI) inhibits colorectal cancer cell growth, migration and invasion via downregulation of AKT. Peer J. 2020, 8, e9400. [Google Scholar] [CrossRef]
- Wang, C.; Liao, Y.; He, W.; Zhang, H.; Zuo, D.; Liu, W.; Yang, Z.; Qiu, J.; Yuan, Y.; Li, K.; et al. Elafin promotes tumor metastasis and attenuates the anti-metastatic effects of erlotinib via binding to EGFR in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2021, 40, 113. [Google Scholar] [CrossRef]
- Jin, Y.; Wang, W.; Wang, Q.; Zhang, Y.; Zahid, K.R.; Raza, U.; Gong, Y. Alpha-1-antichymotrypsin as a novel biomarker for diagnosis, prognosis, and therapy prediction in human diseases. Cancer Cell Int. 2022, 22, 156. [Google Scholar] [CrossRef]
- Tai, C.-S.; Lin, Y.-R.; Teng, T.-H.; Tu, S.-J.; Chou, C.-H.; Huang, Y.-R.; Huang, W.-C.; Weng, S.-L.; Huang, H.-D.; Chen, Y.-L.; et al. Haptoglobin expression correlates with tumor differentiation and five-year overall survival rate in hepatocellular carcinoma. PLoS ONE 2017, 12, e0171269. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Zhi, Q.; Xie, Q.; Wu, X.; Gao, Y.; Chen, X.; Shi, L. Reduced expression of ferroportin1 and ceruloplasmin predicts poor prognosis in adrenocortical carcinoma. J. Trace Elem. Med. Biol. 2019, 56, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.G.; Kumarasinghe, M.P.; Wang, S.M.; Ooi, L.L.; Aw, S.E.; Hui, K.M. Modulation of iron-regulatory genes in human hepatocellular carcinoma and its physiological consequences. Exp. Biol. Med. 2009, 234, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.P.; Lombardi, L.; Stoppacciaro, A.; Melani, C.; Parenza, M.; Bottazzi, B.; Parmiani, G. Granulocyte colony-stimulating factor (G-CSF) gene transduction in murine adenocarcinoma drives neutrophil-mediated tumor inhibition in vivo. Neutrophils discriminate between G-CSF-producing and G-CSF-nonproducing tumor cells. J. Immunol. 1992, 149, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Di Carlo, E.; Forni, G.; Lollini, P.; Colombo, M.P.; Modesti, A.; Musiani, P. The intriguing role of polymorphonuclear neutrophils in antitumor reactions. Blood 2001, 97, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Hicks, A.M.; Riedlinger, G.; Willingham, M.C.; Alexander-Miller, M.A.; von Kap-Herr, C.; Pettenati, M.J.; Sanders, A.M.; Weir, H.M.; Du, W.; Kim, J.; et al. Transferable anticancer innate immunity in spontaneous regression/complete resistance mice. Proc. Natl. Acad. Sci. USA 2006, 103, 7753–7758. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Sagiv, J.Y.; Michaeli, J.; Assi, S.; Mishalian, I.; Kisos, H.; Levy, L.; Damti, P.; Lumbroso, D.; Polyansky, L.; Sionov, R.V.; et al. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep. 2015, 10, 562–573. [Google Scholar] [CrossRef]
- Su, Y.; Qiu, Y.; Qiu, Z.; Qu, P. MicroRNA networks regulate the differentiation, expansion and suppression function of myeloid-derived suppressor cells in tumor microenvironment. J. Cancer 2019, 10, 4350–4356. [Google Scholar] [CrossRef]
- Basso, D.; Gnatta, E.; Padoan, A.; Fogar, P.; Furlanello, S.; Aita, A.; Bozzato, D.; Zambon, C.F.; Arrigoni, G.; Frasson, C.; et al. PDAC-derived exosomes enrich the microenvironment in MDSCs in a SMAD4-dependent manner through a new calcium related axis. Oncotarget 2017, 8, 84928–84944. [Google Scholar] [CrossRef]
- Shang, W.; Tang, Z.; Gao, Y.; Qi, H.; Su, X.; Zhang, Y.; Yang, R. LncRNA RNCR3 promotes Chop expression by sponging miR-185–5p during MDSC differentiation. Oncotarget 2017, 8, 111754–111769. [Google Scholar] [CrossRef] [PubMed]
