

Monitoring Response and Resistance to Treatment in Chronic Lymphocytic Leukemia

 , , ,
, , ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. How to Assess Response to Treatment: Recommendations from the International Guidelines
2.1. BM Evaluation
2.2. Imaging
2.3. Measurable Residual Disease
2.4. Timing of Response Assessment
3. Methodological Considerations of MRD Testing
4. Evaluation of Response
4.1. Continuous Therapies
4.2. Fixed-Duration Therapies
5. Optimizing the Use of MRD Testing: From Fixed-Duration to Time-Limited Therapy
5.1. Fixed-Duration Treatments to Maximize MRD Responses
5.2. Time-Limited MRD-Guided Treatment Strategies
6. Mechanisms of Resistance to Target Therapy in CLL
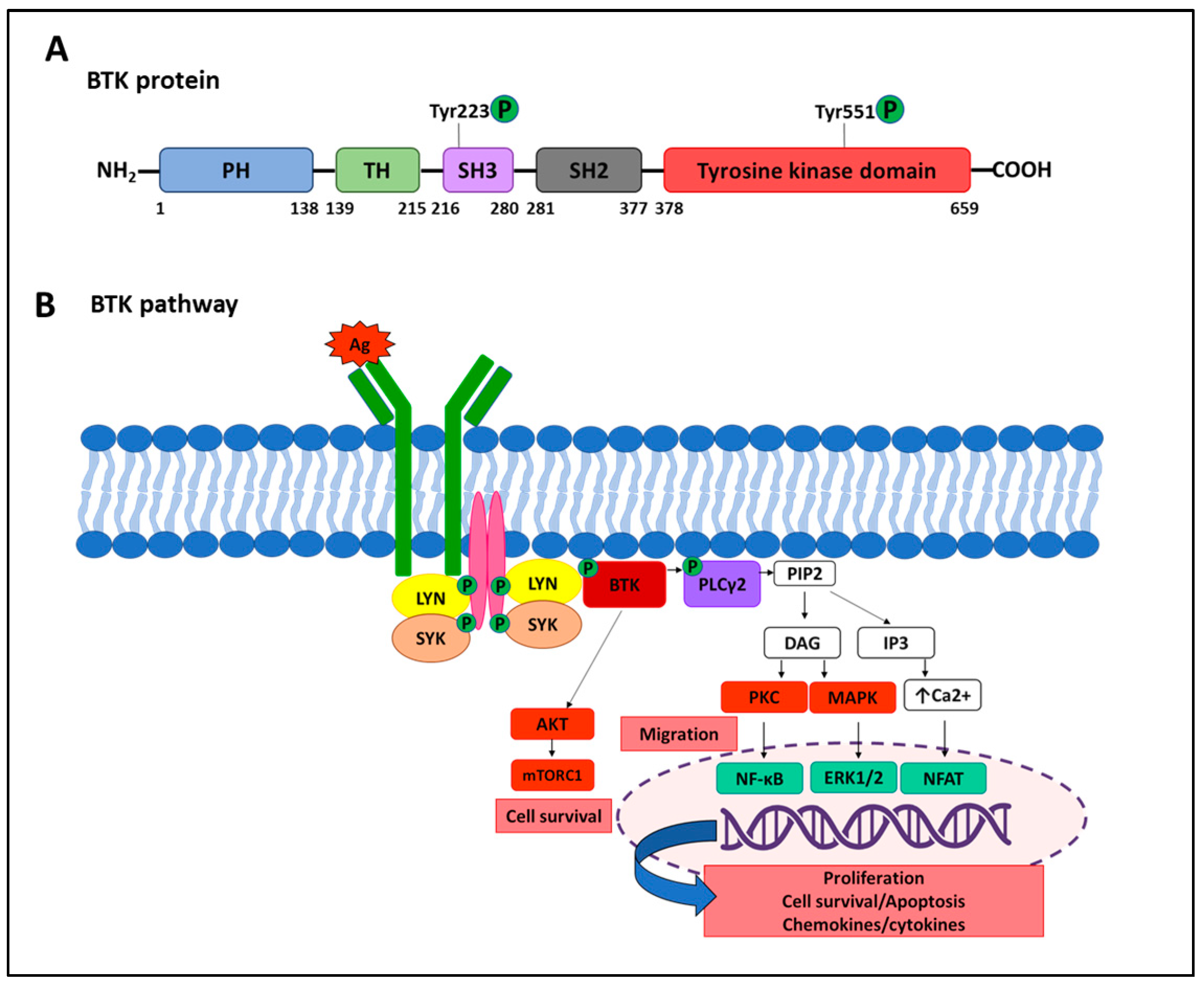
6.1. Resistance to BTK Inhibitors
6.1.1. Target Modification Mediating Resistance to Ibrutinib
6.1.2. Target Modification Mediating Resistance to Acalabrutinib and Zanubrutinib
6.1.3. Resistance to Next-Generation Noncovalent BTK Inhibitors
6.1.4. Gatekeeper Mutations and Super-Resistant Variants
6.1.5. Alternative Genetic Mechanisms of Resistance to BTKi
6.1.6. Non-Genetic Mechanisms of Resistance to BTKi
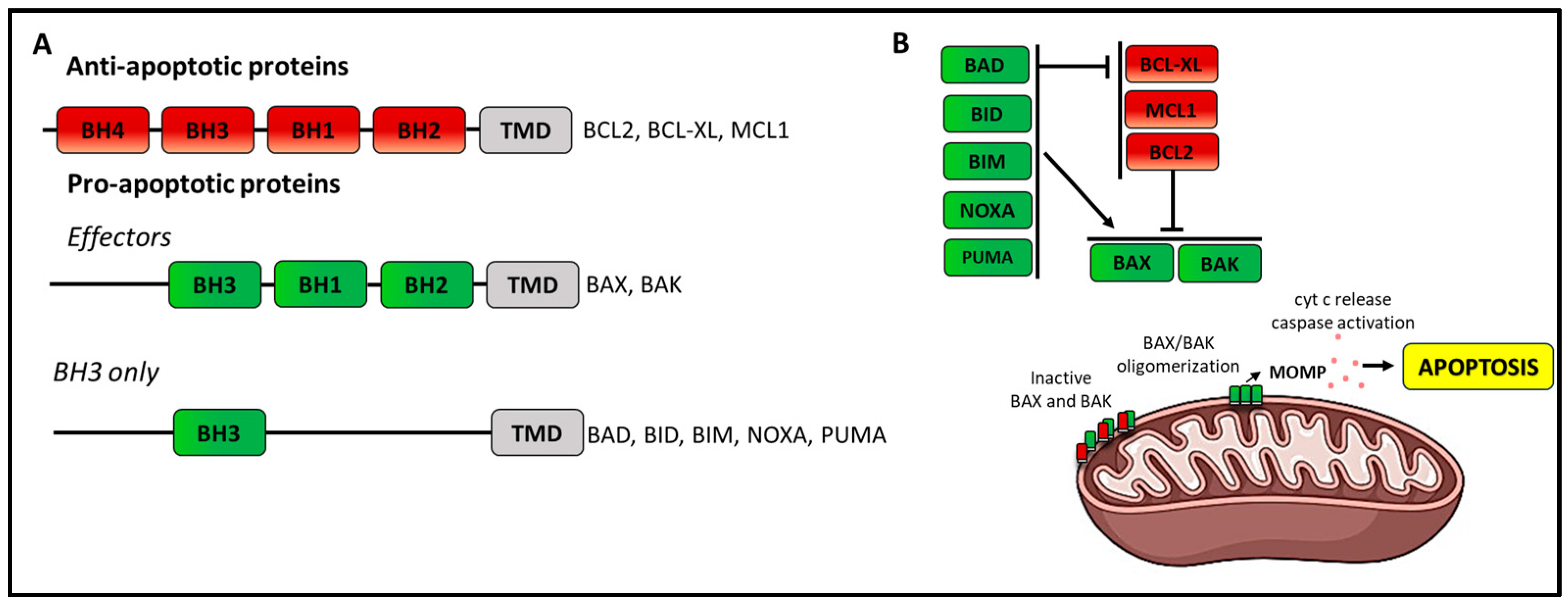
6.2. Resistance to Venetoclax
6.2.1. Target Modification Mediating Resistance to Venetoclax
6.2.2. Mutations of Pro-Apoptotic BCL2 Family Members
6.2.3. Mutations in Other Cancer-Related Genes
6.2.4. Non-Genetic Aberrations of BCL2-Family Proteins
6.2.5. Role of Microenvironment and Bypass Pathways in Resistance
7. Future Directions: Conclusions
- -
- In CLL, the achievement of a CR or even of an uMRD is not always a goal or at least not a goal for everyone, with the availability of continuous treatment with BTK inhibitors;
- -
- The radiologic assessment of response should be limited, especially in continuous treatment approaches;
- -
- In continuous treatment regimens, the timing to assess the best response can be difficult to identify, and clinicians should keep in mind that the achievement of a CR can increase over time;
- -
- For therapies with fixed duration, the duration of treatment is fixed and not dependent on MRD results;
- -
- Even patients with uMRD are not cured and can relapse over time;
- -
- With the introduction of double or triple combinations, the sequencing of therapies or re-treatment is an emerging issue, at least until one of these regimens proves to be curative;
- -
- MRD monitoring outside clinical trials will be soon a reality, but there is a need to select the patients that require it;
- -
- Laboratory standardization and costs still represent an issue;
- -
- Re-treatment remains guided not by MRD conversion but by clinical criteria for disease progression.
