
Novel Pathogenic Variants in Hereditary Cancer Syndromes in a Highly Heterogeneous Cohort of Patients: Insights from Multigene Analysis

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
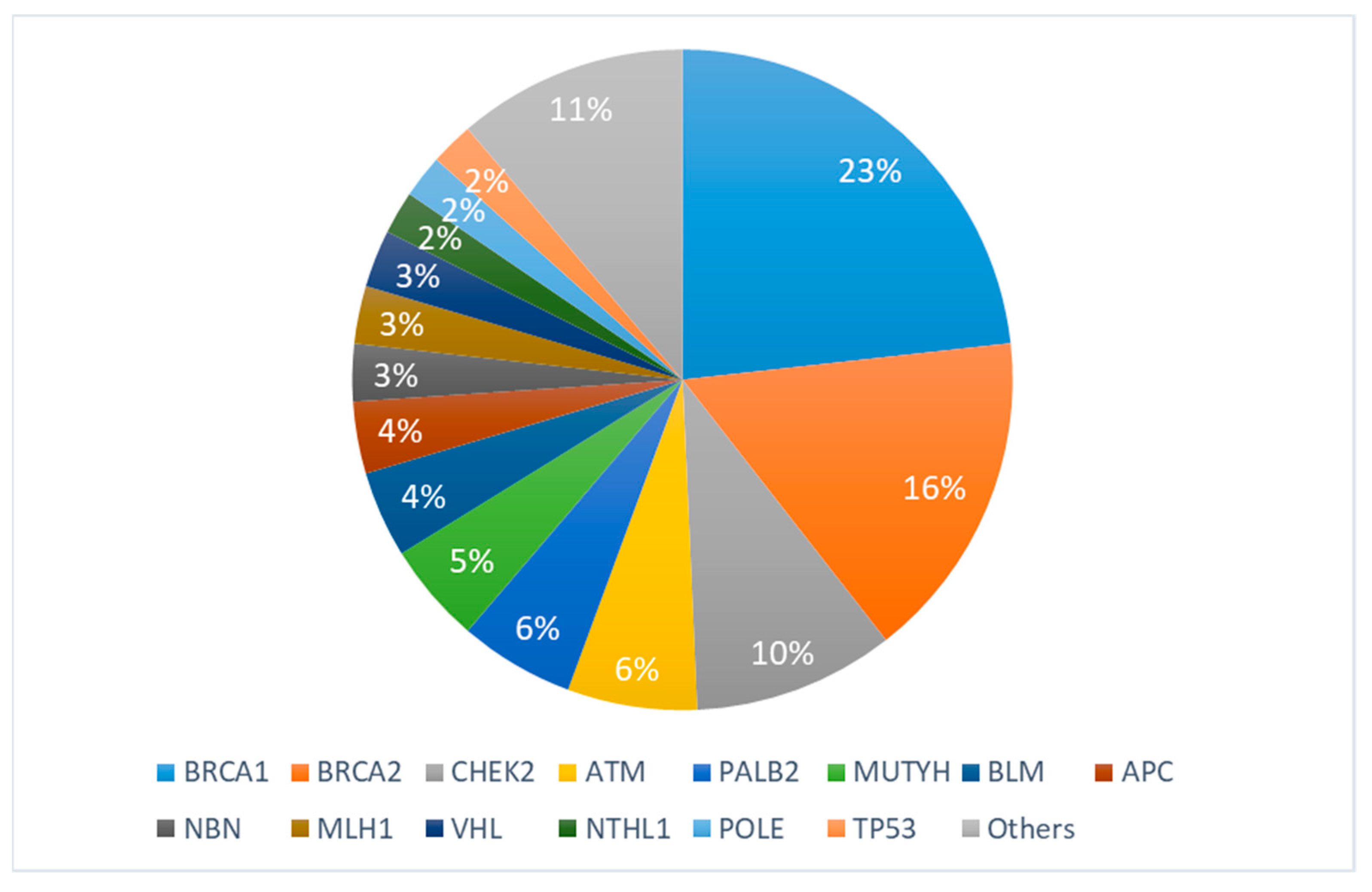
3. Results
4. Discussion
4.1. Novel Undescribed Variants
4.1.1. Mutations in MLH1 Gene
4.1.2. Mutations in EPCAM Gene
4.1.3. Mutations in MLH3 Gene
4.1.4. Mutations in ATM Gene
4.1.5. Mutations in GALNT12 Gene
4.1.6. Mutations in MUTYH Gene
4.1.7. Mutations in POLE Gene
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Garber, J.E.; Offit, K. Hereditary cancer predisposition syndromes. J. Clin. Oncol. 2005, 23, 276–292. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Tsaousis, G.N.; Papadopoulou, E.; Apessos, A.; Agiannitopoulos, K.; Pepe, G.; Kampouri, S.; Diamantopoulos, N.; Floros, T.; Iosifidou, R.; Katopodi, O.; et al. Analysis of hereditary cancer syndromes by using a panel of genes: Novel and multiple pathogenic mutations. BMC Cancer 2019, 19, 535. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.; Lin, N.U.; Kidd, J.; Allen, B.A.; Singh, N.; Wenstrup, R.J.; Hartman, A.R.; Winer, E.P.; Garber, J.E. Frequency of Germline Mutations in 25 Cancer Susceptibility Genes in a Sequential Series of Patients with Breast Cancer. J. Clin. Oncol. 2016, 34, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Guindalini, R.S.C.; Viana, D.V.; Kitajima, J.P.F.W.; Rocha, V.M.; López, R.V.M.; Zheng, Y.; Freitas, É.; Monteiro, F.P.M.; Valim, A.; Schlesinger, D.; et al. Detection of germline variants in Brazilian breast cancer patients using multigene panel testing. Sci. Rep. 2022, 12, 4190. [Google Scholar] [CrossRef] [PubMed]
- Montalban, G.; Bonache, S.; Moles-Fernández, A.; Gisbert-Beamud, A.; Tenés, A.; Bach, V.; Carrasco, E.; López-Fernández, A.; Stjepanovic, N.; Balmaña, J.; et al. Screening of BRCA1/2 deep intronic regions by targeted gene sequencing identifies the first germline BRCA1 variant causing pseudoexon activation in a patient with breast/ovarian cancer. J. Med. Genet. 2019, 56, 63–74. [Google Scholar] [CrossRef]
- Singh, J.; Thota, N.; Singh, S.; Padhi, S.; Mohan, P.; Deshwal, S.; Sur, S.; Ghosh, M.; Agarwal, A.; Sarin, R.; et al. Screening of over 1000 Indian patients with breast and/or ovarian cancer with a multi-gene panel: Prevalence of BRCA1/2 and non-BRCA mutations. Breast Cancer Res. Treat. 2018, 170, 189–196. [Google Scholar] [CrossRef]
- Bono, M.; Fanale, D.; Incorvaia, L.; Cancelliere, D.; Fiorino, A.; Calò, V.; Dimino, A.; Filorizzo, C.; Corsini, L.R.; Brando, C.; et al. Impact of deleterious variants in other genes beyond BRCA1/2 detected in breast/ovarian and pancreatic cancer patients by NGS-based multi-gene panel testing: Looking over the hedge. ESMO Open 2021, 6, 100235. [Google Scholar] [CrossRef]