- Kusmartsev, S.; Nefedova, Y.; Yoder, D.; Gabrilovich, D.I. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J. Immunol. 2004, 172, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.-I.; Collazo, M.; Shalova, I.N.; Biswas, S.K.; Gabrilovich, D.I. Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. J. Leukoc. Biol. 2012, 91, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Ugolini, A.; Tyurin, V.A.; Tyurina, Y.Y.; Tcyganov, E.N.; Donthireddy, L.; Kagan, V.E.; Gabrilovich, D.I.; Veglia, F. Polymorphonuclear myeloid-derived suppressor cells limit antigen cross-presentation by dendritic cells in cancer. JCI Insight 2020, 5, e138581. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Tyurin, V.A.; Mohammadyani, D.; Blasi, M.; Duperret, E.K.; Donthireddy, L.; Hashimoto, A.; Kapralov, A.; Amoscato, A.; Angelini, R.; et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat. Commun. 2017, 8, 2122. [Google Scholar] [CrossRef] [PubMed]
- Al-Khami, A.A.; Zheng, L.; Del Valle, L.; Hossain, F.; Wyczechowska, D.; Zabaleta, J.; Sanchez, M.D.; Dean, M.J.; Rodriguez, P.C.; Ochoa, A.C. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology 2017, 6, e1344804. [Google Scholar] [CrossRef] [PubMed]
- Hossain, F.; Al-Khami, A.A.; Wyczechowska, D.; Hernandez, C.; Zheng, L.; Reiss, K.; Del Valle, L.; Trillo-Tinoco, J.; Maj, T.; Zou, W.; et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol. Res. 2015, 3, 1236–1247. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Tyurin, V.A.; Blasi, M.; De Leo, A.; Kossenkov, A.V.; Donthireddy, L.; To, T.S.J.; Schug, Z.; Basu, S.; Wang, F.; et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature 2019, 569, 73–78. [Google Scholar] [CrossRef]
- Liu, T.W.; Gammon, S.T.; Yang, P.; Ma, W.; Wang, J.; Piwnica-Worms, D. Inhibition of myeloperoxidase enhances immune checkpoint therapy for melanoma. J. Immunother. Cancer 2023, 11, e005837. [Google Scholar] [CrossRef]
- Cui, C.; Chakraborty, K.; Tang, X.A.; Zhou, G.; Schoenfelt, K.Q.; Becker, K.M.; Hoffmann, A.; Chang, Y.-F.; Blank, A.; Reardon, C.A.; et al. Neutrophil elastase selectively kills cancer cells and attenuates tumorigenesis. Cell 2021, 184, 3163–3177. [Google Scholar] [CrossRef]
- Deryugina, E.; Carré, A.; Ardi, V.; Muramatsu, T.; Schmidt, J.; Pham, C.; Quigley, J.P. neutrophil elastase facilitates tumor cell intravasation and early metastatic events. iScience 2020, 23, 101799. [Google Scholar] [CrossRef] [PubMed]
- Hougton, A.M.; Rzymkiewicz, D.M.; Ji, H.; Gregory, A.D.; Egea, E.E.; Metz, H.E.; Stolz, D.B.; Land, S.R.; Marconcini, L.A.; Kliment, C.R.; et al. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat. Med. 2010, 16, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Lerman, I.; Hammes, S.R. Neutrophil elastase in the tumor microenvironment. Steroids 2018, 133, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Tubio-Perez, R.A.; Torres-Durán, M.; Fernández-Villar, A.; Ruano-Raviña, A. Alpha-1 antitrypsin deficiency and risk of lung cancer: A systematic review. Transl. Oncol. 2021, 14, 100914. [Google Scholar] [CrossRef] [PubMed]
- Hiller, A.-M.; Eksröm, M.; Piitulainen, E.; Lindberg, A.; Rönmark, E.; Tanash, H. Cancer risk in severe alpha-1-antitrypsin deficiency. Eur. Respir. J. 2022, 60, 103200. [Google Scholar] [CrossRef] [PubMed]
