
Single-Cell Profiling Reveals the Impact of Genetic Alterations on the Differentiation of Inflammation-Induced Murine Colon Tumors

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Model
2.2. Tumor Digestion and Single-Cell RNA Sequencing
2.3. Cells and Organoids
2.4. Generation of Stable Knockdown Organoids
2.5. RNA Isolation and Gene Expression
2.6. Immunohistochemistry (IHC)
2.7. Immunofluorescence and Imaging
2.8. Statistical Analysis
2.9. Computational Analysis
2.9.1. Single-Cell Data Pre-Processing and QC
2.9.2. Data Integration with Batch Correction
2.9.3. Subsetting and Visualizing Epithelial Data
2.9.4. Gene Ontology Enrichment Analysis
2.9.5. RNA Velocity
2.9.6. Simulated Gene Perturbation
3. Results
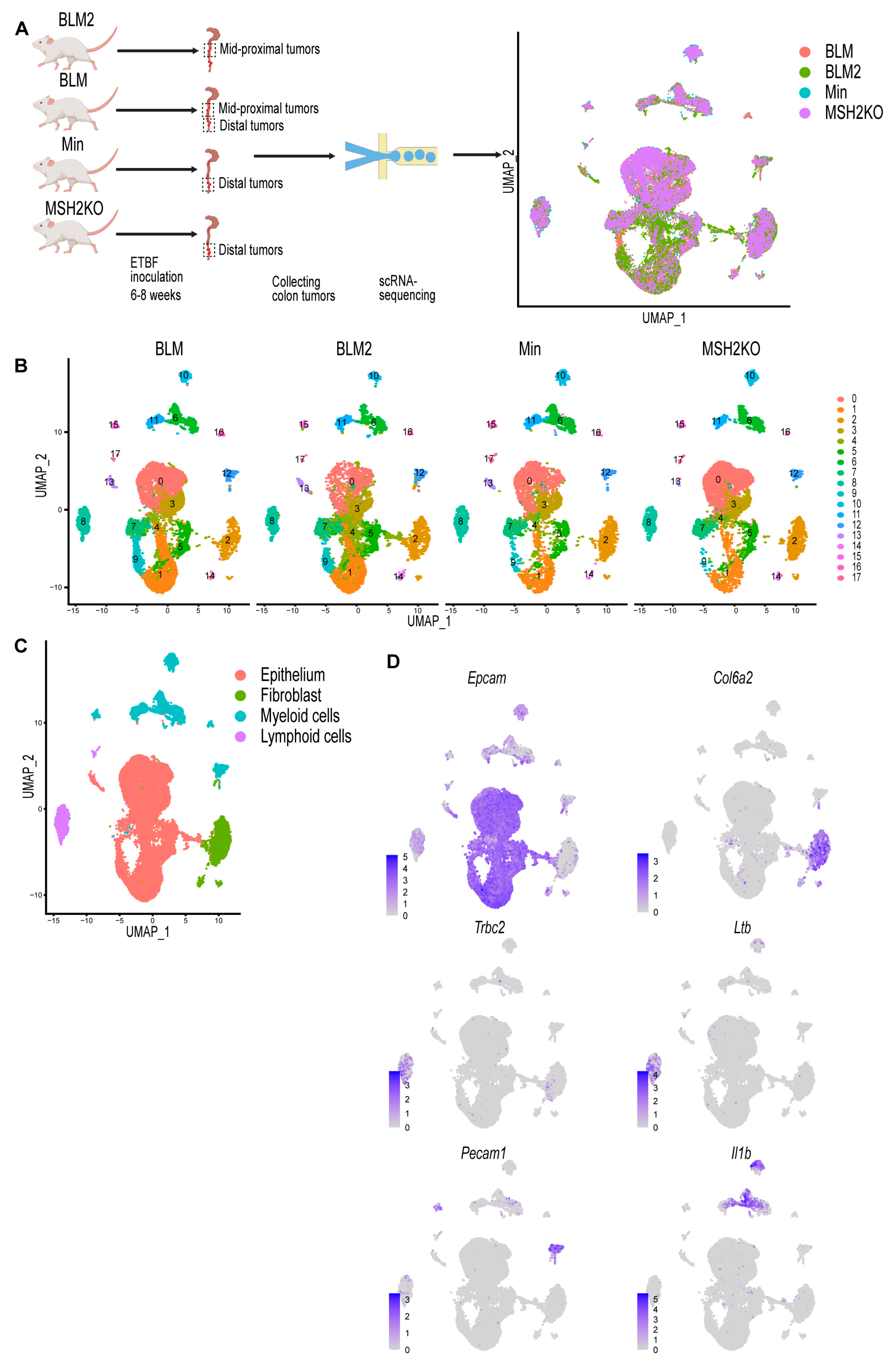
3.1. Single-Cell Profiling Identifies Cell Populations in Colon Tumors
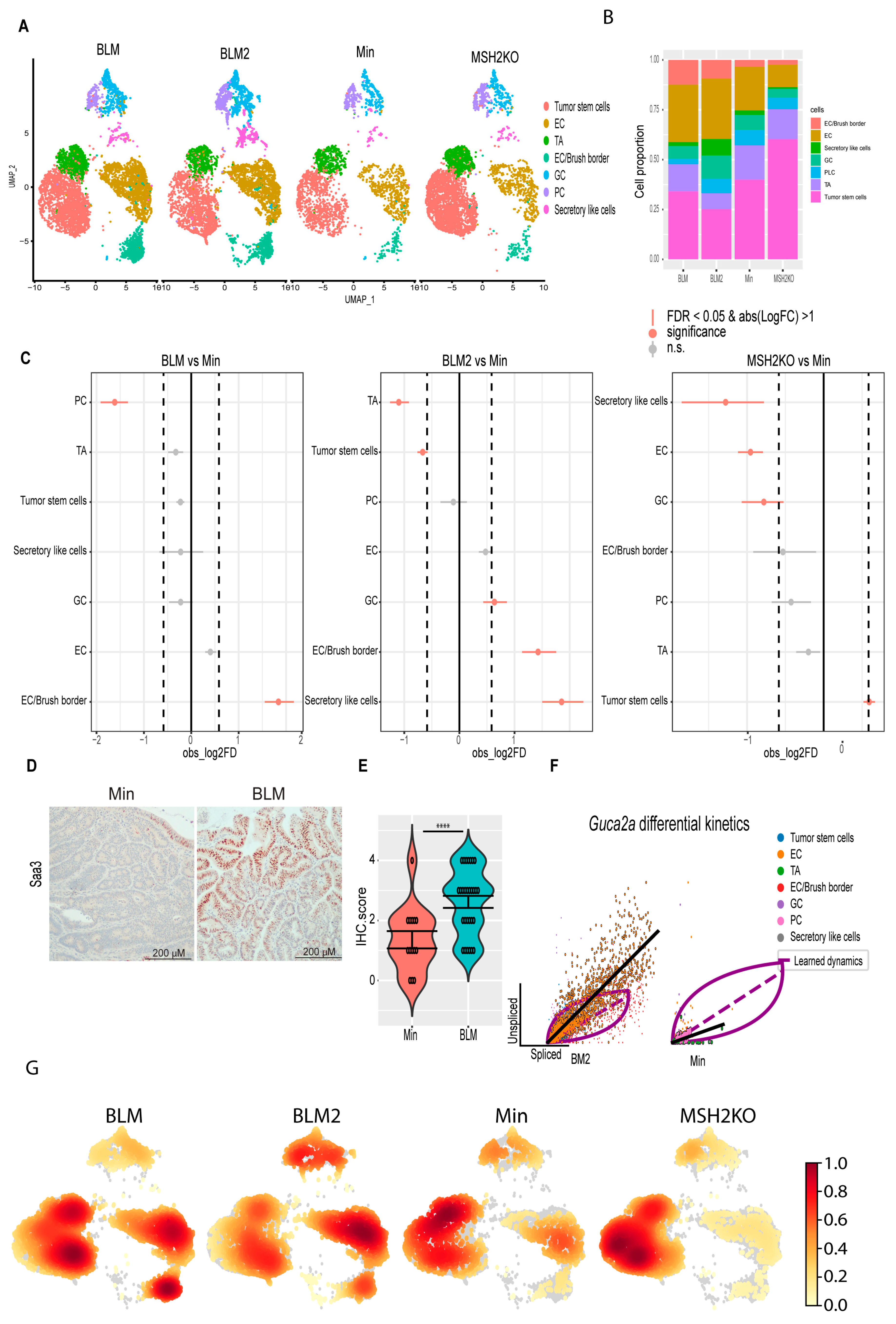
3.2. Single-Cell Survey of Colon Tumor Epithelial Cells
3.3. BRAFV600E Mutation and Msh2 Deletion Alter Colon Tumor Epithelial Cell Composition
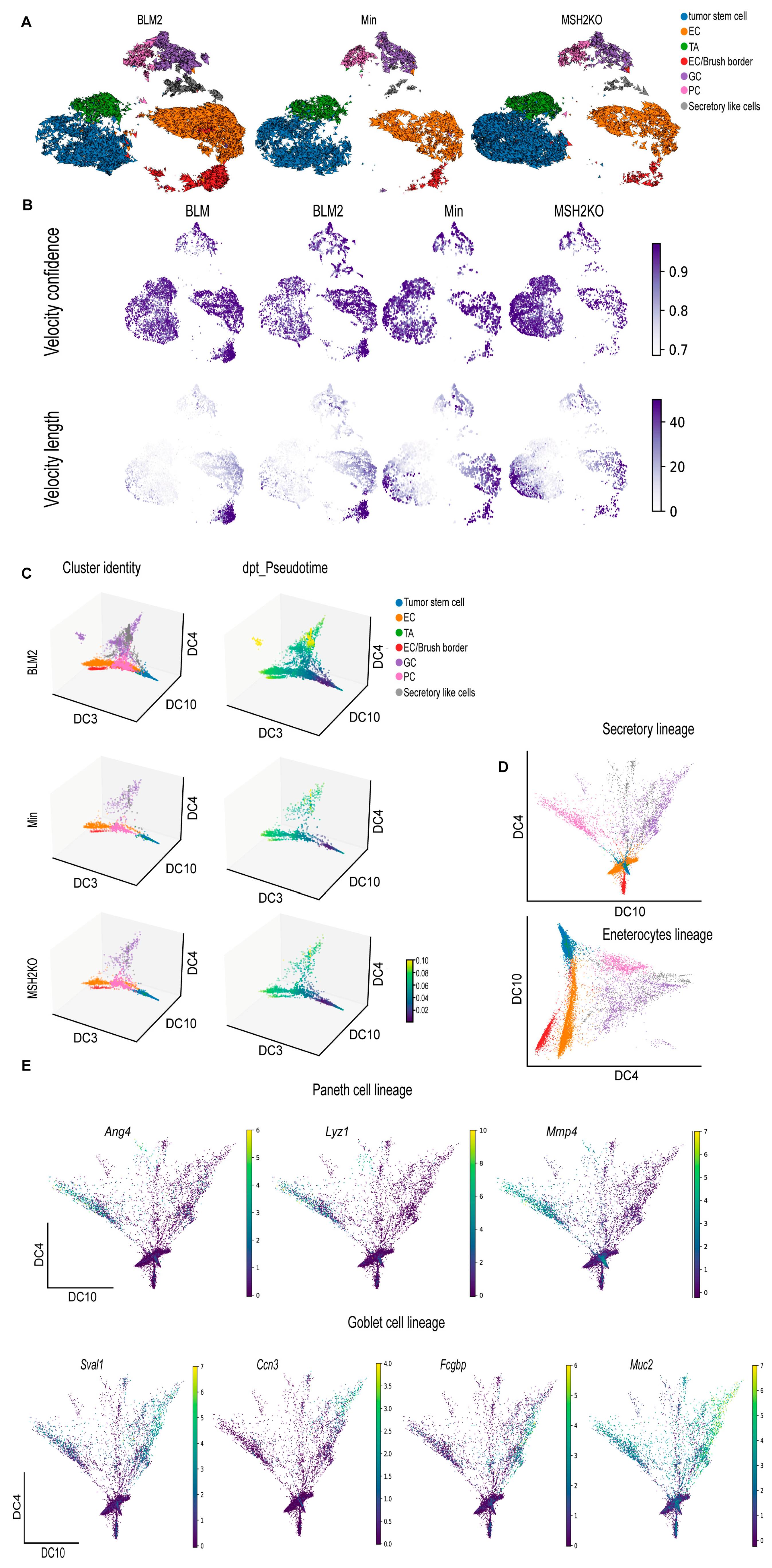
3.4. BRAFV600E Colon Tumors Are More Differentiated Than Min and Msh2 Deleted Tumors
3.5. BLM and MSH2KO Tumors Have Different Tumor Stem Cell Characteristics
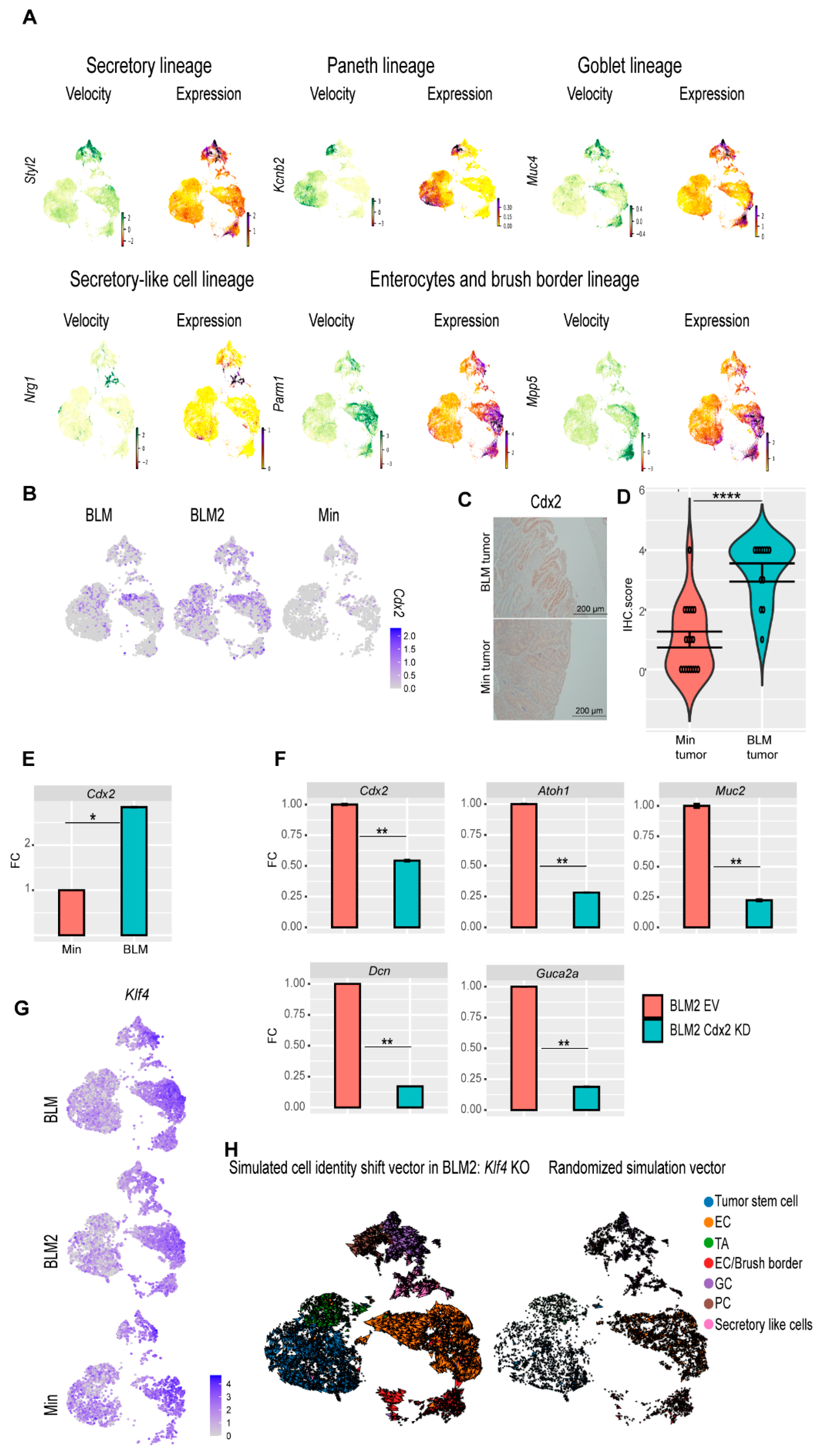
3.6. CDX2 Is Involved in BRAFV600E Colon Tumor epithelial Cell Differentiation
4. Discussion
5. Conclusions
6. Limitations of the Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Marusawa, H.; Ushijima, T. Inflammation-Associated Cancer Development in Digestive Organs: Mechanisms and Roles for Genetic and Epigenetic Modulation. Gastroenterology 2012, 143, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Lucafò, M.; Curci, D.; Franzin, M.; Decorti, G.; Stocco, G. Inflammatory Bowel Disease and Risk of Colorectal Cancer: An Overview From Pathophysiology to Pharmacological Prevention. Front. Pharmacol. 2021, 12, 772101. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Jin, Y.; Chen, G.; Ma, X.; Zhang, L. Gut Microbiota Dysbiosis Drives the Development of Colorectal Cancer. Digestion 2021, 102, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Ulger Toprak, N.; Yagci, A.; Gulluoglu, B.M.; Akin, M.L.; Demirkalem, P.; Celenk, T.; Soyletir, G. A Possible Role of Bacteroides fragilis Enterotoxin in the Aetiology of Colorectal Cancer. Clin. Microbiol. Infect. 2006, 12, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Boleij, A.; Hechenbleikner, E.M.; Goodwin, A.C.; Badani, R.; Stein, E.M.; Lazarev, M.G.; Ellis, B.; Carroll, K.C.; Albesiano, E.; Wick, E.C.; et al. The Bacteroides fragilis Toxin Gene Is Prevalent in the Colon Mucosa of Colorectal Cancer Patients. Clin. Infect. Dis. 2015, 60, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Viljoen, K.S.; Dakshinamurthy, A.; Goldberg, P.; Blackburn, J.M. Quantitative Profiling of Colorectal Cancer-Associated Bacteria Reveals Associations between Fusobacterium spp., Enterotoxigenic Bacteroides fragilis (ETBF) and Clinicopathological Features of Colorectal Cancer. PLoS ONE 2015, 10, e0119462. [Google Scholar] [CrossRef] [PubMed]
