
Synergistic Suppression of NF1 Malignant Peripheral Nerve Sheath Tumor Cell Growth in Culture and Orthotopic Xenografts by Combinational Treatment with Statin and Prodrug Farnesyltransferase Inhibitor PAMAM G4 Dendrimers
 ,
,
Abstract
:Simple Summary
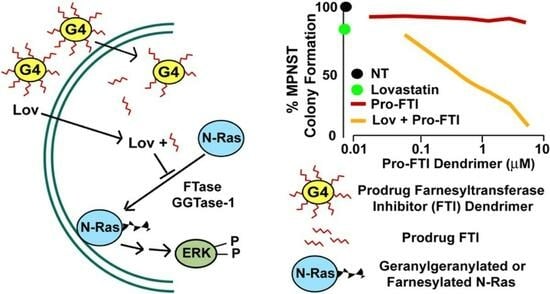
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture
2.3. DEVDase Activity Assay
2.4. Western Blot Analyses
2.5. Colony Formation Assays
2.6. MTT Assay
2.7. Murine Sciatic Nerve Xenograft Tumor Studies and MRI Analyses
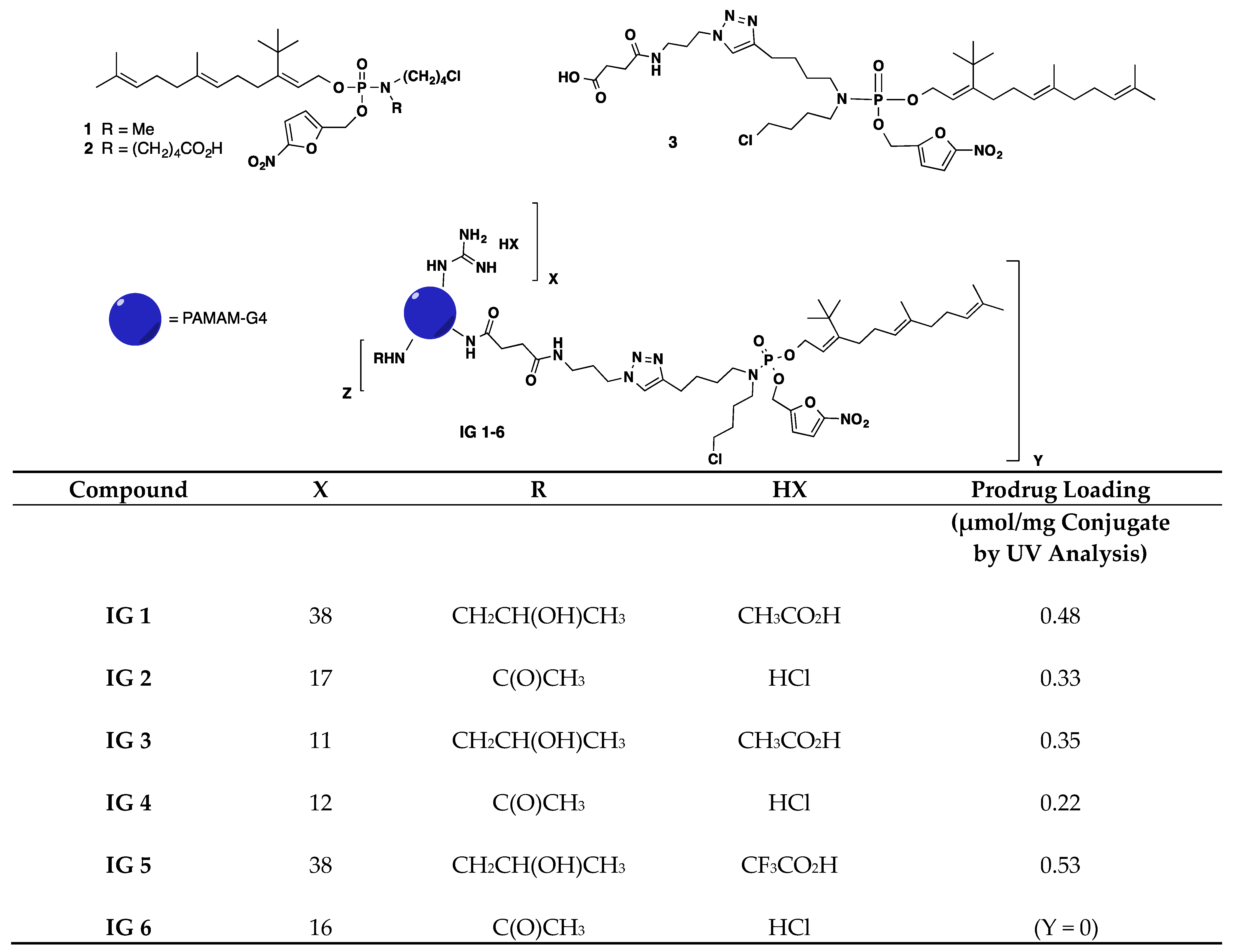
2.8. General Synthetic Methods
2.9. Analyses of Synergism and Statistical Significance
3. Results
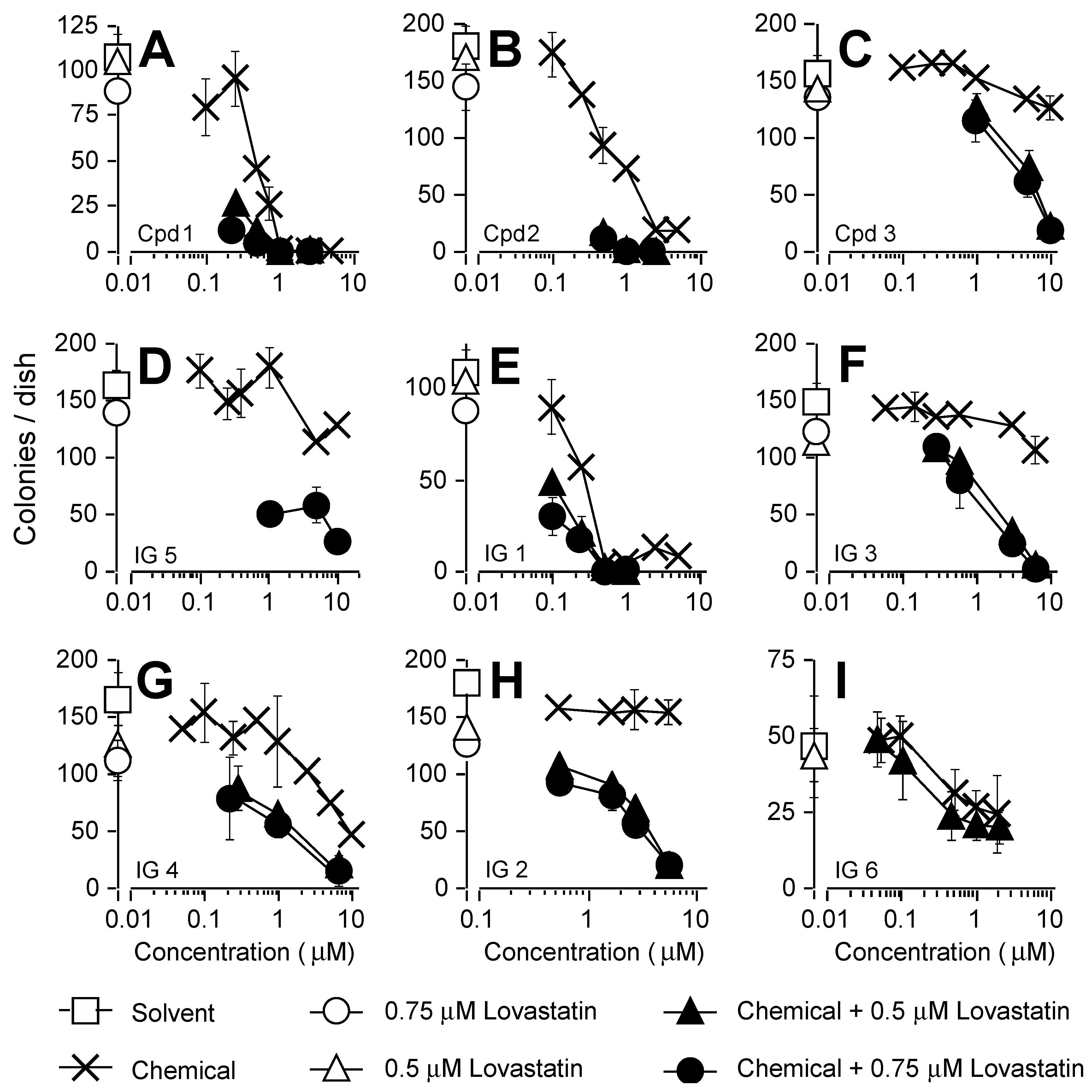
3.1. Antiproliferative Properties of Farnesyl Monophosphate Prodrug Dendrimer Analogs
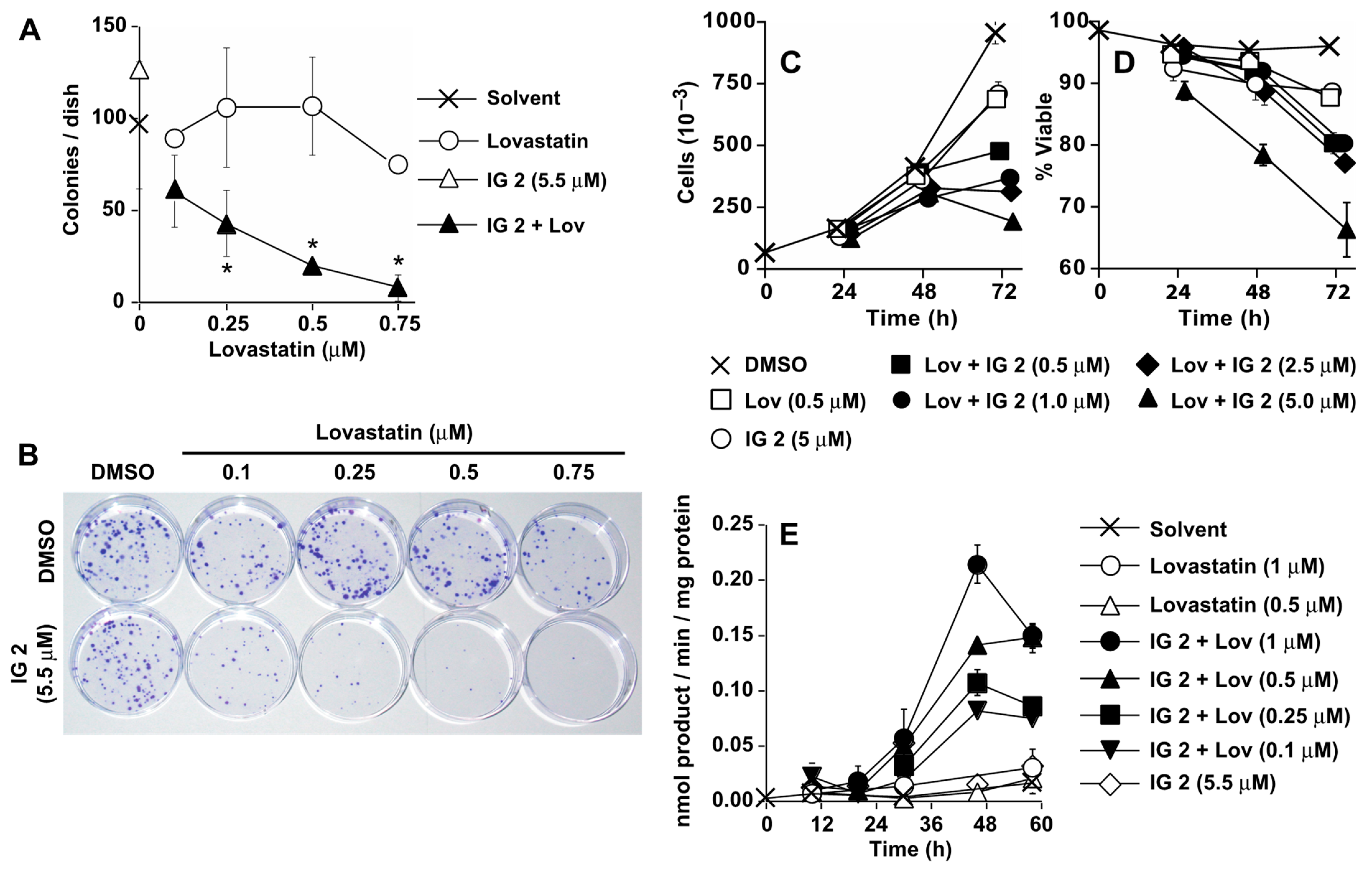
3.2. Analyses of IG 2 Effects on S462TY Proliferation, Viability, and Caspase Activation
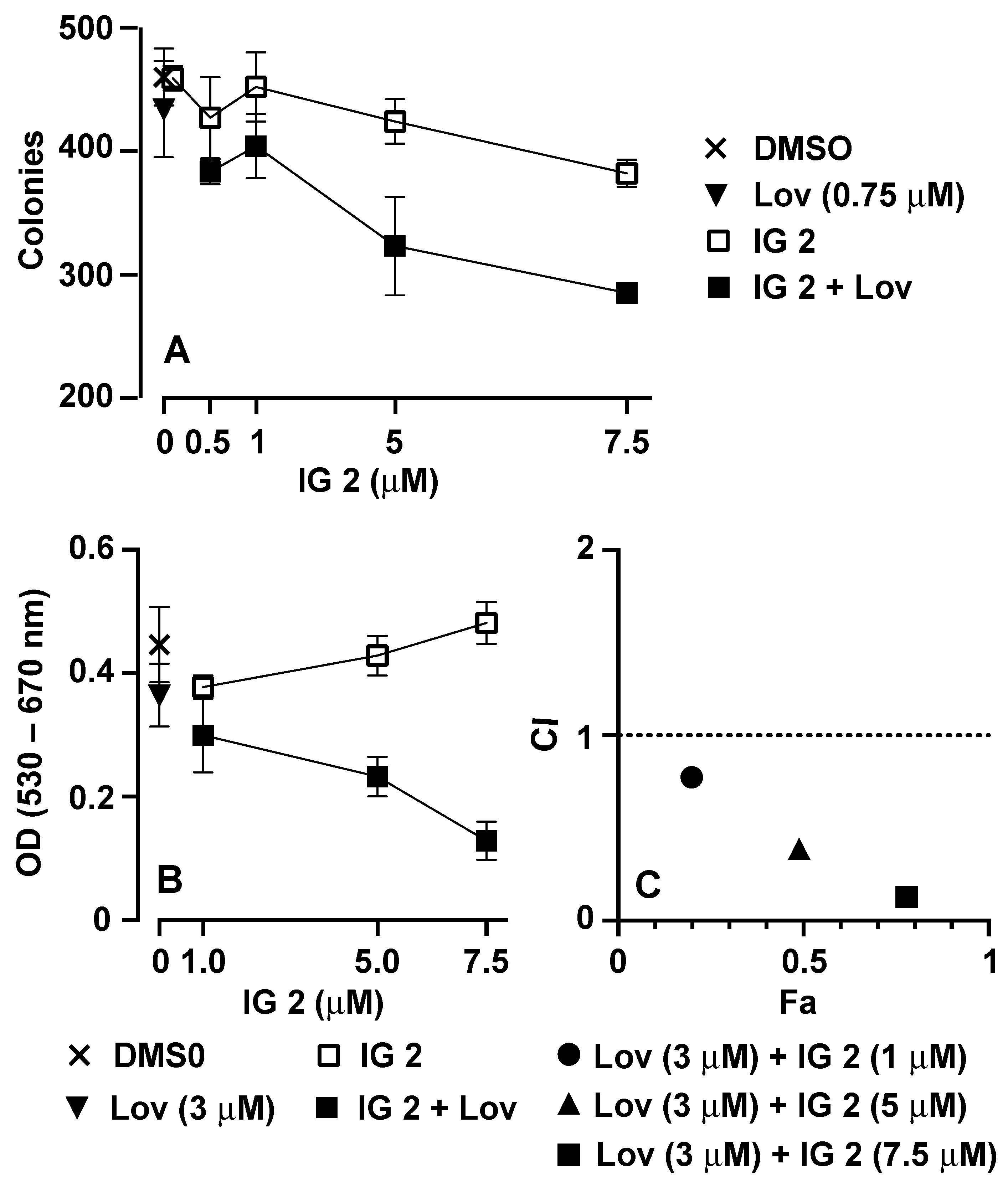
3.3. Analyses of Synergy in Co-Treatment Protocols with S469TY Cells
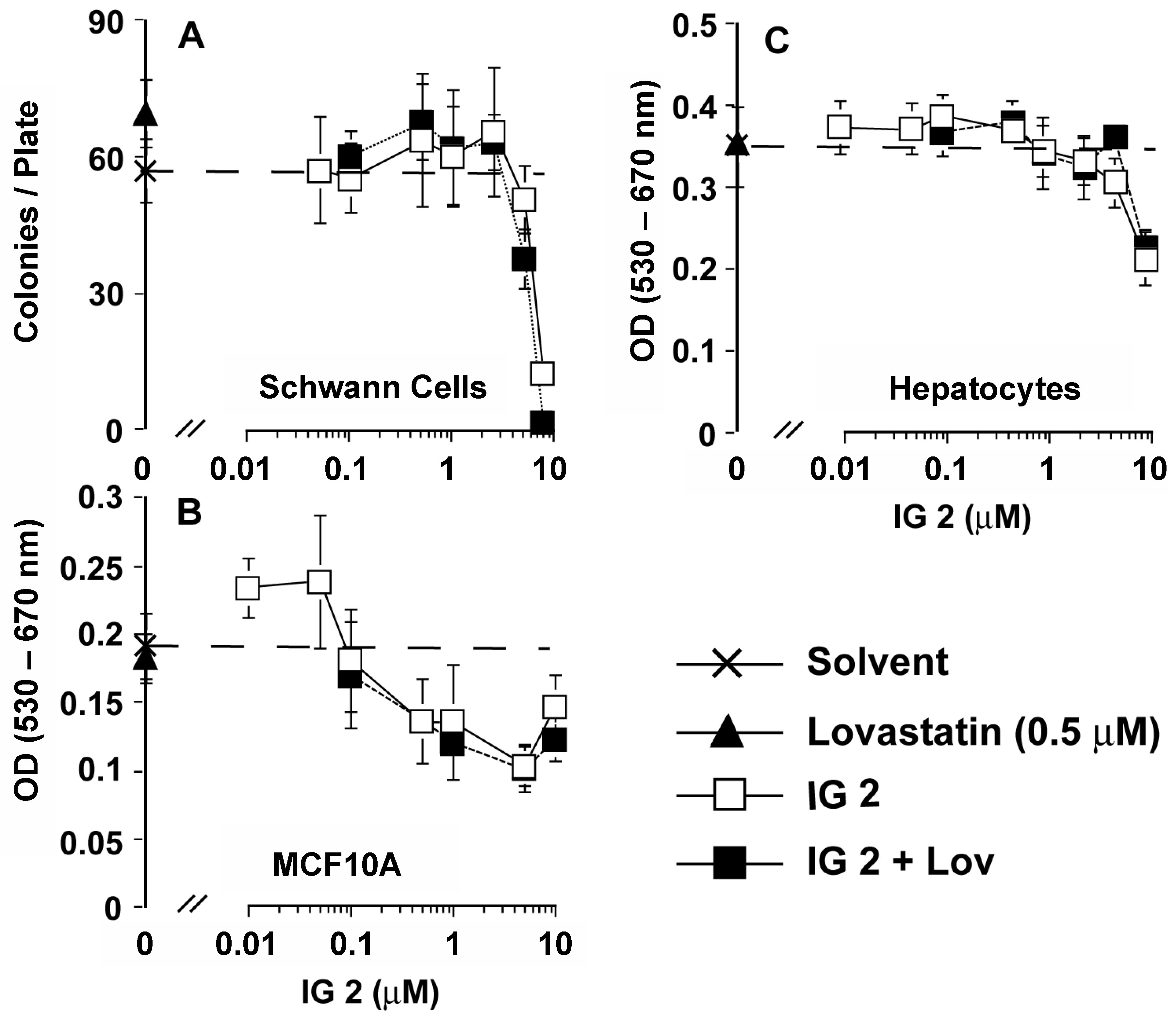
3.4. Analyses of Effects of Co-treatment on Proliferation/Viability in Other MPNST and Normal Cells
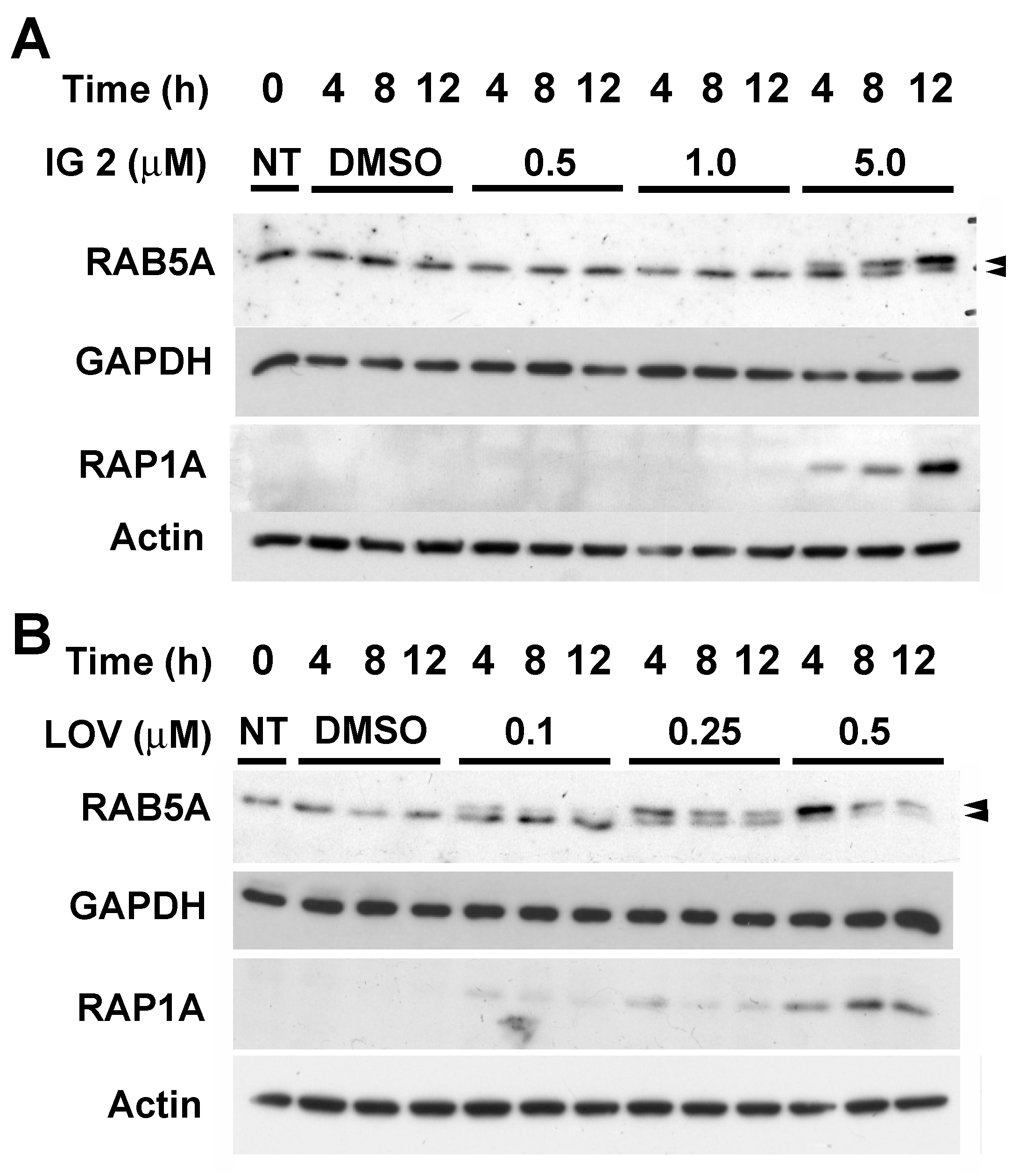
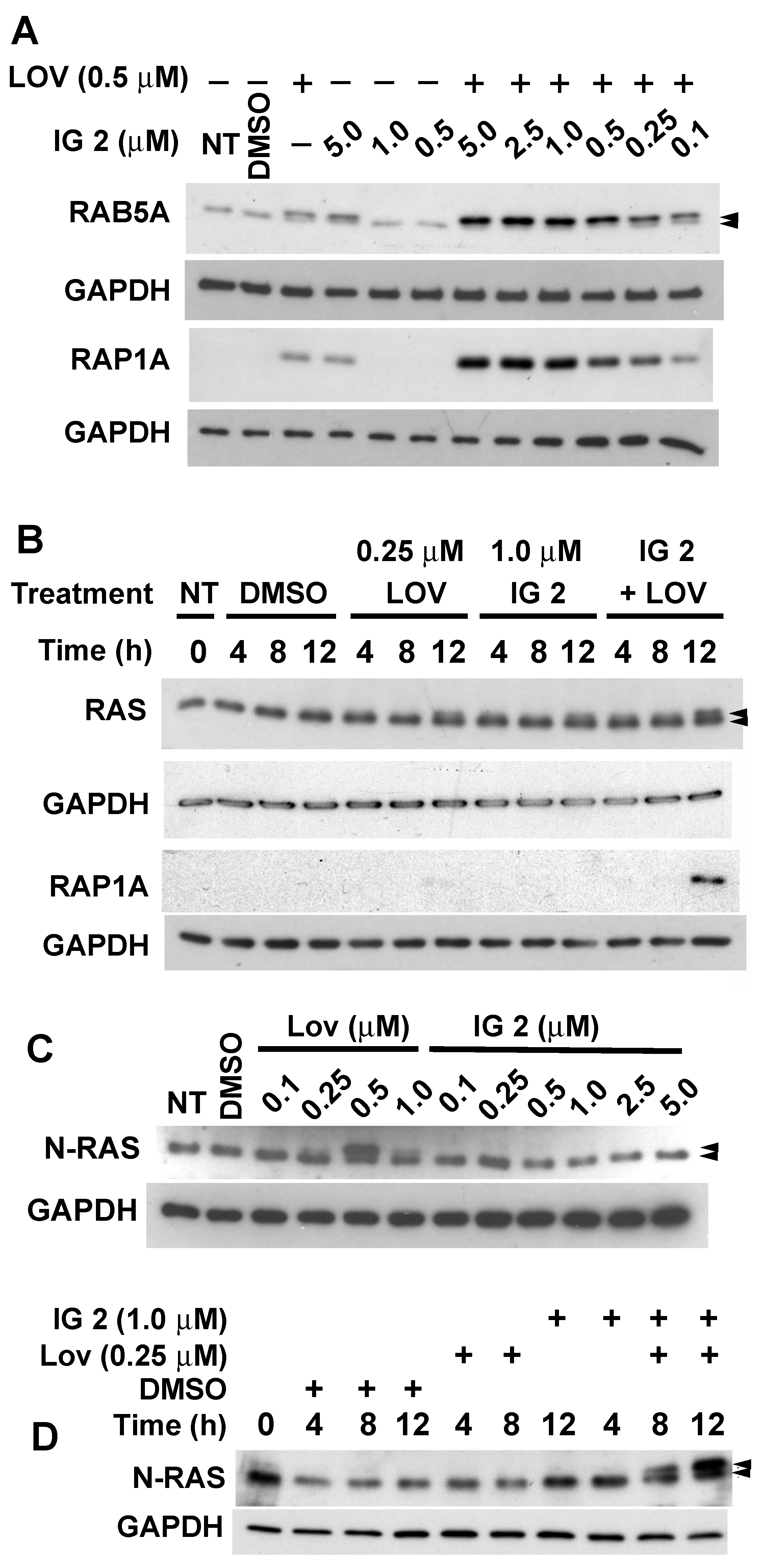
3.5. Lovastatin and FTI Prodrug Dendrimer Effects on Prenylation
3.6. In Vivo Toxicity Studies
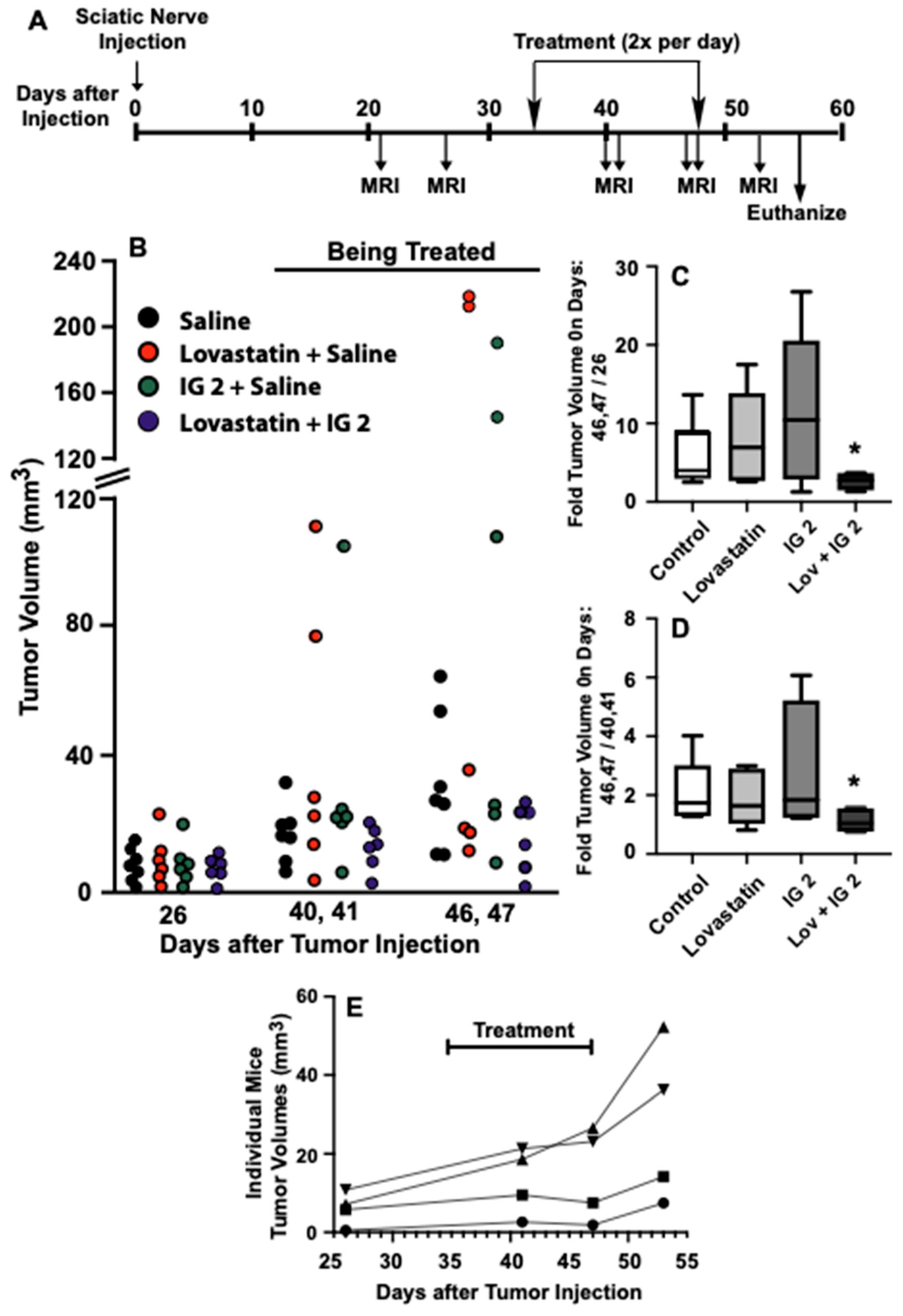
3.7. Orthotopic Sciatic Nerve S469TY Xenograft Studies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Friedman, J.M. Epidemiology of neurofibromatosis type 1. Am. J. Med. Genet. 1999, 89, 1–6. [Google Scholar] [CrossRef]
- Arun, D.; Gutmann, D.H. Recent advances in neurofibromatosis type 1. Curr. Opin. Neurol. 2004, 17, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.M.; Gutmann, D.H. Neurofibromatosis 1. Neurol. Clin. 2002, 20, 841–865. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.; Roth, K.A.; Banerjee, R.; Fuller, C.E.; Gutmann, D.H. NF1 deletions in S-100 protein-positive and negative cells of sporadic and neurofibromatosis 1 (NF1)-associated plexiform neurofibromas and malignant peripheral nerve sheath tumors. Am. J. Pathol. 2001, 159, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Ward, B.A.; Gutmann, D.H. Neurofibromatosis 1: From lab bench to clinic. Pediatr. Neurol. 2005, 32, 221–228. [Google Scholar] [CrossRef] [PubMed]