- Kistowski, M.; Debski, J.; Karczmarski, J.; Paziewska, A.; Oledzki, J.; Mikula, M.; Ostrowski, J.; Dadlez, M. A Strong neutrophil elastase proteolytic fingerprint marks the carcinoma tumor proteome. Mol. Cell Proteom. 2017, 16, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Akizuki, M.; Fukutomi, T.; Takasugi, M.; Takahashi, S.; Sato, T.; Harao, M.; Mizumoto, T.; Yamashita, J.-I. Prognostic significance of immunoreactive neutrophil elastase in human breast cancer: Long-term follow-up results in 313 patients. Neoplasia 2007, 9, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Foekens, J.A.; Ries, C.; Look, M.P.; Gippner-Steppert, C.; Klijn, J.G.; Jochum, M. Elevated expression of polymorphonuclear leukocyte elastase in breast cancer tissue is associated with tamoxifen failure in patients with advanced disease. Br. J. Cancer 2003, 88, 1084–1090. [Google Scholar] [CrossRef]
- Okada, Y.; Nakanishi, I. Activation of matrix metalloproteinase 3 (stromelysin) and matrix metalloproteinase 2 (‘gelatinase’) by human neutrophil elastase and cathepsin G. FEBS Lett. 1989, 249, 353–356. [Google Scholar] [CrossRef]
- Shamanian, P.; Schwartz, J.D.; Pocock, B.J.Z.; Monea, S.; Whiting, D.; Marcus, S.G.; Mignatti, P. Activation of progelatinase (MMP-2) by neutrophil elastase, cathepsin G, and proteinase-3: A role for inflammatory cells in tumor invasion and angiogenesis. J. Cell Physiol. 2001, 189, 197–206. [Google Scholar] [CrossRef]
- Gaggar, A.; Li, Y.; Weathington, N.; Winkler, M.; Kong, M.; Jackson, P.; Blalock, J.E.; Clancy, J.P. Matrix metalloprotease-9 dysregulation in lower airway secretions of cystic fibrosis patients. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L96–L104. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xu, X.; Jiang, Y.; Hansbro, N.G.; Hansbro, P.M.; Xu, J.; Liu, G. Elastin is a key factor of tumor development in colorectal cancer. BMC Cancer 2020, 20, 217. [Google Scholar] [CrossRef] [PubMed]
- Fang, T.; Zhang, L.; Yin, X.; Wang, Y.; Zhang, X.; Bian, X.; Jiang, X.; Yang, S.; Xue, Y. The prognostic marker elastin correlates with epithelial-mesenchymal transition and vimentin-positive fibroblasts in gastric cancer. J. Pathol. Clin. Res. 2023, 9, 56–72. [Google Scholar] [CrossRef]
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular matrix in the tumor microenvironment and its impact on cancer therapy. Front. Mol. Biosci. 2020, 6, 160. [Google Scholar] [CrossRef] [PubMed]
- Taggart, C.; Cervantes-Laurean, D.; Kim, G.; McElvaney, N.G.; Wehr, N.; Moss, J.; Levine, R.L. Oxidation of either methionine 351 or methionine 358 in alpha 1-antitrypsin causes loss of anti-neutrophil elastase activity. J. Biol. Chem. 2000, 275, 27258–27265. [Google Scholar] [CrossRef] [PubMed]
- Dittrich, A.S.; Kühbandner, I.; Gehrig, S.; Rickert-Zacharias, V.; Twigg, M.; Wege, S.; Taggart, C.C.; Herth, F.; Schultz, C.; Mall, M.A. Elastase activity on sputum neutrophils correlates with severity of lung disease in cystic fibrosis. Eur. Respir. J. 2018, 51, 1701910. [Google Scholar] [CrossRef] [PubMed]
- Ramaha, A.; Patston, P.A. Release and degradation of angiotensin I and II from angiotensinogen by neutrophil serine proteases. Arch. Biochem. Biophys. 2002, 397, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Vidotti, D.B.; Casarini, D.E.; Christovam, P.C.; Leite, C.A.; Schor, N.; Boim, M.A. High glucose concentration stimulates intracellular renin activity and angiotensin II generation in rat mesangial cells. Am. J. Physiol. Renal Physiol. 2004, 286, F1039–F1045. [Google Scholar] [CrossRef]
- Penafuerte, C.A.; Gagnon, B.; Sirois, J.; Murphy, J.; MacDonald, N.; Tremblay, M.L. Identification of neutrophil-derived proteases and angiotensin II as biomarkers of cancer cachexia. Br. J. Cancer 2016, 114, 680–687. [Google Scholar] [CrossRef]
