
Microglia in Glioblastomas: Molecular Insight and Immunotherapeutic Potential


Abstract
Simple Summary
Abstract
1. Introduction
2. Role of Microglia in Normal Brain Function
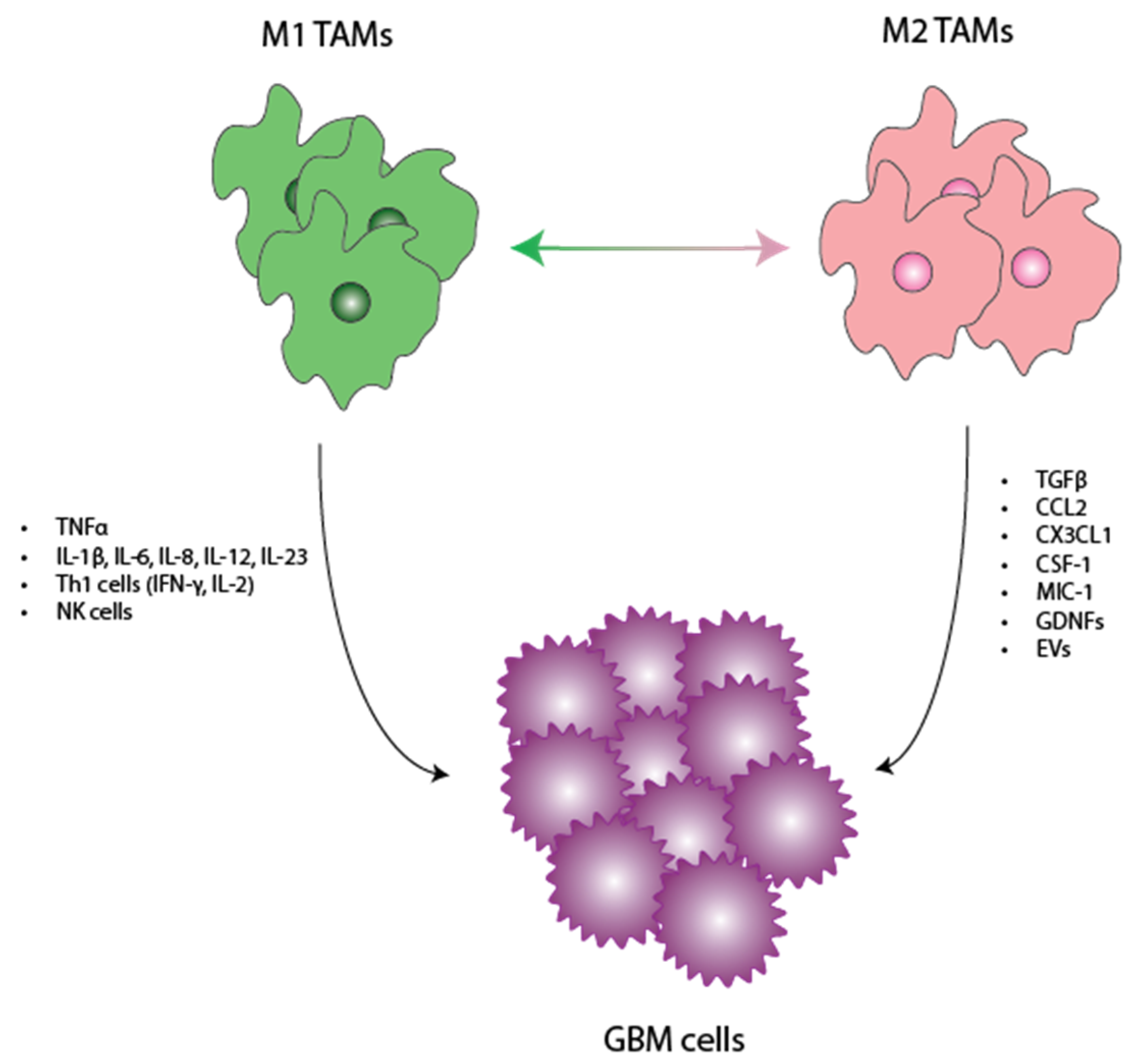
3. Role of Microglia in Glioma Progression
3.1. Tumor-Promoting Role of Microglia
3.1.1. CCL2 and CX3CL1
3.1.2. CSF1 and CSF2
3.1.3. MIC-1
3.1.4. GDNF
3.1.5. Extracellular Vesicles (EVs)
3.2. Tumor-Inhibiting Role of Microglia
4. Targeting Microglia in Novel Immunotherapeutics for Glioblastoma

{kind=link}
| Therapeutic | Targeted Pathway | Type of Studies | Outcome |
|---|---|---|---|
| Minocycline (Tetracycline analog) | Inhibition of M1 polarization [62,63] |
|
|
| PLX3397 | CSF1R inhibition and subsequent microglia depletion |
|
|
| PF-04136309 | CCR2 inhibition |
| |
| C1142 | Chimeric monoclonal antibody that neutralizes CCL2 |
|
|
| Hu5F9-G4 | Antibody that neutralizes CD47 |
|
|
| CpG-Stat3 siRNA | Inhibit STAT3, which is needed for GSC maintenance and regulates TLR9 |
|
|
| WP1066 | Inhibit STAT3 |
|
|
| Peptide R | CXCR4 antagonist |
|
|
| Vanucizumab | Bispecific neutralizing antibody targeting angiopoietin-2 + VEGF |
|
|
5. Conclusions and Future Directions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Nomenclature
| Term | Abbreviation |
| Glioblastoma | GBM |
| World Health Organization | WHO |
| Temozolomide | TMZ |
| Central nervous system | CNS |
| Central nervous system-associated macrophages | CAMs |
| Tumor-associated macrophages | TAMs |
| Glial cell line-derived neurotrophic factor | GDNF |
| G-protein coupled receptors | GPCRs |
| Major histocompatibility complex | MHC |
| Interleukin | IL |
| Tumor necrosis factor alpha | TNFα |
| Macrophage inflammatory protein | MIP |
| Monocyte chemoattractant protein | MCP |
| Insulin growth factor 1 | IGF-1 |
| Alzheimer’s disease | AD |
| Parkinson’s disease | PD |
| Regulatory T-cells | Tregs |
| Myeloid-derived suppressor cells | MDSCs |
| Matrix metalloprotein | MMP |
| C-C motif chemokine ligand 2 | CCL2 |
| C-C motif chemokine receptor 2 | CCR2 |
| C-X3-C motif chemokine ligand 1 | CX3CL1 |
| C-X3-C motif chemokine receptor 1 | CX3CR1 |
| Colony-stimulating factor 1 | CSF1 |
| Macrophage inhibitory cytokine-1 | MIC-1 |
| Extracellular vesicles | EV |
| GBM-derived stem cells | GSC |
| microRNA | miRNA |
| T helper 1 | Th1 |
| Natural killer | NK |
| Toll-like receptors | TLRs |
| Vascular endothelial growth factor | VEGF |
References
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma Multiforme: A Review of Its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma; Exon Publications: Brisbane City, Australia, 2017; pp. 143–153. [Google Scholar] [CrossRef]
- Hertler, C.; Felsberg, J.; Gramatzki, D.; Le Rhun, E.; Clarke, J.; Soffietti, R.; Wick, W.; Chinot, O.; Ducray, F.; Roth, P.; et al. Long-Term Survival with IDH Wildtype Glioblastoma: First Results from the ETERNITY Brain Tumor Funders’ Collaborative Consortium (EORTC 1419). Eur. J. Cancer 2023, 189, 112913. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shui, X.; Sun, R.; Wan, L.; Zhang, B.; Xiao, B.; Luo, Z. Microglial Phenotypic Transition: Signaling Pathways and Influencing Modulators Involved in Regulation in Central Nervous System Diseases. Front. Cell. Neurosci. 2021, 15, 736310. [Google Scholar] [CrossRef] [PubMed]