- DeClue, J.E.; Cohen, B.D.; Lowy, D.R. Identification and characterization of the neurofibromatosis type 1 protein product. Proc. Natl. Acad. Sci. USA 1991, 88, 9914–9918. [Google Scholar] [CrossRef] [PubMed]
- Parada, L.F. Neurofibromatosis type 1. Biochim. Biophys. Acta 2000, 1471, M13–M19. [Google Scholar] [CrossRef] [PubMed]
- Ballester, R.; Marchuk, D.; Boguski, M.; Saulino, A.; Letcher, R.; Wigler, M.; Collins, F. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell 1990, 63, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.F.; O’Connell, P.; Viskochil, D.; Cawthon, R.; Robertson, M.; Culver, M.; Dunn, D.; Stevens, J.; Gesteland, R.; White, R.; et al. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell 1990, 62, 599–608. [Google Scholar] [CrossRef]
- Feldkamp, M.M.; Angelov, L.; Guha, A. Neurofibromatosis type 1 peripheral nerve tumors: Aberrant activation of the Ras pathway. Surg. Neurol. 1999, 51, 211–218. [Google Scholar] [CrossRef]
- Lau, N.; Feldkamp, M.M.; Roncari, L.; Loehr, A.H.; Shannon, P.; Gutmann, D.H.; Guha, A. Loss of neurofibromin is associated with activation of RAS/MAPK and PI3-K/AKT signaling in a neurofibromatosis 1 astrocytoma. J. Neuropathol. Exp. Neurol. 2000, 59, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Hiatt, K.K.; Ingram, D.A.; Zhang, Y.; Bollag, G.; Clapp, D.W. Neurofibromin GTPase-activating protein-related domains restore normal growth in Nf1−/− cells. J. Biol. Chem. 2001, 276, 7240–7245. [Google Scholar] [CrossRef] [PubMed]
- Basso, A.D.; Kirschmeier, P.; Bishop, W.R. Lipid posttranslational modifications. Farnesyl transferase inhibitors. J. Lipid Res. 2006, 47, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Ricca, C.; Fletcher, J.; Glover, T.; Seizinger, B.R.; Manne, V. Farnesyltransferase inhibitors block the neurofibromatosis type I (NF1) malignant phenotype. Cancer Res. 1995, 55, 3569–3575. [Google Scholar] [PubMed]