Author Contributions
Funding
Conflicts of Interest
References
- Hallek, M.; Cheson, B.D.; Catovsky, D.; Caligaris-Cappio, F.; Dighiero, G.; Döhner, H.; Hillmen, P.; Keating, M.; Montserrat, E.; Chiorazzi, N.; et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood 2018, 131, 2745–2760. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; O’Brien, S. Evolution of CLL treatment—From chemoimmunotherapy to targeted and individualized therapy. Nat. Rev. Clin. Oncol. 2018, 15, 510–527. [Google Scholar] [CrossRef] [PubMed]
- Del Giudice, I.; Raponi, S.; Della Starza, I.; De Propris, M.S.; Cavalli, M.; De Novi, L.A.; Cappelli, L.V.; Ilari, C.; Cafforio, L.; Guarini, A.; et al. Minimal Residual Disease in Chronic Lymphocytic Leukemia: A New Goal? Front. Oncol. 2019, 9, 689. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Del Giudice, I.; Foà, R. Measurable residual disease in chronic lymphocytic leukemia. Where do we stand? Leukemia 2023, 37, 2339–2342. [Google Scholar] [CrossRef] [PubMed]
- Eichhorst, B.; Robak, T.; Montserrat, E.; Ghia, P.; Niemann, C.U.; Kater, A.P.; Gregor, M.; Cymbalista, F.; Buske, C.; Hillmen, P.; et al. Chronic lymphocytic leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2021, 32, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Muntañola, A.; Bosch, F.; Arguis, P.; Arellano-Rodrigo, E.; Ayuso, C.; Giné, E.; Crespo, M.; Abrisqueta, P.; Moreno, C.; Cobo, F.; et al. Abdominal computed tomography predicts progression in patients with Rai stage 0 chronic lymphocytic leukemia. J. Clin. Oncol. 2007, 25, 1576–1580. [Google Scholar] [CrossRef] [PubMed]
- Eichhorst, B.F.; Fischer, K.; Fink, A.M.; Elter, T.; Wendtner, C.M.; Goede, V.; Bergmann, M.; Stilgenbauer, S.; Hopfinger, G.; Ritgen, M.; et al. German CLL Study Group (GCLLSG). Limited clinical relevance of imaging techniques in the follow-up of patients with advanced chronic lymphocytic leukemia: Results of a meta-analysis. Blood 2011, 117, 1817–1821. [Google Scholar] [CrossRef] [PubMed]
- Ballotta, L.; Maccaferri, M.; De Paoli, L.; Orsucci, L.; Gottardi, D.; Chiurazzi, F.; Reda, G.; Moia, R.; Cuneo, A.; Foà, R.; et al. Role of chemotherapy in the treatment of chronic lymphocytic leukemia in the era of targeted therapies in Italy. A Campus CLL network report. Hematol. Oncol. 2023, 41, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, S.; Ritgen, M.; Fischer, K.; Stilgenbauer, S.; Busch, R.M.; Fingerle-Rowson, G.; Fink, A.M.; Bühler, A.; Zenz, T.; Wenger, M.K.; et al. Minimal residual disease quantification is an independent predictor of progression-free and overall survival in chronic lymphocytic leukemia: A multivariate analysis from the randomized GCLLSG CLL8 trial. J. Clin. Oncol. 2012, 30, 980–988. [Google Scholar] [CrossRef] [PubMed]
- Kwok, M.; Rawstron, A.C.; Varghese, A.; Evans, P.A.; O’Connor, S.J.; Doughty, C.; Newton, D.J.; Moreton, P.; Hillmen, P. Minimal residual disease is an independent predictor for 10-year survival in CLL. Blood 2016, 128, 2770–2773. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.; Goradia, H.; Martinez-Calle, N.; Patten, P.; Munir, T. The evolving use of measurable residual disease in chronic lymphocytic leukemia clinical trials. Front. Oncol. 2023, 13, 1130617. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Eichhorst, B.; Niemann, C.U.; Kater, A.P.; Fürstenau, M.; von Tresckow, J.; Zhang, C.; Robrecht, S.; Gregor, M.; Juliusson, G.; Thornton, P.; et al. GCLLSG, the HOVON and Nordic CLL Study Groups, the SAKK, the Israeli CLL Association, and Cancer Trials Ireland. First-Line Venetoclax Combinations in Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2023, 388, 1739–1754. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.; Robrecht, S.; Fink, A.M.; Bahlo, J.; Cramer, P.; von Tresckow, J.; Maurer, C.; Langerbeins, P.; Fingerle-Rowson, G.; Ritgen, M.; et al. Minimal Residual Disease Assessment Improves Prediction of Outcome in Patients with Chronic Lymphocytic Leukemia (CLL) Who Achieve Partial Response: Comprehensive Analysis of Two Phase III Studies of the German CLL Study Group. J. Clin. Oncol. 2016, 34, 3758–3765. [Google Scholar] [CrossRef] [PubMed]
- Kater, A.P.; Seymour, J.F.; Hillmen, P.; Eichhorst, B.; Langerak, A.W.; Owen, C.; Verdugo, M.; Wu, J.; Punnoose, E.A.; Jiang, Y.; et al. Fixed Duration of Venetoclax-Rituximab in Relapsed/Refractory Chronic Lymphocytic Leukemia Eradicates Minimal Residual Disease and Prolongs Survival: Post-Treatment Follow-Up of the MURANO Phase III Study. J. Clin. Oncol. 2019, 37, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Kater, A.P.; Wu, J.Q.; Kipps, T.; Eichhorst, B.; Hillmen, P.; D’Rozario, J.; Assouline, S.; Owen, C.; Robak, T.; de la Serna, J.; et al. Venetoclax Plus Rituximab in Relapsed Chronic Lymphocytic Leukemia: 4-Year Results and Evaluation of Impact of Genomic Complexity and Gene Mutations from the MURANO Phase III Study. J. Clin. Oncol. 2020, 38, 4042–4054. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wierda, W.G.; Rawstron, A.; Cymbalista, F.; Badoux, X.; Rossi, D.; Brown, J.R.; Egle, A.; Abello, V.; Cervera Ceballos, E.; Herishanu, Y.; et al. Measurable residual disease in chronic lymphocytic leukemia: Expert review and consensus recommendations. Leukemia 2021, 35, 3059–3072. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rawstron, A.C.; Bottcher, S.; Letestu, R.; Villamor, N.; Fazi, C.; Kartsios, H.; de Tute, R.M.; Shingles, J.; Ritgen, M.; Moreno, C. Improving efficiency and sensitivity: European Research Initiative in CLL (ERIC) update on the international harmonised approach for flow cytometric residual disease monitoring in CLL. Leukemia 2013, 27, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Letestu, R.; Cartron, G.; Lepretre, S.; Le Garff-Tavernier, M.; Solly, F.; Campos, L.; Ticchioni, M.; Quiney, C.; Dahmani, A.; Boubaya, M. Minimal Residual Disease (MRD) By 8-Color Flow Cytometry (Flow-MRD) and IGH Clonospecific Quantitative PCR (ASO RQPCR) Reached Similar Performances for Evaluation of CLL Treatment in a Phase II Clinical Trial: Cross Validation of the Methods. Blood 2014, 124, 3307. [Google Scholar] [CrossRef]
- Rawstron, A.C.; Villamor, N.; Ritgen, M.; Bottcher, S.; Ghia, P.; Zehnder, J.L.; Lozanski, G.; Colomer, D.; Moreno, C.; Geuna, M. International standardized approach for flow cytometric residual disease monitoring in chronic lymphocytic leukaemia. Leukemia 2007, 21, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Rawstron, A.C.; Kreuzer, K.A.; Soosapilla, A.; Spacek, M.; Stehlikova, O.; Gambell, P.; McIver-Brown, N.; Vilamor, N.; Psarra, K.; Arroz, M.; et al. Reproducible diagnosis of chronic lymphocytic leukemia by flow cytometry: A European research initiative on CLL (ERIC) & European society for clinical cell analysis (ESCCA) harmonisation project. Cytom. B Clin. Cytom. 2018, 94, 121–128. [Google Scholar]