- Song, Y.H.; Li, Y.; Du, J.; Mitch, W.E.; Rosenthal, N.; Delafontaine, P. Muscle-specific expression of IGF-1 blocks angiotensin II-induced skeletal muscle wasting. J. Clin. Investig. 2005, 115, 451–458. [Google Scholar] [CrossRef]
- Trobec, K.; von Haehling, S.; Anker, S.D.; Lainsack, M. Growth hormone, insulin-like growth factor 1, and insulin signaling—A pharmacological target in body wasting and cachexia. J. Cachexia Sarcopenia Muscle 2011, 2, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Sanders, P.M.; Russell, S.T.; Tisdale, M.J. Angiotensin II directly induces muscle protein catabolism through the ubiquitin proteasome proteolytic pathway and may play a role in cancer cachexia. Br. J. Cancer 2005, 93, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Lumbers, E.R.; Delforce, S.J.; Pringle, K.G.; Smith, G.R. The lung, the heart, the novel coronavirus, and the renin-angiotensin system; the need for clinical trials. Front. Med. 2020, 7, 248. [Google Scholar] [CrossRef] [PubMed]
- Lamas-Barreiro, J.M.; Alonso-Suárez, M.; Fernández-Martín, J.J.; Saavedra-Alonso, J.A. Angiotensin II suppression in SARS-CoV-2 infection; a therapeutic approach. Nefrologia 2020, 40, 213–216. [Google Scholar] [CrossRef]
- Ino, K.; Shibata, K.; Kajiyama, H.; Yamamoto, E.; Nagasaka, T.; Nawa, A.; Nomura, S.; Kikkawa, F. Angiotensin II type 1 receptor expression in ovarian cancer and its correlation with tumour angiogenesis and patient survival. Br. J. Cancer 2006, 94, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Shirotake, S.; Miyajima, A.; Kosaka, T.; Tanaka, N.; Maeda, T.; Kikuchi, E.; Oya, M. Angiotensin II type 1 receptor expression and microvessel density in human bladder cancer. Urology 2011, 77, 1009.e19–1009.e25. [Google Scholar] [CrossRef]
- Imai, N.; Hashimoto, T.; Kihara, M.; Yoshida, S.; Kawana, I.; Yazawa, T.; Kitamura, H.; Umemura, S. Roles for host and tumor angiotensin II type 1 receptor in tumor growth and tumor-associated angiogenesis. Lab. Investig. 2007, 87, 189–198. [Google Scholar] [CrossRef]
- Oh, E.; Kim, J.Y.; Cho, Y.; An, H.; Lee, N.; Jo, H.; Ban, C.; Seo, J.H. Overexpression of angiotensin II type 1 receptor in breast cancer cells induces epithelial-mesenchymal transition and promotes tumor growth and angiogenesis. Biochim. Biophys. Acta 2016, 1863, 1071–1081. [Google Scholar] [CrossRef]
- Smith, G.R.; Missailidis, S. Cancer, inflammation and the AT1 and AT2 receptors. J. Inflamm. 2004, 1, 3. [Google Scholar] [CrossRef]
- Catarata, M.J.; Ribeiro, R.; Oliviera, M.J.; Cordeiro, C.R.; Medeiros, R. Renin-angiotensin system in lung tumor and microenvironment interactions. Cancers 2020, 12, 1457. [Google Scholar] [CrossRef]
- Carey, R.M.; Whelton, P.K. Prevention, detection, evaluation, and management of high blood pressure in adults: Synopsis of the 2017 American College of Cardiology/American Heart Association Hypertension Guideline. Ann. Int. Med. 2018, 168, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B.M.; Cooper, M.E.; de Zeeuw, D.; Keane, W.F.; Mitch, W.E.; Parving, H.H.; Remuzzi, G.; Snapinn, S.M.; Zhang, Z.; Shanifar, S.; et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N. Engl. J. Med. 2001, 345, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Sipahi, I. Risk of cancer with angiotensin-receptor blockers increases with increasing cumulative exposure: Meta-regression analysis of randomized trials. PLoS ONE 2022, 17, e0263461. [Google Scholar] [CrossRef] [PubMed]