- Kim, H.A.; Ling, B.; Ratner, N. Nf1-deficient mouse Schwann cells are angiogenic and invasive and can be induced to hyperproliferate: Reversion of some phenotypes by an inhibitor of farnesyl protein transferase. Mol. Cell Biol. 1997, 17, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Widemann, B.C.; Dombi, E.; Gillespie, A.; Wolters, P.L.; Belasco, J.; Goldman, S.; Korf, B.R.; Solomon, J.; Martin, S.; Salzer, W.; et al. Phase 2 randomized, flexible crossover, double-blinded, placebo-controlled trial of the farnesyltransferase inhibitor tipifarnib in children and young adults with neurofibromatosis type 1 and progressive plexiform neurofibromas. Neuro Oncol. 2014, 16, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Lerner, E.C.; Zhang, T.T.; Knowles, D.B.; Qian, Y.; Hamilton, A.D.; Sebti, S.M. Inhibition of the prenylation of K-Ras, but not H- or N-Ras, is highly resistant to CAAX peptidomimetics and requires both a farnesyltransferase and a geranylgeranyltransferase I inhibitor in human tumor cell lines. Oncogene 1997, 15, 1283–1288. [Google Scholar] [CrossRef]
- Whyte, D.B.; Kirschmeier, P.; Hockenberry, T.N.; Nunez-Oliva, I.; James, L.; Catino, J.J.; Bishop, W.R.; Pai, J.K. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J. Biol. Chem. 1997, 272, 14459–14464. [Google Scholar] [CrossRef]
- Mattingly, R.R.; Kraniak, J.M.; Dilworth, J.T.; Mathieu, P.; Bealmear, B.; Nowak, J.E.; Benjamins, J.A.; Tainsky, M.A.; Reiners, J.J., Jr. The mitogen-activated protein kinase/extracellular signal-regulated kinase kinase inhibitor PD184352 (CI-1040) selectively induces apoptosis in malignant schwannoma cell lines. J. Pharmacol. Exp. Ther. 2006, 316, 456–465. [Google Scholar] [CrossRef]
- Brossier, N.M.; Prechtl, A.M.; Longo, J.F.; Barnes, S.; Wilson, L.S.; Byer, S.J.; Brosius, S.N.; Carroll, S.L. Classic Ras Proteins Promote Proliferation and Survival via Distinct Phosphoproteome Alterations in Neurofibromin-Null Malignant Peripheral Nerve Sheath Tumor Cells. J. Neuropathol. Exp. Neurol. 2015, 74, 568–586. [Google Scholar] [CrossRef]
- Mattingly, R.R.; Gibbs, R.A.; Menard, R.E.; Reiners, J.J., Jr. Potent suppression of proliferation of a10 vascular smooth muscle cells by combined treatment with lovastatin and 3-allylfarnesol, an inhibitor of protein farnesyltransferase. J. Pharmacol. Exp. Ther. 2002, 303, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Wojtkowiak, J.W.; Gibbs, R.A.; Mattingly, R.R. Working together: Farnesyl transferase inhibitors and statins block protein prenylation. Mol. Cell Pharmacol. 