- Letestu, R.; Dahmani, A.; Boubaya, M.; Baseggio, L.; Campos, L.; Chatelain, B.; Debliquis, A.; Drénou, B.; Jacob, M.C.; Legac, E.; et al. French Innovative Leukemia Organization (FILO). Prognostic value of high-sensitivity measurable residual disease assessment after front-line chemoimmunotherapy in chronic lymphocytic leukemia. Leukemia 2021, 35, 1597–1609. [Google Scholar] [CrossRef] [PubMed]
- Flores-Montero, J.; Sanoja-Flores, L.; Paiva, B.; Puig, N.; García-Sánchez, O.; Böttcher, S.; van der Velden, V.H.J.; Perez-Moran, J.; Vidriales, M.-B.; Garcia-Sanz, R.; et al. Next generation flow for highly sensitive and standardized detection of minimal residual disease in multiple myeloma. Leukemia 2017, 31, 2094–2103. [Google Scholar] [CrossRef] [PubMed]
- Bottcher, S.; Ritgen, M.; Pott, C.; Bruggemann, M.; Raff, T.; Stilgenbauer, S.; Dohner, H.; Dreger, P.; Kneba, M. Comparative analysis of minimal residual disease detection using four-color flow cytometry, consensus IgH-PCR, and quantitative IgH PCR in CLL after allogeneic and autologous stem cell transplantation. Leukemia 2004, 18, 1637–1645. [Google Scholar] [CrossRef] [PubMed]
- Bottcher, S.; Stilgenbauer, S.; Busch, R.; Bruggermann, M.; Raff, T.; Pott, C.; Fischer, K. Standardized MRD flow and ASO IGH RQ-PCR for MRD quantification in CLL patients after rituximab-containing immunochemotherapy: A comparative analysis. Leukemia 2009, 23, 2007–2017. [Google Scholar] [CrossRef] [PubMed]
- Raponi, S.; Della Starza, I.; De Propris, M.S.; Del Giudice, I.; Mauro, F.R.; Marinelli, M.; Di Maio, V.; Piciocchi, A.; Foà, R.; Guarini, A. Minimal residual disease monitoring in chronic lymphocytic leukaemia patients. A comparative analysis of flow cytometry and ASO IgH RQ-PCR. Brit. J. Haematol. 2014, 166, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Della Starza, I.; Cavalli, M.; Del Giudice, I.; Barbero, D.; Mantoan, B.; Genuardi, E.; Urbano, M.; Mannu, C.; Gazzola, A.; Ciabatti, E.; et al. Comparison of two real-time quantitative polymerase chain reaction strategies for minimal residual disease evaluation in lymphoproliferative disorders: Correlation between immunoglobulin gene mutation load and real-time quantitative polymerase chain reaction performance. Hematol. Oncol. 2014, 32, 133–138. [Google Scholar] [PubMed]
- Van der Velden, V.H.; Hochhaus, A.; Cazzaniga, G.; Szczepanski, T.; Gabert, J.; van Dongen, J.J.M. Detection of minimal residual disease in hematologic malignancies by real-time quantitative PCR: Principles, approaches, and laboratory aspects. Leukemia 2003, 17, 1013–1034. [Google Scholar] [CrossRef] [PubMed]
- Van der Velden, V.H.; Cazzaniga, G.; Schrauder, A.; Hancock, J.; Bader, P.; Panzer-Grumayer, E.R.; Flohr, T.; Sutton, R.; Cave, H.; Madsen, H.O.; et al. Analysis of minimal residual disease by Ig/TCR gene rearrangements: Guidelines for interpretation of real-time quantitative PCR data. Leukemia 2007, 21, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Hindson, C.M.; Chevillet, J.R.; Briggs, H.A.; Gallichotte, E.N.; Ruf, I.K.; Hindson, B.J.; Vessella, R.L.; Tewari, M. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat. Methods 2013, 10, 1003–1005. [Google Scholar] [CrossRef]
- Huggett, J.F.; Whale, A. Digital PCR as a novel technology and its potential implications for molecular diagnostics. Clin. Chem. 2013, 59, 1691–1693. [Google Scholar] [CrossRef]
- Drandi, D.; Kubiczkova-Besse, L.; Ferrero, S.; Dani, N.; Passera, R.; Mantoan, B.; Gambella, M.; Monitillo, L.; Saraci, E.; Ghione, P.; et al. Minimal residual disease detection by droplet digital PCR in multiple myeloma, mantle cell lymphoma, and follicular lymphoma: A comparison with Real-Time PCR. J. Mol. Diagn. 2015, 17, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Della Starza, I.; Nunes, V.; Cavalli, M.; De Novi, L.A.; Ilari, C.; Apicella, V.; Vitalle, A.; Testi, A.M.; Del Giudice, I.; Chiaretti, S.; et al. Comparative analysis between RQ-PCR and digital-droplet-PCR of immunoglobulin/T-cell receptor gene rearrangements to monitor minimal residual disease in acute lymphoblastic leukaemia. Br. J. Haematol. 2016, 174, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, M.; De Novi, L.A.; Della Starza, I.; Cappelli, L.V.; Nunes, V.; Pulsoni, A.; Del Giudice, I.; Guarini, A.; Foà, R. Comparative analysis between RQ-PCR and digital droplet PCR of BCL2/IGH gene rearrangement in the peripheral blood and bone marrow of early-stage follicular lymphoma. Br. J. Haematol. 2017, 177, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Drandi, D.; Ferrero, S.; Ladetto, M. Droplet digital PCR for minimal residual disease detection in mature lymphoproliferative disorders. Methods Mol. Biol. 2018, 1768, 229–256. [Google Scholar] [PubMed]
- Assanto, G.M.; Del Giudice, I.; Della Starza, I.; Soscia, R.; Cavalli, M.; Cola, M.; Bellomarino, V.; Di Trani, M.; Guarini, A.; Foà, R. Research Topic: Measurable Residual Disease in Hematologic Malignancies. Can digital droplet PCR improve measurable residual disease monitoring in chronic lymphoid malignancies? Front. Oncol. 2023, 13, 1152467. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Logan, A.C.; Gao, H.; Wang, C.; Sahaf, B.; Jones, C.D.; Marshall, E.L.; Buno, I.; Armstrong, R.; Fire, A.Z.; Weinberg, K.I.; et al. High-throughput VDJ sequencing for quantification of minimal residual disease in chronic lymphocytic leukemia and immune reconstitution assessment. Proc. Natl. Acad. Sci. USA 2011, 108, 21194–21199. [Google Scholar] [CrossRef] [PubMed]
- Logan, A.C.; Zhang, B.; Narasimhan, B.; Carlton, V.; Zheng, J.; Moorhead, M.; Krampf, M.R.; Jones, C.D.; Waqar, A.M.; Faham, M.; et al. Minimal residual disease quantification using consensus primers and high-throughput IGH sequencing predicts post-transplant relapse in chronic lymphocytic leukemia. Leukemia 2013, 27, 1659–1665. [Google Scholar] [CrossRef] [PubMed]
- Ladetto, M.; Brïggemann, M.; Monitillo, L.; Ferrero, S.; Pepin, F.; Drandi, D.; Barbero, D.; Palumbo, A.; Passera, R.; Boccadoro, M.; et al. Next-generation sequencing and real-time quantitative PCR for minimal residual disease detection in B-cell disorders. Leukemia 2014, 28, 1299–1307. [Google Scholar] [CrossRef]
- Stamatopoulos, B.; Timbs, A.; Bruce, D.; Smith, T.; Clifford, R.; Robbe, P.; Burns, A.; Vavoulis, D.V.; Lopez, L.; Antoniou, P.; et al. Targeted deep sequencing reveals clinically relevant subclonal IgHV rearrangements in chronic lymphocytic leukemia. Leukemia 2017, 31, 837–845. [Google Scholar] [CrossRef]