- Armaghany, T.; Wilson, J.D.; Chu, Q.; Mills, G. Genetic Alterations in Colorectal Cancer. Gastrointest. Cancer Res. 2012, 5, 19–27. [Google Scholar] [PubMed]
- Stamos, J.L.; Weis, W.I. The β-Catenin Destruction Complex. Cold Spring Harb. Perspect. Biol. 2013, 5, a007898. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Shay, J.W. Multiple Roles of APC and Its Therapeutic Implications in Colorectal Cancer. J. Natl. Cancer Inst. 2017, 109, djw332. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Vogelstein, B. Lessons from Hereditary Colorectal Cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, A.R.; Peng, M.; Sriramkumar, S.; Kamplain, C.M.; DeStefano Shields, C.E.; Sears, C.L.; O’Hagan, H.M. Mismatch Repair Proteins Initiate Epigenetic Alterations during Inflammation-Driven Tumorigenesis. Cancer Res. 2017, 77, 3467–3478. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.; Rosendahl Huber, A.; Pleguezuelos-Manzano, C.; Puschhof, J.; Wu, S.; Wu, X.; Boot, C.; Saftien, A.; O’Hagan, H.M.; Wang, H.; et al. Colon Tumors in Enterotoxigenic Bacteroides fragilis (ETBF)-Colonized Mice Do Not Display a Unique Mutational Signature but Instead Possess Host-Dependent Alterations in the APC Gene. Microbiol. Spectr. 2022, 10, e01055-22. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Rhee, K.-J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.-R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F.; et al. A Human Colonic Commensal Promotes Colon Tumorigenesis via Activation of T Helper Type 17 T Cell Responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, G.; Wu, W. Recent Advances in Lynch Syndrome. Exp. Hematol. Oncol. 2021, 10, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Poulogiannis, G.; Frayling, I.M.; Arends, M.J. DNA Mismatch Repair Deficiency in Sporadic Colorectal Cancer and Lynch Syndrome. Histopathology 2010, 56, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Brenner, H.; Kloor, M.; Pox, C.P. Colorectal Cancer. Lancet 2014, 383, 1490–1502. [Google Scholar] [CrossRef] [PubMed]
- Lindor, B.N.M.; Burgart, L.J.; Leontovich, O.; Goldberg, R.M.; Cunningham, J.M.; Sargent, D.J.; Walsh-vockley, C.; Petersen, G.M.; Walsh, M.D.; Leggett, B.a.; et al. Testing in Phenotyping Colorectal Tumors. Society 2002, 20, 1043–1048. [Google Scholar]