2009, 1, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wojtkowiak, J.W.; Fouad, F.; LaLonde, D.T.; Kleinman, M.D.; Gibbs, R.A.; Reiners, J.J., Jr.; Borch, R.F.; Mattingly, R.R. Induction of apoptosis in neurofibromatosis type 1 malignant peripheral nerve sheath tumor cell lines by a combination of novel farnesyl transferase inhibitors and lovastatin. J. Pharmacol. Exp. Ther. 2008, 326, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jedrych, M.; Borowska, K.; Galus, R.; Jodlowska-Jedrych, B. The evaluation of the biomedical effectiveness of poly(amido)amine dendrimers generation 4.0 as a drug and as drug carriers: A systematic review and meta-analysis. Int. J. Pharm. 2014, 462, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Joiakim, A.; Dudley, D.T.; Reiners, J.J. Suppression of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-mediated CYP1A1 and CYP1B1 induction by 12-O-tetradecanoylphorbol-13-acetate: Role of transforming growth factor beta and mitogen-activated protein kinases. Biochem. Pharmacol. 2001, 62, 1449–1457. [Google Scholar] [CrossRef] [PubMed]
- Jackson, N.M.; Kocarek, T.A. Suppression of CYP2B induction by alendronate-mediated farnesyl diphosphate synthase inhibition in primary cultured rat hepatocytes. Drug Metab. Dispos. 2008, 36, 2030–2036. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Clark, M.K.; Scott, S.A.; Wojtkowiak, J.; Chirco, R.; Mathieu, P.; Reiners, J.J., Jr.; Mattingly, R.R.; Borch, R.F.; Gibbs, R.A. Synthesis, biochemical, and cellular evaluation of farnesyl monophosphate prodrugs as farnesyltransferase inhibitors. J. Med. Chem. 2007, 50, 3274–3282. [Google Scholar] [CrossRef]
- Wojtkowiak, J.W.; Sane, K.M.; Kleinman, M.; Sloane, B.F.; Reiners, J.J., Jr.; Mattingly, R.R. Aborted autophagy and nonapoptotic death induced by farnesyl transferase inhibitor and lovastatin. J. Pharmacol. Exp. Ther. 2011, 337, 65–74. [Google Scholar] [CrossRef]
- Duarte, D.; Vale, N. Evaluation of synergism in drug combinations and reference models for future orientations in oncology. Curr. Res. Pharmacol. Drug Discov. 2022, 3, 100110. [Google Scholar] [CrossRef]