- Thompson, P.A.; Srivastava, J.; Peterson, C.; Strati, P.; Jorgensen, J.L.; Hether, T.; Keating, M.J.; O’Brien, S.M.; Ferrajoli, A.; Burger, J.A.; et al. Minimal residual disease undetectable by next-generation sequencing predicts improved outcome in CLL after chemoimmunotherapy. Blood 2019, 134, 1951–1959. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Al-Sawaf, O.; Zhang, C.; Lu, T.; Liao, M.Z.; Panchal, A.; Robrecht, S.; Ching, T.; Tandon, M.; Fink, A.M.; Tausch, E.; et al. Minimal Residual Disease Dynamics after Venetoclax-Obinutuzumab Treatment: Extended Off-Treatment Follow-up from the Randomized CLL14 Study. J. Clin. Oncol. 2021, 39, 4049–4060. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Al-Sawaf, O.; Zhang, C.; Jin, H.Y.; Robrecht, S.; Choi, Y.; Balasubramanian, S.; Kotak, A.; Chang, Y.M.; Fink, A.M.; Tausch, E.; et al. Transcriptomic profiles and 5-year results from the randomized CLL14 study of venetoclax plus obinutuzumab versus chlorambucil plus obinutuzumab in chronic lymphocytic leukemia. Nat. Commun. 2023, 14, 2147. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fischer, K.; Al-Sawaf, O.; Bahlo, J.; Fink, A.M.; Tandon, M.; Dixon, M.; Robrecht, S.; Warburton, S.; Humphrey, K.; Samoylova, O.; et al. Venetoclax and Obinutuzumab in Patients with CLL and Coexisting Conditions. N. Engl. J. Med. 2019, 380, 2225–2236. [Google Scholar] [CrossRef] [PubMed]
- Munir, T.; Moreno, C.; Owen, C.; Follows, G.; Benjamini, O.; Janssens, A.; Levin, M.D.; Osterborg, A.; Robak, T.; Simkovic, M.; et al. Impact of Minimal Residual Disease on Progression-Free Survival Outcomes After Fixed-Duration Ibrutinib-Venetoclax Versus Chlorambucil-Obinutuzumab in the GLOW Study. J. Clin. Oncol. 2023, 41, 3689–3699. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wierda, W.G.; Allan, J.N.; Siddiqi, T.; Kipps, T.J.; Opat, S.; Tedeschi, A.; Badoux, X.C.; Kuss, B.J.; Jackson, S.; Moreno, C.; et al. Ibrutinib Plus Venetoclax for First-Line Treatment of Chronic Lymphocytic Leukemia: Primary Analysis Results from the Minimal Residual Disease Cohort of the Randomized Phase II CAPTIVATE Study. J. Clin. Oncol. 2021, 39, 3853–3865. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tam, C.S.; Allan, J.N.; Siddiqi, T.; Kipps, T.J.; Jacobs, R.; Opat, S.; Barr, P.M.; Tedeschi, A.; Trentin, L.; Bannerji, R.; et al. Fixed-duration ibrutinib plus venetoclax for first-line treatment of CLL: Primary analysis of the CAPTIVATE FD cohort. Blood 2022, 139, 3278–3289. [Google Scholar] [CrossRef] [PubMed]
- Lauer, E.M.; Mutter, J.; Scherer, F. Circulating tumor DNA in B-cell lymphoma: Technical advances, clinical applications, and perspectives for translational research. Leukemia 2022, 36, 2151–2164. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.V.; Hanson, C.A.; Tschumper, R.C.; Lesnick, C.E.; Braggio, E.; Paietta, E.M.; O’Brien, S.; Barrientos, J.C.; Leis, J.F.; Zhang, C.C.; et al. Measurable residual disease does not preclude prolonged progression-free survival in CLL treated with ibrutinib. Blood 2021, 138, 2810–2827. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sharman, J.P.; Egyed, M.; Jurczak, W.; Skarbnik, A.; Pagel, J.M.; Flinn, I.W.; Kamdar, M.; Munir, T.; Walewska, R.; Corbett, G.; et al. Efficacy and safety in a 4-year follow-up of the ELEVATE-TN study comparing acalabrutinib with or without obinutuzumab versus obinutuzumab plus chlorambucil in treatment-naïve chronic lymphocytic leukemia. Leukemia 2022, 36, 1171–1175. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Byrd, J.C.; Furman, R.R.; Coutre, S.E.; Flinn, I.W.; Burger, J.A.; Blum, K.A.; Grant, B.; Sharman, J.P.; Coleman, M.; Wierda, W.G.; et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2013, 369, 32–42, Erratum in N. Engl. J. Med. 2014, 370, 786. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Woyach, J.A.; Smucker, K.; Smith, L.L.; Lozanski, A.; Zhong, Y.; Ruppert, A.S.; Lucas, D.; Williams, K.; Zhao, W.; Rassenti, L.; et al. Prolonged lymphocytosis during ibrutinib therapy is associated with distinct molecular characteristics and does not indicate a suboptimal response to therapy. Blood 2014, 123, 1810–1817. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Barr, P.M.; Owen, C.; Robak, T.; Tedeschi, A.; Bairey, O.; Burger, J.A.; Hillmen, P.; Coutre, S.E.; Dearden, C.; Grosicki, S.; et al. Up to 8-year follow-up from RESONATE-2: First-line ibrutinib treatment for patients with chronic lymphocytic leukemia. Blood Adv. 2022, 6, 3440–3450. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mauro, F.R.; Paoloni, F.; Molica, S.; Reda, G.; Trentin, L.; Sportoletti, P.; Marchetti, M.; Pietrasanta, D.; Marasca, R.; Gaidano, G.; et al. Efficacy of Front-Line Ibrutinib and Rituximab Combination and the Impact of Treatment Discontinuation in Unfit Patients with Chronic Lymphocytic Leukemia: Results of the Gimema LLC1114 Study. Cancers 2021, 14, 207. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Moreno, C.; Greil, R.; Demirkan, F.; Tedeschi, A.; Anz, B.; Larratt, L.; Simkovic, M.; Novak, J.; Strugov, V.; Gill, D.; et al. First-line treatment of chronic lymphocytic leukemia with ibrutinib plus obinutuzumab versus chlorambucil plus obinutuzumab: Final analysis of the randomized, phase III iLLUMINATE trial. Haematologica 2022, 107, 2108–2120. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Seymour, J.F.; Kipps, T.J.; Eichhorst, B.F.; D’Rozario, J.; Owen, C.J.; Assouline, S.; Lamanna, N.; Robak, T.; de la Serna, J.; Jaeger, U.; et al. Enduring undetectable MRD and updated outcomes in relapsed/refractory CLL after fixed-duration venetoclax-rituximab. Blood 2022, 140, 839–850. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Thompson, P.A.; Peterson, C.B.; Strati, P.; Jorgensen, J.; Keating, M.J.; O’Brien, S.M.; Ferrajoli, A.; Burger, J.A.; Estrov, Z.; Jain, N.; et al. Serial minimal residual disease (MRD) monitoring during first-line FCR treatment for CLL may direct individualized therapeutic strategies. Leukemia 2018, 32, 2388–2398. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wodarz, D.; Garg, N.; Komarova, N.L.; Benjamini, O.; Keating, M.J.; Wierda, W.G.; Kantarjian, H.; James, D.; O’Brien, S.; Burger, J.A. Kinetics of CLL cells in tissues and blood during therapy with the BTK inhibitor ibrutinib. Blood 2014, 123, 4132–4135. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cervantes-Gomez, F.; Lamothe, B.; Woyach, J.A.; Wierda, W.G.; Keating, M.J.; Balakrishnan, K.; Gandhi, V. Pharmacological and Protein Profiling Suggests Venetoclax (ABT-199) as Optimal Partner with Ibrutinib in Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2015, 21, 3705–3715. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jain, N.; Keating, M.; Thompson, P.; Ferrajoli, A.; Burger, J.; Borthakur, G.; Takahashi, K.; Estrov, Z.; Fowler, N.; Kadia, T.; et al. Ibrutinib and Venetoclax for First-Line Treatment of CLL. N. Engl. J. Med. 2019, 380, 2095–2103. [Google Scholar] [CrossRef] [PubMed]
- Hillmen, P.; Rawstron, A.C.; Brock, K.; Muñoz-Vicente, S.; Yates, F.J.; Bishop, R.; Boucher, R.; MacDonald, D.; Fegan, C.; McCaig, A.; et al. Ibrutinib Plus Venetoclax in Relapsed/Refractory Chronic Lymphocytic Leukemia: The CLARITY Study. J. Clin. Oncol. 2019, 37, 2722–2729, Erratum in J. Clin. Oncol. 2020, 38, 1644. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kater, A.P.; Owen, C.; Moreno, C.; Follows, G.; Munir, T.; Levin, M.D.; Benjamini, O.; Janssens, A.; Osterborg, A.; Robak, T.; et al. Fixed-Duration Ibrutinib-Venetoclax in Patients with Chronic Lymphocytic Leukemia and Comorbidities. NEJM Evid. 2022, 1, EVIDoa2200006. [Google Scholar] [CrossRef] [PubMed]
