Simple Summary
Sufferers of neuroendocrine neoplasms (NENs) of unknown primary origin are a poor prognostic group with largely unmet clinical needs. In the absence of standard therapeutic algorithms, treatment should be based on tumor clinical-pathological characteristics, disease burden, and patient conditions. The aim of this review is to explore the evidence relating to available treatment options for NENs of unknown primary and to offer insights into future perspectives. Particular attention is given to molecular characterization and genomic profiling of NENs with potential therapeutic implications, mainly through the identification of druggable targets for agnostic treatments. Moreover, a treatment algorithm for both well-differentiated and poorly differentiated NENs of unknown primary is proposed.
Abstract
Among neuroendocrine neoplasms (NENs), a non-negligible proportion (9–22%) is represented by sufferers of NENs of unknown primary origin (UPO), a poor prognostic group with largely unmet clinical needs. In the absence of standard therapeutic algorithms, current guidelines suggest that the treatment of UPO-NENs should be based on tumor clinical-pathological characteristics, disease burden, and patient conditions. Chemotherapy represents the backbone for the treatment of high-grade poorly differentiated UPO-NENs, usually providing deep but short-lasting responses. Conversely, the spectrum of available systemic therapy options for well-differentiated UPO-NENs may range from somatostatin analogs in indolent low-grade tumors, to peptide receptor radioligand therapy, tyrosine kinase inhibitors (TKIs), or chemotherapy for more aggressive tumors or in case of high disease burden. In recent years, molecular profiling has provided deep insights into the molecular landscape of UPO-NENs, with both diagnostic and therapeutic implications. Although preliminary, interesting activity data have been provided about upfront chemoimmunotherapy, the use of immune checkpoint inhibitors (ICIs), and the combination of ICIs plus TKIs in this setting. Here, we review the literature from the last 30 years to examine the available evidence about the treatment of UPO-NENs, with a particular focus on future perspectives, including the expanding scenario of targeted agents in this setting.
1. Introduction
Neuroendocrine neoplasms (NENs) are a heterogeneous group of rare malignant neoplasms that arise from diffuse neuroendocrine cells. According to the 2022 World Health Organization (WHO) classification, NENs are classified into well-differentiated neuroendocrine tumors (NETs) and poorly differentiated neuroendocrine carcinomas (NECs) based on morphological features and the proliferation rate. Gastroenteropancreatic (GEP) NENs account for 62–67% of cases and include well-differentiated grade (G) 1–2 NETs (Ki-67 ≤ 20%), G3 NETs (Ki-67 index > 20% and well-differentiated morphology), and G3 NECs (Ki-67 index > 20% and poorly differentiated morphology). Thoracic NENs account for 22–27% of cases, including well-differentiated typical carcinoids of the lung and thymus (<2 mitoses/2 mm2 and absence of necrosis), atypical carcinoids (2–10 mitoses/2 mm2 and/or presence of necrosis), and poorly differentiated small cell (SC) and large cell neuroendocrine carcinomas (LCNECs) (>10 mitoses/2 mm2 and presence of necrosis). In the absence of a clinically or radiologically identifiable primary site despite the use of gold standard diagnostic techniques so far, a non-negligible proportion of histologically documented NENs (9–22%) are of unknown primary origin (UPO) [1,2,3,4,5,6].
Compared to other NENs, UPO-NENs represent a significant diagnostic and therapeutic challenge due to their rarity, their complex clinical presentation, and the lack of definite therapeutic algorithms.
According to the Surveillance, Epidemiology, and End Results (SEER) program, the incidence of UPO NETs was 0.84/100,000 persons per year between 2000 and 2012, with a relatively scant prognosis (median overall survival of 33–48 months) compared to other NET groups [7,8].
UPO-NENs present, by definition, with advanced or metastatic disease, the most frequently involved sites being the liver, followed by the peritoneum, the lymph nodes, the bones, and the lung [9]. Although the gastrointestinal and thoracic origin are the most likely sites of origin of UPO-NENs, unusual locations such as the genitourinary tract or the head and neck district have to be considered [10,11,12,13,14,15,16,17,18,19,20].
If the primary tumor site remains unknown despite extensive workup, the initial treatment strategy should be based on the presumptive site of origin and on tumor clinical-pathological characteristics as suggested by the main international guidelines [21,22]. However, in the absence of definite treatment algorithms based on high-quality evidence from randomized phase III trials, an optimal therapeutic approach to UPO-NENs still represents an unmet clinical need.
This review aims to address current evidence about the treatment landscape of UPO-NENs, and to provide an insight into future perspectives, with a particular focus on the potential therapeutic implications of molecular characterization and genomic profiling of these neoplasms.
2. Materials and Methods
We conducted a comprehensive search on PubMed and the ClinicalTrials.gov website using the following search keywords: “neuroendocrine”, with “tumor-” or “neoplasm-”, combined with “unknown primary”, “unknown origin” and “treatment” or “therapy” or “clinical trial” or “molecular biology”. The reference list of the most important papers and abstract communications from relevant conferences were also examined in order to further check the existing data in the literature. We limited the search to English language publications in the last 30 years. Searches were last updated on December 2023. A total of 379 articles were found. A manual selection of relevant articles based on title and/or abstract content was performed. The full versions of all relevant reports were analyzed, with a total of 103 works included in this review. Figure 1 reports the flow diagram for the identification of relevant manuscripts included in the review.
Figure 1.
Flow diagram of review manuscript.
3. Diagnostic Approach to UPO-NEN
At the first evidence of UPO-NEN, every effort should be made to identify the primary site of origin, as it could lead to surgical treatment with curative intent and/or to the access to systemic treatment strategies for which primary site identification is required by specific registration boundaries [22]. According to available evidence, the resection of midgut and pancreatic primary tumors is independently associated with improved survival outcomes in NET patients with liver metastases. Moreover, primary tumor surgery may reduce the risk of local complications such as occlusion, bleeding, or perforation, especially in the case of small-bowel NETs [23,24,25,26].
A comprehensive assessment for primary site identification should include an extensive clinical evaluation (e.g., pattern of metastatic spread, presence of clinical syndromes), morphological and metabolic imaging, endoscopic procedures, and a thorough immunohistochemical (IHC) evaluation with the possible integration of molecular pathology. Clinical presentation, including the pattern of metastatic organ involvement and the presence of a functional syndrome, might be fundamental as a hint for primary site identification. For example, carcinoid syndrome is usually related to small-bowel NEN, whereas Zollinger–Ellison syndrome and insulinoma and glucagonoma syndromes, should prompt the investigation of the duodenal-pancreatic area, and ectopic ACTH production may suggest lung or thymus primaries [27].
An accurate pathological evaluation is pivotal both to orient primary site identification and to provide tumor grading to guide therapeutic choices. NENs are classified into well differentiated NETs and poorly differentiated NECs based on morphological features and proliferation rate. This classification accounts for critical differences in terms of genomic and biological characteristics, as well as clinical behavior. NETs display a morphological organoid and nesting pattern, with very rare cytological atypia, whereas NECs are characterized by a solid growth pattern with marked atypia and diffuse necrosis. The neuroendocrine phenotype is identified through the assessment of definite immunohistochemical features. NETs usually display Chromogranin A (CgA) and synaptophysin staining, as well as strongly positive somatostatin receptor (SSTR) staining, while NECs retain synaptophysin staining, but may display only focal or absent CgA and SSTR staining. Insulinoma-associated protein-1 (INSM1) is highly specific for NEN independently of primary site and differentiation grade.
The classification of NENs was recently updated in the 2022 WHO classification system. NECs (every site of origin) are poorly differentiated with a high proliferation rate (Ki-67 > 20%, usually >55%) and further distinguished into the small cell and large cell subtypes. The NET nomenclature has been standardized as a three-tiered grading system according to morphological differentiation, grading, and proliferation rate. GEP NETs are distinguished as well-differentiated low-grade G1 NETs (Ki-67 ≤ 2% and mitotic index < 2 mitoses/2 mm2), well-differentiated intermediate-grade G2 NETs (Ki-67 between 3–20% and/or mitotic index between 2 and 20/2 mm2), and well-differentiated G3 NETs (Ki67 > 20% and/or mitotic index > 20/2 mm2). Similarly, thoracic NETs have been categorized into well-differentiated G1 NETs/typical carcinoids (mitotic index < 2/2 mm2 and no necrosis), well-differentiated G2 NETs/atypical carcinoids (mitotic index between 2 and 10/2 mm2 and/or necrosis), and well-differentiated NETs with elevated mitotic counts (atypical carcinoid morphology and >10 mitoses/2 mm2 and/or Ki67 > 30%). The WHO classification is specifically intended for surgical specimens. Therefore, the limitations of diagnoses obtained from core biopsies or cytological specimens should be taken into account. Whereas the diagnosis of NEC on biopsies may be more reproducible, the diagnosis of NET has inherent limitations in terms of accuracy for the evaluation of the Ki-67 labeling index and mitotic index, potentially affecting tumor grading [1].
Comprehensive IHC evaluation encompassing multiple markers may orient primary site identification. For example, CDX2 is a transcription factor associated with gastrointestinal differentiation, and a possible marker for intestinal or pancreatic origin. CDX2 yields a 90% sensitivity for midgut origin, although it is also expressed in 15% of pancreatic primaries. Paired Box (PAX)-8, PAX6 and Islet-1 are markers for both pancreatic and rectal NENs. Even though Islet-1 is a 70%-sensitive pancreatic (p)NEN marker, it is also expressed in rectal NENs and in up to 10% of lung primaries. Pancreatic and duodenal homeobox (PDX)-1, progesteron receptor (Pr), and neuroendocrine secretory protein (NESP)-55 staining are suggestive of pancreatic primary. Conversely, special AT-rich sequence-binding protein (SATB)2 positivity is typical of rectal (96%) and appendiceal (79%) NETs. Thyroid Transcription Factor (TTF)-1 positivity may suggest a thoracic primary, even though its sensitivity is low, whereas CK7 yields high sensitivity but less specificity for pulmonary NENs. Orthopedia Homeobox Protein (OTP), in contrast, represents a highly specific marker for pulmonary carcinoids, with a 60–80% sensitivity [6,28]. Several IHC algorithms have been built for the presumptive primary site identification in case of UPO-NEN [28,29]. Indeed, sequential IHC staining algorithms may help to identify the presumptive primary site with a stepwise approach. Performing baseline CDX2, Pr, PAX8, TTF-1 and SATB2 staining may lead to the identification of a midgut pattern (CDX2 positive, other markers negative), pancreatic pattern (Pr/PAX8 positive, SATB2 negative, CDX2 positive/negative), rectum/appendix pattern (SATB2 positive, TTF1-negative), or lung pattern (TTF-1 positive) which can be further investigated with the addition of other markers (such as Islet-1, PAX6, OTP, PDX-1, NESP-55) [6,28,29].
Although conventional radiology (computed tomography [CT] or magnetic resonance imaging [MRI]) is of utmost importance for patients’ staging, it might fail to detect the primary tumor if the lesions are small, especially in the case of a GEP primary. Therefore, metabolic imaging represents an essential tool in patient’s staging and primary site detection. Less recent reports evaluated the diagnostic impact of In-pentetreotide somatostatin receptor scintigraphy (SRS) in the detection of GEP occult primaries. In a small series of 36 patients by Savelli and Colleagues, SRS was suggestive of the possible site of the primary lesion in 39% of patients and prompted surgical management in 17% of cases [30]. Due to its higher sensitivity, 68-Gallium [68Ga]-labeled somatostatin analogs positron emission tomography (PET) is the gold standard for the detection of low-grade well-differentiated NETs, whereas fluorodeoxyglucose (FDG)-PET is recommended in high-grade poorly differentiated forms and for prognostication. In the case where pheochromocytoma/paraganglioma is suspected, other imaging modalities such as DOPA PET or metaiodobenzylguanidine (MIBG) scanning may also be considered [31,32,33]. According to the available literature, the true positive detection rates for an occult primary with 68Ga-DOTA-D Phe1-Tyr3-Octreotide (DOTATOC) PET imaging ranges from 38% to 61%, with overall sensitivity and specificity of 82–92% and 55–82%, respectively. 68Ga-DOTATOC PET imaging produces an alteration in patient management in 20–50% of cases [34,35,36,37,38,39]. Moreover, the use of radiolabeled somatostatin analogs has been exploited for intra-operative localization of UPO-NETs suspected to be of GEP primary, to improve intra-operative detection rates of small primaries and/or metastatic sites [40].
In cases where morphological or functional imaging has failed to detect the primary site, other investigations should be considered. If a gastrointestinal primary is suspected based on clinical-pathological assessments, endoscopic workout should be performed (including esophagogastroduodenoscopy, colonoscopy, and endoscopic ultrasonography). Since most UPO-NETs are of jejunoileal origin, an accurate study of the small intestine should be performed. Even though data concerning the use of capsule endoscopy (CE) or double balloon enteroscopy (DBE) for UPO-NEN assessment in clinical practice are scant, these assessments may allow the visualization of small intestinal lesions undetectable with conventional radiological imaging. DBE is more invasive when compared with capsule endoscopy; however, it may allow biopsies to be performed to obtain a pathological sample [27]. Moreover, surgical exploration of the abdomen may lead to the identification of the tumor primary in a non-negligible percentage of cases when GEP-NETs are suspected, potentially leading to surgery with radical intent [41].
If other more unusual areas are suspected, a thorough examination of the otolaryngologic and urogenital tracts, as well as full skin, eye, and breast assessments, should be performed [10,11,12,13,14,15,16,17,18,20]. For example, a very aggressive disease with limited therapeutic options is represented by neuroendocrine prostate cancer, which can emerge under the pressure of androgen deprivation treatment or arise de novo in a small percentage of cases [14].
In conclusion, the localization of the primary tumor is of paramount importance for the patient’s management and prognosis. Not only may primary tumor resection have a radical and curative intent, but it has also been associated with improved survival outcomes in midgut and p-NET patients with liver metastases [23,24,25,26]. Moreover, after primary tumor identification and resection, excision or ablation with curative intent of metastatic sites may be pursued as part of a curative strategy (especially in liver-limited disease). Surgery can also decrease the risk of complications related to the primary lesion (such as bleeding, occlusion, or compression of adjacent structures). Finally, identification of the site of origin may allow access to registered systemic treatments which require the primary tumor to be identified [27].
Figure 2 depicts a possible diagnostic algorithm for UPO-NEN.
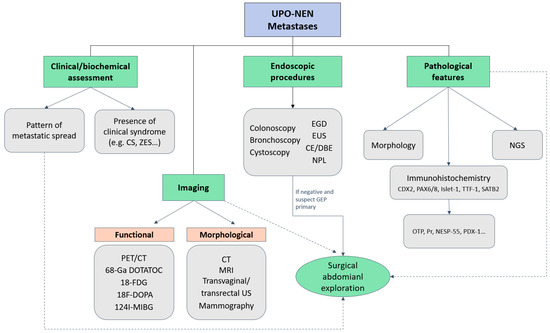
Figure 2.
Diagnostic algorithm for UPO-NEN. CE: capsule endoscopy; CS: Carcinoid syndrome; CT: computed tomography; DBE: double-balloon enteroscopy; EGD: Esophagogastroduodenoscopy; EUS: endoscopic ultrasonography; FDG: fluorodeoxyglucose; GEP: gastroenteropancreatic; MIBG: metaiodobenzylguanidine; MRI: magnetic resonance imaging; NESP-55: neuroendocrine secretory protein-55; NGS: next generation sequencing; NPL: naso-pharyngeal-laryngoscopy; OTP: Orthopedia Homeobox Protein; PAX: paired Box; PDX-1: Pancreatic and duodenal homeobox-1; PET: positron emission tomography; Pr: progesterone receptor; SATB2: special AT-rich sequence-binding protein 2; TTF-1: thyroid transcription factor; UPO-NEN: neuroendocrine neoplasm of unknown primary origin; US: ultrasonography; ZES: Zollinger-Ellison syndrome; 68-Ga DOTATOC: 68Gallium- DOTA-D Phe1-Tyr3-Octreotide.
4. Therapeutic Approach to UPO-NEN
No definite therapeutic algorithms are currently defined for UPO-NENs, mainly due to the lack of high-quality evidence from randomized phase III trials.