- Norreys, P.A.; Sakabe, S.; Tanaka, K.A.; Youssef, A.; Zepf, M.; Yamanaka, T. Tumorigenesis RAF/RAS Oncogenes and Mismatch-Repair Status. Conf. Laser Elec. Opt. 1994, 1, 402–403. [Google Scholar]
- Borowsky, J.; Dumenil, T.; Bettington, M.; Pearson, S.A.; Bond, C.; Fennell, L.; Liu, C.; McKeone, D.; Rosty, C.; Brown, I.; et al. The Role of APC in WNT Pathway Activation in Serrated Neoplasia. Mod. Pathol. 2018, 31, 495–504. [Google Scholar] [CrossRef]
- Tabernero, J.; Ros, J.; Élez, E. The Evolving Treatment Landscape in BRAF-V600E –Mutated Metastatic Colorectal Cancer. Am. Soc. Clin. Oncol. Educ. B 2022, 42, 254–263. [Google Scholar] [CrossRef]
- Samowitz, W.S.; Sweeney, C.; Herrick, J.; Albertsen, H.; Levin, T.R.; Murtaugh, M.A.; Wolff, R.K.; Slattery, M.L. Poor Survival Associated with the BRAF V600E Mutation in Microsatellite-Stable Colon Cancers. Cancer Res. 2005, 65, 6063–6070. [Google Scholar] [CrossRef]
- Cannon, T.L.; Randall, J.N.; Sokol, E.S.; Alexander, S.M.; Wadlow, R.C.; Winer, A.A.; Barnett, D.M.; Rayes, D.L.; Nimeiri, H.S.; McGregor, K.A. Concurrent BRAFV600E and BRCA Mutations in MSS Metastatic Colorectal Cancer: Prevalence and Case Series of MCRC Patients with Prolonged OS. Cancer Treat. Res. Commun. 2022, 32, 100569. [Google Scholar] [CrossRef]
- Lochhead, P.; Kuchiba, A.; Imamura, Y.; Liao, X.; Yamauchi, M.; Nishihara, R.; Qian, Z.R.; Morikawa, T.; Shen, J.; Meyerhardt, J.A.; et al. Microsatellite Instability and Braf Mutation Testing in Colorectal Cancer Prognostication. J. Natl. Cancer Inst. 2013, 105, 1151–1156. [Google Scholar] [CrossRef]
- Phipps, A.I.; Buchanan, D.D.; Makar, K.W.; Burnett-Hartman, A.N.; Coghill, A.E.; Passarelli, M.N.; Baron, J.A.; Ahnen, D.J.; Win, A.K.; Potter, J.D.; et al. BRAF Mutation Status and Survival after Colorectal Cancer Diagnosis According to Patient and Tumor Characteristics. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1792–1798. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Shima, K.; Meyerhardt, J.A.; McCleary, N.J.; Ng, K.; Hollis, D.; Saltz, L.B.; Mayer, R.J.; Schaefer, P.; Whittom, R.; et al. Predictive and Prognostic Roles of BRAF Mutation in Stage III Colon Cancer: Results from Intergroup Trial CALGB 89803. Clin. Cancer Res. 2012, 18, 890–900. [Google Scholar] [CrossRef]
- Destefano Shields, C.E.; White, J.R.; Chung, L.; Wenzel, A.; Hicks, J.L.; Tam, A.J.; Chan, J.L.; Dejea, C.M.; Fan, H.; Michel, J.; et al. Bacterial-Driven Inflammation and Mutant Braf Expression Combine to Promote Murine Colon Tumorigenesis That Is Sensitive to Immune Checkpoint Therapy. Cancer Discov. 2021, 11, 1792–1807. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. The Intestinal Crypt, a Prototype Stem Cell Compartment. Cell 2013, 154, 274. [Google Scholar] [CrossRef] [PubMed]