- Bingle, L.; Brown, N.J.; Lewis, C.E. The Role of Tumour-Associated Macrophages in Tumour Progression: Implications for New Anticancer Therapies. J. Pathol. 2002, 196, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Dumas, A.A.; Pomella, N.; Rosser, G.; Guglielmi, L.; Vinel, C.; Millner, T.O.; Rees, J.; Aley, N.; Sheer, D.; Wei, J.; et al. Microglia Promote Glioblastoma via MTOR-Mediated Immunosuppression of the Tumour Microenvironment. EMBO J. 2020, 39, e103790. [Google Scholar] [CrossRef]
- Ku, M.C.; Wolf, S.A.; Respondek, D.; Matyash, V.; Pohlmann, A.; Waiczies, S.; Waiczies, H.; Niendorf, T.; Synowitz, M.; Glass, R.; et al. GDNF Mediates Glioblastoma-Induced Microglia Attraction but Not Astrogliosis. Acta Neuropathol. 2013, 125, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Popovich, P.G.; Longbrake, E.E. Can the Immune System Be Harnessed to Repair the CNS? Nat. Rev. Neurosci. 2008, 9, 481–493. [Google Scholar] [CrossRef]
- Li, W.W.; Setzu, A.; Zhao, C.; Franklin, R.J.M. Minocycline-Mediated Inhibition of Microglia Activation Impairs Oligodendrocyte Progenitor Cell Responses and Remyelination in a Non-Immune Model of Demyelination. J. Neuroimmunol. 2005, 158, 58–66. [Google Scholar] [CrossRef]
- Burnstock, G.; Krügel, U.; Abbracchio, M.P.; Illes, P. Purinergic Signalling: From Normal Behaviour to Pathological Brain Function. Prog. Neurobiol. 2011, 95, 229–274. [Google Scholar] [CrossRef]
- Inoue, K. Purinergic Systems in Microglia. Cell. Mol. Life Sci. 2008, 65, 3074–3080. [Google Scholar] [CrossRef] [PubMed]
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.B.; Julius, D. The P2Y12 Receptor Regulates Microglial Activation by Extracellular Nucleotides. Nat. Neurosci. 2006, 9, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- Bilimoria, P.M.; Stevens, B. Microglia Function during Brain Development: New Insights from Animal Models. Brain Res. 2015, 1617, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.C.; Hu, S.; Peterson, P.K. Modulation of Human Microglial Cell Superoxide Production by Cytokines. J. Leukoc. Biol. 1995, 58, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Sierra, A.; Abiega, O.; Shahraz, A.; Neumann, H.; Leist, M.; Linden, R.; Basque, A. Janus-Faced Microglia: Beneficial and Detrimental Consequences of Microglial Phagocytosis. Front. Cell. Neurosci. 2013, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Podleśny-Drabiniok, A.; Marcora, E.; Goate, A.M. Microglial Phagocytosis: A Disease-Associated Process Emerging from Alzheimer’s Disease Genetics. Trends Neurosci. 2020, 43, 965–979. [Google Scholar] [CrossRef]
- Jha, M.K.; Lee, W.H.; Suk, K. Functional Polarization of Neuroglia: Implications in Neuroinflammation and Neurological Disorders. Biochem. Pharmacol. 2016, 103, 1–16. [Google Scholar] [CrossRef]
- Streit, W.J.; Xue, Q.S.; Tischer, J.; Bechmann, I. Microglial Pathology. Acta Neuropathol. Commun. 2014, 2, 142. [Google Scholar] [CrossRef]