Therapeutic decision making for UPO-NENs is essentially based on tumor histology and grading, presumptive site of origin (based on histopathological and immunohistochemical characteristics), SSTR status, functionality, tumor burden, and progression rate, as well as the patient’s general condition and comorbidities [21].
4.1. Treatment of Poorly Differentiated NEC of Unknown Origin
4.1.1. First-Line Setting
The frontline therapeutic approach to poorly differentiated UPO-NECs is primarily based on the use of platinum-based chemotherapy regimens. In the first-line setting, cisplatin plus etoposide represents the preferred regimen, providing response rates up to ~40–70%, progression-free survival (PFS) rates up to 9 months, and overall survival (OS) rates up to 19 months in historical series [42,43,44,45]. In an analogy with the treatment of SCLC, the combination of platinum agents and irinotecan may be evaluated as a possible alternative to cisplatin and etoposide with some differences in clinical outcomes by ethnicity (Asiatic versus non-Asiatic populations) [46]. A recent study by Zhang and colleagues assessed the non-inferiority of cisplatin plus etoposide vs. cisplatin plus irinotecan in terms of safety and efficacy as a first-line treatment in advanced NEC patients, including UPO-NEC (eight patients), with different toxicity profiles. The overall response rate (ORR) was 42.4% in both arms, with median PFS of 6.4 months and 5.8 months (p = 0.81), and median OS of 11.3 months and 10.2 months (p = 0.37) for platinum–etoposide and platinum-irinotecan, respectively [47]. The NORDIC-NEC trial retrospectively evaluated the prognostic and predictive factors for survival and treatment outcomes in 305 patients with G3 NEN (32% of whom had UPO-NEN) receiving palliative chemotherapy (including cisplatin-etoposide and carboplatin–etoposide with or without vincristine). ORR to first-line chemotherapy was 31%, with a disease stabilization rate of 33%. The response rates did not differ among different platinum-based regimens. Patients with Ki-67 < 55% had a lower response rate to chemotherapy (15% vs. 42%, p < 0.001), but displayed better OS compared to patients with Ki-67 ≥ 55% (14 vs. 10 months, p < 0.001) [48]. On the basis of these results, platinum–etoposide containing regimens may not be the most appropriate chemotherapeutic approach for NEN G3 with Ki-67 <55% and alternative regimens (e.g., fluoropyrimidines, temozolomide, and oxaliplatin-containing regimens) should be considered. Recently, the randomized phase II EA2142 trial was designed to assess the efficacy and activity of a regimen with capecitabine and temozolomide (CAPTEM) compared to cisplatin plus etoposide in patients with previously untreated unresectable or metastatic G3 NEN with a Ki-67 labeling index 20–100%. A total of 67 patients with tumors of suspected GEP origin were enrolled. The study was prematurely closed due to futility at 57.7% information time, showing median PFS of 2.43 vs. 5.36 months, OS of 12.6 vs. 13.6 months, and response rate of 9% vs. 10% with CAPTEM and platinum–etoposide, respectively. CAPTEM did not appear to be superior to platinum–etoposide chemotherapy, but was associated with a more favorable toxicity profile [49]. Overall, prospective data assessing G3 NETs independently of G3 NECs are needed to establish the optimal front-line treatment strategy.
Due to the poor prognosis of these tumors, treatment intensification with three-drug therapeutic regimens has been attempted. The combination of carboplatin, etoposide, and paclitaxel was evaluated in two trials including patients with UPO-NECs, yielding ORR of 47–53% and median OS of 13.4–14.5 months, at the cost of moderate toxicity [50,51]. The FOLFIRINEC randomized phase II trial, with the aim of assessing the efficacy and activity of the FOLFIRINOX (5-fluorouracil, irinotecan, oxaliplatin) regimen compared to platinum plus etoposide in a population of patients with metastatic GEP or UPO NEC, is currently ongoing (NCT04325425) [52].
The combination of frontline chemotherapy with immunotherapy has been recently explored with the aim of improving outcomes in this poor-prognosis population. The non-randomized open-label phase II NICE-NEC trial (EudraCT: 2019-001546-18) evaluated the combination of carboplatin–etoposide with nivolumab as a first-line treatment for patients with advanced or metastatic G3 GEP and UPO NEN. Overall, 38 patients were enrolled. With a median follow-up of 18.6 months (range: 2.2–24.6), ORR was 54.1%, the disease control rate (DCR) was 83.8%, median PFS was 5.7 months (95% confidence interval [CI]: 5.1–9), and median OS was 13.9 months (with 32.4% of patients surviving more than 18 months) [53].
4.1.2. Second-Line Setting
For cases of UPO-NEC progressing on platinum–etoposide, regimens containing irinotecan, fluoropyrimidines, temozolomide, or oxaliplatin (e.g., CAPTEM; FOLFIRI [5-fluorouracil-irinotecan]; FOLFOX [5-fluorouracil-oxaliplatin]) may be considered, mostly based on retrospective data showing ORR rates of ~30% [54,55,56,57].
The randomized, multicenter, non-comparative, open-label, phase 2 BEVANEC trial evaluated the efficacy of FOLFIRI plus bevacizumab, or FOLFIRI alone in patients with advanced NEC (including UPO-NEC) progressing on first-line platinum–etoposide-based chemotherapy. Of the 126 patients included in the intent-to-treat population, 18% had UPO-NEC. After a median follow-up of 25.7 months, no clinically significant survival benefit was evidenced with the addition of bevacizumab to the FOLFIRI backbone, with a 6-month OS rate of 53% in the FOLFIRI plus bevacizumab group and 60% in the FOLFIRI group. Moreover, one fatal toxicity (ischemic stroke) occurred in the FOLFIRI-bevacizumab cohort [58]. The NET-02 randomized, non-comparative, phase II trial evaluated the efficacy of liposomal irinotecan (nal-IRI) plus 5-fluorouracil (arm A) or docetaxel (arm B) in patients with poorly differentiated extrapulmonary NECs progressing on first-line platinum–etoposide chemotherapy. Of the 58 enrolled patients, 10% had UPO-NECs. The trial met its primary endpoint in arm A, with a 6-month PFS rate of 29.6% (95% lower confidence limit: 15.7%) that exceeded the prespecified threshold for efficacy, but not in arm B (6 months PFS rate of 13.8%). ORR was 11.1% in arm A and 10.3% in arm B, with similar median PFS (3 months and 2 months) and OS (6 months and 6 months) in patients receiving nal-IRI plus 5-fluorouracil and docetaxel, respectively. According to the authors, 5-fluorouracil plus nal-IRI may represent a viable therapeutic option in this setting, whereas the poor performance of docetaxel, with the poor associated tolerability profile, should discourage further investigation of this regimen in this setting [59]. Another recent phase II trial aimed at evaluating the activity of temozolomide monotherapy in patients with extrapulmonary NECs progressing on first-line platinum–etoposide treatment. The trial enrolled 13 patients, 1 of whom had UPO-NEC. ORR was 15.4%, with median PFS of 1.8 months (95% CI, 1.0–2.7) and median OS of 7.8 months (95% CI, 6.0–9.5). O6-methylguanine-DNA methyltransferase (MGMT) deficiency was observed in one patient, who displayed partial response as the best response [57]. Of note, in the absence of other viable therapeutic options, some authors recently evaluated the opportunity of etoposide rechallenge in patients with a relapse-free interval of ≥3 months after first-line platinum–etoposide treatment. The retrospective RBNEC trial including 121 NEC patients (12% of whom with UPO-NEC) reported DCR of 62%, and median PFS and OS of 3.2 and 11.7 months, respectively, among the 31 patients receiving this treatment strategy [60].
Besides standard chemotherapy regimens, other treatment strategies employing immunotherapeutic agents and small molecules have been recently evaluated in order to expand the therapeutic armamentarium in high-grade NENs including UPO-NENs.
As immunotherapy has produced a paradigm shift in the treatment landscape of specific NENs such as Merkel cell carcinoma, immune checkpoint inhibitors (ICIs) have been recently tested in this setting [61,62]. Anti-programmed death (PD)- (ligand [L])1 monotherapy has shown limited activity in molecularly unselected G3 NENs including UPO-NECs, with ORR of 0–7%, median PFS range of 1.8–2.0 months, and median OS range of 4.2–7.8 months [63,64,65,66,67]. The combination of pembrolizumab plus mono-chemotherapy (weekly paclitaxel or weekly irinotecan) in high-grade pretreated extrapulmonary NECs (including 23% of UPO-NECs) also showed unsatisfactory activity (ORR 9%) and poor survival outcomes (median PFS: 2 months, median OS: 4 months) in unselected patients [63,68].
Conversely, dual anti-Cytotoxic T-Lymphocyte Antigen (CTLA)-4/anti-PD-1 blockade yielded more clinically significant results in high-grade NEN patients. The high-grade NEN cohort of the phase II DART-SWOG S1609 trial assessed the activity of the combination of the anti-CTLA-4 agent ipilimumab at the dose of 1 mg/kg every 6 weeks plus anti-PD-1 nivolumab in microsatellite-stable advanced G3 NEN patients (mostly with poorly differentiated tumors). A total of 19 patients were included, 4 (21%) of whom had UPO-NEC. Most of the included patients were pretreated for metastatic disease with a median of 1 (0–3) prior line. ORR was 26% (95% CI, 11–45%), with median PFS of 2.0 months and median OS of 8.7 months. Among the four patients with UPO-NEC, two achieved stable disease as the best response [69]. Another study, the NEN subgroup analysis of the CA209-538 trial of ipilimumab (at the dose of 1 mg/kg every 3 weeks for a total of four doses) and nivolumab in rare cancers, evaluated the outcome of 29 patients with advanced NEN (90% pretreated patients, 45% high grade NEN, 7% UPO-NEN). ORR was 24%, with DCR of 72% in the entire cohort. ORR was 31% and 23% in patients with G3 and G2 NEN, respectively. No responses were observed in the two enrolled patients with UPO-NEC. Median PFS was 4.82 (95% CI 2.71–10.53) months and median OS was 14.78 (95% CI: 4.07–21.25) months [70]. No unexpected toxicities have been documented in both trials combining ipilimumab and nivolumab.
Another ICI combination, durvalumab plus tremelimumab, was evaluated in advanced pretreated NENs in the multicohort phase II DUNE trial, showing only limited activity in advanced G3 GEP and UPO-NENs progressing to platinum-based first-line chemotherapy (cohort 4). In this cohort, ORR was 9.1%, 9-month OS rate was 36.1%, and median OS was 5.9 (95% CI: 2–9.7) months. Even though the prespecified futility threshold for OS was surpassed, response rates and survival outcomes were poor. PD-L1 expression by combined positive score (CPS) did not correlate with treatment activity. In analyzed patients, no microsatellite instability (MSI)-high status was reported [71].
Targeted agents with well-known activity in well-differentiated NENs have also been evaluated in the setting of high-grade progressive NENs. The EVINEC phase II trial evaluated the safety and efficacy of second-line everolimus in NEN G3 patients progressing on platinum chemotherapy. Of the 36 enrolled patients, 13 (36%) had NET G3, 14 (39%) had NEC, and 9 (25%) had mixed-neuroendocrine/non-neuroendocrine neoplasm (MiNEN) with a NEC G3 component. Six patients (16%) had UPO-NEN. No unexpected safety events occurred. Efficacy was promising (ORR 7.7%, median PFS and OS of 5.2 and 23.9 months, respectively) in the NET G3 group. However, results were poor in NECs (ORR 0%, median PFS and OS of 1.8 and 5.6 months, respectively), and in MiNENs (ORR 0%, median PFS and OS of 2.2 and 7.0 months, respectively) [72].
An alternative treatment strategy that is currently being explored in ongoing trials is the combination of small molecules (tyrosine kinase inhibitors-TKIs) and ICIs. Weber and colleagues recently reported the activity and safety of cabozantinib (a c-MET, vascular endothelial growth factor receptor [VEGFR]2, RET, KIT, AXL, and Fms Related Tyrosine Kinase 3 inhibitor) in combination with avelumab in patients with G3 NEN, reporting ORR of 21% and median PFS of 48.1 weeks, with a manageable toxicity profile [73]. Conversely, results of the CABATEN/GETNE-T1914 trial, evaluating the activity of cabozantinib plus the anti-PD-L1 agent atezolizumab, showed limited activity and poor survival outcomes in progressive NENs including high grade NECs [74].
Surufatinib (a VEGFR 1, 2, 3, fibroblast growth factor receptor [FGFR]-1, and colony-stimulating-factor-1 receptor inhibitor), combined with the anti PD-1 sintilimab and the anti-CTLA-4 IBI310, is currently being evaluated for the treatment of high-grade NENs (NCT05165407) [75].
Of note, most of the aforementioned trials included patients with high-grade (G3) NEN, without specifically addressing tumor differentiation. In the future, subgroup analyses assessing NECs independently of G3 NETs are warranted to establish the activity of novel treatment options in these different populations.
4.2. Treatment of Well-Differentiated NETs of Unknown Origin
Therapeutic options for well-differentiated UPO-NETs potentially encompass the available agents registered for site-specific disease, ranging from somatostatin analogs (SSAs) to targeted agents (such as everolimus or TKIs), peptide-receptor radio-ligand therapy (PRRT) and chemotherapy. In this setting, therapeutic choices should be tailored according to tumor grading and functionality, SSTR status, tumor burden, and progression rate, as well as the patient’s general condition and comorbidities.
SSA monotherapy may be considered for patients with well-differentiated, low-grade NETs expressing SSTRs, with low tumor burden and/or indolent disease behavior. Moreover, its use must always be considered in functioning tumors for symptomatic control.
UPO-NETs (13%) were included in the pivotal phase III randomized double-blind, placebo-controlled CLARINET trial, evaluating the efficacy of lanreotide in patients with well-differentiated advanced or metastatic, nonfunctioning, SSTR–positive NETs with Ki-67 < 10%. Progression-free survival was significantly improved in the lanreotide group compared to the placebo arm (median PFS not reached [NR] vs. 18.0 months, respectively; hazard ratio [HR] 0.47; 95% CI, 0.30–0.73; p < 0.001). Overall survival did not significantly differ between the two arms, although the results’ interpretation may have been impacted by crossover from the placebo to the lanreotide group that occurred in the extension study (CLARINET OLE) and by the long life expectancy of patients with indolent disease [76,77].
Faiss and Colleagues conducted a prospective randomized trial assessing the efficacy of subcutaneous lanreotide (at the dose of 1 mg three times a day), interferon alpha, or their combination in 80 patients with advanced progressive treatment-naïve NETs, including 11 (14%) UPO-NETs, showing a 32% DCR in the lanreotide group [78].
More recently, a phase II study in Japanese patients investigated lanreotide autogel in 28 patients with well-differentiated G1–2 NETs including 5 patients (18%) with UPO-NETs, reporting DCR of 64.3%, ORR of 3.6%, and median PFS of 36.3 weeks (95% CI: 24.1–53.1) [79].
PRRT is a valuable therapeutic option for NETs expressing SSTRs, with a good safety profile and limited acute and medium-term toxicities. PRRT employs radiolabeled, beta-emitting SSAs (90Yttrium[Y]-DOTATOC and 177Lutetium-[DOTA°, Tyr3] octreotate [177Lu-Dotatate]) that bind to SSTRs on the surface of tumor cells, with consequent radiopeptide internalization and cell death [80]. The landmark NETTER-1 phase III trial demonstrated the superiority of PRRT with 177Lu-Dotatate (in association with octreotide long-acting release [LAR] 30 mg every 28 days) versus high-dose octreotide LAR (60 mg every 28 days) in SSTR-positive midgut NET patients progressing on octreotide treatment, with a significant improvement in terms of PFS and ORR, and a clinically meaningful (~11 months) trend to improved OS in the PRRT arm [81,82]. Even though the phase III NETTER-1 trial only enrolled patients with well-differentiated midgut NETs, the activity of PRRT in other GEP NETs and UPO-NETs has been explored in several retrospective studies. In a meta-analysis of 18 studies considering 1920 patients with unresectable metastatic NETs treated with 177Lu-Dotatate, PRRT exhibited a pooled disease response rate of ~30% and a combined DCR of 74–81% [83]. Another systematic review, including one publication addressing eight patients with UPO-NEN, reported ORR of 38% and DCR of 88%, with median PFS of 17.5 months (95% CI 7–34) and median OS of 43 months (95% CI 15 months–NR) [84]. Moreover, data from non-randomized trials of PRRT have consistently shown high response rates and long-term PFS outcomes in heterogeneous patient populations, including UPO-NETs [85,86,87]. PRRT may be the treatment of choice for patients with high STTR expression and/or high tumor burden, in whom tumor shrinkage and symptomatic response, rather than tumor stabilization, represent the therapeutic goal. Moreover, PRRT may also be considered in selected cases in the setting of NEN G3. Data deriving from retrospective studies of PRRT in patients with G3 NEN (including UPO-NEN) highlight disease control rates of 30–80%, median PFS ranging from 9 to 23 months, and median OS ranging from 19 to 53 months. However, reported outcomes were unsatisfactory in patients with Ki-67 > 55% [88,89,90,91].