- Schatoff, E.M.; Leach, B.I.; Dow, L.E. Wnt Signaling and Colorectal Cancer. Curr. Colorectal Cancer Rep. 2017, 13, 101–110. [Google Scholar] [CrossRef]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of Stem Cells in Small Intestine and Colon by Marker Gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef]
- Maiuri, A.R.; Li, H.; Stein, B.D.; Tennessen, J.M.; O’Hagan, H.M. Inflammation-Induced DNA Methylation of DNA Polymerase Gamma Alters the Metabolic Profile of Colon Tumors. Cancer Metab. 2018, 6, 9. [Google Scholar] [CrossRef]
- Ghobashi, A.H.; Vuong, T.T.; Kimani, J.W.; Ladaika, C.A.; Hollenhorst, P.C.; O’Hagan, H.M. Activation of AKT Induces EZH2-Mediated β-Catenin Trimethylation in Colorectal Cancer. iScience 2023, 26, 107630. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated Analysis of Multimodal Single-Cell Data. Cell 2021, 184, 3573–3587. [Google Scholar] [CrossRef] [PubMed]
- McGinnis, C.S.; Murrow, L.M.; Gartner, Z.J. DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 2019, 8, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.A.; Policastro, R.A.; Sriramkumar, S.; Lai, T.; Huntington, T.D.; Ladaika, C.A.; Kim, D.; Hao, C.; Zentner, G.E.; O’Hagan, H.M. LSD1 and Aberrant DNA Methylation Mediate Persistence of Enteroendocrine Progenitors That Support BRAF-Mutant Colorectal Cancer. Cancer Res. 2021, 81, 3791–3805. [Google Scholar] [CrossRef] [PubMed]
- Borcherding, N.; Vishwakarma, A.; Voigt, A.P.; Bellizzi, A.; Kaplan, J.; Nepple, K.; Salem, A.K.; Jenkins, R.W.; Zakharia, Y.; Zhang, W. Mapping the Immune Environment in Clear Cell Renal Carcinoma by Single-Cell Genomics. Commun. Biol. 2021, 4, 122. [Google Scholar] [CrossRef] [PubMed]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-Scale Single-Cell Gene Expression Data Analysis. Genome Biol. 2018, 19, 15. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape Provides a Biologist-Oriented Resource for the Analysis of Systems-Level Datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- La Manno, G.; Soldatov, R.; Zeisel, A.; Braun, E.; Hochgerner, H.; Petukhov, V.; Lidschreiber, K.; Kastriti, M.E.; Lönnerberg, P.; Furlan, A.; et al. RNA Velocity of Single Cells. Nature 2018, 560, 494–498. [Google Scholar] [CrossRef]
- Bergen, V.; Lange, M.; Peidli, S.; Wolf, F.A.; Theis, F.J. Generalizing RNA Velocity to Transient Cell States through Dynamical Modeling. Nat. Biotechnol. 2020, 38, 1408–1414. [Google Scholar] [CrossRef]