- Dámaso, E.; González-Acosta, M.; Vargas-Parra, G.; Navarro, M.; Balmaña, J.; Ramon, Y.; Cajal, T.; Tuset, N.; Thompson, B.A.; Marín, F.; et al. Comprehensive Constitutional Genetic and Epigenetic Characterization of Lynch-Like Individuals. Cancers 2020, 12, 1799. [Google Scholar] [CrossRef]
- Pearlman, R.; Frankel, W.L.; Swanson, B.; Zhao, W.; Yilmaz, A.; Miller, K.; Bacher, J.; Bigley, C.; Nelsen, L.; Goodfellow, P.J. Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations among Patients with Early-Onset Colorectal Cancer. JAMA Oncol. 2017, 3, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, A.; Bean, G.R.; Hagemann, I.S.; Lin, C.Y. Molecular Testing in Breast Cancer: Current Status and Future Directions. J. Mol. Diagn. 2021, 23, 1422–1432. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, S.; Chiyoda, T.; Nakamura, K.; Sakai, K.; Yoshihama, T.; Nishio, H.; Kobayashi, Y.; Iwata, T.; Banno, K.; Yamagami, W.; et al. Clinical availability and characteristics of multigene panel testing for recurrent/advanced gynecologic cancer. Int. J. Clin. Oncol. 2023, 28, 1554–1562. [Google Scholar] [CrossRef] [PubMed]
- Shirts, B.H.; Casadei, S.; Jacobson, A.L.; Lee, M.K.; Gulsuner, S.; Bennett, R.L.; Miller, M.; Hall, S.A.; Hampel, H.; Hisama, F.M.; et al. Improving performance of multigene panels for genomic analysis of cancer predisposition. Genet. Med. 2016, 18, 974–981. [Google Scholar] [CrossRef] [PubMed]
- LaDuca, H.; Stuenkel, A.J.; Dolinsky, J.S.; Keiles, S.; Tandy, S.; Pesaran, T.; Chen, E.; Gau, C.L.; Palmaer, E.; Shoaepour, K.; et al. Utilization of multigene panels in hereditary cancer predisposition testing: Analysis of more than 2,000 patients. Genet. Med. 2014, 16, 830–837. [Google Scholar] [CrossRef] [PubMed]
- Bilyalov, A.; Nikolaev, S.; Shigapova, L.; Khatkov, I.; Danishevich, A.; Zhukova, L.; Smolin, S.; Titova, M.; Lisica, T.; Bodunova, N.; et al. Application of Multigene Panels Testing for Hereditary Cancer Syndromes. Biology 2022, 11, 1461. [Google Scholar] [CrossRef] [PubMed]
- Vasimuddin, M.; Misra, S.; Li, H.; Aluru, S. Efficient Architecture-Aware Acceleration of BWA-MEM for Multicore Systems. In Proceedings of the 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS), Rio de Janeiro, Brazil, 20–24 May 2019; pp. 314–324. [Google Scholar]
- Broadinstitute/Picard: A Set of Command Line Tools (in Java) for Manipulating High-Throughput Sequencing (HTS) Data and Formats Such as SAM/BAM/CRAM and VCF. 2019. Available online: https://github.com/broadinstitute/picard (accessed on 1 November 2023).
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 2017, 201178. [Google Scholar] [CrossRef]