Targeted therapies with demonstrated efficacy in NEN treatment encompass the mammalian target of rapamycin (mTOR) inhibitor everolimus and TKIs. Few data are currently available about the use of targeted therapies in UPO-NENs.
Yao and Colleagues evaluated the activity of everolimus (at the dose of either 5 mg/day or 10 mg/day) in combination with octreotide LAR in 60 patients with well-differentiated NETs, including 5 patients with UPO-NETs (8%). In the intent to-treat population, ORR was 20% (13% in the 5 mg cohort and 30% in the 10 mg cohort). Median PFS was 60 weeks (95% CI, 54–66 weeks), with a 3-year OS rate of 78% [92]. The Italian Trials in Medical Oncology (ITMO) trial enrolled 50 patients with treatment-naïve NETs (including 14 patients with UPO-NETs) to receive octreotide LAR plus everolimus. ORR was 18%, with a complete response rate of 2%, a partial response rate of 16% (including three patients with UPO-NETs), and a disease stabilization rate of 74%. In all patients experiencing a clinical benefit, disease control lasted more than 6 months. In the 5-year updated analysis of this study, 17 (34%) of patients had received treatment for more than 2 years, with a median time to progression of 33.6 months (95% CI 18.7–41.2) and a median OS of 61.0 months (95% CI 49.8-NR) [93].
A subgroup analysis of the RADIANT-4 trial of everolimus vs. placebo in G1–G2 advanced non-functional NETs, specifically focusing on patients with gastrointestinal carcinoids (175 patients) and UPO-NETs (36 patients), showed a clinically meaningful PFS advantage (13.6 months versus 7.5 months) for everolimus vs. placebo (HR 0.60; 95% CI, 0.24–1.51) in the UPO-NET setting [94].
The SANET-ep trial evaluated the efficacy and safety of surufatinib in patients with well-differentiated advanced extra-pancreatic NETs, including 27 (14%) UPO-NETs, evidencing a PFS advantage of surufatinib over placebo (9.2 vs. 3.8 months, HR 0.33; 95% CI, 0.22–0.49, p < 0.0001). A specific subgroup analysis of patients with UPO-NETs or NETs of uncommon tumor origin showed median PFS of 13.9 vs. 7.4 months (HR 0.5, 95% CI 0.24–1.06, p: 0.069) in the surufatinib and placebo groups, respectively [95].
More recently, the double-blinded phase III Alliance A021602-CABINET trial evaluated the efficacy of cabozantinib versus placebo in patients with advanced NETs progressing on prior therapy. In the extra-pancreatic NET cohort including patients with UPO-NETs, despite modest ORR rates (4% vs. 1% for cabozantinib versus placebo), a statistically significant improvement in PFS (8.2 vs. 3.2 months, HR 0.41, 95% CI 0.27–0.62, p < 0.0001) was evidenced in the cabozantinib group over the placebo arm [96].
Cytotoxic chemotherapy also remains an option for patients with well-differentiated UPO-NETs and may be the preferred treatment strategy in patients with high disease burden, higher Ki-67, poor 68GaPET, and/or significant FDG-PET uptake, or in patients for whom rapid tumor shrinkage is a desirable goal. In fit patients, polychemotherapy is a preferred option over mono-chemotherapy in terms of activity. Regimens containing alkylating agents (streptozotocin, temozolomide), fluoropyrimidines (5-fluorouracil, capecitabine), oxaliplatin, and irinotecan proved active in NETs.
Chan and colleagues evaluated the activity of temozolomide in association with bevacizumab in 34 patients with carcinoids, including 7 UPO-NETs (21%), and pNETs. ORR was 15% in the whole study population, even though all responses occurred in the pNET cohort and none in the carcinoid group. However, 12 patients in the carcinoid cohort (63%) experienced some degree of tumor shrinkage. Median PFS was 7.3 months (95% CI, 3.9-NR) and median OS was 18.8 months (95% CI, 8.5–36.1) for carcinoid tumors [97]. Chauhan and colleagues reported median PFS of 10.8 and 7 months, respectively, on CAPTEM in G2 and G3 UPO-NETs [98]. A retrospective real-world experience on 170 patients with GEP, lung, and UPO NETs treated with the CAPTEM combination or temozolomide monotherapy showed clinically meaningful ORR (15.4%) and median PFS (16.9 months, 95% CI 6.0–30.4) and OS (35.7 months, 95% CI 16.2–63.0) results among the 16 (9%) patients with UPO-NETs (4 of whom were diagnosed with NET G3) [99]. Although higher response rates have been reported in patients with MGMT deficiency or promoter methylation treated with temozolomide, the use of MGMT status for preselection of patients remains controversial [99,100,101]. Recently, Walter and colleagues showed increased ORR, PFS, and OS outcomes in patients with methylated compared to unmethylated MGMT NETs (including UPO-NETs) treated with alkylating agents, but not oxaliplatin-based chemotherapy [102].
The NET-01 study randomized 86 patients with advanced GEP and UPO NETs to receive capecitabine and streptozotocin with or without cisplatin. ORR was similar between the two arms (12% vs. 16%), as well as median PFS (10.2 vs. 9.7 months) and OS (26.7 vs. 27.5 months) for the doublet vs. triplet arm, respectively. However, patients enrolled in the triplet group experienced a non-negligible proportion of G ≥ 3 adverse events (68%), compared to the lower rate of high-grade toxicities of patients receiving capecitabine and streptozotocin (44%) [103].
Another valuable treatment option in the setting of well-differentiated NETs is represented by platinum derivates (oxaliplatin) in combination with fluoropyrimidines, with reported ORRs of ~13–30% and median PFS outcomes of 8–20 months [104,105,106].
Immunotherapy has yielded poor results in well-differentiated NENs, with only modest signals of activity in thoracic NENs. Translational studies are needed in order to identify subgroup of patients who are more likely to respond to this treatment strategy [66,70,107,108]. Only the anti-PD-1 agent toripalimab yielded promising response rates (ORR 20%) in Asian patients with G2–3 pretreated NEN. The subgroups of PD-L1-positive, tumor mutational burden (TMB)-high, or ARID1A-mutated tumors were enriched in responders [109].
Figure 3 depicts the clinical and pathological determinants of therapeutic choices in UPO-NEN.
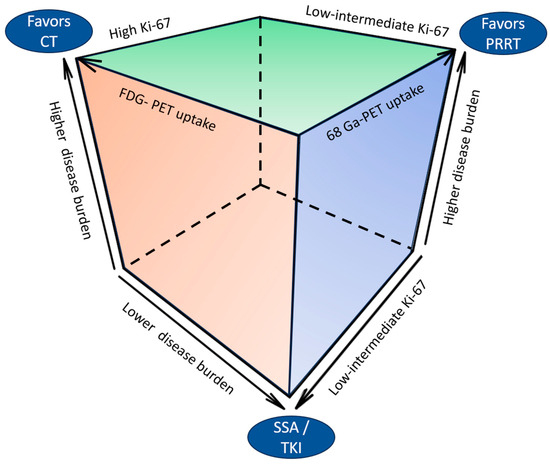
Figure 3.
Clinical and pathological determinants of therapeutic choices in UPO-NEN. CT: chemotherapy: FDG: fluorodeoxyglucose; PET: positron emission tomography; PRRT: peptide receptor radioligand therapy; SSA: somatostatin analog; TKI: tyrosine kinase inhibitor.
Whenever possible, due to the exiguity of the therapeutic armamentarium and country-specific restrictions in the use of approved treatment options, patients should be considered for enrollment in clinical trials. Table 1 reports ongoing clinical trials including patients with UPO-NEN.

Table 1.
Ongoing clinical trials including UPO-NEN.
4.3. Special Situations: Liver-Limited Disease
Even in the case of UPO-NETs, locoregional treatments may be employed as part of the therapeutic strategy. In particular, liver-directed locoregional therapies, alone or in combination with systemic treatments, may be considered in selected cases to improve local disease control, reduce hepatic tumor burden, and possibly improve patient prognosis [110]. Kim and colleagues conducted a phase 1b dose-finding study of pasireotide, everolimus, and selective internal radioembolization therapy (SIRT) in 13 patients with NET and secondary liver involvement (including 2 patients with UPO-NETs). No dose-limiting toxicities were reported; median PFS was 18.6 months and OS was 46.3 months [111]. Another study of systemic 5-fluorouracil combined with 90Y-SIRT in 34 NEN patients with liver metastases showed radiologic ORR of 50% and mean survival of 27.6 months. Among enrolled patients with UPO-NEN, 3 out of 8 (37.5%) experienced a radiologic response [112].
In patients with liver-limited disease, several potentially curative treatment options may be considered, including liver resections or liver transplant. Few cases of liver transplant in patients with UPO-NET have been reported, with alternate outcomes. More data are needed in order to possibly extend the transplant strategy in the setting of UPO-NEN, provided an accurate selection based on patient features, disease behavior, and characteristics [113,114].
5. Unraveling Molecular Characterization of UPO-NENs: Time for an Agnostic Approach?
Molecular biology and gene expression profiling represent an area of increasing interest for the characterization of UPO-NENs, with both diagnostic and potential therapeutic implications. In recent years, a deeper insight has been achieved in subtype-specific NEN molecular landscape characterization. These findings, if appropriately integrated with clinical and pathological data, may help to determine tumor origin in UPO-NENs and/or identify druggable molecular targets.
With regard to GEP-NENs, a recent comprehensive genomic profiling analysis confirmed the differences in terms of genomic background of high grade versus low grade GEP-NENs. Among low-grade tumors, the most frequently mutated genes are ATRX (13%), ARID1A (10%), and MEN1 (10%), whereas high-grade tumors exhibit TP53 (51%), KRAS (30%), APC (27%), and ARID1A (23%) mutations and a higher prevalence of BRAF (5.4–70%) alterations. Moreover, immune-related biomarkers such as the MSI-high status (4–12% vs. 0–3%), PD-L1 overexpression (6% vs. 1%), and high TMB (7% vs. 1%) are prevalent in high-grade compared to low-grade tumors [115,116]. Among NENs of pancreatic primary, loss of DAXX or ATRX protein expression defines well-differentiated NETs, whereas abnormal p53, Rb, and SMAD4 define poorly differentiated NECs [117].
Similarly to GEP-NENs, genomic profiling provides further insight into thoracic NENs biology. Carcinoids generally display low TMB (<1 mutations/megabase [Mb]) and few recurrently mutated genes, including alterations in chromatin remodeling genes and genes in the phosphoinositide 3-kinase (PI3K)/Akt/mTOR pathway. Indeed, mutations in genes involved in chromatin remodeling are detected with a high frequency (~40%) in carcinoids, the most frequent being MEN1 (11–22%), genes of the SWI/SNF complex mostly affecting ARID1A (6–7% of cases) and KMT2C/MLL3 (8%). Other frequently mutated genes in carcinoids include TP53 (10%), NOTCH2 (5%), and PIK3CA. Of note, atypical carcinoids usually carry more alterations in the MEN1 (22% vs. 6%) and PIK3CA genes (39% vs. 13%) compared to typical carcinoids. Furthermore, amplification of MYCL, BCL2, and SRC is almost exclusively described in atypical carcinoids [118]. Small cell NECs and LCNECs have higher TMB rates (8.5–10.5 mutations/Mb) compared to carcinoids. The most frequently described LCNEC alterations lie in tumor suppression genes such as TP53, RB1, and STK11, and genes of the RAS pathway. Indeed, two mutually exclusive genomic subtypes have been identified in LCNECs: the first, which is similar to SCLC, shows concurrent mutation of TP53 and RB1, whereas the other subtype, more similar to non-small cell lung cancer, is predominantly RB1 wild-type, harboring STK11/KEAP1 alteration [119]. Preliminary data indicate that RB1 wild-type tumors may have better outcomes when receiving NSCLC-type chemotherapy (platinum–gemcitabine or paclitaxel) than platinum–etoposide SCLC-like chemotherapy [118].
Thus, genomic profiling may provide insights into tumor biological characteristics that segregate with the anatomic primary site and guide therapeutic choices. For example, pulmonary and pancreatic NENs are frequently mutated in chromatin-remodeling genes, whereas midgut NETs exhibit cell cycle and Wnt pathway mutations, such as CTNNB1, MEN1, or APC alterations [29]. Recently, commercially available gene-expression profiling platforms have been implemented with the aim to assist clinicians in the prediction of tumor origin in cases of diagnostic uncertainty [120]. A validated real-time polymerase chain reaction 92-gene cancer ID analysis, able to categorize NENs according to putative primary site and differentiation, was recently shown to yield accuracy in primary site identification of metastatic UPO-NENs, with a likely impact on patient treatment and outcome [120,121]. As previously pointed out, NETs have a lower number of somatic mutations compared to epithelial tumors, while epigenetic modifications due to the mutation or loss of expression of chromatin modifiers are more common. Methylation array data have recently been used in order to create an algorithm for predicting the tissue of origin in UPO-NENs, based on a training set of 97 pNENs and small intestinal NENs [6].
Extensive molecular characterization through commercially available next generation sequencing (NGS) platforms may also lead to the identification of druggable targets for approved “agnostic” treatments. Potentially targetable rare molecular alterations in NENs, including UPO-NENs, encompass BRAF and KRAS mutations, RET, Anaplastic Lymphoma Kinase (ALK) and Neurotrophic tyrosine receptor kinase (NTRK) rearrangements, Delta-like ligand 3 (DLL-3) expression, and MSI-high or TMB-high status.
BRAF V600E mutations, although rare overall in NENs, are usually enriched in high-grade NENs of GEP origin (being reported in up to 70% of right-colon NECs) and have been associated with promising activity of combined BRAF-MEK inhibition [116]. The Food and Drug Administration (FDA) recently approved combined BRAF/MEK inhibition with dabrafenib and trametinib for the treatment of patients with advanced solid tumors harboring BRAF V600E mutations, based on the outcomes of basket trials enrolling patients with 24 tumor types (including 2 patients with MiNEN and 2 patients with colon NEC) [116]. Different case series have reported clinically significant responses in pretreated NEC patients harboring BRAF V600E mutations with BRAF/MEK targeted agents, even though no specific information about UPO-NENs has been reported so far to our knowledge [122,123,124]. Moreover, in a recent report, a LCNEC patient harboring a non-V600E BRAF activating mutation (G469R) showed durable disease control (>15 months) with the combination of dabrafenib and trametinib [125].
Since the development of the KRAS G12C inhibitors sotorasib and adagrasib, few case reports have addressed the potential use of these molecules in the setting of NENs harboring KRAS G12C mutations. One case, showing some clinical benefit of sotorasib in a patient with KRAS G12C mutant atypical lung carcinoid, has been recently reported [126,127]. More data are required to address the impact of KRAS inhibitors in the setting of NENs, and patients whose tumors harbor KRAS-actionable mutations should be referred for enrollment in clinical trials.
ALK fusions seem characteristic of thoracic NENs, with a reported incidence of ~0.9–3%, and correlate with high-grade and advanced stages. Few case series have reported significant disease responses of crizotinib and alectinib in lung NECs harboring ALK fusion, with a manageable toxicity profile [128,129,130,131].