- Zhang, B.; Wei, Y.Z.; Wang, G.Q.; Li, D.D.; Shi, J.S.; Zhang, F. Targeting MAPK Pathways by Naringenin Modulates Microglia M1/M2 Polarization in Lipopolysaccharide-Stimulated Cultures. Front. Cell. Neurosci. 2019, 12, 531. [Google Scholar] [CrossRef]
- Frank-Cannon, T.C.; Alto, L.T.; McAlpine, F.E.; Tansey, M.G. Does Neuroinflammation Fan the Flame in Neurodegenerative Diseases? Mol. Neurodegener. 2009, 4, 47. [Google Scholar] [CrossRef] [PubMed]
- Song, G.J.; Suk, K. Pharmacological Modulation of Functional Phenotypes of Microglia in Neurodegenerative Diseases. Front. Aging Neurosci. 2017, 9, 139. [Google Scholar] [CrossRef] [PubMed]
- Correale, J. The Role of Microglial Activation in Disease Progression. Mult. Scler. 2014, 20, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Puchalski, R.B.; Shah, N.; Miller, J.; Dalley, R.; Nomura, S.R.; Yoon, J.G.; Smith, K.A.; Lankerovich, M.; Bertagnolli, D.; Bickley, K.; et al. An Anatomic Transcriptional Atlas of Human Glioblastoma. Science 2018, 360, 660–663. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.B.; Yagnik, G.; Di Lullo, E.; Malatesta, M.; et al. Single-Cell Profiling of Human Gliomas Reveals Macrophage Ontogeny as a Basis for Regional Differences in Macrophage Activation in the Tumor Microenvironment. Genome Biol. 2017, 18, 234. [Google Scholar] [CrossRef] [PubMed]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef] [PubMed]
- Bouhlel, M.A.; Derudas, B.; Rigamonti, E.; Dièvart, R.; Brozek, J.; Haulon, S.; Zawadzki, C.; Jude, B.; Torpier, G.; Marx, N.; et al. PPARγ Activation Primes Human Monocytes into Alternative M2 Macrophages with Anti-Inflammatory Properties. Cell Metab. 2007, 6, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; Dechant, C.; Harrison Farber, S.; Elsamadicy, A.A.; Cui, X.; Koyama, S.; Jackson, C.; et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin. Cancer Res. 2018, 24, 4175–4186. [Google Scholar] [CrossRef] [PubMed]
- Woroniecka, K.I.; Rhodin, K.E.; Chongsathidkiet, P.; Keith, K.A.; Fecci, P.E. T-Cell Dysfunction in Glioblastoma: Applying a New Framework. Clin. Cancer Res. 2018, 24, 3792–3802. [Google Scholar] [CrossRef]
- Quintero-Fabián, S.; Arreola, R.; Becerril-Villanueva, E.; Torres-Romero, J.C.; Arana-Argáez, V.; Lara-Riegos, J.; Ramírez-Camacho, M.A.; Alvarez-Sánchez, M.E. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front. Oncol. 2019, 9, 1370. [Google Scholar] [CrossRef]
- Wang, G.; Zhong, K.; Wang, Z.; Zhang, Z.; Tang, X.; Tong, A.; Zhou, L. Tumor-Associated Microglia and Macrophages in Glioblastoma: From Basic Insights to Therapeutic Opportunities. Front. Immunol. 2022, 13, 964898. [Google Scholar] [CrossRef] [PubMed]
- Sincevičiūtė, R.; Vaitkienė, P.; Urbanavičiūtė, R.; Steponaitis, G.; Tamašauskas, A.; Skiriutė, D. MMP2 Is Associated with Glioma Malignancy and Patient Outcome. Int. J. Clin. Exp. Pathol. 2018, 11, 3010. [Google Scholar] [PubMed]