- Kurian, A.W.; Li, Y.; Hamilton, A.S.; Ward, K.C.; Hawley, S.T.; Morrow, M.; McLeod, M.C.; Jagsi, R.; Katz, S.J. Gaps in Incorporating Germline Genetic Testing into Treatment Decision-Making for Early-Stage Breast Cancer. J. Clin. Oncol. 2017, 35, 2232–2239. [Google Scholar] [CrossRef]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548. [Google Scholar] [CrossRef]
- Bonadona, V.; Bonaïti, B.; Olschwang, S.; Grandjouan, S.; Huiart, L.; Longy, M.; Guimbaud, R.; Buecher, B.; Bignon, Y.J.; Caron, O.; et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 2011, 305, 2304–2310. [Google Scholar] [CrossRef]
- Idos, G.; Valle, L. Lynch Syndrome. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Wallace, S.E., Bean, L.J.H., Eds.; University of Washington: Seattle, WA, USA, 2004. [Google Scholar]
- Møller, P.; Seppälä, T.T.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Gareth Evans, D.; Lindblom, A.; Macrae, F.; Blanco, I.; Sijmons, R.H.; et al. Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: A report from the Prospective Lynch Syndrome Database. Gut 2018, 67, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Herlyn, M.; Steplewski, Z.; Herlyn, D.; Koprowski, H. Colorectal carcinoma-specific antigen: Detection by means of monoclonal antibodies. Proc. Natl. Acad. Sci. USA 1979, 76, 1438–1442. [Google Scholar] [CrossRef] [PubMed]
- Schnell, U.; Cirulli, V.; Giepmans, B.N. EpCAM: Structure and function in health and disease. Biochim. Biophys. Acta 2013, 1828, 1989–2001. [Google Scholar] [CrossRef] [PubMed]
- Sivagnanam, M.; Mueller, J.L.; Lee, H.; Chen, Z.; Nelson, S.F.; Turner, D.; Zlotkin, S.H.; Pencharz, P.B.; Ngan, B.Y.; Libiger, O.; et al. Identification of EpCAM as the gene for congenital tufting enteropathy. Gastroenterology 2008, 135, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Ligtenberg, M.J.; Kuiper, R.P.; Chan, T.L.; Goossens, M.; Hebeda, K.M.; Voorendt, M.; Lee, T.Y.; Bodmer, D.; Hoenselaar, E.; Hendriks-Cornelissen, S.J.; et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3’ exons of TACSTD1. Nat. Genet. 2009, 41, 112–117. [Google Scholar] [CrossRef]
- Ligtenberg, M.J.; Kuiper, R.P.; Geurts van Kessel, A.; Hoogerbrugge, N. EPCAM deletion carriers constitute a unique subgroup of Lynch syndrome patients. Fam. Cancer 2013, 12, 169–174. [Google Scholar] [CrossRef]
- Wu, Y.; Berends, M.J.; Sijmons, R.H.; Mensink, R.G.; Verlind, E.; Kooi, K.A.; van der Sluis, T.; Kempinga, C.; van dDer Zee, A.G.; Hollema, H.; et al. A role for MLH3 in hereditary nonpolyposis colorectal cancer. Nat. Genet. 2001, 29, 137–138. [Google Scholar] [CrossRef]
- Ou, J.; Rasmussen, M.; Westers, H.; Andersen, S.D.; Jager, P.O.; Kooi, K.A.; Niessen, R.C.; Eggen, B.J.; Nielsen, F.C.; Kleibeuker, J.H.; et al. Biochemical characterization of MLH3 missense mutations does not reveal an apparent role of MLH3 in Lynch syndrome. Genes Chromosomes Cancer 2009, 48, 340–350. [Google Scholar] [CrossRef]