In a large dataset of 2417 NEN patients, the incidence of NTRK rearrangements was 0.3% (including 2 patients with UPO-NEN) in the absence of site-specific prevalence [132]. Recently, entrectinib and larotrectinib received agnostic FDA approval for advanced adult or pediatric tumors bearing NTRK fusions based on results of the ALKA-372-001, STARTRK-1, and STARTRK-2 trials. ORR for the five patients with NENs enrolled in these trials was 40% [116,133]. Updated results of entrectinib in 121 patients with NTRK-rearranged solid tumors showed ORR of 61.2%, median PFS of 13.8 months (95% CI 10.1–19.9), median OS of 33.8 months (95% CI, 23.4–46.4), and intracranial ORR of 63.6% in the whole population. Among the five patients with NEN, ORR was 40%, median PFS was 15.6 months (95% CI 0.9-not estimable), and median OS was 40.5 months (95% CI 28.6–40.5) [134]. In a phase I trial of taletrectinib, a ROS1/NTRK inhibitor, 1 partial response (8.3%) and 7/12 (58%) disease stabilizations were observed among 12 molecularly unselected NEN patients, with median PFS of 10.2 months [135].
RET alterations include mutations (typical of medullary thyroid cancer and MEN2-related tumors) and rearrangements. The NCT03157128 and NCT03037385 studies of solid tumors (including NENs) harboring RET alterations are currently evaluating the safety and activity of the selective inhibitors selpercatinib and pralsetinib, respectively. Pralsetinib is a RET kinase inhibitor, including RET fusion proteins. The phase I/II ARROW trial reported the activity of pralsetinib in patients with RET-fusion-positive solid tumors, evidencing 67% ORR among the three enrolled patients with NEN [136]. Other case reports described the clinical activity of selpercatinib and cabozantinib in patients with NECs harboring RET alterations, underlying the possibility of an agnostic approach with anti-RET drugs in patients with RET-positive NEN [137,138].
DLL3, a negative regulator of Notch signaling, is frequently overexpressed in poorly differentiated NECs, but not in well-differentiated NETs [139]. A DLL3-targeted antibody-drug conjugate linked to a pyrrolobenzodiazepine dimer toxin, Rovalpituzumab tesirine, achieved a 17% ORR in 35 DLL3-overexpressing NEN (NEC and NET) patients (including UPO-NEN) in a phase I/II trial. Median PFS was 4.3 (2.7–6.1) months and median OS was 7.4 (5.6–13.1) months. [140] Further data are, however, required to fully elucidate the effectiveness of Rovalpituzumab tesirine in this setting, as this drug failed to demonstrate an OS benefit over placebo in SCLC patients as maintenance after platinum-based therapy [141] and over topotecan in second-line setting in phase III trials, at the cost of significant toxicity [142]. The DLL3-targeting T Cell engager BI 764532 is currently being evaluated in the DAREON™-5 open-label, multicenter phase II dose-selection trial, in patients with relapsed/refractory neuroendocrine carcinomas (NCT05882058).
With regard to immunotherapeutic agents, few data are currently available about prognostic and/or predictive immune-response biomarkers. In recent years, the anti-PD-1 agent pembrolizumab received agnostic FDA approval for pretreated solid tumors bearing the MSI-H phenotype or a high TMB status (≥10 mutations/Mb). The prevalence of the MSI-H phenotype has been reported in up to 12% of NEN cases, being enriched in cases in gastrointestinal NECs and MiNEN [143].
The updated results of cohort K of the phase II Keynote 158 trial considering 351 patients with MSI-high/deficient mismatch repair (dMMR) non-colorectal cancers, including 12 (3.4%) patients with NEN, showed 30.8% ORR, with a median duration of response of 47.5 months, median PFS of 3.5 (95% CI 2.3–4.2) months, and median OS of 20.1 (95% CI 14.1–27.1) months in the entire cohort, with a manageable safety profile. No separate data on the 12 NEN patients have been specifically reported [144]. These results were recently confirmed in the tumor-agnostic cohort of the DRUP trial (NCT02925234), evaluating the activity of the anti-PD-L1 agent durvalumab in heavily pretreated patients with dMMR/MSI-H solid tumors, including one patient with NEN. ORR was 29%, median PFS was 5 months (95% CI 2-NR), and median OS was 26 months (95% CI 9-NR) in the entire cohort [145]. The Keynote-158 (NCT02628067) phase II study and the NCT04272034 phase I study are currently evaluating the activity and safety of the anti-PD-1 agent pembrolizumab and the anti-PDL-1 inhibitor INCB099318, respectively, in patients with MSI-H solid tumors (potentially including NENs).
High TMB has been detected in up to 45.6% of LCNEC, 11.8% of colon NEN, and 5.9% of patients with small intestinal NEN [146]. Agnostic approval of pembrolizumab in this molecular subset was based on the results of a pre-planned retrospective analysis of the Keynote-158 trial in patients with TMB ≥ 10 mutations/Mb, showing ORR of 29% among the 102 patients included in the efficacy analysis. Notably, among the five patients with NEN (primary site not specified), ORR was 40% [116,147]. Table 2 summarized potential agnostic targets for the treatment of UPO-NEN.
Table 2.
Novel biomarkers in UPO-NENs and the relative potential therapeutic strategies.
6. Discussion
In recent decades, the incidence and prevalence of NENs has dramatically risen across all primary sites, stages, and grades, mainly as a result of increased detection rates and advances in systemic therapies. In this scenario, a non-negligible proportion (up to 22% of cases) is represented by sufferers of UPO-NENs, a poor prognostic group with largely unmet clinical needs [7]. UPO NENs present, by definition, with advanced or metastatic disease. No standard therapeutic algorithms are defined, as this population is usually poorly represented in registration randomized phase III trials. Current guidelines suggest that treatment of UPO-NENs should be based on the presumptive site of origin, tumor clinical-pathological characteristics, disease burden, and the patient’s conditions and comorbidities, thus requiring a case-by-case therapeutic individualization [21,22].
The differentiation between well- and poorly differentiated UPO-NEN represents one of the main criteria to orient therapeutic choices. This dichotomous morphological classification reflects underlying differences in terms of genomic characteristics and clinical and biological behavior. Chemotherapy still represents the backbone for the treatment of high-grade, poorly differentiated UPO-NECs, an approach that usually provides deep but short-lasting responses with poor survival outcomes. Attempts to improve survival in this particularly poor prognostic group include treatment intensification with three-drug chemotherapeutic regimens [50,51,52], chemo-immunotherapy combinations [53,63,68], ICIs, or a combination of ICIs and TKIs [63,64,65,66,67,68,69,70,71,73,74,75]. ICI monotherapy has provided unsatisfactory results in unselected patients’ populations in recent clinical trials [63,64,65,66,67]. Although preliminary, interesting activity data have been recently provided about upfront chemoimmunotherapy [53], the use of anti-CTLA-4 plus anti-PD-L1 agents [69,70], and ICI plus TKI combinations [73]. However, such treatment strategies have not been directly compared with standard platinum-based chemotherapy and their development should entail more accurate patient selection based on predictive molecular biomarkers. Indeed, the differences observed in ICI activity between low- and high-grade NENs may rely on the higher TMB and neoantigen burden, and the higher PD-L1 expression in the latter group. To date, conflicting data are available about the role of PD-L1 expression in predicting response to ICIs in UPO-NEN [64,66,109], and the DART-SWOG and CA209–538 trials did not provide activity data of the ipilimumab/nivolumab combination stratified by PD-L1 expression [69,70]. Conversely, MSI-high and TMB-high status in addition to POLε alterations are more reproducible biomarkers of ICI-related benefits across different tumor types [139,146,147].
In recent years, molecular profiling has provided deep insights into the molecular landscape of UPO-NENs, with both diagnostic and therapeutic implications. Overall, about 20% of high-grade NECs may harbor one druggable molecular alteration (including BRAF and KRAS mutations, RET, ALK, and NTRK 1/2/3 rearrangements, and MSI-high or TMB-high status), so that comprehensive NGS analysis should be advocated in this poor-prognosis subset to orient agnostic target therapies [116].
Although well differentiated UPO-NETs harbor few somatic druggable alterations, molecular profiling platforms providing primary-site specific genomic profiles have been recently developed, with potential clinical applications in the near future [6,120,121]. The integration of molecular biology with standard pathology and imaging approaches may significantly contribute to primary site identification, potentially leading to surgical approaches with radical intent.
The spectrum of available systemic therapy options for well-differentiated UPO- NETs may range from SSA monotherapy in indolent low-grade NETs, to TKIs, PRRT, or chemotherapy for more aggressive tumors or in the case of a symptomatic disease burden.
Novel TKIs have shown promising activity and efficacy in the setting of well-differentiated UPO-NETs. Cabozantinib and surufatinib are multitargeted small molecules with high-spectrum biological activity against multiple and non-redundant oncogenic pathways responsible for tumor growth and neoangiogenesis, which yielded improved PFS outcomes compared to placebo in phase III trials including UPO-NETs. In the CABINET and SANET-ep trials, the PFS advantage was evidenced despite poor objective response rates, suggesting that TKI monotherapy may represent a valuable therapeutic option whenever tumor stabilization rather than tumor shrinkage is the therapeutic goal [95,96]. In the case of high tumor burden or symptomatic disease, PRRT or chemotherapy represent the treatments of choice, depending on tumor grade and SSTR expression. Chemotherapy, for example, is a valuable choice for G3 NETs or for NETs with low SSTR on functional imaging [85,86,87,88,89,90,91,97,98,99,100,101,102,103,104,105,106].
Even though immunotherapy showed limited efficacy in well-differentiated NETs, the anti-CTLA-4/anti PD-1 combination or the anti PD-L1 plus TKI combination showed signs of activity in G3 NETs, a subset of tumors with a higher biological aggressiveness and in which multidrug immune modulation may represent a promising therapeutic approach [69,70,73].
Figure 4 depicts a possible future therapeutic algorithm for UPO-NENs, integrating molecular biology profiling, agnostic, and multimodal treatment strategies.
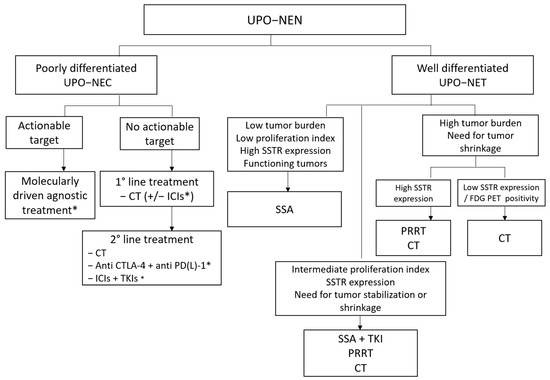
Figure 4.
Possible therapeutic algorithm for UPO-NEN. CT: chemotherapy; CTLA-4: anti-Cytotoxic T-Lymphocyte Antigen 4; ICI: immune checkpoint inhibitor; FDG PET: fluorodeoxyglucose positron emission tomography; NEC: neuroendocrine carcinoma; NEN: neuroendocrine neoplasm; NET: neuroendocrine tumor; PD-(L)-1: programmed death (Ligand)-1; PRRT: peptide receptor radioligand therapy; SSA: somatostatin analog; SSTR: somatostatin receptor; TKI: Tyrosine kinase inhibitor; UPO: unknown primary origin. *: under investigation.
7. Conclusions and Future Directions
In conclusion, UPO-NENs represent a rare and heterogeneous disease with limited treatment options. Due to their rarity, we claim the possibility of a change in UPO-NEN management, moving from morphologically driven therapeutic choices, or those presuming the site of origin, to the integration of molecular biology. This opportunity, coupled with agnostic treatments, may pave the way to the definition of personalized therapeutic strategies, particularly when lacking clinical trials specifically drawn for this rare entity. In-development treatment approaches include multidrug combinations (e.g., combinations of ICIs, TKIs, and PRRT) and the implementation of genomic profiling in order to identify druggable molecular alterations, especially in high-grade disease. Improvement in diagnostic and surgical techniques may lead to enhanced locoregional treatments with radical intent. Multidisciplinarity and referral in high-volume centers is of utmost importance to optimize patients’ management.
Author Contributions
Conceptualization, F.C., R.E.R. and S.P.; methodology, F.C., R.E.R., P.C., G.P., A.T. and S.P.; validation, F.C., R.E.R., P.C., G.P., A.T., S.P., A.G., R.L. and D.L.C.; formal analysis, F.C., R.E.R., P.C., G.P. and A.T.; investigation, F.C., R.E.R., P.C., G.P., A.T., S.P., J.C., S.O., A.G., R.L. and D.L.C.; resources: not applicable; data curation, F.C., R.E.R., P.C., G.P., A.T. and S.P.; writing—original draft preparation, F.C., R.E.R., P.C., G.P., A.T. and S.P.; writing—review and editing, F.C., R.E.R., P.C., G.P., A.T., S.P., J.C., S.O., A.G., R.L. and D.L.C.; visualization, F.C., R.E.R., P.C., G.P., A.T., S.P., J.C., S.O., A.G., R.L. and D.L.C.; supervision, F.C., R.E.R., S.P., J.C., R.L. and D.L.C.; project administration, F.C., R.E.R., S.P. and D.L.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
F.C. received travel grants from Ipsen, Advanced Accelerator Applications, Istituto Gentili, Wave Pharma outside the submitted work; S.P. received honoraria from Novartis, Ipsen, Pfizer, Merck Serono, and Advanced Accelerator Applications (AAA), received grants from Ipsen and Pfizer, and received personal fees from AAA, Novartis, and Merck outside the submitted work; D.L.C. received honoraria as a speaker bureau/scientific advisor from MSD, BMS, Astra Zeneca, Sanofi Genzyme, Seagen, Novartis, Amgen, Roche, and Takeda outside the submitted work. The remaining authors declare no conflicts of interest.