- Kamimoto, K.; Stringa, B.; Hoffmann, C.M.; Jindal, K.; Solnica-Krezel, L.; Morris, S.A. Dissecting Cell Identity via Network Inference and in Silico Gene Perturbation. Nature 2023, 614, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Reigstad, C.S.; Bäckhed, F. Microbial Regulation of SAA3 Expression in Mouse Colon and Adipose Tissue. Gut Microbes 2010, 1, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Gorman, H.; Moreau, F.; Dufour, A.; Chadee, K. IgGFc-Binding Protein and MUC2 Mucin Produced by Colonic Goblet-like Cells Spatially Interact Non-Covalently and Regulate Wound Healing. Front. Immunol. 2023, 14, 1211336. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Stetler-Stevenson, W.G. Matrix Metalloproteinases and Metastasis. Cancer Chemother. Pharmacol. 1999, 43, S42–S51. [Google Scholar] [CrossRef] [PubMed]
- Tong, K.; Pellón-Cárdenas, O.; Sirihorachai, V.R.; Warder, B.N.; Kothari, O.A.; Perekatt, A.O.; Fokas, E.E.; Fullem, R.L.; Zhou, A.; Thackray, J.K.; et al. Degree of Tissue Differentiation Dictates Susceptibility to BRAF-Driven Colorectal Cancer. Cell Rep. 2017, 21, 3833–3845. [Google Scholar] [CrossRef] [PubMed]
- Haghverdi, L.; Büttner, M.; Wolf, F.A.; Buettner, F.; Theis, F.J. Diffusion Pseudotime Robustly Reconstructs Lineage Branching. Nat. Methods 2016, 13, 845–848. [Google Scholar] [CrossRef] [PubMed]
- Ayyaz, A.; Kumar, S.; Sangiorgi, B.; Ghoshal, B.; Gosio, J.; Ouladan, S.; Fink, M.; Barutcu, S.; Trcka, D.; Shen, J.; et al. Single-Cell Transcriptomes of the Regenerating Intestine Reveal a Revival Stem Cell. Nature 2019, 569, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Tape, C.J. Plastic Persisters: Revival Stem Cells in Colorectal Cancer. Trends Cancer 2024, 10, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Cardoso Rodriguez, F.; Sufi, J.; Vlckova, P.; Claus, J.; Tape, C.J. An Oncogenic Phenoscape of Colonic Stem Cell Polarization. Cell 2023, 186, 5554–5568.e18. [Google Scholar] [CrossRef]
- Lo, Y.-H.; Chung, E.; Li, Z.; Wan, Y.-W.; Mahe, M.M.; Chen, M.-S.; Noah, T.K.; Bell, K.N.; Yalamanchili, H.K.; Klisch, T.J.; et al. Transcriptional Regulation by ATOH1 and Its Target SPDEF in the Intestine. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 51–71. [Google Scholar] [CrossRef]
- Chang, S.K.; Dohrman, A.F.; Basbaum, C.B.; Ho, S.B.; Tsuda, T.; Toribara, N.W.; Gum, J.R.; Kim, Y.S. Localization of Mucin (MUC2 and MUC3) Messenger RNA and Peptide Expression in Human Normal Intestine and Colon Cancer. Gastroenterology 1994, 107, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Xie, J.; Liu, H.; Zhao, R.; Zhang, W.; Wang, H.; Pan, H.; Zhou, Y.; Han, W. STIM1 Deficiency In Intestinal Epithelium Attenuates Colonic Inflammation and Tumorigenesis by Reducing ER Stress of Goblet Cells. Cell. Mol. Gastroenterol. Hepatol. 2022, 14, 193–217. [Google Scholar] [CrossRef] [PubMed]