- Liu, H.X.; Zhou, X.L.; Liu, T.; Werelius, B.; Lindmark, G.; Dahl, N.; Lindblom, A. The role of hMLH3 in familial colorectal cancer. Cancer Res. 2003, 63, 1894–1899. [Google Scholar]
- Taylor, N.P.; Powell, M.A.; Gibb, R.K.; Rader, J.S.; Huettner, P.C.; Thibodeau, S.N.; Mutch, D.G.; Goodfellow, P.J. MLH3 mutation in endometrial cancer. Cancer Res. 2006, 66, 7502–7508. [Google Scholar] [CrossRef]
- Song, H.; Ramus, S.J.; Quaye, L.; DiCioccio, R.A.; Tyrer, J.; Lomas, E.; Shadforth, D.; Hogdall, E.; Hogdall, C.; McGuire, V.; et al. Common variants in mismatch repair genes and risk of invasive ovarian cancer. Carcinogenesis 2006, 27, 2235–2242. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F.; Scott, S.; Gueven, N.; Kozlov, S.; Peng, C.; Chen, P. Functional consequences of sequence alterations in the ATM gene. DNA Repair 2004, 3, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Swift, M.; Morrell, D.; Cromartie, E.; Chamberlin, A.R.; Skolnick, M.H.; Bishop, D.T. The incidence and gene frequency of ataxia-telangiectasia in the United States. Am. J. Hum. Genet. 1986, 39, 573–583. [Google Scholar] [PubMed]
- Taylor, A.M.; Byrd, P.J. Molecular pathology of ataxia telangiectasia. J. Clin. Pathol. 2005, 58, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Mavrou, A.; Tsangaris, G.T.; Roma, E.; Kolialexi, A. The ATM gene and ataxia telangiectasia. Anticancer Res. 2008, 28, 401–405. [Google Scholar] [PubMed]
- Choi, M.; Kipps, T.; Kurzrock, R. ATM Mutations in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 1781–1791. [Google Scholar] [CrossRef]
- Bernstein, J.L.; Teraoka, S.; Southey, M.C.; Jenkins, M.A.; Andrulis, I.L.; Knight, J.A.; John, E.M.; Lapinski, R.; Wolitzer, A.L.; Whittemore, A.S.; et al. Population-based estimates of breast cancer risks associated with ATM gene variants c.7271T>G and c.1066-6T>G (IVS10-6T>G) from the Breast Cancer Family Registry. Hum. Mutat. 2006, 27, 1122–1128. [Google Scholar] [CrossRef]
- Hsu, F.C.; Roberts, N.J.; Childs, E.; Porter, N.; Rabe, K.G.; Borgida, A.; Ukaegbu, C.; Goggins, M.G.; Hruban, R.H.; Zogopoulos, G.; et al. Risk of Pancreatic Cancer Among Individuals With Pathogenic Variants in the ATM Gene. JAMA Oncol. 2021, 7, 1664–1668. [Google Scholar] [CrossRef]
- Nanda, N.; Roberts, N.J. ATM Serine/Threonine Kinase and its Role in Pancreatic Risk. Genes 2020, 11, 108. [Google Scholar] [CrossRef]
- Moslemi, M.; Moradi, Y.; Dehghanbanadaki, H.; Afkhami, H.; Khaledi, M.; Sedighimehr, N.; Fathi, J.; Sohrabi, E. The association between ATM variants and risk of breast cancer: A systematic review and meta-analysis. BMC Cancer 2021, 21, 27. [Google Scholar] [CrossRef]