References
- Rindi, G.; Mete, O.; Uccella, S.; Basturk, O.; La Rosa, S.; Brosens, L.A.A.; Ezzat, S.; de Herder, W.W.; Klimstra, D.S.; Papotti, M.; et al. Overview of the 2022 WHO Classification of Neuroendocrine Neoplasms. Endocr. Pathol. 2022, 33, 115–154. [Google Scholar] [CrossRef] [PubMed]
- Taal, B.; Visser, O. Epidemiology of neuroendocrine tumours. Neuroendocrinology 2004, 80 (Suppl. S1), 3–7. [Google Scholar] [CrossRef] [PubMed]
- Polish, A.; Vergo, M.T.; Agulnik, M. Management of neuroendocrine tumors of unknown origin. J. Natl. Compr. Cancer Netw. 2011, 9, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- Perren, A.; Couvelard, A.; Scoazec, J.-Y.; Costa, F.; Borbath, I.; Fave, G.D.; Gorbounova, V.; Gross, D.; Grossman, A.; Jensen, R.T.; et al. ENETS consensus guidelines for the standards of care in neuroendocrine tumors: Pathology-diagnosis and prognostic stratification. Neuroendocrinology 2017, 105, 196–200. [Google Scholar] [CrossRef]
- Fernandez, C.J.; Agarwal, M.; Pottakkat, B.; Haroon, N.N.; George, A.S.; Pappachan, J.M. Gastroenteropancreatic neuroendocrine neoplasms: A clinical snapshot. World J. Gastrointest. Surg. 2021, 13, 231–255. [Google Scholar] [CrossRef] [PubMed]
- Berner, A.M.; Pipinikas, C.; Ryan, A.; Dibra, H.; Moghul, I.; Webster, A.; Luong, T.V.; Thirlwell, C. Diagnostic Approaches to Neuroendocrine Neoplasms of Unknown Primary Site. Neuroendocrinology 2020, 110, 563–573. [Google Scholar] [CrossRef]
- Dasari, A.; Shen, C.; Halperin, D.; Zhao, B.; Zhou, S.; Xu, Y.; Shih, T.; Yao, J.C. Trends in the Incidence, Prevalence, and Survival Outcomes in Patients with Neuroendocrine Tumors in the United States. JAMA Oncol. 2017, 3, 1335–1342. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Carbonero, R.; Capdevila, J.; Crespo-Herrero, G.; Díaz-Pérez, J.A.; del Prado, M.P.M.; Orduña, V.A.; Sevilla-García, I.; Villabona-Artero, C.; Beguiristain-Gómez, A.; Llanos-Muñoz, M.; et al. Incidence, patterns of care and prognostic factors for outcome of gastroenteropancreatic neuroendocrine tumors (GEP-NETs): Results from the National Cancer Registry of Spain (RGETNE). Ann. Oncol. 2010, 21, 1794–1803. [Google Scholar] [CrossRef]
- Bhosale, P.; Shah, A.; Wei, W.; Varadhachary, G.; Johnson, V.; Shah, V.; Kundra, V. Carcinoid tumours: Predicting the location of the primary neoplasm based on the sites of metastases. Eur. Radiol. 2013, 23, 400–407. [Google Scholar] [CrossRef]
- Kurl, S.; Rytkönen, H.; Farin, P.; Ala-Opas, M.; Soimakallio, S. A primary carcinoid tumor of the kidney: A case report and review of the literature. Abdom. Imaging 1996, 21, 464–467. [Google Scholar] [CrossRef]
- Stroosma, O.B.; Delaere, K.P. Carcinoid tumours of the testis. BJU Int. 2008, 101, 1101–1105. [Google Scholar] [CrossRef] [PubMed]
- Radfar, L.; Fatahzadeh, M. Neuroendocrine carcinoma of the oral cavity: A case report and review of the literature. Gen. Dent. 2008, 56, 714–718. [Google Scholar] [PubMed]
- Widmeier, E.; Fuellgraf, H.; Waller, C.F. Complete remission of Cdx-2 positive primary testicular carcinoid tumor: 10-years follow-up and literature review. BMC Urol. 2020, 20, 197. [Google Scholar] [CrossRef] [PubMed]
- Okasho, K.; Ogawa, O.; Akamatsu, S. Narrative review of challenges in the management of advanced neuroendocrine prostate cancer. Transl. Androl. Urol. 2021, 10, 3953–3962. [Google Scholar] [CrossRef] [PubMed]
- Jagiella-Lodise, O.; Jagiella, V.; Weitman, E. An abdominal wall neuroendocrine tumor of unknown primary origin: A case report and review of the literature. Cancer Rep. 2022, 5, e1610. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; van Zante, A.; Granados, M.L. Combined Neuroendocrine and Squamous Cell Carcinoma of the Sinonasal Tract: A Morphologic and Immunohistochemical Analysis and Review of Literature. Head Neck Pathol. 2022, 16, 1019–1033. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Dai, S.; Xu, J.; Liu, L.; Yu, J.; Sun, T. Primary Neuroendocrine Tumor of the Breast: Current Understanding and Future Perspectives. Front. Oncol. 2022, 12, 848485. [Google Scholar] [CrossRef] [PubMed]
- Miquelestorena-Standley, E.; Dujardin, F.; Arbion, F.; Touzé, A.; Machet, L.; Velut, S.; Guyétant, S. Recurrent primary cutaneous mucinous carcinoma with neuroendocrine differentiation: Case report and review of the literature. J. Cutan. Pathol. 2014, 41, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Haq, S.; Batra, V.V.; Majumdar, K.; Javed, A.; Agarwal, A.K.; Sakhuja, P. Signet ring cell neuroendocrine tumor liver with mesenteric metastasis: Description of a rare phenomenon, with literature review. J. Cancer Res. Ther. 2015, 11, 658. [Google Scholar] [CrossRef]
- La Salvia, A.; Persano, I.; Trevisi, E.; Parlagreco, E.; Muratori, L.; Scagliotti, G.V.; Brizzi, M.P. Ocular metastases from neuroendocrine tumors: A literature review. Semin. Oncol. 2020, 47, 144–147. [Google Scholar] [CrossRef]
- Pavel, M.; O’Toole, D.; Costa, F.; Capdevila, J.; Gross, D.; Kianmanesh, R.; Krenning, E.; Knigge, U.; Salazar, R.; Pape, U.-F.; et al. ENETS Consensus Guidelines Update for the Management of Distant Metastatic Disease of Intestinal, Pancreatic, Bronchial Neuroendocrine Neoplasms (NEN) and NEN of Unknown Primary Site. Neuroendocrinology 2016, 103, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Pavel, M.; de Herder, W.W. ENETS Consensus Guidelines for the Standard of Care in Neuroendocrine Tumors. Neuroendocrinology 2017, 105, 193–195. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Turner, G.; King, B.; Jones, L.; Culliford, D.; McCance, D.; Ardill, J.; Johnston, B.T.; Poston, G.; Rees, M.; et al. Midgut neu-roendocrine tumours with liver metastases: Results of the UKINETS study. Endocr. Relat. Cancer 2009, 16, 885–894. [Google Scholar] [CrossRef]
- Citterio, D.; Pusceddu, S.; Facciorusso, A.; Coppa, J.; Milione, M.; Buzzoni, R.; Bongini, M.; Debraud, F.; Mazzaferro, V. Primary tumour resection may improve survival in functional well-differentiated neuroendocrine tumours metastatic to the liver. Eur. J. Surg. Oncol. (EJSO) 2016, 43, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Capurso, G.; Rinzivillo, M.; Bettini, R.; Boninsegna, L.; Fave, G.D.; Falconi, M. Systematic review of resection of primary midgut carcinoid tumour in patients with unresectable liver metastases. Br. J. Surg. 2012, 99, 1480–1486. [Google Scholar] [CrossRef] [PubMed]
- Keutgen, X.M.; Nilubol, N.; Glanville, J.; Sadowski, S.M.; Liewehr, D.J.; Venzon, D.J.; Steinberg, S.M.; Kebebew, E. Resection of primary tumor site is associated with prolonged survival in metastatic nonfunctioning pancreatic neuroendocrine tumors. Surgery 2016, 159, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Rossi, R.E.; Corti, F.; Pusceddu, S.; Milione, M.; Coppa, J.; Masoni, B.; Oldani, S.; Sabella, G.; Cafaro, P.; Repici, A. Multidisciplinary Approach to the Diagnosis of Occult Primary Neuroendocrine Neoplasm: A Clinical Challenge. J. Clin. Med. 2023, 12, 5537. [Google Scholar] [CrossRef]
- Bellizzi, A.M. Immunohistochemistry in the diagnosis and classification of neuroendocrine neoplasms: What can brown do for you? Hum. Pathol. 2020, 96, 8–33. [Google Scholar] [CrossRef] [PubMed]
- Juhlin, C.C.; Zedenius, J.; Höög, A. Metastatic Neuroendocrine Neoplasms of Unknown Primary: Clues from Pathology Workup. Cancers 2022, 14, 2210. [Google Scholar] [CrossRef] [PubMed]
- Savelli, G.; Lucignani, G.; Seregni, E.; Marchian, A.; Serafini, G.; Aliberti, G.; Villano, C.; Maccauro, M.; Bombardieri, E. Feasibility of somatostatin receptor scintigraphy in the detection of occult primary gastro-entero-pancreatic (GEP) neuroendocrine tumours. Nucl. Med. Commun. 2004, 25, 445–449. [Google Scholar] [CrossRef]
- Santhanam, P.; Chandramahanti, S.; Kroiss, A.; Yu, R.; Ruszniewski, P.; Kumar, R.; Taïeb, D. Nuclear imaging of neuroendocrine tumors with unknown primary: Why, when and how? Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 1144–1155. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, V.; Kunikowska, J.; Baudin, E.; Bodei, L.; Bouvier, C.; Capdevila, J.; Cremonesi, M.; de Herder, W.W.; Dromain, C.; Falconi, M.; et al. Consensus on molecular imaging and theranostics in neuroendocrine neoplasms. Eur. J. Cancer 2021, 146, 56–73. [Google Scholar] [CrossRef] [PubMed]
- Balogova, S.; Talbot, J.-N.; Nataf, V.; Michaud, L.; Huchet, V.; Kerrou, K.; Montravers, F. 18F-Fluorodihydroxyphenylalanine vs other radiopharmaceuticals for imaging neuroendocrine tumours according to their type. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 943–966. [Google Scholar] [CrossRef] [PubMed]
- Menda, Y.; O’Dorisio, T.M.; Howe, J.R.; Schultz, M.; Dillon, J.S.; Dick, D.; Watkins, G.L.; Ginader, T.; Bushnell, D.L.; Sunderland, J.J.; et al. Localization of Unknown Primary Site with 68Ga-DOTATOC PET/CT in Patients with Metastatic Neuroendocrine Tumor. J. Nucl. Med. 2017, 58, 1054–1057. [Google Scholar] [CrossRef] [PubMed]
- Graham, M.M.; Gu, X.; Ginader, T.; Breheny, P.; Sunderland, J.J. 68Ga-DOTATOC Imaging of Neuroendocrine Tumors: A Systematic Review and Metaanalysis. J. Nucl. Med. 2017, 58, 1452–1458. [Google Scholar] [CrossRef]
- Sanli, Y.; Garg, I.; Kandathil, A.; Zanetti, M.J.B.; Kuyumcu, S.; Subramaniam, R.M. Neuroendocrine Tumor Diagnosis and Man-agement: 68Ga-DOTATATE PET/CT. AJR Am. J. Roentgenol. 2018, 211, 267–277. [Google Scholar] [CrossRef]
- De Dosso, S.; Treglia, G.; Pascale, M.; Tamburello, A.; Santhanam, P.; Kroiss, A.S.; Mestre, R.P.; Saletti, P.; Giovanella, L. Detection rate of unknown primary tumour by using somatostatin receptor PET/CT in patients with metastatic neuroendocrine tumours: A meta-analysis. Endocrine 2019, 64, 456–468. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.; McNeely, P.; Menda, Y. Nuclear Imaging of Neuroendocrine Tumors. Surg. Oncol. Clin. N. Am. 2020, 29, 209–221. [Google Scholar] [CrossRef]
- Ma, H.; Kan, Y.; Yang, J.-G. Clinical value of 68Ga-DOTA-SSTR PET/CT in the diagnosis and detection of neuroendocrine tumors of unknown primary origin: A systematic review and meta-analysis. Acta Radiol. 2021, 62, 1217–1228. [Google Scholar] [CrossRef]
- Hubalewska-Dydejczyk, A.; Kulig, J.; Szybinski, P.; Mikolajczak, R.; Pach, D.; Sowa-Staszczak, A.; Fröss-Baron, K.; Huszno, B. Radio-guided surgery with the use of [99mTc-EDDA/HYNIC]octreotate in intra-operative detection of neuroendocrine tumours of the gastrointestinal tract. Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 1545–1555. [Google Scholar] [CrossRef]
- Wang, Y.Z.; Chauhan, A.; Rau, J.; Diebold, A.E.; Opoku-Boateng, A.; Ramcharan, T.; Boudreaux, J.P.; Woltering, E.A. Neuroendocrine tumors (NETs) of unknown primary: Is early surgical exploration and aggressive debulking justifiable? Chin. Clin. Oncol. 2016, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Moertel, C.G.; Kvols, L.K.; O’Connell, M.J.; Rubin, J. Treatment of neuroendocrine carcinomas with combined etoposide and cisplatin. Evidence of major therapeutic activity in the anaplastic variants of these neoplasms. Cancer 1991, 68, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Mitry, E.; Baudin, E.; Ducreux, M.; Sabourin, J.-C.; Rufié, P.; Aparicio, T.; Lasser, P.; Elias, D.; Duvillard, P.; Schlumberger, M.; et al. Treatment of poorly differentiated neuroendocrine tumours with etoposide and cisplatin. Br. J. Cancer 1999, 81, 1351–1355. [Google Scholar] [CrossRef] [PubMed]