- Gendron, F.P.; Mongrain, S.; Laprise, P.; McMahon, S.; Dubois, C.M.; Blais, M.; Asselin, C.; Rivard, N. The CDX2 Transcription Factor Regulates Furin Expression during Intestinal Epithelial Cell Differentiation. Am. J. Physiol.-Gastrointest. Liver Physiol. 2006, 290, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Tsai, Y.-H.; Chen, L.; Zhou, A.; Banerjee, K.K.; Saxena, M.; Huang, S.; Toke, N.H.; Xing, J.; Shivdasani, R.A.; et al. The Lineage-Specific Transcription Factor CDX2 Navigates Dynamic Chromatin to Control Distinct Stages of Intestine Development. Development 2019, 146, dev172189. [Google Scholar] [CrossRef]
- Yu, T.; Chen, X.; Zhang, W.; Li, J.; Xu, R.; Wang, T.C.; Ai, W.; Liu, C. Krüppel-like Factor 4 Regulates Intestinal Epithelial Cell Morphology and Polarity. PLoS ONE 2012, 7, e32492. [Google Scholar] [CrossRef] [PubMed]
- Watari, K.; Shibata, T.; Nabeshima, H.; Shinoda, A.; Fukunaga, Y.; Kawahara, A.; Karasuyama, K.; Fukushi, J.I.; Iwamoto, Y.; Kuwano, M.; et al. Impaired Differentiation of Macrophage Lineage Cells Attenuates Bone Remodeling and Inflammatory Angiogenesis in Ndrg1 Deficient Mice. Sci. Rep. 2016, 6, 19470. [Google Scholar] [CrossRef] [PubMed]
- Cai, K.; El-Merahbi, R.; Loeffler, M.; Mayer, A.E.; Sumara, G. Ndrg1 Promotes Adipocyte Differentiation and Sustains Their Function. Sci. Rep. 2017, 7, 7191. [Google Scholar] [CrossRef]
- Haber, A.L.; Biton, M.; Rogel, N.; Herbst, R.H.; Shekhar, K.; Smillie, C.; Burgin, G.; Delorey, T.M.; Howitt, M.R.; Katz, Y.; et al. A Single-Cell Survey of the Small Intestinal Epithelium. Nature 2017, 551, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Hickey, J.W.; Becker, W.R.; Nevins, S.A.; Horning, A.; Perez, A.E.; Zhu, C.; Zhu, B.; Wei, B.; Chiu, R.; Chen, D.C.; et al. Organization of the Human Intestine at Single-Cell Resolution. Nature 2023, 619, 572–584. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour Heterogeneity and Resistance to Cancer Therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Waarts, M.R.; Stonestrom, A.J.; Park, Y.C.; Levine, R.L. Targeting Mutations in Cancer. J. Clin. Investig. 2022, 132, e154943. [Google Scholar] [CrossRef] [PubMed]
- Riemer, P.; Sreekumar, A.; Reinke, S.; Rad, R.; Schäfer, R.; Sers, C.; Bläker, H.; Herrmann, B.G.; Morkel, M. Transgenic Expression of Oncogenic BRAF Induces Loss of Stem Cells in the Mouse Intestine, Which Is Antagonized by β-Catenin Activity. Oncogene 2015, 34, 3164–3175. [Google Scholar] [CrossRef]