- Guo, J.M.; Zhang, Y.; Cheng, L.; Iwasaki, H.; Wang, H.; Kubota, T.; Tachibana, K.; Narimatsu, H. Molecular cloning and characterization of a novel member of the UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferase family, pp-GalNAc-T12. FEBS Lett. 2002, 524, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Guda, K.; Moinova, H.; He, J.; Jamison, O.; Ravi, L.; Natale, L.; Lutterbaugh, J.; Lawrence, E.; Lewis, S.; Willson, J.K.; et al. Inactivating germ-line and somatic mutations in polypeptide N-acetylgalactosaminyltransferase 12 in human colon cancers. Proc. Natl. Acad. Sci. USA 2009, 106, 12921–12925. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.R.; Venkitachalam, S.; Revoredo, L.; Dohey, A.T.; Clarke, E.; Pennell, J.J.; Powell, A.E.; Quinn, E.; Ravi, L.; Gerken, T.A.; et al. Evidence for GALNT12 as a moderate penetrance gene for colorectal cancer. Hum. Mutat. 2018, 39, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Arun, B.; Schnabel, F.R.; Chun, J.; Heeke, A.L.; Smith, J.A.; Roses, D.F.; Kurz, E.; Landry, K.; Barrera, A.G.; Wood, M.; et al. Non-BRCA hereditary gene mutations and breast cancer phenotype: An ISC-RAM Consortia study. J. Clin. Oncol. 2018, 36, 1540. [Google Scholar] [CrossRef]
- Cheadle, J.P.; Sampson, J.R. Exposing the MYtH about base excision repair and human inherited disease. Hum. Mol. Genet. 2003, 12, R159–R165. [Google Scholar] [CrossRef] [PubMed]
- Sieber, O.M.; Lipton, L.; Crabtree, M.; Heinimann, K.; Fidalgo, P.; Phillips, R.K.; Bisgaard, M.L.; Orntoft, T.F.; Aaltonen, L.A.; Hodgson, S.V.; et al. Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N. Engl. J. Med. 2003, 348, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Emmerson, P.; Maynard, J.; Best, J.M.; Jordan, S.; Williams, G.T.; Sampson, J.R.; Cheadle, J.P. Biallelic germline mutations in MYH predispose to multiple colorectal adenoma and somatic G:C-->T:A mutations. Hum. Mol. Genet. 2002, 11, 2961–2967. [Google Scholar] [CrossRef]
- Sampson, J.R.; Dolwani, S.; Jones, S.; Eccles, D.; Ellis, A.; Evans, D.G.; Frayling, I.; Jordan, S.; Maher, E.R.; Mak, T.; et al. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet 2003, 362, 39–41. [Google Scholar] [CrossRef]
- Farrington, S.M.; Tenesa, A.; Barnetson, R.; Wiltshire, A.; Prendergast, J.; Porteous, M.; Campbell, H.; Dunlop, M.G. Germline susceptibility to colorectal cancer due to base-excision repair gene defects. Am. J. Hum. Genet. 2005, 77, 112–119. [Google Scholar] [CrossRef]
- Rennert, G.; Lejbkowicz, F.; Cohen, I.; Pinchev, M.; Rennert, H.S.; Barnett-Griness, O. MutYH mutation carriers have increased breast cancer risk. Cancer 2012, 118, 1989–1993. [Google Scholar] [CrossRef]