- Fjällskog, M.L.; Granberg, D.P.; Welin, S.L.; Eriksson, C.; Oberg, K.E.; Janson, E.T.; Eriksson, B.K. Treatment with cisplatin and etoposide in patients with neuroendocrine tumors. Cancer 2001, 92, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Brandi, G.; Paragona, M.; Campana, D. Good performance of platinum-based chemotherapy for high-grade gastroentero-pancreatic and unknown primary neuroendocrine neoplasms. J. Chemother. 2018, 30, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Lobins, R.; Floyd, J. Small cell carcinoma of unknown primary. Semin. Oncol. 2007, 34, 39–42. [Google Scholar] [CrossRef]
- Zhang, P.; Li, J.; Li, J.; Zhang, X.; Zhou, J.; Wang, X.; Peng, Z.; Shen, L.; Lu, M. Etoposide and cisplatin versus irinotecan and cisplatin as the first-line therapy for patients with advanced, poorly differentiated gastroenteropancreatic neuroendocrine carcinoma: A randomized phase 2 study. Cancer 2020, 126 (Suppl. S9), 2086–2092. [Google Scholar] [CrossRef] [PubMed]
- Sorbye, H.; Welin, S.; Langer, S.W.; Vestermark, L.W.; Holt, N.; Osterlund, P.; Dueland, S.; Hofsli, E.; Guren, M.G.; Ohrling, K.; et al. Predictive and prognostic factors for treatment and survival in 305 patients with advanced gastrointestinal neuroendocrine carcinoma (WHO G3): The NORDIC NEC study. Ann. Oncol. 2012, 24, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Eads, J.R.; Catalano, P.J.; Fisher, G.A.; Rubin, D.; Iagaru, A.; Klimstra, D.S.; Konda, B.; Kwong, M.S.; Chan, J.A.; De Jesus-Acosta, A.; et al. Randomized phase II study of platinum and etoposide (EP) versus temozolomide and capecitabine (CAPTEM) in patients (pts) with advanced G3 non-small cell gastroenteropancreatic neuroendocrine neoplasms (GEPNENs): ECOG-ACRIN EA2142. J. Clin. Oncol. 2022, 40 (Suppl. S16), 4020. [Google Scholar] [CrossRef]
- Hainsworth, J.D.; Erland, J.B.; Kalman, L.A.; Schreeder, M.T.; Greco, F.A. Carcinoma of unknown primary site: Treatment with 1-hour paclitaxel, carboplatin, and extended-schedule etoposide. J. Clin. Oncol. 1997, 15, 2385–2393. [Google Scholar] [CrossRef]
- Hainsworth, J.D.; Spigel, D.R.; Litchy, S.; Greco, F.A. Phase II Trial of Paclitaxel, Carboplatin, and Etoposide in Advanced Poorly Differentiated Neuroendocrine Carcinoma: A Minnie Pearl Cancer Research Network Study. J. Clin. Oncol. 2006, 24, 3548–3554. [Google Scholar] [CrossRef] [PubMed]
- Hadoux, J.; Afchain, P.; Walter, T.; Tougeron, D.; Hautefeuille, V.; Monterymard, C.; Lorgis, V.; Thuillier, F.; Baudin, E.; Scoazec, J.Y.; et al. FOLFIRINEC: A randomized phase II trial of mFOLFIRINOX vs platinum-etoposide for metastatic neuroendocrine carcinoma of gastroenteropancreatic or unknown origin. Dig. Liver Dis. 2021, 53, 824–829. [Google Scholar] [CrossRef] [PubMed]
- Riesco Martinez, M.C.; Capdevila Castillon, J.; Alonso, V.; Jimenez-Fonseca, P.; Teule, A.; Grande, E.; Sevilla, I.; Benavent, M.; Alonso-Gordoa, T.; Custodio, A.; et al. Final overall survival results from the NICE-NEC trial (GETNE-T1913): A phase II study of nivolumab and platinum-doublet chemotherapy (CT) in untreated advanced G3 neuroendocrine neoplasms (NENs) of gastro-enteropancreatic (GEP) or unknown (UK) origin. Ann. Oncol. 2022, 33 (Suppl. S7), S769. [Google Scholar] [CrossRef]
- Hentic, O.; Hammel, P.; Couvelard, A.; Rebours, V.; Zappa, M.; Palazzo, M.; Maire, F.; Goujon, G.; Gillet, A.; Lévy, P.; et al. FOLFIRI regimen: An effective second-line chemotherapy after failure of etoposide–platinum combination in patients with neuroendocrine carcinomas grade 3. Endocr. Relat. Cancer 2012, 19, 751–757. [Google Scholar] [CrossRef]
- Welin, S.; Sorbye, H.; Sebjornsen, S.; Knappskog, S.; Busch, C.; Öberg, K. Clinical effect of temozolomide-based chemotherapy in poorly differentiated endocrine carcinoma after progression on first-line chemotherapy. Cancer 2011, 117, 4617–4622. [Google Scholar] [CrossRef] [PubMed]
- Hadoux, J.; Malka, D.; Planchard, D.; Scoazec, J.Y.; Caramella, C.; Guigay, J.; Boige, V.; Leboulleux, S.; Burtin, P.; Berdelou, A.; et al. Post-first-line FOLFOX chemotherapy for grade 3 neuroendocrine carcinoma. Endocr. Relat. Cancer 2015, 22, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Takeda, Y.; Okubo, N.; Suzuki, A.; Tokuhisa, M.; Hiroshima, Y.; Ichikawa, Y. Phase II study of temozolomide monotherapy in patients with extrapulmonary neuroendocrine carcinoma. Cancer Sci. 2021, 112, 1936–1942. [Google Scholar] [CrossRef]
- Walter, T.; Lievre, A.; Coriat, R.; Malka, D.; Elhajbi, F.; Di Fiore, F.; Hentic, O.; Smith, D.; Hautefeuille, V.; Roquin, G.; et al. Bevacizumab plus FOLFIRI after failure of platinum-etoposide first-line chemotherapy in patients with advanced neuroendocrine carcinoma (PRODIGE 41-BEVANEC): A randomised, multicentre, non-comparative, open-label, phase 2 trial. Lancet Oncol. 2023, 24, 297–306. [Google Scholar] [CrossRef]
- McNamara, M.G.; Swain, J.; Craig, Z.; Sharma, R.; Faluyi, O.; Wadsley, J.; Morgan, C.; Wall, L.R.; Chau, I.; Reed, N.; et al. NET-02: A randomised, non-comparative, phase II trial of nal-IRI/5-FU or docetaxel as second-line therapy in patients with progressive poorly differentiated extra-pulmonary neuroendocrine carcinoma. eClinicalMedicine 2023, 60, 102015. [Google Scholar] [CrossRef]
- Hadoux, J.; Walter, T.; Kanaan, C.; Hescot, S.; Hautefeuille, V.; Perrier, M.; Tauveron, I.; Laboureau, S.; Cao, C.D.; Petorin, C.; et al. Second-line treatment and prognostic factors in neuroendocrine carcinoma: The RBNEC study. Endocr. Relat. Cancer 2022, 29, 569–580. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Russell, J.; Hamid, O.; Bhatia, S.; Terheyden, P.; D’Angelo, S.P.; Shih, K.C.; Lebbé, C.; Linette, G.P.; Milella, M.; et al. Avelumab in Patients with Chemotherapy-Refractory Metastatic Merkel Cell Carcinoma: A Multicentre, Single-Group, Open-Label, Phase 2 Trial. Lancet Oncol. 2016, 17, 1374–1385. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.P.; Russell, J.; Lebbé, C.; Chmielowski, B.; Gambichler, T.; Grob, J.J.; Kiecker, F.; Rabinowits, G.; Terheyden, P.; Zwiener, I.; et al. Efficacy and Safety of First-Line Avelumab Treatment in Patients with Stage IV Metastatic Merkel Cell Carcinoma: A Pre-planned Interim Analysis of a Clinical Trial. JAMA Oncol. 2018, 4, e180077. [Google Scholar] [CrossRef] [PubMed]
- Raj, N.; Chan, J.A.; Wang, S.J.; Aggarwal, R.R.; Calabrese, S.; DeMore, A.; Fong, L.; Grabowsky, J.; Hope, T.A.; Kolli, K.P.; et al. Pembrolizumab alone and pembrolizumab plus chemotherapy in previously treated, extrapulmonary poorly differentiated neuroendocrine carcinomas. Br. J. Cancer 2023, 129, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Vijayvergia, N.; Dasari, A.; Deng, M.; Litwin, S.; Al-Toubah, T.; Alpaugh, R.K.; Dotan, E.; Hall, M.J.; Ross, N.M.; Runyen, M.M.; et al. Pembrolizumab monotherapy in patients with previously treated metastatic high-grade neuroendocrine neoplasms: Joint analysis of two prospective, non-randomised trials. Br. J. Cancer 2020, 122, 1309–1314. [Google Scholar] [CrossRef] [PubMed]
- Fottner, C.; Apostolidis, L.; Ferrata, M.; Krug, S.; Michl, P.; Schad, A.; Roth, W.; Jaeger, D.; Galle, P.R.; Weber, M.M. A phase II, open label, multicenter trial of avelumab in patients with advanced, metastatic high-grade neuroendocrine carcinomas NEC G3 (WHO 2010) progressive after first-line chemotherapy (AVENEC). J. Clin. Oncol. 2019, 37 (Suppl. S15), 4103. [Google Scholar] [CrossRef]
- Yao, J.C.; Strosberg, J.; Fazio, N.; Pavel, M.E.; Bergsland, E.; Ruszniewski, P.; Halperin, D.M.; Li, D.; Tafuto, S.; Raj, N.; et al. Spartalizumab in metastatic, well/poorly-differentiated neuroendocrine neoplasms. Endocr.-Relat. Cancer 2021, 28, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.L.; Rodriguez-Freixinos, V.; Doherty, M.; Wasson, K.; Iscoe, N.; Raskin, W.; Hallet, J.; Myrehaug, S.; Law, C.; Thawer, A.; et al. Avelumab in unresectable/metastatic, progressive, grade 2-3 neuroendocrine neoplasms (NENs): Combined results from NET-001 and NET-002 trials. Eur. J. Cancer 2022, 169, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.A.; Raj, N.P.; Aggarwal, R.R.; Calabrese, S.; DeMore, A.; Dhawan, M.S.; Fattah, D.; Fong, L.; Grabowsky, J.; Hope, T.A.; et al. Phase II study of pembrolizumab-based therapy in previously treated extrapulmonary poorly differentiated neuroendocrine carcinomas: Results of Part B (pembrolizumab + chemotherapy). J. Clin. Oncol. 2021, 39 (Suppl. S15), 4148. [Google Scholar] [CrossRef]
- Patel, S.P.; Mayerson, E.; Chae, Y.K.; Strosberg, J.; Wang, J.; Konda, B.; Hayward, J.; McLeod, C.M.; Chen, H.X.; Sharon, E.; et al. A phase II basket trial of Dual Anti-CTLA-4 and Anti-PD-1 Blockade in Rare Tumors (DART) SWOG S1609: High-grade neuroendocrine neoplasm cohort. Cancer 2021, 127, 3194–3201. [Google Scholar] [CrossRef]
- Klein, O.; Kee, D.; Markman, B.; Michael, M.; Underhill, C.; Carlino, M.S.; Jackett, L.; Lum, C.; Scott, C.; Nagrial, A.; et al. Immunotherapy of ipilimumab and nivolumab in patients with advanced neuroendocrine tumours: A subgroup analysis of the CA209–538 clinical trial for rare cancers. Clin. Cancer Res. 2020, 26, 4454–4459. [Google Scholar] [CrossRef]
- Capdevila, J.; Hernando, J.; Teule, A.; Lopez, C.; Garcia-Carbonero, R.; Benavent, M.; Custodio, A.; Garcia-Alvarez, A.; Cubillo, A.; Alonso, V.; et al. Durvalumab plus tremelimumab for the treatment of advanced neuroendocrine neoplasms of gastroentero-pancreatic and lung origin. Nat. Commun. 2023, 14, 2973. [Google Scholar] [CrossRef]
- Pavel, M.E.; Fischer von Weikersthal, L.; Klöppel, G.; Krause, K.; Apostolidis, L. Safety and efficacy of everolimus (EVE) as second–line treatment in neuroendocrine neoplasms G3 (NEN G3)—An AIO phase II study (EVINEC). Ann. Oncol. 2023, 34 (Suppl. S2), S702. [Google Scholar] [CrossRef]
- Weber, M.; Apostolidis, L.; Krug, S.; Rinke, A.; Gruen, B.; Michl, P.; Gress, T.; Wagner, D.; Roth, W.; Mettler, E.; et al. Activity and safety of avelumab alone or in combination with cabozantinib in patients with advanced high grade neuroendocrine neoplasias (NEN G3) progressing after chemotherapy. The phase II, open-label, multicenter AVENEC and CABOAVENEC trials. Ann. Oncol. 2023, 34 (Suppl. S2), S702. [Google Scholar] [CrossRef]
- Capdevila Castillon, J.; Molina-Cerrillo, J.; Benavent Viñuales, M.; Garcia-Carbonero, R.; Teule, A.; Custodio, A.; Jimenez-Fonseca, P.; Lopez, C.; Hierro, C.; Carmona-Bayonas, A.; et al. Cabozantinib plus Atezolizumab in Advanced and Progressive Neoplasms of the Endocrine System: A multi-cohort Basket Phase II Trial (CABATEN/GETNE-T1914). Ann. Oncol. 2023, 34 (Suppl. S2), S498. [Google Scholar] [CrossRef]
- Lu, M.; Wang, Y.; Zhang, P.; Shen, L. Surufatinib combined with sintilimab and IBI310 in the treatment of high-grade advanced-neuroendocrine neoplasm: A single arm, open-label, multicenter study. Ann. Oncol. 2022, 33 (Suppl. S7), S770. [Google Scholar] [CrossRef]
- Caplin, M.E.; Pavel, M.; Ćwikła, J.B.; Phan, A.T.; Raderer, M.; Sedláčková, E.; Cadiot, G.; Wolin, E.M.; Capdevila, J.; Wall, L.; et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N. Engl. J. Med. 2014, 371, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Caplin, M.E.; Pavel, M.; Phan, A.T.; Ćwikła, J.B.; Sedláčková, E.; Thanh, X.T.; Wolin, E.M.; Ruszniewski, P.; CLARINET Investigators. Lanreotide autogel/depot in advanced enteropancreatic neuroendocrine tumours: Final results of the CLARINET openlabel ex-tension study. Endocrine 2021, 71, 502–551. [Google Scholar] [CrossRef] [PubMed]