- Touil, Y.; Igoudjil, W.; Corvaisier, M.; Dessein, A.-F.; Vandomme, J.; Monté, D.; Stechly, L.; Skrypek, N.; Langlois, C.; Grard, G.; et al. Colon Cancer Cells Escape 5FU Chemotherapy-Induced Cell Death by Entering Stemness and Quiescence Associated with the c-Yes/YAP Axis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Jen, J.; Vogelstein, B.; Hamilton, S.R. Clinical and Pathological Characteristics of Sporadic Colorectal Carcinomas with DNA Replication Errors in Microsatellite Sequences. Am. J. Pathol. 1994, 145, 148–156. [Google Scholar] [PubMed]
- Xiao, H.; Yoon, Y.S.; Hong, S.-M.; Roh, S.A.; Cho, D.-H.; Yu, C.S.; Kim, J.C. Poorly Differentiated Colorectal Cancers: Correlation of Microsatellite Instability with Clinicopathologic Features and Survival. Am. J. Clin. Pathol. 2013, 140, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Jho, E.; Zhang, T.; Domon, C.; Joo, C.-K.; Freund, J.-N.; Costantini, F. Wnt/β-Catenin/Tcf Signaling Induces the Transcription of Axin2, a Negative Regulator of the Signaling Pathway. Mol. Cell. Biol. 2002, 22, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Ming, T.; Tang, S.; Ren, S.; Yang, H.; Liu, M.; Tao, Q.; Xu, H. Wnt Signaling in Colorectal Cancer: Pathogenic Role and Therapeutic Target. Mol. Cancer 2022, 21, 144. [Google Scholar] [CrossRef]
- Pelka, K.; Hofree, M.; Chen, J.H.; Sarkizova, S.; Pirl, J.D.; Jorgji, V.; Bejnood, A.; Dionne, D.; Ge, W.H.; Xu, K.H.; et al. Spatially Organized Multicellular Immune Hubs in Human Colorectal Cancer. Cell 2021, 184, 4734–4752. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, N.; Feng, Y.; Stolfi, C.; Kurosu, Y.; Green, M.; Lin, J.; Green, M.E.; Sentani, K.; Yasui, W.; McMahon, M.; et al. BRAFV600E Cooperates with CDX2 Inactivation to Promote Serrated Colorectal Tumorigenesis. Elife 2017, 6, e20331. [Google Scholar] [CrossRef]
- Yang, L.; Tu, L.; Bisht, S.; Mao, Y.; Petkovich, D.; Thursby, S.-J.; Liang, J.; Patel, N.; Yen, R.-W.C.; Largent, T.; et al. Tissue-Location-Specific Transcription Programs Drive Tumor Dependencies in Colon Cancer. Nat. Commun. 2024, 15, 1384. [Google Scholar] [CrossRef]
- Kim, J.H.; Rhee, Y.-Y.; Bae, J.M.; Cho, N.-Y.; Kang, G.H. Loss of CDX2/CK20 Expression Is Associated with Poorly Differentiated Carcinoma, the CpG Island Methylator Phenotype, and Adverse Prognosis in Microsatellite-Unstable Colorectal Cancer. Am. J. Surg. Pathol. 2013, 37, 1532–1541. [Google Scholar] [CrossRef] [PubMed]
- Landau, M.S.; Kuan, S.F.; Chiosea, S.; Pai, R.K. BRAF-Mutated Microsatellite Stable Colorectal Carcinoma: An Aggressive Adenocarcinoma with Reduced CDX2 and Increased Cytokeratin 7 Immunohistochemical Expression. Hum. Pathol. 2014, 45, 1704–1712. [Google Scholar] [CrossRef] [PubMed]