- Maurer-Stroh, S.; Koranda, M.; Benetka, W.; Schneider, G.; Sirota, F.L.; Eisenhaber, F. Towards complete sets of farnesylated and geranylgeranylated proteins. PLoS Comput. Biol. 2007, 3, e66. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Naeem, A.; Xiao, S.; Hu, L.; Zhang, J.; Zheng, Q. Safety Challenges and Application Strategies for the Use of Dendrimers in Medicine. Pharmaceutics 2022, 14, 1292. [Google Scholar] [CrossRef] [PubMed]
- Janaszewska, A.; Lazniewska, J.; Trzepinski, P.; Marcinkowska, M.; Klajnert-Maculewicz, B. Cytotoxicity of Dendrimers. Biomolecules 2019, 9, 330. [Google Scholar] [CrossRef] [PubMed]
- Kisakova, L.A.; Apartsin, E.K.; Nizolenko, L.F.; Karpenko, L.I. Dendrimer-Mediated Delivery of DNA and RNA Vaccines. Pharmaceutics 2023, 15, 1106. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.Y.; DeVault, A.R.; Wang-Iverson, D.; Ivashkiv, E.; Swanson, B.N.; Sugerman, A.A. Comparative pharmacokinetics and pharmacodynamics of pravastatin and lovastatin. J. Clin. Pharmacol. 1990, 30, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.K.; Oza, A.M.; Siu, L.L. The statins as anticancer agents. Clin. Cancer Res. 2003, 9, 10–19. [Google Scholar] [PubMed]
- Zhang, H.; Seabra, M.C.; Deisenhofer, J. Crystal structure of Rab geranylgeranyltransferase at 2.0 A resolution. Structure 2000, 8, 241–251. [Google Scholar] [CrossRef]
- Adamson, P.; Marshall, C.J.; Hall, A.; Tilbrook, P.A. Post-translational modifications of p21rho proteins. J. Biol. Chem. 1992, 267, 20033–20038. [Google Scholar] [CrossRef]
- Armstrong, S.A.; Hannah, V.C.; Goldstein, J.L.; Brown, M.S. CAAX geranylgeranyl transferase transfers farnesyl as efficiently as geranylgeranyl to RhoB. J. Biol. Chem. 1995, 270, 7864–7868. [Google Scholar] [CrossRef]
- Yokoyama, K.; McGeady, P.; Gelb, M.H. Mammalian protein geranylgeranyltransferase-I: Substrate specificity, kinetic mechanism, metal requirements, and affinity labeling. Biochemistry 1995, 34, 1344–1354. [Google Scholar] [CrossRef]
- Owen, D.J.; Alexandrov, K.; Rostkova, E.; Scheidig, A.J.; Goody, R.S.; Waldmann, H. Chemo-Enzymatic Synthesis of Fluorescent Rab 7 Proteins: Tools to Study Vesicular Trafficking in Cells. Angew. Chem. Int. Ed. 1999, 38, 509–512. [Google Scholar] [CrossRef]
- Thoma, N.H.; Iakovenko, A.; Owen, D.; Scheidig, A.S.; Waldmann, H.; Goody, R.S.; Alexandrov, K. Phosphoisoprenoid binding specificity of geranylgeranyltransferase type II. Biochemistry 2000, 39, 12043–12052. [Google Scholar] [CrossRef] [PubMed]