- Komohara, Y.; Horlad, H.; Ohnishi, K.; Fujiwara, Y.; Bai, B.; Nakagawa, T.; Suzu, S.; Nakamura, H.; Kuratsu, J.I.; Takeya, M. Importance of Direct Macrophage-Tumor Cell Interaction on Progression of Human Glioma. Cancer Sci. 2012, 103, 2165–2172. [Google Scholar] [CrossRef] [PubMed]
- Prosniak, M.; Harshyne, L.A.; Andrews, D.W.; Kenyon, L.C.; Bedelbaeva, K.; Apanasovich, T.V.; Heber-Katz, E.; Curtis, M.T.; Cotzia, P.; Hooper, D.C. Glioma Grade Is Associated with the Accumulation and Activity of Cells Bearing M2 Monocyte Markers. Clin. Cancer Res. 2013, 19, 3776–3786. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, M.D.; Dahlrot, R.H.; Boldt, H.B.; Hansen, S.; Kristensen, B.W. Tumour-Associated Microglia/Macrophages Predict Poor Prognosis in High-Grade Gliomas and Correlate with an Aggressive Tumour Subtype. Neuropathol. Appl. Neurobiol. 2018, 44, 185–206. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Xu, S. Tumor-Associated CD204-Positive Macrophage Is a Prognostic Marker in Clinical Stage I Lung Adenocarcinoma. Biomed. Res. Int. 2018, 2018, 8459193. [Google Scholar] [CrossRef] [PubMed]
- Déry, L.; Charest, G.; Guérin, B.; Akbari, M.; Fortin, D. Chemoattraction of Neoplastic Glial Cells with CXCL10, CCL2 and CCL11 as a Paradigm for a Promising Therapeutic Approach for Primary Brain Tumors. Int. J. Mol. Sci. 2021, 22, 12150. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.L.; Miska, J.; Wainwright, D.A.; Dey, M.; Rivetta, C.V.; Yu, D.; Kanojia, D.; Pituch, K.C.; Qiao, J.; Pytel, P.; et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer Res. 2016, 76, 5671–5682. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sarkar, S.; Cua, R.; Zhou, Y.; Hader, W.; Wee Yong, V. A Dialog between Glioma and Microglia That Promotes Tumor Invasiveness through the CCL2/CCR2/Interleukin-6 Axis. Carcinogenesis 2012, 33, 312–319. [Google Scholar] [CrossRef]
- Sciumé, G.; Soriani, A.; Piccoli, M.; Frati, L.; Santoni, A.; Bernardini, G. CX3CR1/CX3CL1 Axis Negatively Controls Glioma Cell Invasion and Is Modulated by Transforming Growth Factor-Β1. Neuro Oncol. 2010, 12, 701–710. [Google Scholar] [CrossRef]
- Lee, S.; Latha, K.; Manyam, G.; Yang, Y.; Rao, A.; Rao, G. Role of CX3CR1 Signaling in Malignant Transformation of Gliomas. Neuro Oncol. 2020, 22, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- De, I.; Steffen, M.D.; Clark, P.A.; Patros, C.J.; Sokn, E.; Bishop, S.M.; Litscher, S.; Maklakova, V.I.; Kuo, J.S.; Rodriguez, F.J.; et al. CSF1 Overexpression Promotes High-Grade Glioma Formation without Impacting the Polarization Status of Glioma-Associated Microglia and Macrophages. Cancer Res. 2016, 76, 2552–2560. [Google Scholar] [CrossRef]
- Komohara, Y.; Ohnishi, K.; Kuratsu, J.; Takeya, M. Possible Involvement of the M2 Anti-Inflammatory Macrophage Phenotype in Growth of Human Gliomas. J. Pathol. 2008, 216, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Wei, J.; Kong, L.Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma Cancer Stem Cells Induce Immunosuppressive Macrophages/Microglia. Neuro Oncol. 