- Faiss, S.; Pape, U.F.; Böhmig, M.; Dörffel, Y.; Mansmann, U.; Golder, W.; Riecken, E.O.; Wiedenmann, B.; International Lanreotide and Interferon Alfa Study Group. Prospective, randomized, multicenter trial on the antiproliferative effect of lanreotide, interferon alfa, and their combination for therapy of metastatic neuroendocrine gastroenteropancreatic tumors—The International Lanreo-tide and Interferon Alfa Study Group. J. Clin. Oncol. 2003, 21, 2689–2696. [Google Scholar]
- Ito, T.; Honma, Y.; Hijioka, S.; Kudo, A.; Fukutomi, A.; Nozaki, A.; Kimura, Y.; Motoi, F.; Isayama, H.; Komoto, I.; et al. Phase II study of lanreotide autogel in Japanese patients with unresectable or metastatic well-differentiated neuroendocrine tumors. Investig. New Drugs 2017, 35, 499–508. [Google Scholar] [CrossRef]
- Kwekkeboom, D.J.; Mueller-Brand, J.; Paganelli, G.; Anthony, L.B.; Pauwels, S.; Kvols, L.K.; O’Dorisio, T.M.; Valkema, R.; Bodei, L.; Chinol, M.; et al. Overview of results of peptide receptor radionuclide therapy with 3 radiolabeled somatostatin analogs. J. Nucl. Med. 2005, 46, 62S–66S. [Google Scholar]
- Strosberg, J.; El-Haddad, G.; Wolin, E.; Hendifar, A.; Yao, J.; Chasen, B.; Mittra, E.; Kunz, P.L.; Kulke, M.H.; Jacene, H.; et al. Phase 3 Trial of 177Lu-Dotatate for Midgut Neuroendocrine Tumors. N. Engl. J. Med. 2017, 376, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Strosberg, J.R.; Caplin, M.E.; Kunz, P.L.; Kunz, P.L.; Ruszniewski, P.B.; Bodei, L.; Hendifar, A.; Mittra, E.; Wolin, E.M.; Yao, J.C.; et al. 177Lu-Dotatate plus long-acting octreotide versus high-dose long-acting octreotide in patients with midgut neuroendocrine tumours (NETTER-1): Final overall survival and long-term safety results from an open-label, randomised, controlled, phase 3 trial. Lancet Oncol. 2021, 22, 1752–1763. [Google Scholar] [CrossRef] [PubMed]
- Saravana-Bawan, B.; Bajwa, A.; Paterson, J.; McEwan, A.J.B.; McMullen, T.P.W. Efficacy of 177Lu Peptide Receptor Radionuclide Therapy for the Treatment of Neuroendocrine Tumors: A Meta-analysis. Clin. Nucl. Med. 2019, 44, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Hertelendi, M.; Belguenani, O.; Cherfi, A.; Folitar, I.; Kollar, G.; Polack, B.D. Efficacy and Safety of [177Lu]Lu-DOTA-TATE in Adults with Inoperable or Metastatic Somatostatin Receptor-Positive Pheochromocytomas/Paragangliomas, Bronchial and Unknown Origin Neuroendocrine Tumors, and Medullary Thyroid Carcinoma: A Systematic Literature Review. Biomedicines 2023, 11, 1024. [Google Scholar] [CrossRef] [PubMed]
- Forrer, F.; Waldherr, C.; Maecke, H.R.; Mueller-Brand, J. Targeted radionuclide therapy with 90Y-DOTATOC in patients with neuroendocrine tumors. J. Anticancer Res. 2006, 26, 703–707. [Google Scholar]
- Seregni, E.; Maccauro, M.; Chiesa, C.; Mariani, L.; Pascali, C.; Mazzaferro, V.; De Braud, F.; Buzzoni, R.; Milione, M.; Lorenzoni, A.; et al. Treatment with tandem [90Y]DOTATATE and [177Lu]DOTA-TATE of neuroendocrine tumours refractory to conventional therapy. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Frilling, A.; Weber, F.; Saner, F.; Bockisch, A.; Hofmann, M.; Mueller-Brand, J.; Broelsch, C.E. Treatment with (90)Y- and (177)Lu-DOTATOC in patients with metastatic neuroendocrine tumors. Surgery 2006, 140, 968–977; Discussion 976–977. [Google Scholar] [CrossRef] [PubMed]
- Thang, S.P.; Lung, M.S.; Kong, G.; Hofman, M.S.; Callahan, J.; Michael, M.; Hicks, R.J. Peptide receptor radionuclidetherapy (PRRT) in European Neuroendocrine Tumour Society (ENETS)grade 3 (G3) neuroendocrine neoplasia (NEN)–a single-institution retro-spective analysis. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 262–277. [Google Scholar] [CrossRef]
- Carlsen, E.A.; Fazio, N.; Granberg, D.; Grozinsky-Glasberg, S.; Ahmadzadehfar, H.; Grana, C.M.; Zandee, W.T.; Cwikla, J.; Walter, M.A.; Oturai, P.S.; et al. Peptide receptor radionuclidetherapy in gastroenteropancreatic NEN G3: A multicenter cohortstudy. Endocr. Relat. Cancer 2019, 26, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, S.; Severi, S.; Ianniello, A.; Sansovini, M.; Ambrosetti, A.; Bongiovanni, A.; Scarpi, E.; Di Mauro, F.; Rossi, A.; Matteucci, F.; et al. Investigation of receptor radionuclide therapy with (177)Lu-DOTATATE in patients with GEP-NEN and a high Ki-67 proliferation index. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 923–930. [Google Scholar] [CrossRef]
- Zhang, J.; Kulkarni, H.R.; Singh, A.; Niepsch, K.; Müller, D.; Baum, R.P. Peptide receptor radionuclide therapy in grade 3 neuro-endocrine neoplasms: Safety and survival analysis in 69 patients. J. Nucl. Med. 2019, 60, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.C.; Phan, A.T.; Chang, D.Z.; Wolff, R.A.; Hess, K.; Gupta, S.; Jacobs, C.; Mares, J.E.; Landgraf, A.N.; Rashid, A.; et al. Efficacy of RAD001 (everolimus) and octreotide LAR in advanced low- to intermediate-grade neuroendocrine tumors: Results of a phase II trial. J. Clin. Oncol. 2008, 26, 4311–4318. [Google Scholar] [CrossRef] [PubMed]
- Bajetta, E.; Catena, L.; Pusceddu, S.; Spada, F.; Iannacone, C.; Sarno, I.; Di Menna, G.; Dottorini, L.; Marte, A.M. Everolimus in Combination with Octreotide Long-Acting Repeatable in a First-Line Setting for Patients with Neuroendocrine Tumors: A 5-Year Update. Neuroendocrinology 2018, 106, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Carnaghi, C.; Buzzoni, R.; Pommier, R.F.; Raderer, M.; Tomasek, J.; Lahner, H.; Valle, J.W.; Voi, M.; Bubuteishvili-Pacaud, L.; et al. Everolimus in Neuroendocrine Tumors of the Gastrointestinal Tract and Unknown Primary. Neuroendocrinology 2018, 106, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Shen, L.; Zhou, Z.; Li, J.; Bai, C.; Chi, Y.; Li, Z.; Xu, N.; Li, E.; Liu, T.; et al. Surufatinib in advanced extrapancreatic neuroendocrine tumours (SANET-ep): A randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 1500–1512. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.A.; Geyer, S.; Ou, F.S.; Knopp, M.; Behr, S.; Zemla, T.; Acoba, J.; Shergill, A.; Wolin, E.M.; Halfdanarson, T.R.; et al. Alliance A021602: Phase III, double-blinded study of cabozantinib versus placebo for advanced neuroendocrine tumors (NET) after progression on prior therapy (CABINET). Ann. Oncol. 2023, 34 (Suppl. S2), S1292. [Google Scholar] [CrossRef]
- Chan, J.A.; Stuart, K.; Earle, C.C.; Clark, J.W.; Bhargava, P.; Miksad, R.; Blaszkowsky, L.; Enzinger, P.C.; Meyerhardt, J.A.; Zheng, H.; et al. Prospective study of bevacizumab plus temozolomide in patients with advanced neuroendocrine tumors. J. Clin. Oncol. 2012, 30, 2963–2968. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Farooqui, Z.; Murray, L.A.; Weiss, H.L.; Myint, Z.W.; Raajasekar, A.K.A.; Evers, B.M.; Arnold, S.; Anthony, L. Capecitabine and Temozolomide in Neuroendocrine Tumor of Unknown Primary. J. Oncol. 2018, 2018, 3519247. [Google Scholar] [CrossRef] [PubMed]
- Spada, F.; Maisonneuve, P.; Fumagalli, C.; Marconcini, R.; Gelsomino, F.; Antonuzzo, L.; Campana, D.; Puliafito, I.; Rossi, G.; Faviana, P.; et al. Temozolomide alone or in combination with capecitabine in patients with advanced neuroendocrine neoplasms: An Italian multicenter real-world analysis. Endocrine 2020, 72, 268–278. [Google Scholar] [CrossRef]
- Cives, M.; Ghayouri, M.; Morse, B.; Brelsford, M.; Black, M.; Rizzo, A.; Meeker, A.; Strosberg, J. Analysis of potential response pre-dictors to capecitabine/temozolomide in metastatic pancreatic neuroendocrine tumors. Endocr. Relat. Cancer 2016, 23, 759–767. [Google Scholar] [CrossRef]
- Kulke, M.H.; Hornick, J.L.; Frauenhoffer, C.; Hooshmand, S.; Ryan, D.P.; Enzinger, P.C.; Meyerhardt, J.A.; Clark, J.W.; Stuart, K.; Fuchs, C.S.; et al. O 6-methylguanine DNA methyltransferase deficiency and response to temozolomide-based therapy in patients with neuroendocrine tumors. Clin. Cancer Res. 2009, 15, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Walter, T.; Lecomte, T.; Hadoux, J.; Niccoli, P.; Saban-Roche, L.; Gaye, E.; Guimbaud, R.; Baconnier, M.; Hautefeuille, V.; Cao, C.D.; et al. Alkylating agent-based vs oxaliplatin-based chemotherapy in neuroendocrine tumours according to the O6-methylguanine-DNA methyltransferase (MGMT) status: A randomized phase II study (MGMT-NET) on behalf of the French Group of Endocrine Tumors (GTE) and ENDOCAN-RENATEN network. Ann. Oncol. 2023, 34 (Suppl. S2), S1292–S1293. [Google Scholar]
- Meyer, T.; Qian, W.; Caplin, M.E.; Armstrong, G.; Lao-Sirieix, S.-H.; Hardy, R.; Valle, J.W.; Talbot, D.C.; Cunningham, D.; Reed, N.; et al. Capecitabine and streptozocin±cisplatin in advanced gastroenteropancreatic neuroendocrine tumours. Eur. J. Cancer 2014, 50, 902–911. [Google Scholar] [CrossRef] [PubMed]
- Bajetta, E.; Catena, L.; Procopio, G.; De Dosso, S.; Bichisao, E.; Ferrari, L.; Martinetti, A.; Platania, M.; Verzoni, E.; Formisano, B.; et al. Are capecitabine and oxaliplatin (XELOX) suitable treatments for progressing low-grade and high-grade neuroendocrine tumours? Cancer Chemother. Pharmacol. 2007, 59, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Spada, F.; Antonuzzo, L.; Marconcini, R.; Radice, D.; Antonuzzo, A.; Ricci, S.; Di Costanzo, F.; Fontana, A.; Gelsomino, F.; Luppi, G.; et al. Oxaliplatin-Based Chemotherapy in Advanced Neuroendocrine Tumors: Clinical Outcomes and Preliminary Correlation with Biological Factors. Neuroendocrinology 2016, 103, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Ollivier, S.; Fonck, M.; Bécouarn, Y.; Brunet, R. Dacarbazine, fluorouracil, and leucovorin in patients with advanced neuroen-docrine tumors: A phase II trial. Am. J. Clin. Oncol. 1998, 21, 237–240. [Google Scholar] [CrossRef]
- Strosberg, J.R.; Mizuno, N.; Doi, T.; Grande, E.; Delord, J.P.; Shapira-Frommer, R.; Bergsland, E.; Shah, M.; Fakih, M.; Takahashi, S.; et al. Efficacy and safety of pembrolizumab in previously treated advanced neuroendocrine tumors: Results from the phase 2 KEYNOTE-158 study. Clin. Cancer Res. 2020, 26, 2124–2130. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.P.; Othus, M.; Chae, Y.K.; Giles, F.J.; Hansel, D.E.; Singh, P.P.; Fontaine, A.; Shah, M.H.; Kasi, A.; Baghdadi, T.A.; et al. A phase II basket trial of dual anti–CTLA-4 and anti–PD-1 blockade in rare tumors (DART SWOG 1609) in patients with non-pancreatic neuroendocrine tumors. Clin. Cancer Res. 2020, 26, 2290–2296. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Zhang, P.; Zhang, Y.; Li, Z.; Gong, J.; Li, J.; Li, J.; Li, Y.; Zhang, X.; Lu, Z.; et al. Efficacy, safety, and biomarkers of toripalimab in patients with recurrent or metastatic neuroendocrine neoplasms: A multiple-center phase Ib trial. Clin. Cancer Res. 2020, 26, 2337–2345. [Google Scholar] [CrossRef]
- Cavalcoli, F.; Rausa, E.; Conte, D.; Nicolini, A.F.; Massironi, S. Is there still a role for the hepatic locoregional treatment of meta-static neuroendocrine tumors in the era of systemic targeted therapies? World J. Gastroenterol. 2017, 23, 2640–2650. [Google Scholar] [CrossRef]