- Lackner, M.R.; Kindt, R.M.; Carroll, P.M.; Brown, K.; Cancilla, M.R.; Chen, C.; de Silva, H.; Franke, Y.; Guan, B.; Heuer, T.; et al. Chemical genetics identifies Rab geranylgeranyl transferase as an apoptotic target of farnesyl transferase inhibitors. Cancer Cell 2005, 7, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Lobell, R.B.; Omer, C.A.; Abrams, M.T.; Bhimnathwala, H.G.; Brucker, M.J.; Buser, C.A.; Davide, J.P.; deSolms, S.J.; Dinsmore, C.J.; Ellis-Hutchings, M.S.; et al. Evaluation of farnesyl:protein transferase and geranylgeranyl:protein transferase inhibitor combinations in preclinical models. Cancer Res. 2001, 61, 8758–8768. [Google Scholar] [PubMed]
- Mellman, I.; Yarden, Y. Endocytosis and cancer. Cold Spring Harb. Perspect. Biol. 2013, 5, a016949. [Google Scholar] [CrossRef] [PubMed]
- Jung, N.H.; Egert-Schwender, S.; Schossow, B.; Kehl, V.; Wahllander, U.; Brich, L.; Janke, V.; Blankenstein, C.; Zenker, M.; Mall, V. Improvement of synaptic plasticity and cognitive function in RASopathies-a monocentre, randomized, double-blind, parallel-group, placebo-controlled, cross-over clinical trial (SynCoRAS). Trials 2023, 24, 383. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, N.J.; Payne, J.M.; Walsh, K.S.; Cutter, G.; Packer, R.; North, K.; Rey-Casserly, C.; Consortium, N.F.C.T. Visual spatial learning outcomes for clinical trials in neurofibromatosis type 1. Ann. Clin. Transl. Neurol. 2020, 7, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Payne, J.M.; Hearps, S.J.C.; Walsh, K.S.; Paltin, I.; Barton, B.; Ullrich, N.J.; Haebich, K.M.; Coghill, D.; Gioia, G.A.; Cantor, A.; et al. Reproducibility of cognitive endpoints in clinical trials: Lessons from neurofibromatosis type 1. Ann. Clin. Transl. Neurol. 2019, 6, 2555–2565. [Google Scholar] [CrossRef] [PubMed]
- van der Vaart, T.; Rietman, A.B.; Plasschaert, E.; Legius, E.; Elgersma, Y.; Moll, H.A.; Group, N.S.S. Behavioral and cognitive outcomes for clinical trials in children with neurofibromatosis type 1. Neurology 2016, 86, 154–160. [Google Scholar] [CrossRef]