2010, 12, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Shnaper, S.; Desbaillets, I.; Brown, D.A.; Murat, A.; Migliavacca, E.; Schluep, M.; Ostermann, S.; Hamou, M.F.; Stupp, R.; Breit, S.N.; et al. Elevated Levels of MIC-1/GDF15 in the Cerebrospinal Fluid of Patients Are Associated with Glioblastoma and Worse Outcome. Int. J. Cancer 2009, 125, 2624–2630. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Walling, J.; Ahn, S.; Kotliarov, Y.; Su, Q.; Quezado, M.; Oberholtzer, J.C.; Park, J.; Zenklusen, J.C.; Fine, H.A. Unsupervised Analysis of Transcriptomic Profiles Reveals Six Glioma Subtypes. Cancer Res. 2009, 69, 2091–2099. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Wang, S.; Guo, X.; Wang, J.; Zhang, Z.; Qiu, W.; Gao, X.; Chen, Z.; Xu, J.; Zhao, R.; et al. Hypoxic Glioma-Derived Exosomes Deliver MicroRNA-1246 to Induce M2 Macrophage Polarization by Targeting TERF2IP via the STAT3 and NF-ΚB Pathways. Oncogene 2020, 39, 428–442. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Xu, B.; Ren, J.; Liu, Z.; Cai, L.; Zhang, X.; Wang, W.; Li, S.; Jin, L.; Ding, L. The Importance of M1-and M2-Polarized Macrophages in Glioma and as Potential Treatment Targets. Brain Sci. 2023, 13, 1269. [Google Scholar] [CrossRef]
- Qiu, W.; Guo, X.; Li, B.; Wang, J.; Qi, Y.; Chen, Z.; Zhao, R.; Deng, L.; Qian, M.; Wang, S.; et al. Exosomal MiR-1246 from Glioma Patient Body Fluids Drives the Differentiation and Activation of Myeloid-Derived Suppressor Cells. Mol. Ther. 2021, 29, 3449–3464. [Google Scholar] [CrossRef]
- Yang, Q.; Guo, N.; Zhou, Y.; Chen, J.; Wei, Q.; Han, M. The Role of Tumor-Associated Macrophages (TAMs) in Tumor Progression and Relevant Advance in Targeted Therapy. Acta Pharm. Sin. B 2020, 10, 2156. [Google Scholar] [CrossRef]
- Georgieva, P.B.; Mathivet, T.; Alt, S.; Giese, W.; Riva, M.; Balcer, M.; Gerhardt, H. Long-Lived Tumor-Associated Macrophages in Glioma. Neurooncol. Adv. 2020, 2, vdaa127. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Iwakabe, K.; Sekimoto, M.; Ohmi, Y.; Yahata, T.; Nakui, M.; Sato, T.; Habu, S.; Tashiro, H.; Sato, M.; et al. Distinct Role of Antigen-Specific T Helper Type 1 (Th1) and Th2 Cells in Tumor Eradication In Vivo. J. Exp. Med. 1999, 190, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhou, H.; Xu, J.; Lu, Y.; Ji, X.; Yao, Y.; Chao, H.; Zhang, J.; Zhang, X.; Yao, S.; et al. Different T-Cell Subsets in Glioblastoma Multiforme and Targeted Immunotherapy. Cancer Lett. 2021, 496, 134–143. [Google Scholar] [CrossRef]
- Morimoto, T.; Nakazawa, T.; Maeoka, R.; Nakagawa, I.; Tsujimura, T.; Matsuda, R. Natural Killer Cell-Based Immunotherapy against Glioblastoma. Int. J. Mol. Sci. 2023, 24, 2111. [Google Scholar] [CrossRef] [PubMed]
- Gwalani, L.A.; Orange, J.S. Single Degranulations in NK Cells Can Mediate Target Cell Killing. J. Immunol. 2018, 200, 3231–3243. [Google Scholar] [CrossRef]
- Andersen, R.S.; Anand, A.; Harwood, D.S.L.; Kristensen, B.W. Tumor-Associated Microglia and Macrophages in the Glioblastoma Microenvironment and Their Implications for Therapy. Cancers 2021, 13, 4255. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The Role of Microglia and Macrophages in Glioma Maintenance and Progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef]