- Choudhari, S.K.; Choudhari, M.; Bagde, S.; Gadbail, A.R.; Joshi, V. Nitric oxide and cancer: A review. World J. Surg. Oncol. 2013, 11, 18. [Google Scholar] [CrossRef] [PubMed]
- Niu, F.; Yu, Y.; Li, Z.; Ren, Y.; Li, Z.; Ye, Q.; Liu, P.; Ji, C.; Qian, L.; Xiong, Y. Arginase: A emerging and promising therapeutic target for cancer treatment. Biomed. Pharmacother. 2022, 149, 112840. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.W.; Chi, C.W.; Lin, E.C.; Lui, W.Y.; P’Eng, F.K.; Wang, S.R. Serum arginase level in patients with gastric cancer. J. Clin. Gastroenterol. 1994, 18, 84–85. [Google Scholar] [CrossRef]
- Leu, S.Y.; Wang, S.R. Clinical significance of arginase in colorectal cancer. Cancer 1992, 70, 733–736. [Google Scholar] [CrossRef]
- Chrzanowska, A.; Graboń, W.; Mielczarek-Puta, M.; Barańczyk-Kuźma, A. Significance of arginase determination in body fluids of patients with hepatocellular carcinoma and liver cirrhosis before and after surgical treatment. Clin. Biochem. 2014, 47, 1056–1059. [Google Scholar] [CrossRef]
- Roci, I.; Watrous, J.D.; Lagerborg, K.A.; Lafranchi, L.; Lindqvist, A.; Jain, M.; Nilsson, R. Mapping metabolic events in the cancer cell cycle reveals arginine catabolism in the committed SG2M phase. Cell Rep. 2019, 26, 1691–1700.e5. [Google Scholar] [CrossRef]
- Srivastava, S.; Ghosh, S.K. Modulation of L-arginine-arginase metabolic pathway enzymes: Immunocytochemistry and mRNA expression in peripheral blood and tissue levels in head and neck squamous cell carcinomas in North East India. Asian Pac. J. Cancer Prev. 2015, 16, 7031–7038. [Google Scholar] [CrossRef]
- Munder, M.; Engelhardt, M.; Knies, D.; Medenhoff, S.; Wabnitz, G.; Luckner-Minden, C.; Feldmeyer, N.; Voss, R.H.; Kropf, P.; Müller, I.; et al. Cytotoxicity of tumor antigen specific human T cells is impaired by arginine depletion. PLoS ONE 2013, 5, e63521. [Google Scholar] [CrossRef]
- Segal, A.W.; Jones, O.T.G. Novel cytochrome b system in phagocytic vacuoles from human granulocytes. Nature 1978, 276, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Wientjes, F.B.; Segal, A.W. NADPH oxidase and the respiratory burst. Semin. Cell Biol. 1995, 6, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.S.; Ballou, D.P.; Palmer, G.; Massey, V. The reaction of xanthine oxidase with molecular oxygen. J. Biol. Chem. 1974, 249, 4350–4362. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.F.; Hille, R.; Massey, V. The radical chemistry of milk xanthine oxidase as studied by radiation chemistry technique. J. Biol. Chem. 1986, 261, 15870–15876. [Google Scholar] [CrossRef] [PubMed]
- Carrell, R.W.; Winterbourn, C.C.; Rachmilewitz, E.A. Activated oxygen and hemolysis. Br. J. Hematol. 1975, 30, 259–264. [Google Scholar] [CrossRef]
- Harel, S.; Kanner, J. Hemoglobin and myoglobin as inhibitors of hydroxyl radical generation in a model system of “iron redox” cycle. Free Radic. Res. Commun. 1989, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nozik-Grayck, E.; Suliman, H.B.; Piantadosi, C.A. Extracellular superoxide dismutase. Int. J. Biochem. Cell Biol. 2005, 37, 2466–2471. [Google Scholar] [CrossRef]
- Teoh-Fitzgerald, M.L.; Fitzgerald, M.P.; Jensen, T.J.; Futscher, B.W.; Domann, F.E. Genetic and epigenetic inactivation of extracellular superoxide dismutase promotes an invasive phenotype in human lung cancer by disrupting ECM homeostasis. Mol. Cancer Res. 2012, 10, 40–51. [Google Scholar] [CrossRef]