- Ghia, P.; Allan, J.N.; Siddiqi, T.; Kipps, T.J.; Jacobs, R.; Opat, S.; Barr, P.M.; Tedeschi, A.; Trentin, L.; Bennerji, R.; et al. Fixed-duration (FD) first-line treatment (tx) with ibrutinib (I) plus venetoclax (V) for chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL): Primary analysis of the FD cohort of the phase 2 captivate study [Abstract]. J. Clin. Oncol. 2021, 39 (Suppl. S15), 7501. [Google Scholar] [CrossRef]
- Jain, N.; Keating, M.; Thompson, P.; Ferrajoli, A.; Burger, J.A.; Borthakur, G.; Takahashi, K.; Estrov, Z.; Sasaki, K.; Fowler, N.; et al. Ibrutinib Plus Venetoclax for First-line Treatment of Chronic Lymphocytic Leukemia: A Nonrandomized Phase 2 Trial. JAMA Oncol. 2021, 7, 1213–1219. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Niemann, C.U.; Munir, T.; Moreno, C.; Owen, C.; Follows, G.A.; Benjamini, O.; Janssens, A.; Levin, M.D.; Robak, T.; Simkovic, M.; et al. Fixed-duration ibrutinib-venetoclax versus chlorambucil-obinutuzumab in previously untreated chronic lymphocytic leukaemia (GLOW): 4-year follow-up from a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2023, 24, 1423–1433. [Google Scholar] [CrossRef] [PubMed]
- Fürstenau, M.; Ritgen, M.; Robrecht, S.; Von Tresckow, J.; Zhang, C.; Schilhabel, A.; Gregor, M.; Thornton, P.; Staber, P.B.; Tadmor, T.; et al. First-Line Venetoclax Combinations in Fit Patients with CLL: 4-Year Follow-up and NGS-Based MRD Analysis from the Phase 3 GAIA/CLL13 Trial. Blood 2023, 142 (Suppl. S1), 635. [Google Scholar] [CrossRef]
- Rogers, K.A.; Huang, Y.; Ruppert, A.S.; Abruzzo, L.V.; Andersen, B.L.; Awan, F.T.; Bhat, S.A.; Dean, A.; Lucas, M.; Banks, C.; et al. Phase II Study of Combination Obinutuzumab, Ibrutinib, and Venetoclax in Treatment-Naïve and Relapsed or Refractory Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2020, 38, 3626–3637. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Huber, H.; Edenhofer, S.; von Tresckow, J.; Robrecht, S.; Zhang, C.; Tausch, E.; Schneider, C.; Bloehdorn, J.; Fürstenau, M.; Dreger, P.; et al. Obinutuzumab (GA-101), ibrutinib, and venetoclax (GIVe) frontline treatment for high-risk chronic lymphocytic leukemia. Blood 2022, 139, 1318–1329. [Google Scholar] [CrossRef] [PubMed]
- Davids, M.S.; Lampson, B.L.; Tyekucheva, S.; Wang, Z.; Lowney, J.C.; Pazienza, S.; Montegaard, J.; Patterson, V.; Weinstock, M.; Crombie, J.L.; et al. Acalabrutinib, venetoclax, and obinutuzumab as frontline treatment for chronic lymphocytic leukaemia: A single-arm, open-label, phase 2 study. Lancet Oncol. 2021, 22, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Soumerai, J.D.; Mato, A.R.; Dogan, A.; Seshan, V.E.; Joffe, E.; Flaherty, K.; Carter, J.; Hochberg, E.; Barnes, J.A.; Hamilton, A.M.; et al. Zanubrutinib, obinutuzumab, and venetoclax with minimal residual disease-driven discontinuation in previously untreated patients with chronic lymphocytic leukaemia or small lymphocytic lymphoma: A multicentre, single-arm, phase 2 trial. Lancet Haematol. 2021, 8, e879–e890. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kater, A.P.; Levin, M.D.; Dubois, J.; Kersting, S.; Enggaard, L.; Veldhuis, G.J.; Mous, R.; Mellink, C.H.M.; van der Kevie-Kersemaekers, A.F.; Dobber, J.A.; et al. Minimal residual disease-guided stop and start of venetoclax plus ibrutinib for patients with relapsed or refractory chronic lymphocytic leukaemia (HOVON141/VISION): Primary analysis of an open-label, randomised, phase 2 trial. Lancet Oncol. 2022, 23, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Scarfò, L.; Heltai, S.; Albi, E.; Scarano, E.; Schiattone, L.; Farina, L.; Moia, R.; Deodato, M.; Ferrario, A.; Motta, M.; et al. Minimal residual disease-driven treatment intensification with sequential addition of ibrutinib to venetoclax in R/R CLL. Blood 2022, 140, 2348–2357. [Google Scholar] [CrossRef] [PubMed]
- Ghia, P.; Wierda, W.G.; Barr, P.M.; Kipps, T.J.; Siddiqi, T.; Allan, J.N.; Hunter, Z.; Zhou, C.; Szoke, A.; Dean, J.P.; et al. Relapse after First-Line Fixed Duration Ibrutinib + Venetoclax: High Response Rates to Ibrutinib Retreatment and Absence of BTK Mutations in Patients with Chronic Lymphocytic Leukemia (CLL)/Small Lymphocytic Lymphoma (SLL) with up to 5 Years of Follow-up in the Phase 2 Captivate Study. Blood 2023, 142 (Suppl. S1), 633. [Google Scholar] [CrossRef]
- Hillmen, P.; Cairns, D.A.; Bloor, A.J.C.; Allsup, D.; Cwynarski, K.; Pettitt, A.; Paneesha, S.; Fox, C.P.; Eyre, T.A.; Forconi, F.; et al. Ibrutinib Plus Venetoclax with MRD-Directed Duration of Treatment Is Superior to FCR and Is a New Standard of Care for Previously Untreated CLL: Report of the Phase III UK NCRI FLAIR Study. Blood 2023, 142 (Suppl. S1), 631. [Google Scholar] [CrossRef]
- Ryan, C.E.; Davids, M.S.; Hermann, R.; Shahkarami, M.; Biondo, J.; Abhyankar, S.; Alhasani, H.; Sharman, J.P.; Mato, A.R.; Roeker, L.E. MAJIC: A phase III trial of acalabrutinib + venetoclax versus venetoclax + obinutuzumab in previously untreated chronic lymphocytic leukemia or small lymphocytic lymphoma. Future Oncol. 2022, 18, 3689–3699, Erratum in Future Oncol. 2023, 19, 271. [Google Scholar] [CrossRef] [PubMed]
- Wen, T.; Wang, J.; Shi, Y.; Qian, H.; Liu, P. Inhibitors targeting Bruton’s tyrosine kinase in cancers: Drug development advances. Leukemia 2021, 35, 312–332. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pal Singh, S.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer. 2018, 17, 57, Published correction appears in Mol. Cancer 2019, 18, 79. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, R.W.; Yuvaraj, S.; Kil, L.P. Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat. Rev. Cancer. 2014, 14, 219–232. [Google Scholar] [CrossRef]
- Doyle, S.L.; Jefferies, C.A.; Feighery, C.; O’Neill, L.A. Signaling by Toll-like receptors 8 and 9 requires Bruton’s tyrosine kinase. J. Biol. Chem. 2007, 282, 36953–36960. [Google Scholar] [CrossRef]
- Spaargaren, M.; Beuling, E.A.; Rurup, M.L.; Meijer, H.P.; Klok, M.D.; Middendorp, M.; Hendriks, R.W.; Pals, S.T. The B cell antigen receptor controls integrin activity through Btk and PLCgamma2. J. Exp. Med. 2003, 198, 1539–1550. [Google Scholar] [CrossRef] [PubMed]
- de Rooij, M.F.; Kuil, A.; Geest, C.R.; Eldering, E.; Chang, B.Y.; Buggy, J.J.; Pals, S.T.; Spaargaren, M. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood 2012, 119, 2590–2594. [Google Scholar] [CrossRef] [PubMed]
- Cheng, E.H.; Wei, M.C.; Weiler, S.; Flavell, R.A.; Mak, T.W.; Lindsten, T.; Korsmeyer, S.J. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol. Cell 2001, 8, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Shamas-Din, A.; Brahmbhatt, H.; Leber, B.; Andrews, D.W. BH3-only proteins: Orchestrators of apoptosis. Biochim. Biophys. Acta 2011, 1813, 508–520. [Google Scholar] [CrossRef] [PubMed]
- Honigberg, L.A.; Smith, A.M.; Sirisawad, M.; Verner, E.; Loury, D.; Chang, B.; Li, S.; Pan, Z.; Thamm, D.; Miller, R.A.; et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. USA 2010, 107, 13075–13080. [Google Scholar] [CrossRef] [PubMed]