- Wasielewski, M.; Out, A.A.; Vermeulen, J.; Nielsen, M.; van den Ouweland, A.; Tops, C.M.; Wijnen, J.T.; Vasen, H.F.; Weiss, M.M.; Klijn, J.G.; et al. Increased MUTYH mutation frequency among Dutch families with breast cancer and colorectal cancer. Breast Cancer Res. Treat. 2010, 124, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Win, A.K.; Reece, J.C.; Dowty, J.G.; Buchanan, D.D.; Clendenning, M.; Rosty, C.; Southey, M.C.; Young, J.P.; Cleary, S.P.; Kim, H.; et al. Risk of extracolonic cancers for people with biallelic and monoallelic mutations in MUTYH. Int. J. Cancer 2016, 139, 1557–1563. [Google Scholar] [CrossRef] [PubMed]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [PubMed]
- Thibodeau, M.L.; Zhao, E.Y.; Reisle, C.; Ch’ng, C.; Wong, H.L.; Shen, Y.; Jones, M.R.; Lim, H.J.; Young, S.; Cremin, C.; et al. Base excision repair deficiency signatures implicate germline and somatic MUTYH aberrations in pancreatic ductal adenocarcinoma and breast cancer oncogenesis. Cold Spring Harb. Mol. Case Stud. 2019, 5, a003681. [Google Scholar] [CrossRef] [PubMed]
- Chaffee, K.G.; Oberg, A.L.; McWilliams, R.R.; Majithia, N.; Allen, B.A.; Kidd, J.; Singh, N.; Hartman, A.R.; Wenstrup, R.J.; Petersen, G.M. Prevalence of germ-line mutations in cancer genes among pancreatic cancer patients with a positive family history. Genet. Med. 2018, 20, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Asahara, H.; Patel, V.S.; Zhou, S.; Linn, S. Purification, cDNA cloning, and gene mapping of the small subunit of human DNA polymerase epsilon. J. Biol. Chem. 1997, 272, 32337–32344. [Google Scholar] [CrossRef] [PubMed]
- Pachlopnik Schmid, J.; Lemoine, R.; Nehme, N.; Cormier-Daire, V.; Revy, P.; Debeurme, F.; Debré, M.; Nitschke, P.; Bole-Feysot, C.; Legeai-Mallet, L.; et al. Polymerase ε1 mutation in a human syndrome with facial dysmorphism, immunodeficiency, livedo, and short stature (“FILS syndrome”). J. Exp. Med. 2012, 209, 2323–2330. [Google Scholar] [CrossRef]
- Thiffault, I.; Saunders, C.; Jenkins, J.; Raje, N.; Canty, K.; Sharma, M.; Grote, L.; Welsh, H.I.; Farrow, E.; Twist, G.; et al. A patient with polymerase E1 deficiency (POLE1): Clinical features and overlap with DNA breakage/instability syndromes. BMC Med. Genet. 2015. [CrossRef]
- Logan, C.V.; Murray, J.E.; Parry, D.A.; Robertson, A.; Bellelli, R.; Tarnauskaitė, Ž.; Challis, R.; Cleal, L.; Borel, V.; Fluteau, A.; et al. DNA Polymerase Epsilon Deficiency Causes IMAGe Syndrome with Variable Immunodeficiency. Am. J. Hum. Genet. 2018, 103, 1038–1044. [Google Scholar] [CrossRef]
- Palles, C.; Cazier, J.B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Salguero, I.; et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013, 45, 136–144. [Google Scholar] [CrossRef]
- Elsayed, F.A.; Kets, C.M.; Ruano, D.; van den Akker, B.; Mensenkamp, A.R.; Schrumpf, M.; Nielsen, M.; Wijnen, J.T.; Tops, C.M.; Ligtenberg, M.J.; et al. Germline variants in POLE are associated with early onset mismatch repair deficient colorectal cancer. Eur. J. Hum. Genet. 2015, 23, 1080–1084. [Google Scholar] [CrossRef] [PubMed]