- Mainberger, F.; Jung, N.H.; Zenker, M.; Wahllander, U.; Freudenberg, L.; Langer, S.; Berweck, S.; Winkler, T.; Straube, A.; Heinen, F.; et al. Lovastatin improves impaired synaptic plasticity and phasic alertness in patients with neurofibromatosis type 1. BMC Neurol. 2013, 13, 131. [Google Scholar] [CrossRef]
- Tilija Pun, N.; Jeong, C.-H. Statin as a Potential Chemotherapeutic Agent: Current Updates as a Monotherapy, Combination Therapy, and Treatment for Anti-Cancer Drug Resistance. Pharmaceuticals 2021, 14, 470. [Google Scholar] [CrossRef]
- Xia, L.; Ding, S.; Wang, X.; Zhang, X.; Zhu, L.; Zhang, H.; Li, H. Advances in ovarian cancer treatment using a combination of statins with other drugs. Front. Pharmacol. 2022, 13, 1048484. [Google Scholar] [CrossRef]
- Issat, T.; Nowis, D.; Bil, J.; Winiarska, M.; Jakobisiak, M.; Golab, J. Antitumor effects of the combination of cholesterol reducing drugs. Oncol. Rep. 2011, 26, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Zaky, M.Y.; Fan, C.; Zhang, H.; Sun, X.F. Unraveling the Anticancer Potential of Statins: Mechanisms and Clinical Significance. Cancers 2023, 15, 4787. [Google Scholar] [CrossRef] [PubMed]
- El-Refai, S.M.; Brown, J.D.; Arnold, S.M.; Black, E.P.; Leggas, M.; Talbert, J.C. Epidemiologic Analysis Along the Mevalonate Pathway Reveals Improved Cancer Survival in Patients Who Receive Statins Alone and in Combination with Bisphosphonates. JCO Clin. Cancer Inform. 2017, 1, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ruiz, C.; Morales, A.; Fernandez-Checa, J.C. Statins and protein prenylation in cancer cell biology and therapy. Anticancer Agents Med. Chem. 2012, 12, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Sarrabayrouse, G.; Pich, C.; Teiti, I.; Tilkin-Mariame, A.F. Regulatory properties of statins and rho gtpases prenylation inhibitiors to stimulate melanoma immunogenicity and promote anti-melanoma immune response. Int. J. Cancer 2017, 140, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Brock, E.J.; Ji, K.; Reiners, J.J.; Mattingly, R.R. How to Target Activated Ras Proteins: Direct Inhibition vs. Induced Mislocalization. Mini Rev. Med. Chem. 2016, 16, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.E.; Pleier, A.K. Intramolecular hydroamination of alkynes catalysed by late transition metals. J. Chem. Soc. Dalton Trans. 1999, 583–588. [Google Scholar] [CrossRef]