- Markovic, D.S.; Vinnakota, K.; van Rooijen, N.; Kiwit, J.; Synowitz, M.; Glass, R.; Kettenmann, H. Minocycline Reduces Glioma Expansion and Invasion by Attenuating Microglial MT1-MMP Expression. Brain Behav. Immun. 2011, 25, 624–628. [Google Scholar] [CrossRef]
- Daginakatte, G.C.; Gutmann, D.H. Neurofibromatosis-1 (Nf1) Heterozygous Brain Microglia Elaborate Paracrine Factors That Promote Nf1-Deficient Astrocyte and Glioma Growth. Hum. Mol. Genet. 2007, 16, 1098–1112. [Google Scholar] [CrossRef]
- Toonen, J.A.; Solga, A.C.; Ma, Y.; Gutmann, D.H. Estrogen Activation of Microglia Underlies the Sexually Dimorphic Differences in Nf1 Optic Glioma-Induced Retinal Pathology. J. Exp. Med. 2017, 214, 17–25. [Google Scholar] [CrossRef]
- Abraham, J.; Fox, P.D.; Condello, C.; Bartolini, A.; Koh, S. Minocycline Attenuates Microglia Activation and Blocks the Long-Term Epileptogenic Effects of Early-Life Seizures. Neurobiol. Dis. 2012, 46, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Afshari, A.R.; Mollazadeh, H.; Sahebkar, A. Minocycline in Treating Glioblastoma Multiforme: Far beyond a Conventional Antibiotic. J. Oncol. 2020, 2020, 8659802. [Google Scholar] [CrossRef] [PubMed]
- Frazier, J.L.; Wang, P.P.; Case, D.; Tyler, B.M.; Pradilla, G.; Weingart, J.D.; Brem, H. Local Delivery of Minocycline and Systemic BCNU Have Synergistic Activity in the Treatment of Intracranial Glioma. J. Neurooncol 2003, 64, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R Inhibition Alters Macrophage Polarization and Blocks Glioma Progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Butowski, N.; Colman, H.; De Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally Administered Colony Stimulating Factor 1 Receptor Inhibitor PLX3397 in Recurrent Glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium Phase II Study. Neuro Oncol. 2016, 18, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Kuntzel, T.; Bagnard, D. Manipulating Macrophage/Microglia Polarization to Treat Glioblastoma or Multiple Sclerosis. Pharmaceutics 2022, 14, 344. [Google Scholar] [CrossRef] [PubMed]
- Noel, M.; O’Reilly, E.M.; Wolpin, B.M.; Ryan, D.P.; Bullock, A.J.; Britten, C.D.; Linehan, D.C.; Belt, B.A.; Gamelin, E.C.; Ganguly, B.; et al. Phase 1b Study of a Small Molecule Antagonist of Human Chemokine (C-C Motif) Receptor 2 (PF-04136309) in Combination with Nab-Paclitaxel/Gemcitabine in First-Line Treatment of Metastatic Pancreatic Ductal Adenocarcinoma. Investig. New Drugs 2020, 38, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Nywening, T.M.; Wang-Gillam, A.; Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; Worley, L.A.; Yano, M.; et al. Targeting Tumour-Associated Macrophages with CCR2 Inhibition in Combination with FOLFIRINOX in Patients with Borderline Resectable and Locally Advanced Pancreatic Cancer: A Single-Centre, Open-Label, Dose-Finding, Non-Randomised, Phase 1b Trial. Lancet Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Fujita, M.; Snyder, L.A.; Okada, H. Systemic Delivery of Neutralizing Antibody Targeting CCL2 for Glioma Therapy. J. Neurooncol 2011, 104, 83–92. [Google Scholar] [CrossRef]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D.; van Rooijen, N.; Weissman, I.L. CD47 Is an Adverse Prognostic Factor and Therapeutic Antibody Target on Human Acute Myeloid Leukemia Stem Cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef]