- Bellido, F.; Pineda, M.; Aiza, G.; Valdés-Mas, R.; Navarro, M.; Puente, D.A.; Pons, T.; González, S.; Iglesias, S.; Darder, E.; et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: Review of reported cases and recommendations for genetic testing and surveillance. Genet. Med. 2016, 18, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Rohlin, A.; Zagoras, T.; Nilsson, S.; Lundstam, U.; Wahlström, J.; Hultén, L.; Martinsson, T.; Karlsson, G.B.; Nordling, M. A mutation in POLE predisposing to a multi-tumour phenotype. Int. J. Oncol. 2014, 45, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.F.; Johansen, J.; Bjørnevoll, I.; Sylvander, A.E.; Steinsbekk, K.S.; Sætrom, P.; Sandvik, A.K.; Drabløs, F.; Sjursen, W. A novel POLE mutation associated with cancers of colon, pancreas, ovaries and small intestine. Fam. Cancer 2015, 14, 437–448. [Google Scholar]
- Mur, P.; García-Mulero, S.; Del Valle, J.; Magraner-Pardo, L.; Vidal, A.; Pineda, M.; Cinnirella, G.; Martín-Ramos, E.; Pons, T.; López-Doriga, A.; et al. Role of POLE and POLD1 in familial cancer. Genet. Med. 2020, 22, 2089–2100. [Google Scholar] [CrossRef]

{kind=link}
| Genes | Gastric Cancer | Colorectal Cancer | Pancreatic Cancer | Breast Cancer | Ovarian Cancer | Multiple Primary Tumors | P/LP | Sum | Gender (Male/Female) | Age of Manifestation (Mean ± SD) |
|---|---|---|---|---|---|---|---|---|---|---|
| BRCA1 | 1 | 24 | 7 | 1 | 30/3 | 33 | 2/31 | 43.9 ± 11.2 | ||
| BRCA2 | 2 | 14 | 5 | 2 | 21/2 | 23 | 1/22 | 48.4 ± 11.6 | ||
| CHEK2 | 1 | 1 | 11 | 1 | 8/6 | 14 | 2/12 | 42.5 ± 12.4 | ||
| ATM | 1 | 3 | 5 | 7/2 | 9 | 2/7 | 43.6 ± 10.2 | |||
| PALB2 | 1 | 7 | 6/2 | 8 | 2/6 | 48.6 ± 8.2 | ||||
| MUTYH | 1 | 2 | 3 | 1 | 3/4 | 7 | 2/5 | 41.6 ± 10 | ||
| BLM | 5 | 1 | 7/0 | 6 | 2/5 | 44 ± 12.7 | ||||
| APC | 3 | 2 | 5/0 | 5 | 4/5 | 37.1 ± 14.7 | ||||
| NBN | 1 | 2 | 1 | 4/0 | 4 | 1/3 | 57 ± 8 | |||
| MLH1 | 3 | 1 | 3/1 | 4 | 0/4 | 39.2 ± 6.1 | ||||
| VHL | 1 | 3 | 0/4 | 4 | 0/4 | 41 ± 12.6 | ||||
| NTHL1 | 1 | 1 | 1 | 3/0 | 3 | 0/3 | 46.6 ± 6 | |||
| POLE | 1 | 1 | 1 | 0/3 | 3 | 0/3 | 32.3 ± 12.5 | |||
| TP53 | 3 | 2/1 | 3 | 0/3 | 37.6 ± 15.9 | |||||
| PMS2 | 1 | 1 | 2/0 | 2 | 2/0 | 49 ± 5.6 | ||||
| MSH3 | 1 | 1 | 2/0 | 2 | 1/1 | 37.5 ± 3.5 | ||||
| BARD1 | 2 | 2/0 | 2 | 0/2 | 44 ± 1.4 | |||||
| MSH2 | 1 | 1 | 1/1 | 2 | 1/1 | 50 ± 4.2 | ||||
| MEN1 | 1 | 0/1 | 1 | 1/0 | 47 | |||||
| MSH6 | 1 | 0/1 | 1 | 1/0 | 31 | |||||
| CDKN2A | 1 | 1/0 | 1 | 1/0 | 62 | |||||
| EPCAM | 1 | 0/1 | 1 | 0/1 | 49 | |||||
| TSC2 | 1 | 0/1 | 1 | 1/0 | 42 | |||||
| GALNT12 | 1 | 0/1 | 1 | 0/1 | 40 | |||||
| MLH3 | 1 | 0/1 | 1 | 0/1 | 54 | |||||