- Teoh-Fitzgerald, M.L.; Fitzgerald, M.P.; Zhong, W.; Askeland, R.W.; Domann, F.E. Epigenetic reprogramming governs EcSOD expression during human mammary epithelial cell differentiation, tumorigenesis and metastasis. Oncogene 2014, 33, 358–368. [Google Scholar] [CrossRef]
- Laukkanen, M.O. Extracellular superoxide dismutase: Growth promotor or tumor suppressor? Oxid. Med. Cell Longev. 2016, 2016, 3612589. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lu, X.; Zhang, Y.; Zhao, D.; Gong, H.; Du, Y.; Sun, H. The effect of extracellular superoxide dismutase (SOD3) gene in lung cancer. Front. Oncol. 2022, 12, 722646. [Google Scholar] [CrossRef] [PubMed]
- Mira, E.; Carmona-Rodriguez, L.; Pérez-Villamil, B.; Casas, J.; Fernández-Aceñero, M.J.; Martinez-Rey, D.; Martin-González, P.; Heras-Murillo, I.; Paz-Cabezas, M.; Tardáguila, M.; et al. SOD3 improves the tumor response to chemotherapy by stabilizing endothelial HIF-2alpha. Nat. Commun. 2018, 9, 575. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Rodriguez, L.; Martinez-Rey, D.; Fernández-Aceñero, M.J.; González-Martin, A.; Paz-Cabezas, M.; Rodriguez-Rodriguez, N.; Pérez-Villamil, B.; Sáez, M.E.; Diaz-Rubio, E.; Mira, E.; et al. SOD3 induces a HIF-2a-dependent program in endothelial cells that provides a selective signal for tumor infiltration by T cells. J. Immunother. Cancer 2020, 8, e000432. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.D.; Pryor, W.A. Cigarette smoking. emphysema and damage to alpha 1-proteinase inhibitor. Am. J. Physiol. Lung Cell Mol. Physiol. 1994, 266, L593–L611. [Google Scholar] [CrossRef]
- Martinez-Ray, D.; Carmona-Rodriguez, L.; Fernández-Aceñero, M.J.; Mira, E.; Mañes, S. Extracellular superoxide dismutase, the endothelial basement membrane, and the WNT pathway: New players in vascular normalization and tumor infiltration by T cells. Front. Immunol. 2020, 11, 579552. [Google Scholar] [CrossRef]
- Teoh, M.L.T.; Fitzgerald, M.P.; Oberley, L.W.; Domann, F.E. Overexpression of extracellular superoxide dismutase attenuates heparanase expression and inhibits breast carcinoma cell growth and invasion. Cancer Res. 2009, 69, 6355–6363. [Google Scholar] [CrossRef]
- Mueller, B.M.; Schraufstatter, I.U.; Goncharova, V.; Povaliy, T.; DiScipio, R.; Khaldoyanidi, S.K. Hyaluronan inhibits postchemotherapy tumor regrowth in a colon carcinoma xenograft model. Mol. Cancer Ther. 2010, 9, 3024–3032. [Google Scholar] [CrossRef]
- Tian, X.; Azpurua, J.; Hine, C.; Vaidya, A.; Myakishev-Rempel, M.; Ablaeva, J.; Mao, Z.; Nevo, E.; Gorbunova, V.; Seluanov, A. High molecular weight hyaluronan mediates the cancer resistance of the naked mole-rat. Nature 2013, 499, 346–349. [Google Scholar] [CrossRef]
- Toole, B.P. Hyaluronan: From extracellular glue to pericellular cue. Nat. Rev. Cancer 2004, 4, 528–539. [Google Scholar] [CrossRef]
- Liu, M.; Tolg, C.; Turley, E. Dissecting the dual nature of hyaluronan in the tumor microenvironment. Front. Immunol. 2019, 10, 947. [Google Scholar] [CrossRef] [PubMed]
- Frangociannis, N.G. Transforming growth factor-β in tissue fibrosis. J. Exp. Med. 2020, 217, e20190103. [Google Scholar] [CrossRef] [PubMed]
- Budi, E.H.; Schaub, J.R.; Decaris, M.; Turner, S.; Derynck, R. TGF-β as a driver of fibrosis: Physiological roles and therapeutic opportunities. J. Pathol. 2021, 254, 358–373. [Google Scholar] [CrossRef] [PubMed]