- Lu, B.; Li, C.; Zhang, L. Gold-Catalyzed Highly Regioselective Oxidation of C−C Triple Bonds without Acid Additives: Propargyl Moieties as Masked α,β-Unsaturated Carbonyls. J. Am. Chem. Soc. 2010, 132, 14070–14072. [Google Scholar] [CrossRef]
- Knor, S.; Modlinger, A.; Poethko, T.; Schottelius, M.; Wester, H.J.; Kessler, H. Synthesis of novel 1,4,7,10-tetraazacyclodecane-1,4,7,10-tetraacetic acid (DOTA) derivatives for chemoselective attachment to unprotected polyfunctionalized compounds. Chemistry 2007, 13, 6082–6090. [Google Scholar] [CrossRef]
- Theodossiou, T.A.; Pantos, A.; Tsogas, I.; Paleos, C.M. Guanidinylated dendritic molecular transporters: Prospective drug delivery systems and application in cell transfection. ChemMedChem 2008, 3, 1635–1643. [Google Scholar] [CrossRef]
- Feichtinger, K.; Zapf, C.; Sings, H.L.; Goodman, M. Diprotected Triflylguanidines: A New Class of Guanidinylation Reagents. J. Org. Chem. 1998, 63, 3804–3805. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reiners, J.J., Jr.; Mathieu, P.A.; Gargano, M.; George, I.; Shen, Y.; Callaghan, J.F.; Borch, R.F.; Mattingly, R.R. Synergistic Suppression of NF1 Malignant Peripheral Nerve Sheath Tumor Cell Growth in Culture and Orthotopic Xenografts by Combinational Treatment with Statin and Prodrug Farnesyltransferase Inhibitor PAMAM G4 Dendrimers. Cancers 2024, 16, 89. https://doi.org/10.3390/cancers16010089
Reiners JJ Jr., Mathieu PA, Gargano M, George I, Shen Y, Callaghan JF, Borch RF, Mattingly RR. Synergistic Suppression of NF1 Malignant Peripheral Nerve Sheath Tumor Cell Growth in Culture and Orthotopic Xenografts by Combinational Treatment with Statin and Prodrug Farnesyltransferase Inhibitor PAMAM G4 Dendrimers. Cancers. 2024; 16(1):89. https://doi.org/10.3390/cancers16010089
Chicago/Turabian StyleReiners, John J., Jr., Patricia A. Mathieu, Mary Gargano, Irene George, Yimin Shen, John F. Callaghan, Richard F. Borch, and Raymond R. Mattingly. 2024. "Synergistic Suppression of NF1 Malignant Peripheral Nerve Sheath Tumor Cell Growth in Culture and Orthotopic Xenografts by Combinational Treatment with Statin and Prodrug Farnesyltransferase Inhibitor PAMAM G4 Dendrimers" Cancers 16, no. 1: 89. https://doi.org/10.3390/cancers16010089