| BRIP1 | 1 | 1/0 | 1 | 0/1 | 37 |
| Gene | Transcript | Chromosomal Change | Coding | Protein | ACMG | Diagnosis |
|---|---|---|---|---|---|---|
| ATM | NM_000051.4 | chr11:108256317delTC | c.2227_2228del | p.Ser743ArgfsTer21 | LP | Pancreatic cancer |
| chr11:108315875insGCTGT | c.6060_6064dup | p.Gly2022AlafsTer27 | LP | Breast cancer | ||
| BRCA1 | NM_007294 | chr17:43070934insT | c.4980dup | p.Glu1661ArgfsTer18 | LP | Breast cancer |
| chr17:43094515delTT | c.1015_1016del | p.Lys339GlyfsTer6 | LP | Breast cancer | ||
| BRCA2 | NM_000059 | chr13:32336925insTT | c.2570_2571insTT | p.Arg858Ter | LP | Pancreatic cancer |
| chr13:32340800delATTA | c.6446_6449del | p.Ile2149LysfsTer18 | LP | Breast cancer | ||
| EPCAM | NM_002354 | chr2:47373571G>A | c.184+1G>A | - | LP | Breast cancer |
| GALNT12 | NM_024642 | chr9:98807865delCGCGCCCCGGGCGG | c.171_184del | p.Pro58AlafsTer42 | LP | Breast cancer |
| MLH1 | NM_000249 | chr3:36996662delGGAGGCC | c.160_166del | p.Gly54Ter | LP | Colorectal cancer |
| MLH3 | NM_001040108 | chr14:75048112delG | c.1544del | p.Pro515HisfsTer11 | LP | Ovarian cancer |
| MSH2 | NM_000251 | chr2:47414369delA | c.893del | p.Gln298ArgfsTer3 | LP | Ovarian cancer |
| chr2:47471032delA | c.1729del | p.Ile577LeufsTer13 | LP | Multiple primary tumors | ||
| MUTYH | NM_001048174 | chr1:45332310C>A | c.705G>T | p.Trp235Cys | LP | Pancreatic cancer |
| POLE | NM_006231 | chr12:132624986delCA | c.6665_6666del | p.Leu2222GlnfsTer81 | LP | Ovarian cancer |
| chr12:132676655T>C | c.802-2A>G | - | LP | Colorectal cancer | ||
| chr12:132677365G>A | c.799C>T | p.Pro267Ser | LP | Pancreatic cancer |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bilyalov, A.; Danishevich, A.; Nikolaev, S.; Vorobyov, N.; Abramov, I.; Pismennaya, E.; Terehova, S.; Kosilova, Y.; Primak, A.; Stanoevich, U.; et al. Novel Pathogenic Variants in Hereditary Cancer Syndromes in a Highly Heterogeneous Cohort of Patients: Insights from Multigene Analysis. Cancers 2024, 16, 85. https://doi.org/10.3390/cancers16010085
Bilyalov A, Danishevich A, Nikolaev S, Vorobyov N, Abramov I, Pismennaya E, Terehova S, Kosilova Y, Primak A, Stanoevich U, et al. Novel Pathogenic Variants in Hereditary Cancer Syndromes in a Highly Heterogeneous Cohort of Patients: Insights from Multigene Analysis. Cancers. 2024; 16(1):85. https://doi.org/10.3390/cancers16010085
Chicago/Turabian StyleBilyalov, Airat, Anastasiia Danishevich, Sergey Nikolaev, Nikita Vorobyov, Ivan Abramov, Ekaterina Pismennaya, Svetlana Terehova, Yuliya Kosilova, Anastasiia Primak, Uglesha Stanoevich, and et al. 2024. "Novel Pathogenic Variants in Hereditary Cancer Syndromes in a Highly Heterogeneous Cohort of Patients: Insights from Multigene Analysis" Cancers 16, no. 1: 85. https://doi.org/10.3390/cancers16010085