- Taleb, S.; Cancello, R.; Clément, K.; Lacasa, D. Cathepsin S promotes human preadipocyte differentiation: Possible involvement of fibronectin degradation. Endocrinology 2006, 147, 4950–4959. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Zhang, G.; Fu, J.; Wu, Q. Matrix metalloproteinase-14 (MMP-14) down-regulation inhibits esophageal squamous cell carcinoma cell migration, invasion, and proliferation. Thorac. Cancer 2020, 11, 3168–3174. [Google Scholar] [CrossRef] [PubMed]
- Aguilera-Montilla, N.; Bailón, E.; Uceda-Castro, R.; Ugarte-Bercal, E.; Santos, A.; Guitérrez-González, A.; Pérez-Sánchez, C.; Van den Steen, P.E.; Opdenakker, G.; Garcia-Marco, J.E.; et al. MMP-9 affects gene expression in chronic lymphocytic leukemia revealing CD99 as an MMP-9 target and a novel partner in malignant cell migration/arrest. Oncogene 2019, 38, 4605–4619. [Google Scholar] [CrossRef] [PubMed]
- Cabral-Pacheco, G.A.; Garca-Veloz, I.; Castruita-De la Rosa, C.; Ramirez, J.M.; Perez-Romero, B.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The role of matrix metalloproteinases and their inhibitors in human diseases. Int. J. Mol. Sci. 2020, 21, 9739. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Zhao, K.; Jose, I.; Hoogenraad, N.J.; Osellame, L.D. Biomarkers for cancer cachexia: A mini review. Int. J. Mol. Sci. 2021, 22, 4501. [Google Scholar] [CrossRef]
- Prokopchuk, O.; Grünwald, B.; Nitsche, U.; Jäger, C.; Prokopchuk, O.L.; Schubert, E.C.; Friess, H.; Martignoni, M.E.; Krüger, A. Elevated systemic levels of the matrix metalloproteinase inhibitor TIMP-1 correlate with clinical markers of cachexia in patients with chronic pancreatitis and pancreatic cancer. BMC Cancer 2018, 18, 128. [Google Scholar] [CrossRef]
- Narasimhan, A.; Shahda, S.; Kays, J.K.; Perkins, S.M.; Cheng, L.; Schloss, K.N.H.; Schloss, D.E.I.; Koniaris, L.G.; Zimmers, T.A. Identification of potential serum protein biomarkers and pathways for pancreatic cancer cachexia using an aptamer-based discovery platform. Cancers 2020, 12, 3787. [Google Scholar] [CrossRef]
- Bachman, K.E.; Herman, J.G.; Corn, P.G.; Merlo, A.; Costello, J.F.; Cavenee, W.K.; Baylin, S.B.; Graff, J.R. Methylation-associated silencing of the tissue inhibitor of metalloproteinase-3 gene suggest a suppressor role in kidney, brain, and other human cancers. Cancer Res. 1999, 59, 798–802. [Google Scholar] [PubMed]
- Ahonen, M.; Baker, A.H.; Kahari, V.M. Adenovirus-mediated gene delivery of tissue inhibitor of metalloproteinases-3 inhibits invasion and induces apoptosis in melanoma cells. Cancer Res. 1998, 58, 2310–2315. [Google Scholar] [PubMed]
- Baker, A.H.; George, S.J.; Zaltsman, A.B.; Murphy, G.; Newby, A.C. Inhibition of invasion and induction of apoptotic cell death of cancer cell lines by overexpression of TIMP-3. Br. J. Cancer 1999, 79, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Nagao, Y.; Hisaoka, M.; Matsuyama, A.; Kanemitsu, S.; Hamada, T.; Fukuyama, T.; Nakano, R.; Uchiyama, A.; Kawamoto, M.; Yamaguchi, K.; et al. Association of microRNA-21 expression with its targets, PDCD4 and TIMP3, in pancreatic ductal adenocarcinoma. Mod. Pathol. 2012, 25, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Fatheree, J.P.; Guarin, J.R.; McGinn, R.A.; Naber, S.P.; Oudin, M.J. Chemotherapy-induced collagen IV drives cancer cell motility through activation of Src and focal adhesion kinase. Cancer Res. 2022, 82, 2031–2044. [Google Scholar] [CrossRef] [PubMed]
- Jansson, M.; Lindberg, J.; Rask, G.; Svensson, J.; Billing, O.; Nazemroaya, A.; Berglund, A.; Wärnberg, F.; Sund, M. Prognostic value of stromal type IV collagen expression in small invasive breast cancers. Front. Mol. Biosci. 2022, 9, 904526. [Google Scholar] [CrossRef]