- Woyach, J.A.; Furman, R.R.; Liu, T.M.; Ozer, H.G.; Zapatka, M.; Ruppert, A.S.; Xue, L.; Li, D.H.; Steggerda, S.M.; Versele, M.; et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N. Engl. J. Med. 2014, 370, 2286–2294. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Furman, R.R.; Cheng, S.; Lu, P.; Setty, M.; Perez, A.R.; Guo, A.; Racchumi, J.; Xu, G.; Wu, H.; Ma, J.; et al. Ibrutinib resistance in chronic lymphocytic leukemia. N. Engl. J. Med. 2014, 370, 2352–2354. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kanagal-Shamanna, R.; Jain, P.; Patel, K.P.; Routbort, M.; Bueso-Ramos, C.; Alhalouli, T.; Khoury, J.D.; Luthra, R.; Ferrajoli, A.; Keating, M.; et al. Targeted multigene deep sequencing of Bruton tyrosine kinase inhibitor-resistant chronic lymphocytic leukemia with disease progression and Richter transformation. Cancer 2019, 125, 559–574. [Google Scholar] [CrossRef] [PubMed]
- Landau, D.A.; Sun, C.; Rosebrock, D.; Herman, S.E.M.; Fein, J.; Sivina, M.; Underbayev, C.; Liu, D.; Hoellenriegel, J.; Ravichandran, S.; et al. The evolutionary landscape of chronic lymphocytic leukemia treated with ibrutinib targeted therapy. Nat. Commun. 2017, 8, 2185. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sharma, S.; Galanina, N.; Guo, A.; Lee, J.; Kadri, S.; Van Slambrouck, C.; Long, B.; Wang, W.; Ming, M.; Furtado, L.V.; et al. Identification of a structurally novel BTK mutation that drives ibrutinib resistance in CLL. Oncotarget 2016, 7, 68833–68841. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ahn, I.E.; Underbayev, C.; Albitar, A.; Herman, S.E.; Tian, X.; Maric, I.; Arthur, D.C.; Wake, L.; Pittaluga, S.; Yuan, C.M.; et al. Clonal evolution leading to ibrutinib resistance in chronic lymphocytic leukemia. Blood 2017, 129, 1469–1479. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Quinquenel, A.; Fornecker, L.M.; Letestu, R.; Ysebaert, L.; Fleury, C.; Lazarian, G.; Dilhuydy, M.S.; Nollet, D.; Guieze, R.; Feugier, P.; et al. French Innovative Leukemia Organization (FILO) CLL Group. Prevalence of BTK and PLCG2 mutations in a real-life CLL cohort still on ibrutinib after 3 years: A FILO group study. Blood 2019, 134, 641–644. [Google Scholar] [CrossRef] [PubMed]
- Maddocks, K.J.; Ruppert, A.S.; Lozanski, G.; Heerema, N.A.; Zhao, W.; Abruzzo, L.; Lozanski, A.; Davis, M.; Gordon, A.; Smith, L.L.; et al. Etiology of Ibrutinib Therapy Discontinuation and Outcomes in Patients with Chronic Lymphocytic Leukemia. JAMA Oncol. 2015, 1, 80–87. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hamasy, A.; Wang, Q.; Blomberg, K.E.; Mohammad, D.K.; Yu, L.; Vihinen, M.; Berglöf, A.; Smith, C.I. Substitution scanning identifies a novel, catalytically active ibrutinib-resistant BTK cysteine 481 to threonine (C481T) variant. Leukemia 2017, 31, 177–185. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Estupiñán, H.Y.; Wang, Q.; Berglöf, A.; Schaafsma, G.C.P.; Shi, Y.; Zhou, L.; Mohammad, D.K.; Yu, L.; Vihinen, M.; Zain, R.; et al. BTK gatekeeper residue variation combined with cysteine 481 substitution causes super-resistance to irreversible inhibitors acalabrutinib, ibrutinib and zanubrutinib. Leukemia 2021, 35, 1317–1329. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chopra, N.; Wales, T.E.; Joseph, R.E.; Boyken, S.E.; Engen, J.R.; Jernigan, R.L.; Andreotti, A.H. Dynamic Allostery Mediated by a Conserved Tryptophan in the Tec Family Kinases. PLoS Comput. Biol. 2016, 12, e1004826. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gángó, A.; Alpár, D.; Galik, B.; Marosvári, D.; Kiss, R.; Fésüs, V.; Aczél, D.; Eyüpoglu, E.; Nagy, N.; Nagy, Á.; et al. Dissection of subclonal evolution by temporal mutation profiling in chronic lymphocytic leukemia patients treated with ibrutinib. Int. J. Cancer 2020, 146, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.; Woyach, J.A.; Zhao, W.; Caruthers, S.; Tu, H.; Coleman, J.; Byrd, J.C.; Johnson, A.J.; Lozanski, G. PLCG2 C2 domain mutations co-occur with BTK and PLCG2 resistance mutations in chronic lymphocytic leukemia undergoing ibrutinib treatment. Leukemia 2017, 31, 1645–1647. [Google Scholar] [CrossRef] [PubMed]
- Kadri, S.; Lee, J.; Fitzpatrick, C.; Galanina, N.; Sukhanova, M.; Venkataraman, G.; Sharma, S.; Long, B.; Petras, K.; Theissen, M.; et al. Clonal evolution underlying leukemia progression and Richter transformation in patients with ibrutinib-relapsed CLL. Blood Adv. 2017, 1, 715–727. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, T.M.; Woyach, J.A.; Zhong, Y.; Lozanski, A.; Lozanski, G.; Dong, S.; Strattan, E.; Lehman, A.; Zhang, X.; Jones, J.A.; et al. Hypermorphic mutation of phospholipase C, γ2 acquired in ibrutinib-resistant CLL confers BTK independency upon B-cell receptor activation. Blood 2015, 126, 61–68. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Woyach, J.A.; Ruppert, A.S.; Guinn, D.; Lehman, A.; Blachly, J.S.; Lozanski, A.; Heerema, N.A.; Zhao, W.; Coleman, J.; Jones, D.; et al. BTKC481S-Mediated Resistance to Ibrutinib in Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2017, 35, 1437–1443. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Woyach, J.; Huang, Y.; Rogers, K.; Bhat, S.A.; Grever, M.R.; Lozanski, A.; Doong, T.J.; Blachly, J.S.; Lozanski, G.; Jones, D.; et al. Resistance to Acalabrutinib in CLL Is Mediated Primarily by BTK Mutations. Blood 2019, 134 (Suppl. S1), 504. [Google Scholar] [CrossRef]
- Woyach, J.A.; Jones, D.; Jurczak, W.; Robak, T.; Illés, A.; Kater, A.P.; Ghia, P.; Byrd, J.C.; Seymour, J.F.; Long, S.; et al. Characterization of Mechanisms of Resistance in Previously Treated Chronic Lymphocytic Leukemia (CLL) From a Head-to-Head Trial of Acalabrutinib Versus Ibrutinib. Hematol. Oncol. 2023, 41, 249–250. [Google Scholar] [CrossRef]
- Handunnetti, S.M.; Tang, C.P.S.; Nguyen, T.; Zhou, X.; Thompson, E.; Sun, H.; Xing, H.; Zhang, B.; Guo, Y.; Sutton, L.A.; et al. BTK Leu528Trp—A Potential Secondary Resistance Mechanism Specific for Patients with Chronic Lymphocytic Leukemia Treated with the Next Generation BTK Inhibitor Zanubrutinib. Blood 2019, 134 (Suppl. S1), 170. [Google Scholar] [CrossRef]
- Blombery, P.; Thompson, E.R.; Lew, T.E.; Tiong, I.S.; Bennett, R.; Cheah, C.Y.; Lewis, K.L.; Handunnetti, S.M.; Tang, C.P.S.; Roberts, A.; et al. Enrichment of BTK Leu528Trp mutations in patients with CLL on zanubrutinib: Potential for pirtobrutinib cross-resistance. Blood Adv. 2022, 6, 5589–5592. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xu, B.; Liang, L.; Jiang, Y.; Zhao, Z. Investigating the ibrutinib resistance mechanism of L528W mutation on Bruton’s tyrosine kinase via molecular dynamics simulations. J. Mol. Graph. Model 2023, 126, 108623. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Mi, X.; Thompson, M.C.; Montoya, S.; Notti, R.Q.; Afaghani, J.; Durham, B.H.; Penson, A.; Witkowski, M.T.; Lu, S.X.; et al. Mechanisms of Resistance to Noncovalent Bruton’s Tyrosine Kinase Inhibitors. N. Engl. J. Med. 2022, 386, 735–743. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Naeem, A.; Utro, F.; Wang, Q.; Cha, J.; Vihinen, M.; Martindale, S.; Zhou, Y.; Ren, Y.; Tyekucheva, S.; Kim, A.S.; et al. Pirtobrutinib targets BTK C481S in ibrutinib-resistant CLL but second-site BTK mutations lead to resistance. Blood Adv. 2023, 7, 1929–1943. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Montoya, S.; Bourcier, J.; Thompson, M.C.; Noviski, M.; Tan, M.; Wang, E.; Mi, X.; Brathaban, N.; Barrientos Risso, C.; Tsai, D.; et al. Kinase Dead BTK Mutations Confer Resistance to Covalent and Noncovalent BTK Inhibitors but Are Susceptible to Clinical Stage BTK Degraders. Blood 2022, 140 (Suppl. S1), 1811–1813. [Google Scholar] [CrossRef]