- Aasebø, K.; Dragomir, A.; Sundström, M.; Mezheyeuski, A.; Edqvist, P.H.; Eide, G.E.; Ponten, F.; Pfeiffer, P.; Glimelius, B.; Sorbye, H. CDX2: A Prognostic Marker in Metastatic Colorectal Cancer Defining a Better BRAF Mutated and a Worse KRAS Mutated Subgroup. Front. Oncol. 2020, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.J.; Funakoshi, S.; Lee, H.H.; Kong, J.; Lynch, J.P. The Intestine-Specific Transcription Factor Cdx2 Inhibits β-Catenin/TCF Transcriptional Activity by Disrupting the β-Catenin-TCF Protein Complex. Carcinogenesis 2010, 31, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Hoffsten, A.; Lilja, H.E.; Mobini-Far, H.; Sindelar, R.; Markasz, L. Paneth Cell Proteins DEFA6 and GUCA2A as Tissue Markers in Necrotizing Enterocolitis. Eur. J. Pediatr. 2023, 182, 2775–2784. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Bozadjieva-Kramer, N.; Seeley, R.J. Reg3γ: Current Understanding and Future Therapeutic Opportunities in Metabolic Disease. Exp. Mol. Med. 2023, 55, 1672–1677. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Liu, J.; Wu, L.; Fan, Y.; Sun, L.; Qian, F.; Chen, D.; Ye, R.D. Elevated Expression of Serum Amyloid A 3 Protects Colon Epithelium against Acute Injury through TLR2-Dependent Induction of Neutrophil IL-22 Expression in a Mouse Model of Colitis. Front. Immunol. 2018, 9, 1503. [Google Scholar] [CrossRef] [PubMed]
- Joshi, V.; Lakhani, S.R.; McCart Reed, A.E. NDRG1 in Cancer: A Suppressor, Promoter, or Both? Cancers 2022, 14, 5739. [Google Scholar] [CrossRef]
- Chekmarev, J.; Azad, M.G.; Richardson, D.R. The Oncogenic Signaling Disruptor, NDRG1: Molecular and Cellular Mechanisms of Activity. Cells 2021, 10, 2382. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghobashi, A.H.; Lanzloth, R.; Ladaika, C.A.; Masood, A.; O’Hagan, H.M. Single-Cell Profiling Reveals the Impact of Genetic Alterations on the Differentiation of Inflammation-Induced Murine Colon Tumors. Cancers 2024, 16, 2040. https://doi.org/10.3390/cancers16112040
Ghobashi AH, Lanzloth R, Ladaika CA, Masood A, O’Hagan HM. Single-Cell Profiling Reveals the Impact of Genetic Alterations on the Differentiation of Inflammation-Induced Murine Colon Tumors. Cancers. 2024; 16(11):2040. https://doi.org/10.3390/cancers16112040
Chicago/Turabian StyleGhobashi, Ahmed H., Rosie Lanzloth, Christopher A. Ladaika, Ashiq Masood, and Heather M. O’Hagan. 2024. "Single-Cell Profiling Reveals the Impact of Genetic Alterations on the Differentiation of Inflammation-Induced Murine Colon Tumors" Cancers 16, no. 11: 2040. https://doi.org/10.3390/cancers16112040
APA StyleGhobashi, A. H., Lanzloth, R., Ladaika, C. A., Masood, A., & O’Hagan, H. M. (2024). Single-Cell Profiling Reveals the Impact of Genetic Alterations on the Differentiation of Inflammation-Induced Murine Colon Tumors. Cancers, 16(11), 2040. https://doi.org/10.3390/cancers16112040