- Kim, H.S.; Shaib, W.L.; Zhang, C.; Nagaraju, G.P.; Wu, C.; Alese, O.B.; Chen, Z.; Brutcher, E.; Renfroe, M.; El-Rayes, B.F. Phase 1b study of pasireotide, everolimus, and selective internal radioembolization therapy for unresectable neuroendocrine tumors with hepatic metastases. Cancer 2018, 124, 1992–2000. [Google Scholar] [CrossRef] [PubMed]
- King, J.; Quinn, R.; Glenn, D.M.; Janssen, J.; Tong, D.; Liaw, W.; Morris, D.L. Radioembolization with selective internal radiation microspheres for neuroendocrine liver metastases. Cancer 2008, 113, 921–929. [Google Scholar] [CrossRef]
- Blonski, W.C.; Reddy, K.R.; Shaked, A.; Siegelman, E.; Metz, D.C. Liver transplantation for metastatic neuroendocrine tumor: A case report and review of the literature. World J. Gastroenterol. 2005, 11, 7676–7683. [Google Scholar] [CrossRef] [PubMed]
- Moradi, A.; Lamsehchi, N.; Khaki, S.; Nasiri-Toosi, M.; Jafarian, A. Liver Transplant for Patients with Neuroendocrine Tumor: A Report of 2 Exceptional Cases and Literature Review. Exp. Clin. Transplant. 2023, 21, 578–585. [Google Scholar]
- Puccini, A.; Poorman, K.; Salem, M.E.; Soldato, D.; Seeber, A.; Goldberg, R.M.; Shields, A.F.; Xiu, J.; Battaglin, F.; Berger, M.D.; et al. Comprehensive Genomic Profiling of Gastroenteropancreatic Neuroendocrine Neoplasms (GEP-NENs). Clin. Cancer Res. 2020, 26, 5943–5951. [Google Scholar] [CrossRef]
- Garcia-Carbonero, R.; Anton-Pascual, B.; Modrego, A.; Del Carmen Riesco-Martinez, M.; Lens-Pardo, A.; Carretero-Puche, C.; Ru-bio-Cuesta, B.; Soldevilla, B. Advances in the Treatment of Gastroenteropancreatic Neuroendocrine Carcinomas: Are we Moving Forward? Endocr. Rev. 2023, 44, 724–736. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.H.; Basturk, O.; Sue, J.J.B.; Klimstra, D.S. A Practical Approach to the Classification of WHO Grade 3 (G3) Well-differentiated Neuroendocrine Tumor (WD-NET) and Poorly Differentiated Neuroendocrine Carcinoma (PD-NEC) of the Pancreas. Am. J. Surg. Pathol. 2016, 40, 1192–1202. [Google Scholar] [CrossRef]
- Derks, J.L.; Leblay, N.; Lantuejoul, S.; Dingemans, A.C.; Speel, E.M.; Fernandez-Cuesta, L. New Insights into the Molecular Characteristics of Pulmonary Carcinoids and Large Cell Neuroendocrine Carcinomas, and the Impact on Their Clinical Management. J. Thorac. Oncol. 2018, 13, 752–766. [Google Scholar] [CrossRef] [PubMed]
- Pusceddu, S.; Corti, F.; Milione, M.; Centonze, G.; Prinzi, N.; Torchio, M.; de Braud, F. Are Cyclin-Dependent Kinase 4/6 Inhibitors Without Future in Neuroendocrine Tumors? Oncologist 2020, 25, e1257–e1258. [Google Scholar] [CrossRef]
- Hendifar, A.E.; Ramirez, R.A.; Anthony, L.B.; Liu, E. Current Practices and Novel Techniques in the Diagnosis and Management of Neuroendocrine Tumors of Unknown Primary. Pancreas 2019, 48, 1111–1118. [Google Scholar] [CrossRef]
- Saller, J.J.; Haider, M.; Al-Diffalha, S.; Coppola, D. Benefit of Gene Expression Profiling in Gastrointestinal Neuroendocrine Tumors of Unknown Primary Origin. Anticancer Res. 2022, 42, 1381–1396. [Google Scholar] [CrossRef] [PubMed]
- Klempner, S.J.; Gershenhorn, B.; Tran, P.; Lee, T.K.; Erlander, M.G.; Gowen, K.; Schrock, A.B.; Morosini, D.; Ross, J.S.; Miller, V.A.; et al. BRAFV600E Mutations in High-Grade Colorectal Neuroendocrine Tumors May Predict Responsiveness to BRAF-MEK Combination Therapy. Cancer Discov. 2016, 6, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Burkart, J.; Owen, D.; Shah, M.H.; Abdel-Misih, S.R.Z.; Roychowdhury, S.; Wesolowski, R.; Haraldsdottir, S.; Reeser, J.W.; Samorodnitsky, E.; Smith, A.; et al. Targeting BRAF Mutations in High-Grade Neuroendocrine Carcinoma of the Colon. J. Natl. Compr. Cancer Netw. 2018, 16, 1035–1040. [Google Scholar] [CrossRef] [PubMed]
- Ricco, G.; Seminerio, R.; Andrini, E.; Malvi, D.; Gruppioni, E.; Altimari, A.; Zagnoni, S.; Campana, D.; Lamberti, G. BRAF V600E-mutated large cell neuroendocrine carcinoma responding to targeted therapy: A case report and review of the literature. Anti-Cancer Drugs 2023, 34, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Tamragouri, K.B.; Chung, J.; Lin, X.; Miller, V.; Ali, S.M.; Giles, F.J. Large-Cell Neuroendocrine Carcinoma of the Lung: A Focused Analysis of BRAF Alterations and Case Report of a BRAF Non-V600–Mutated Tumor Responding to Targeted Therapy. JCO Precis. Oncol. 2018, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Balbach, M.L.; Eisenberg, R.; Iams, W.T. De Novo KRAS G12C-Mutant SCLC: A Case Report. JTO Clin. Res. Rep. 2022, 3, 100306. [Google Scholar] [CrossRef] [PubMed]
- Saiki, M.; Omori, C.; Morikawa, H.; Shinohara, K.; Shimamura, S.; Ohkoshi, H.; Uchida, Y.; Inoue, T.; Kondo, T.; Ikemura, S.; et al. The First Case Report of Effective Treatment with Sotorasib for Metastatic Atypical Lung Carcinoid Harboring KRAS G12C Mutation and Aggressive Disseminated Lung Metastasis: A Case Report. JTO Clin. Res. Rep. 2023, 5, 100620. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Zheng, M.; Jin, Y.; Shen, X.; Shan, L.; Shen, L.; Sun, Y.; Chen, H.; Li, Y. ALK-Rearrangement Neuroendocrine Carcinoma of the Lung: A Comprehensive Study of a Rare Case Series and Review of Literature. OncoTargets Ther. 2018, 11, 4991–4998. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, N.; Fujita, A.; Saikai, T.; Takabatake, H.; Sotoshiro, M.; Sekine, K.; Kawana, A. Large Cell Neuroendocrine Carcinoma Harboring an Anaplastic Lymphoma Kinase (ALK) Rearrangement with Response to Alectinib. Intern. Med. 2018, 57, 713–716. [Google Scholar] [CrossRef]
- Shimizu, N.; Akashi, Y.; Fujii, T.; Shiono, H.; Yane, K.; Kitahara, T.; Ohta, Y.; Kakudo, K.; Wakasa, T. Use of ALK Immunohistochemistry for Optimal Therapeutic Strategy of Pulmonary Large-Cell Neuroendocrine Carcinoma and Identification of a Novel KIF5B–ALK Fusion Oncokinase. Anticancer Res. 2019, 39, 413–420. [Google Scholar] [CrossRef]
- Nakajima, M.; Uchiyama, N.; Shigemasa, R.; Matsumura, T.; Matsuoka, R.; Nomura, A. Atypical Carcinoid Tumor with Anaplastic Lymphoma Kinase (ALK) Rearrangement Successfully Treated by an ALK Inhibitor. Intern. Med. 2016, 55, 3151–3153. [Google Scholar] [CrossRef] [PubMed]
- Sigal, D.S.; Bhangoo, M.S.; Hermel, J.A.; Pavlick, D.C.; Frampton, G.; Miller, V.A.; Ross, J.S.; Ali, S.M. Comprehensive Genomic Profiling Identifies Novel NTRK Fusions in Neuroendocrine Tumors. Oncotarget 2018, 9, 35809–35812. [Google Scholar] [CrossRef] [PubMed]
- Bazhenova, L.; Liu, S.; Lin, J.; Lu, S.; Drilon, A.; Chawla, S.; Fakih, M.; Krzakowski, M.; Paz-Ares, L.; Blakely, C.; et al. Efficacy and safety of entrectinib in patients with locally advanced/metastatic NTRK fusion-positive (NTRK-FP) solid tumours. Ann. Oncol. 2021, 32 (Suppl. S5), S598–S599. [Google Scholar] [CrossRef]
- Demetri, G.D.; De Braud, F.; Drilon, A.; Siena, S.; Patel, M.R.; Cho, B.C.; Liu, S.; Ahn, M.-J.; Chiu, C.-H.; Lin, J.J.; et al. Updated Integrated Analysis of the Efficacy and Safety of Entrectinib in Patients With NTRK Fusion-Positive Solid Tumors. Clin. Cancer Res. 2022, 28, 1302–1312. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, K.P.; Borazanci, E.; Shaw, A.T.; Katayama, R.; Shimizu, Y.; Zhu, V.W.; Sun, T.Y.; Wakelee, H.A.; Madison, R.; Schrock, A.B.; et al. Phase I First-In-Human Study of Taletrectinib (DS-6051b/AB-106), a ROS1/TRK Inhibitor, in Patients With Advanced Solid Tumors. Clin. Cancer Res. 2020, 26, 4785–4794. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Cassier, P.A.; Siena, S.; Garralda, E.; Paz-Ares, L.; Garrido, P.; Nadal, E.; Vuky, J.; Lopes, G.; Kalemkerian, G.P.; et al. Pan-cancer efficacy of pralsetinib in patients with RET fusion-positive solid tumors from the phase 1/2 ARROW trial. Nat. Med. 2022, 28, 1640–1645. [Google Scholar] [CrossRef] [PubMed]
- Geiger, J.L.; Chiosea, S.I.; Challinor, S.M.; Nikiforova, M.N.; Bauman, J.E. Primary RET-mutated lung neuroendocrine carcinoma in MEN2B: Response to RET-targeted therapy. Endocr. Relat. Cancer 2015, 22, L19–L22. [Google Scholar] [CrossRef] [PubMed]
- Arora, A.; Zaemes, J.; Ozdemirli, M.; Kim, C. Response to selpercatinib in a patient with RET fusion-positive pulmonary large-cell neuroendocrine carcinoma: A case report. Front. Oncol. 2023, 13, 1134151. [Google Scholar] [CrossRef] [PubMed]
- Prisciandaro, M.; Antista, M.; Raimondi, A.; Corti, F.; Morano, F.; Centonze, G.; Sabella, G.; Mangogna, A.; Randon, G.; Pagani, F.; et al. Biomarker Landscape in Neuroendocrine Tumors with High-Grade Features: Current Knowledge and Future Perspective. Front. Oncol. 2022, 12, 780716. [Google Scholar] [CrossRef]
- Mansfield, A.S.; Hong, D.S.; Hann, C.L.; Farago, A.F.; Beltran, H.; Waqar, S.N.; Hendifar, A.E.; Anthony, L.B.; Taylor, M.H.; Bryce, A.H.; et al. A phase I/II study of rovalpituzumab tesirine in delta-like 3—Expressing advanced solid tumors. npj Precis. Oncol. 2021, 5, 74. [Google Scholar] [CrossRef]
- Johnson, M.L.; Zvirbule, Z.; Laktionov, K.; Helland, A.; Cho, B.C.; Gutierrez, V.; Colinet, B.; Lena, H.; Wolf, M.; Gottfried, M.; et al. Rovalpituzumab tesirine as a maintenance therapy after first-line platinum-based chemotherapy in patients with exten-sive-stage-SCLC: Results from the phase 3 MERU study. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2021, 16, 1570–1581. [Google Scholar]
- Blackhall, F.; Jao, K.; Greillier, L.; Cho, B.C.; Penkov, K.; Reguart, N.; Majem, M.; Nackaerts, K.; Syrigos, K.; Hansen, K.; et al. Efficacy and safety of rovalpituzumab tesirine compared with topotecan as second-line therapy in DLL3-high SCLC: Results from the phase 3 TAHOE study. J. Thorac. Oncol. 2021, 16, 1547–1558. [Google Scholar] [CrossRef] [PubMed]
- Sahnane, N.; Furlan, D.; Monti, M.; Romualdi, C.; Vanoli, A.; Vicari, E.; Solcia, E.; Capella, C.; Sessa, F.; La Rosa, S. Microsatellite Unstable Gastrointestinal Neuroendocrine Carcinomas: A New Clinicopathologic Entity. Endocr.-Relat. Cancer 2014, 22, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Maio, M.; Ascierto, P.A.; Manzyuk, L.; Motola-Kuba, D.; Penel, N.; Cassier, P.A.; Bariani, G.M.; Acosta, A.D.J.; Doi, T.; Longo, F.; et al. Pembrolizumab in microsatellite instability high or mismatch repair deficient cancers: Updated analysis from the phase II KEYNOTE-158 study. Ann. Oncol. 2022, 33, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Geurts, B.; Battaglia, T.; Henegouwen, J.v.B.; Zeverijn, L.; Hoes, L.; van der Wijngaart, H.; de Wit, G.; Roepman, P.; Jansen, A.; de Leng, W.; et al. Durvalumab in advanced, pre-treated microsatellite instability-high solid tumors: Results of a tumor-agnostic DRUP trial cohort. Ann. Oncol. 2022, 33 (Suppl. S7), S594. [Google Scholar] [CrossRef]
- Shao, C.; Li, G.; Huang, L.; Pruitt, S.; Castellanos, E.; Frampton, G.; Carson, K.R.; Snow, T.; Singal, G.; Fabrizio, D.; et al. Prevalence of High Tumor Mutational Burden and Association with Survival in Patients With Less Common Solid Tumors. JAMA Netw. Open 2020, 3, e2025109. [Google Scholar] [CrossRef]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H., Jr.; et al. Association of Tumour Mutational Burden with Outcomes in Patients with Advanced Solid Tumours Treated with Pembrolizumab: Prospective Biomarker Analysis of the Multicohort, Open-Label, Phase 2 KEYNOTE-158 Study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]
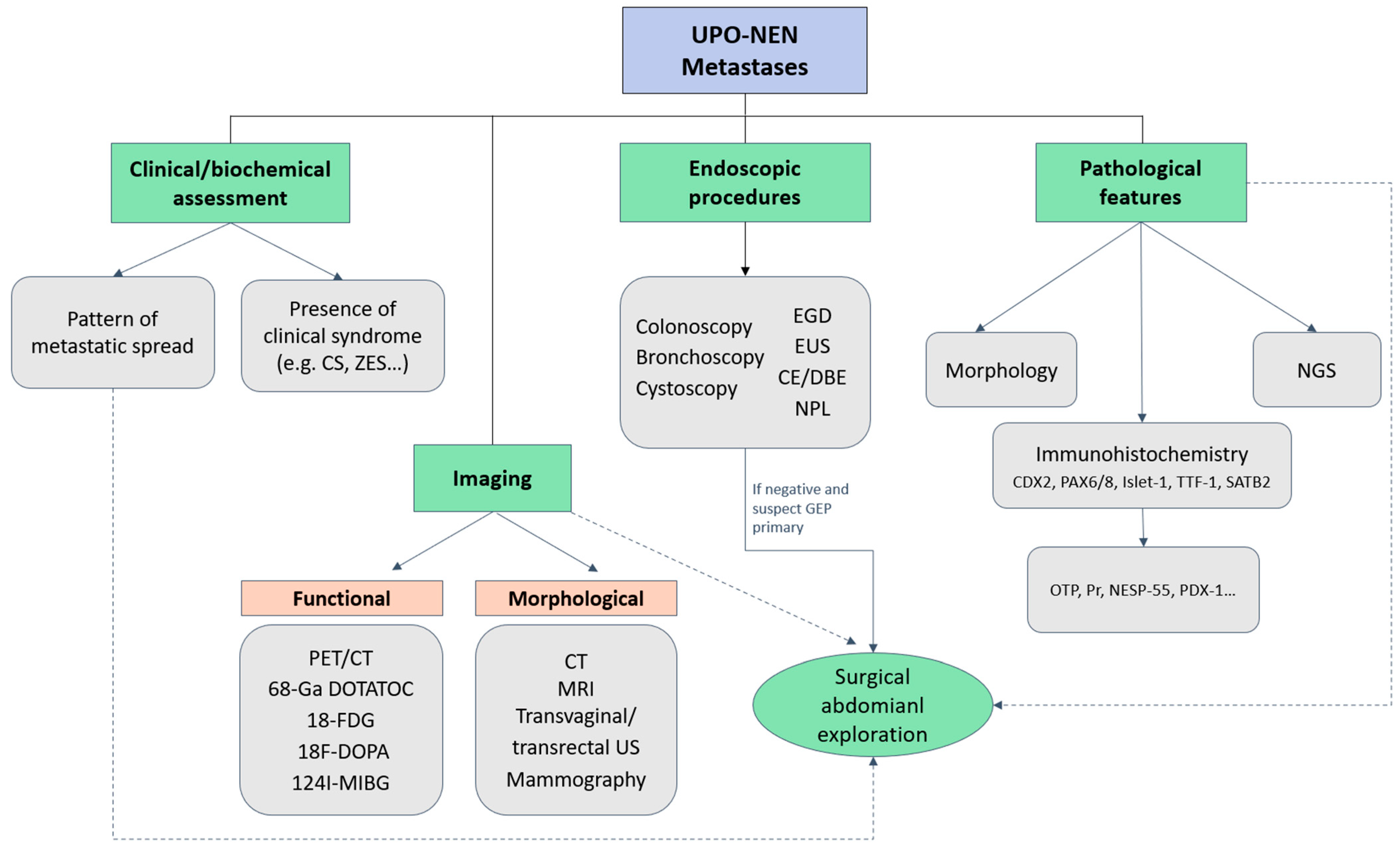
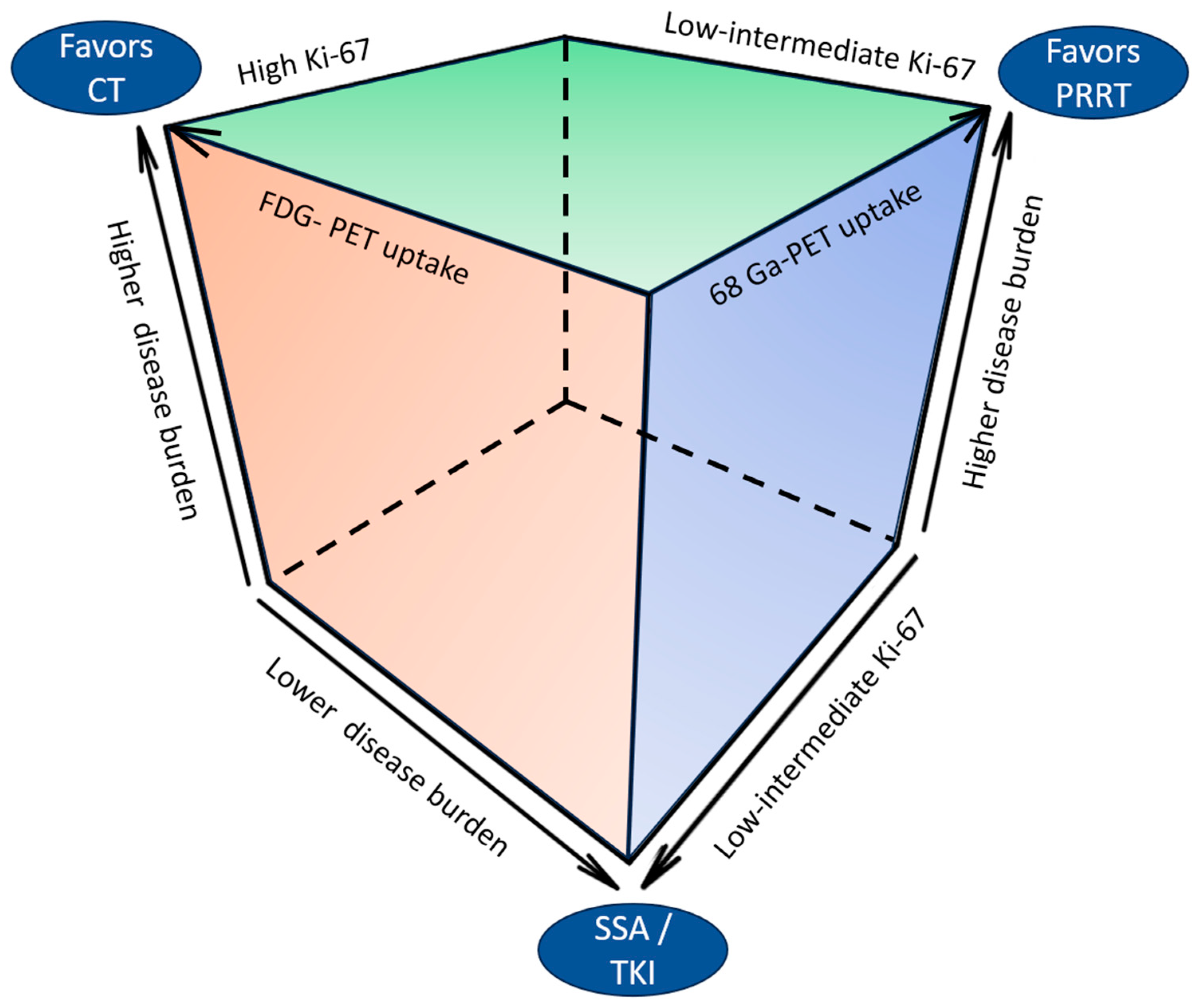
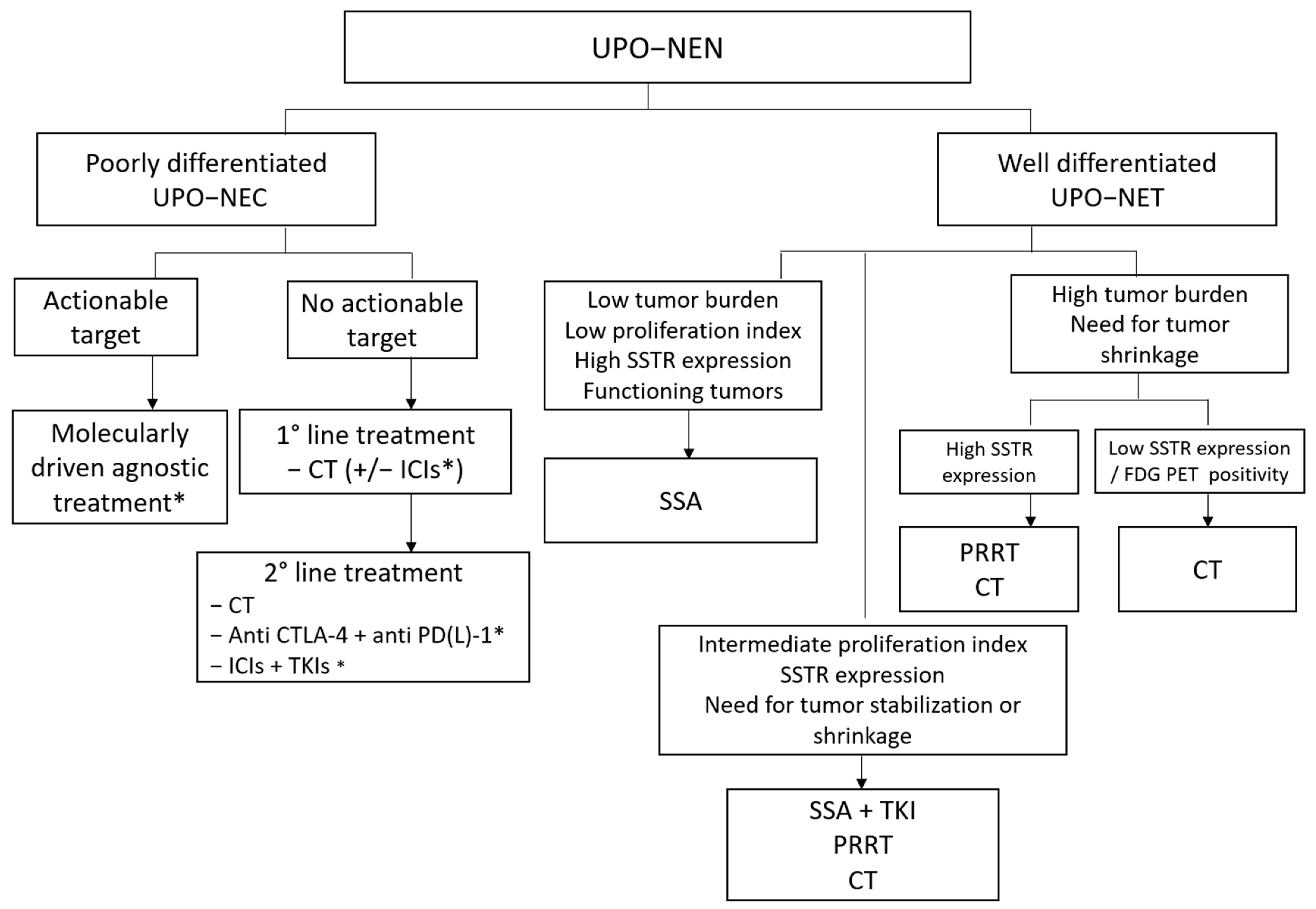
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).