- Brown, J.R.; Desikan, S.P.; Nguyen, B.; Won, H.; Tantawy, S.; Mcneely, S.C.; Marella, N.; Ebata, K.; Woyach, J.; Patel, K.; et al. S146: Genomic Evolution and Resistance to Pirtobrutinib in covalent Btk-Inhibitor (cBTKi) Pre-Treated Chronic Lymphocytic Leukemia (Cll) Patients: Results From The Phase I/Ii Bruin Study. Hemasphere 2023, 7 (Suppl. S3), e6233246. [Google Scholar] [CrossRef] [PubMed Central]
- Bonfiglio, S.; Sutton, L.A.; Ljungström, V.; Capasso, A.; Pandzic, T.; Weström, S.; Foroughi-Asl, H.; Skaftason, A.; Gellerbring, A.; Lyander, A.; et al. BTK and PLCG2 remain unmutated in one-third of patients with CLL relapsing on ibrutinib. Blood Adv. 2023, 7, 2794–2806. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cosson, A.; Chapiro, E.; Bougacha, N.; Lambert, J.; Herbi, L.; Cung, H.-A.; Algrin, C.; Keren, B.; Damm, F.; Gabillaud, C.; et al. Gain in the short arm of chromosome 2 (2p+) induces gene overexpression and drug resistance in chronic lymphocytic leukemia: Analysis of the central role of XPO1. Leukemia 2017, 31, 1625–1629. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Landau, D.A.; Taylor-Weiner, A.; Bozic, I.; Zhang, H.; Sarosiek, K.; Wang, L.; Stewart, C.; Fan, J.; Hoellenriegel, J.; et al. Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition. Nat. Commun. 2016, 7, 11589. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, A.L., 3rd; Phelan, J.D.; Wang, J.Q.; Huang, D.; Wright, G.W.; Kasbekar, M.; Choi, J.; Young, R.M.; Webster, D.E.; Yang, Y.; et al. Overcoming Acquired Epigenetic Resistance to BTK Inhibitors. Blood Cancer Discov. 2021, 2, 630–647. [Google Scholar] [CrossRef] [PubMed]
- Del Papa, B.; Baldoni, S.; Dorillo, E.; De Falco, F.; Rompietti, C.; Cecchini, D.; Cantelmi, M.G.; Sorcini, D.; Nogarotto, M.; Adamo, F.M.; et al. Decreased NOTCH1 Activation Correlates with Response to Ibrutinib in Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2019, 25, 7540–7553. [Google Scholar] [CrossRef] [PubMed]
- Niemann, C.U.; Herman, S.E.; Maric, I.; Gomez-Rodriguez, J.; Biancotto, A.; Chang, B.Y.; Martyr, S.; Stetler-Stevenson, M.; Yuan, C.M.; Calvo, K.R.; et al. Disruption of in vivo Chronic Lymphocytic Leukemia Tumor-Microenvironment Interactions by Ibrutinib—Findings from an Investigator-Initiated Phase II Study. Clin. Cancer Res. 2016, 22, 1572–1582. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Guo, A.; Lu, P.; Coffey, G.; Conley, P.; Pandey, A.; Wang, Y.L. Dual SYK/JAK inhibition overcomes ibrutinib resistance in chronic lymphocytic leukemia: Cerdulatinib, but not ibrutinib, induces apoptosis of tumor cells protected by the microenvironment. Oncotarget 2017, 8, 12953–12967. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bibikova, E.; Law, B.; Clevenger, T.; Cheung, J.; De Jesus, G.; Gulrajani, M.; Yamaguchi, K.; Do, P.; Burke, K.; Brock, G.; et al. High Surface Expression of CD49d (VLA-4) and CD79b Correlates with Acalabrutinib Resistance in Patients with Chronic Lymphocytic Leukemia (CLL). Blood 2019, 134 (Suppl. S1), 2571. [Google Scholar] [CrossRef]
- Burke, K.; Nuttall, B.; Karl, D.L.; Callahan, M.; Mendler, K.; Criscione, S.; Naumenko, S.; Bibikova, E.; Bloecher, A.; Dougherty, B.; et al. Novel Mechanisms of Acalabrutinib Resistance in Patients with Chronic Lymphocytic Leukemia By Whole Genome Methylome Sequencing. Blood 2021, 138 (Suppl. S1), 4361. [Google Scholar] [CrossRef]
- Pekarsky, Y.; Balatti, V.; Croce, C.M. BCL2 and miR-15/16: From gene discovery to treatment. Cell Death Differ. 2018, 25, 21–26. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rozovski, U.; Wu, J.Y.; Harris, D.M.; Liu, Z.; Li, P.; Hazan-Halevy, I.; Ferrajoli, A.; Burger, J.A.; O’Brien, S.; Jain, N.; et al. Stimulation of the B-cell receptor activates the JAK2/STAT3 signaling pathway in chronic lymphocytic leukemia cells. Blood 2014, 123, 3797–3802. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hanada, M.; Delia, D.; Aiello, A.; Stadtmauer, E.; Reed, J.C. bcl-2 gene hypomethylation and high-level expression in B-cell chronic lymphocytic leukemia. Blood 1993, 82, 1820–1828. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Ferracin, M.; Cimmino, A.; Di Leva, G.; Shimizu, M.; Wojcik, S.E.; Iorio, M.V.; Visone, R.; Sever, N.I.; Fabbri, M.; et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N. Engl. J. Med. 2005, 353, 1793–1801, Erratum in N. Engl. J. Med. 2006, 355, 533. [Google Scholar] [CrossRef] [PubMed]
- Diepstraten, S.T.; Anderson, M.A.; Czabotar, P.E.; Lessene, G.; Strasser, A.; Kelly, G.L. The manipulation of apoptosis for cancer therapy using BH3-mimetic drugs. Nat. Rev. Cancer. 2022, 22, 45–64. [Google Scholar] [CrossRef] [PubMed]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Cory, S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 2018, 25, 27–36. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Blombery, P.; Anderson, M.A.; Gong, J.N.; Thijssen, R.; Birkinshaw, R.W.; Thompson, E.R.; Teh, C.E.; Nguyen, T.; Xu, Z.; Flensburg, C.; et al. Acquisition of the Recurrent Gly101Val Mutation in BCL2 Confers Resistance to Venetoclax in Patients with Progressive Chronic Lymphocytic Leukemia. Cancer Discov. 2019, 9, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Tausch, E.; Close, W.; Dolnik, A.; Bloehdorn, J.; Chyla, B.; Bullinger, L.; Döhner, H.; Mertens, D.; Stilgenbauer, S. Venetoclax resistance and acquired BCL2 mutations in chronic lymphocytic leukemia. Haematologica 2019, 104, e434–e437. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Birkinshaw, R.W.; Gong, J.N.; Luo, C.S.; Lio, D.; White, C.A.; Anderson, M.A.; Blombery, P.; Lessene, G.; Majewski, I.J.; Thijssen, R.; et al. Structures of BCL-2 in complex with venetoclax reveal the molecular basis of resistance mutations. Nat. Commun. 2019, 10, 2385. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lucas, F.; Larkin, K.; Gregory, C.T.; Orwick, S.; Doong, T.J.; Lozanski, A.; Lozanski, G.; Misra, S.; Ngankeu, A.; Ozer, H.G.; et al. Novel BCL2 mutations in venetoclax-resistant, ibrutinib-resistant CLL patients with BTK/PLCG2 mutations. Blood 2020, 135, 2192–2195. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Blombery, P.; Thompson, E.R.; Nguyen, T.; Birkinshaw, R.W.; Gong, J.N.; Chen, X.; McBean, M.; Thijssen, R.; Conway, T.; Anderson, M.A.; et al. Multiple BCL2 mutations cooccurring with Gly101Val emerge in chronic lymphocytic leukemia progression on venetoclax. Blood 2020, 135, 773–777. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chyla, B.J.; Popovic, R.; Lu, C.; Dunbar, F.; Quarless, D.; Robinson, K.; Warder, S.; Jacobson, A.; Zhou, L.; Souers, A.; et al. Identification of Recurrent Genomic Alterations in the Apoptotic Machinery in CLL Patients Treated with Venetoclax Monotherapy. Blood 2019, 134 (Suppl. S1), 172. [Google Scholar] [CrossRef]