- Hutter, G.; Theruvath, J.; Graef, C.M.; Zhang, M.; Schoen, M.K.; Manz, E.M.; Bennett, M.L.; Olson, A.; Azad, T.D.; Sinha, R.; et al. Microglia Are Effector Cells of CD47-SIRPα Antiphagocytic Axis Disruption against Glioblastoma. Proc. Natl. Acad. Sci. USA 2019, 116, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, D.H.; Kettenmann, H. Microglia/Brain Macrophages as Central Drivers of Brain Tumor Pathobiology. Neuron 2019, 104, 442–449. [Google Scholar] [CrossRef]
- Li, F.; Lv, B.; Liu, Y.; Hua, T.; Han, J.; Sun, C.; Xu, L.; Zhang, Z.; Feng, Z.; Cai, Y.; et al. Blocking the CD47-SIRPα Axis by Delivery of Anti-CD47 Antibody Induces Antitumor Effects in Glioma and Glioma Stem Cells. Oncoimmunology 2017, 7, e1391973. [Google Scholar] [CrossRef]
- Gholamin, S.; Mitra, S.S.; Feroze, A.H.; Liu, J.; Kahn, S.A.; Zhang, M.; Esparza, R.; Richard, C.; Ramaswamy, V.; Remke, M.; et al. Disrupting the CD47-SIRPα Anti-Phagocytic Axis by a Humanized Anti-CD47 Antibody Is an Efficacious Treatment for Malignant Pediatric Brain Tumors. Sci. Transl. Med. 2017, 9, eaaf2968. [Google Scholar] [CrossRef]
- Hu, F.; Dzaye, O.D.A.; Hahn, A.; Yu, Y.; Scavetta, R.J.; Dittmar, G.; Kaczmarek, A.K.; Dunning, K.R.; Ricciardelli, C.; Rinnenthal, J.L.; et al. Glioma-Derived Versican Promotes Tumor Expansion via Glioma-Associated Microglial/Macrophages Toll-like Receptor 2 Signaling. Neuro Oncol. 2015, 17, 200–210. [Google Scholar] [CrossRef]
- Fehri, E.; Ennaifer, E.; Bel Haj Rhouma, R.; Ardhaoui, M.; Boubaker, S. TLR9 and Glioma: Friends or Foes? Cells 2022, 12, 152. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhang, Q.; Lubas, M.; Yuan, Y.; Yalcin, F.; Efe, I.E.; Xia, P.; Motta, E.; Buonfiglioli, A.; Lehnardt, S.; et al. Synergistic Toll-like Receptor 3/9 Signaling Affects Properties and Impairs Glioma-Promoting Activity of Microglia. J. Neurosci. 2020, 40, 6428–6443. [Google Scholar] [CrossRef]
- Reilly, M.; Miller, R.M.; Thomson, M.H.; Patris, V.; Ryle, P.; Mcloughlin, L.; Mutch, P.; Gilboy, P.; Miller, C.; Broekema, M.; et al. Randomized, Double-Blind, Placebo-Controlled, Dose-Escalating Phase I, Healthy Subjects Study of Intravenous OPN-305, a Humanized Anti-TLR2 Antibody. Clin. Pharmacol. Ther. 2013, 94, 593–600. [Google Scholar] [CrossRef]
- Tiwari, R.K.; Singh, S.; Gupta, C.L.; Pandey, P.; Singh, V.K.; Sayyed, U.; Shekh, R.; Bajpai, P. Repolarization of Glioblastoma Macrophage Cells Using Non-Agonistic Dectin-1 Ligand Encapsulating TLR-9 Agonist: Plausible Role in Regenerative Medicine against Brain Tumor. Int. J. Neurosci. 2021, 131, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Ginzkey, C.; Eicker, S.O.; Marget, M.; Krause, J.; Brecht, S.; Westphal, M.; Hugo, H.H.; Mehdorn, H.M.; Steinmann, J.; Hamel, W. Increase in Tumor Size Following Intratumoral Injection of Immunostimulatory CpG-Containing Oligonucleotides in a Rat Glioma Model. Cancer Immunol. Immunother. 2010, 59, 541–551. [Google Scholar] [CrossRef]