- Lipton, A.; Leitzel, K.; Ali, S.M.; Polimera, H.V.; Nagabhairu, V.; Marks, E.; Richardson, A.E.; Krecko, L.; Ali, A.; Koestler, W.; et al. High turnover of extracellular matrix reflected by specific protein fragments measured in serum Is associated with poor outcomes in two metastatic breast cancer cohorts. Int. J. Cancer 2018, 143, 3027–3034. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, M.; Jansson, M.; Tavelin, B.; Dirix, L.; Vermeulen, P.; Nyström, H. Type IV collagen as a potential biomarker of metastatic breast cancer. Clin. Exp. Metastasis 2021, 38, 175–185. [Google Scholar] [CrossRef]
- Lee, S.W.; Kim, H.K.; Naidansuren, P.; Ham, K.A.; Choi, H.S.; Ahn, H.Y.; Kim, M.; Kang, D.H.; Kang, S.W.; Joe, A.E. Peroxidasin is essential for endothelial cell survival and growth signaling by sulflimine crosslink-dependent matrix assembly. FASEB J. 2020, 34, 10228–10241. [Google Scholar] [CrossRef]
- He, C.; Song, W.; Weston, T.A.; Tran, C.; Kurtz, I.; Zuckerman, J.E.; Guagliardo, P.; Miner, J.H.; Ivanov, S.V.; Bougoure, J.; et al. Peroxidasin-mediated bromine enrichment of basement membranes. Proc. Natl. Acad. Sci. USA 2020, 117, 15827–15836. [Google Scholar] [CrossRef]
- Medfai, H.; Khalil, A.; Rousseau, A.; Nuyens, V.; Paumann-Page, M.; Sevcnikar, B.; Furtmüller, P.G.; Obinger, C.; Moguilevsky, N.; Peulen, O.; et al. Human peroxidasin 1 promotes angiogenesis through ERK1/2, Akt, and FAK pathways. Cardiovasc. Res. 2019, 115, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Paumann-Page, M.; Kienzl, N.F.; Motwani, J.; Bathish, B.; Paton, L.N.; Magon, N.J.; Sevcnikar, B.; Furtmüller, P.G.; Traxlmayr, M.W.; Obinger, C.; et al. Peroxidasin protein expression and enzymatic activity in metastatic melanoma cell lines are associated with invasive potential. Redox Biol. 2021, 46, 102090. [Google Scholar] [CrossRef] [PubMed]
- Wyllie, K.; Panagopoulos, V.; Cox, T.R. The role of peroxidasin in solid cancer progression. Biochem. Soc. Trans. 2023, 51, 1881–1895. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhai, Z.; Chen, C.; Tian, X.; Xing, Z.; Xing, P.; Yang, Y.; Zhang, J.; Wang, C.; Dong, L. Air pollution particles hijack peroxidasin to disrupt immunosurveillance and promote lung cancer. eLife 2022, 11, e75345. [Google Scholar] [CrossRef]
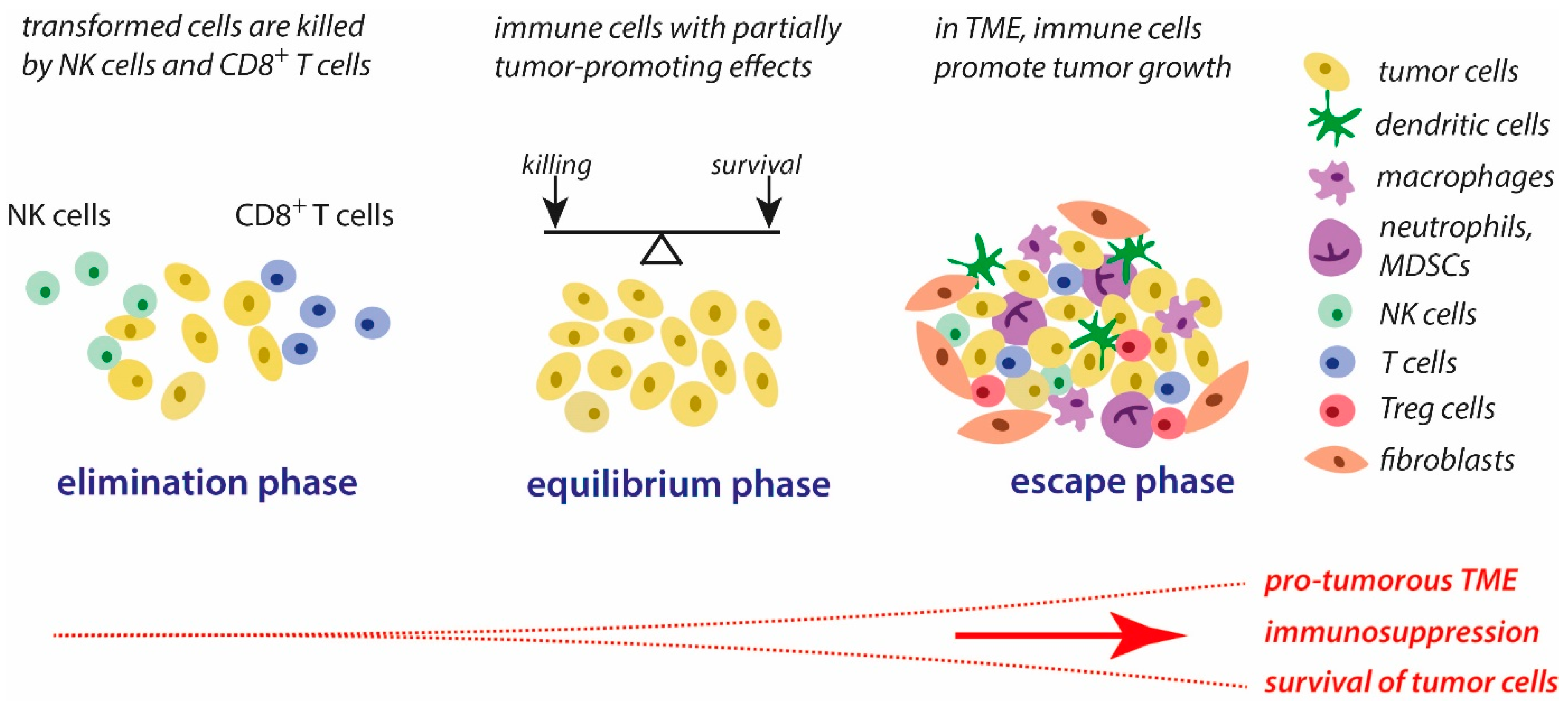



| Antioxidative System | Functions | Effects in Cancer Cells | Cancer Types | References |
|---|---|---|---|---|
| Transferrin receptor | Iron uptake to cells | Upregulation | Liver, breast, colon, and lung cancer | [275,280] |
| Ferritin | Cellular iron storage protein | Upregulation | Esophageal adenocarcinoma, glioblastoma, and breast and colorectal cancer | [276,281] |
| Ferroportin | Iron export from cells | Downregulation | Prostate and breast cancers | [277,278,279] |
| Heme oxygenase 1 | Removal of free heme | Upregulation | Multiple types of cancer | [282] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arnhold, J. Inflammation-Associated Cytotoxic Agents in Tumorigenesis. Cancers 2024, 16, 81. https://doi.org/10.3390/cancers16010081
Arnhold J. Inflammation-Associated Cytotoxic Agents in Tumorigenesis. Cancers. 2024; 16(1):81. https://doi.org/10.3390/cancers16010081
Chicago/Turabian StyleArnhold, Jürgen. 2024. "Inflammation-Associated Cytotoxic Agents in Tumorigenesis" Cancers 16, no. 1: 81. https://doi.org/10.3390/cancers16010081
APA StyleArnhold, J. (2024). Inflammation-Associated Cytotoxic Agents in Tumorigenesis. Cancers, 16(1), 81. https://doi.org/10.3390/cancers16010081