- Popovic, R.; Dunbar, F.; Lu, C.; Robinson, K.; Quarless, D.; Warder, S.E.; Mukherjee, N.; Pesko, J.; Souers, A.J.; Waring, J.F.; et al. Identification of recurrent genomic alterations in the apoptotic machinery in chronic lymphocytic leukemia patients treated with venetoclax monotherapy. Am. J. Hematol. 2022, 97, E47–E51. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Blombery, P.; Thompson, E.R.; Chen, X.; Nguyen, T.; Anderson, M.A.; Westerman, D.A.; Seymour, J.F.; Tam, C.S.; Dengler, M.A.; Huang, D.C.S.; et al. BAX-Mutated Clonal Hematopoiesis in Patients on Long-Term Venetoclax for Relapsed/Refractory Chronic Lymphocytic Leukemia. Blood 2020, 136 (Suppl. S1), 9–10. [Google Scholar] [CrossRef]
- Blombery, P.; Lew, T.E.; Dengler, M.A.; Thompson, E.R.; Lin, V.S.; Chen, X.; Nguyen, T.; Panigrahi, A.; Handunnetti, S.M.; Carney, D.A.; et al. Clonal hematopoiesis, myeloid disorders and BAX-mutated myelopoiesis in patients receiving venetoclax for CLL. Blood 2022, 139, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Nahta, R.; Yuan, L.X.; Fiterman, D.J.; Zhang, L.; Symmans, W.F.; Ueno, N.T.; Esteva, F.J. B cell translocation gene 1 contributes to antisense Bcl-2-mediated apoptosis in breast cancer cells. Mol. Cancer Ther. 2006, 5, 1593–1601. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.L.; Duncavage, E.J.; Mathew, R.; den Besten, W.; Pei, D.; Naeve, D.; Yamamoto, T.; Cheng, C.; Sherr, C.J.; Roussel, M.F. Arf induces p53-dependent and -independent antiproliferative genes. Cancer Res. 2003, 63, 1046–1053. [Google Scholar] [PubMed]
- Herling, C.D.; Abedpour, N.; Weiss, J.; Schmitt, A.; Jachimowicz, R.D.; Merkel, O.; Cartolano, M.; Oberbeck, S.; Mayer, P.; Berg, V.; et al. Clonal dynamics towards the development of venetoclax resistance in chronic lymphocytic leukemia. Nat. Commun. 2018, 9, 727. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Leverson, J.D.; Sampath, D.; Souers, A.J.; Rosenberg, S.H.; Fairbrother, W.J.; Amiot, M.; Konopleva, M.; Letai, A. Found in Translation: How Preclinical Research Is Guiding the Clinical Development of the BCL2-Selective Inhibitor Venetoclax. Cancer Discov. 2017, 7, 1376–1393. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Guièze, R.; Liu, V.M.; Rosebrock, D.; Jourdain, A.A.; Hernández-Sánchez, M.; Martinez Zurita, A.; Sun, J.; Ten Hacken, E.; Baranowski, K.; Thompson, P.A.; et al. Mitochondrial Reprogramming Underlies Resistance to BCL-2 Inhibition in Lymphoid Malignancies. Cancer Cell. 2019, 36, 369–384.e13. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Thijssen, R.; Tian, L.; Anderson, M.A.; Flensburg, C.; Jarratt, A.; Garnham, A.L.; Jabbari, J.S.; Peng, H.; Lew, T.E.; Teh, C.E.; et al. Single-cell multiomics reveal the scale of multilayered adaptations enabling CLL relapse during venetoclax therapy. Blood 2022, 140, 2127–2141. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Thomalla, D.; Beckmann, L.; Grimm, C.; Oliverio, M.; Meder, L.; Herling, C.D.; Nieper, P.; Feldmann, T.; Merkel, O.; Lorsy, E.; et al. Deregulation and epigenetic modification of BCL2-family genes cause resistance to venetoclax in hematologic malignancies. Blood 2022, 140, 2113–2126. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jayappa, K.D.; Portell, C.A.; Gordon, V.L.; Capaldo, B.J.; Bekiranov, S.; Axelrod, M.J.; Brett, L.K.; Wulfkuhle, J.D.; Gallagher, R.I.; Petricoin, E.F.; et al. Microenvironmental agonists generate de novo phenotypic resistance to combined ibrutinib plus venetoclax in CLL and MCL. Blood Adv. 2017, 1, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Ghia, E.M.; Rassenti, L.Z.; Choi, M.Y.; Quijada-Álamo, M.; Chu, E.; Widhopf, G.F., 2nd; Kipps, T.J. High expression level of ROR1 and ROR1-signaling associates with venetoclax resistance in chronic lymphocytic leukemia. Leukemia 2022, 36, 1609–1618. [Google Scholar] [CrossRef] [PubMed]
- Jayappa, K.D.; Gordon, V.L.; Morris, C.G.; Wilson, B.; Shetty, B.D.; Cios, K.J.; Arora, P.C.; Isaac, K.M.; Saha, S.; Bender, T.P.; et al. Extrinsic interactions in the microenvironment in vivo activate an antiapoptotic multidrug-resistant phenotype in CLL. Blood Adv. 2021, 5, 3497–3510. [Google Scholar] [CrossRef] [PubMed]
- Fiorcari, S.; Maffei, R.; Atene, C.G.; Mesini, N.; Maccaferri, M.; Leonardi, G.; Martinelli, S.; Paolini, A.; Nasillo, V.; Debbia, G.; et al. Notch2 Increases the Resistance to Venetoclax-Induced Apoptosis in Chronic Lymphocytic Leukemia B Cells by Inducing Mcl-1. Front. Oncol. 2022, 11, 777587. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chong, S.J.F.; Zhu, F.; Dashevsky, O.; Mizuno, R.; Lai, J.X.; Hackett, L.; Ryan, C.E.; Collins, M.C.; Iorgulescu, J.B.; Guièze, R.; et al. Hyperphosphorylation of BCL-2 family proteins underlies functional resistance to venetoclax in lymphoid malignancies. J. Clin. Investig. 2023, 133, e170169. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acquired BTK Mutations | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| C481S/R/F/Y/G | L528W | T474I/S | M477I | M437R | A428D | V416L | T316A | E41K/V | R28S | |
| Ibrutinib | selected | selected | selected | not selected | not selected | not selected | not selected | selected | selected | selected |
| Acalabrutinib | selected | not selected | selected | not selected | not selected | not selected | not selected | not selected | selected | not selected |
| Zanubrutinib | selected | selected | not selected | not selected | not selected | not selected | not selected | not selected | not selected | not selected |
| Pirtobrutinib | not selected | selected | selected | selected | selected | selected | selected | not selected | not selected | not selected |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Del Giudice, I.; Della Starza, I.; De Falco, F.; Gaidano, G.; Sportoletti, P. Monitoring Response and Resistance to Treatment in Chronic Lymphocytic Leukemia. Cancers 2024, 16, 2049. https://doi.org/10.3390/cancers16112049
Del Giudice I, Della Starza I, De Falco F, Gaidano G, Sportoletti P. Monitoring Response and Resistance to Treatment in Chronic Lymphocytic Leukemia. Cancers. 2024; 16(11):2049. https://doi.org/10.3390/cancers16112049
Chicago/Turabian StyleDel Giudice, Ilaria, Irene Della Starza, Filomena De Falco, Gianluca Gaidano, and Paolo Sportoletti. 2024. "Monitoring Response and Resistance to Treatment in Chronic Lymphocytic Leukemia" Cancers 16, no. 11: 2049. https://doi.org/10.3390/cancers16112049
APA StyleDel Giudice, I., Della Starza, I., De Falco, F., Gaidano, G., & Sportoletti, P. (2024). Monitoring Response and Resistance to Treatment in Chronic Lymphocytic Leukemia. Cancers, 16(11), 2049. https://doi.org/10.3390/cancers16112049