- Coniglio, S.J.; Eugenin, E.; Dobrenis, K.; Stanley, E.R.; West, B.L.; Symons, M.H.; Segall, J.E. Microglial Stimulation of Glioblastoma Invasion Involves Epidermal Growth Factor Receptor (EGFR) and Colony Stimulating Factor 1 Receptor (CSF-1R) Signaling. Mol. Med. 2012, 18, 519–527. [Google Scholar] [CrossRef]
- Patel, S.; Player, M. Colony-Stimulating Factor-1 Receptor Inhibitors for the Treatment of Cancer and Inflammatory Disease. Curr. Top. Med. Chem. 2009, 9, 599–610. [Google Scholar] [CrossRef]
- Quail, D.F.; Bowman, R.L.; Akkari, L.; Quick, M.L.; Schuhmacher, A.J.; Huse, J.T.; Holland, E.C.; Sutton, J.C.; Joyce, J.A. The Tumor Microenvironment Underlies Acquired Resistance to CSF-1R Inhibition in Gliomas. Science 2016, 352, aad3018. [Google Scholar] [CrossRef]
- Fujiwara, T.; Yakoub, M.A.; Chandler, A.; Christ, A.B.; Yang, G.; Ouerfelli, O.; Rajasekhar, V.K.; Yoshida, A.; Kondo, H.; Hata, T.; et al. CSF1/CSF1R Signaling Inhibitor Pexidartinib (PLX3397) Reprograms Tumor-Associated Macrophages and Stimulates T-Cell Infiltration in the Sarcoma Microenvironment. Mol. Cancer Ther. 2021, 20, 1388–1399. [Google Scholar] [CrossRef]
- Mercurio, L.; Ajmone-Cat, M.A.; Cecchetti, S.; Ricci, A.; Bozzuto, G.; Molinari, A.; Manni, I.; Pollo, B.; Scala, S.; Carpinelli, G.; et al. Targeting CXCR4 by a Selective Peptide Antagonist Modulates Tumor Microenvironment and Microglia Reactivity in a Human Glioblastoma Model. J. Exp. Clin. Cancer Res. 2016, 35, 55. [Google Scholar] [CrossRef]
- Wang, Z.; Zhong, H.; Liang, X.; Ni, S. Targeting Tumor-Associated Macrophages for the Immunotherapy of Glioblastoma: Navigating the Clinical and Translational Landscape. Front. Immunol. 2022, 13, 1024921. [Google Scholar] [CrossRef]
- Kloepper, J.; Riedemann, L.; Amoozgar, Z.; Seano, G.; Susek, K.; Yu, V.; Dalvie, N.; Amelung, R.L.; Datta, M.; Song, J.W.; et al. Ang-2/VEGF Bispecific Antibody Reprograms Macrophages and Resident Microglia to Anti-Tumor Phenotype and Prolongs Glioblastoma Survival. Proc. Natl. Acad. Sci. USA 2016, 113, 4476–4481. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nusraty, S.; Boddeti, U.; Zaghloul, K.A.; Brown, D.A. Microglia in Glioblastomas: Molecular Insight and Immunotherapeutic Potential. Cancers 2024, 16, 1972. https://doi.org/10.3390/cancers16111972
Nusraty S, Boddeti U, Zaghloul KA, Brown DA. Microglia in Glioblastomas: Molecular Insight and Immunotherapeutic Potential. Cancers. 2024; 16(11):1972. https://doi.org/10.3390/cancers16111972
Chicago/Turabian StyleNusraty, Sabrina, Ujwal Boddeti, Kareem A. Zaghloul, and Desmond A. Brown. 2024. "Microglia in Glioblastomas: Molecular Insight and Immunotherapeutic Potential" Cancers 16, no. 11: 1972. https://doi.org/10.3390/cancers16111972
APA StyleNusraty, S., Boddeti, U., Zaghloul, K. A., & Brown, D. A. (2024). Microglia in Glioblastomas: Molecular Insight and Immunotherapeutic Potential. Cancers, 16(11), 1972. https://doi.org/10.3390/cancers16111972