

Hu14.18K.322A Causes Direct Cell Cytotoxicity and Synergizes with Induction Chemotherapy in High-Risk Neuroblastoma

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Media
2.2. Cell Viability
2.3. Proliferation Assay
2.4. Colony Formation Assay
2.5. Cell Cycle Analysis
2.6. GD2 Surface Expression Quantitation
2.7. Western Blot Analysis
2.8. RNA Isolation and Real-Time PCR
2.9. Apotracker Assay for Apoptosis
2.10. Lipid Peroxidation Analysis
2.11. RNA-Seq Data Analysis
2.12. Synergy and Drug Sensitization Assays
2.13. Statistical Analysis
2.14. Data Availability
3. Results
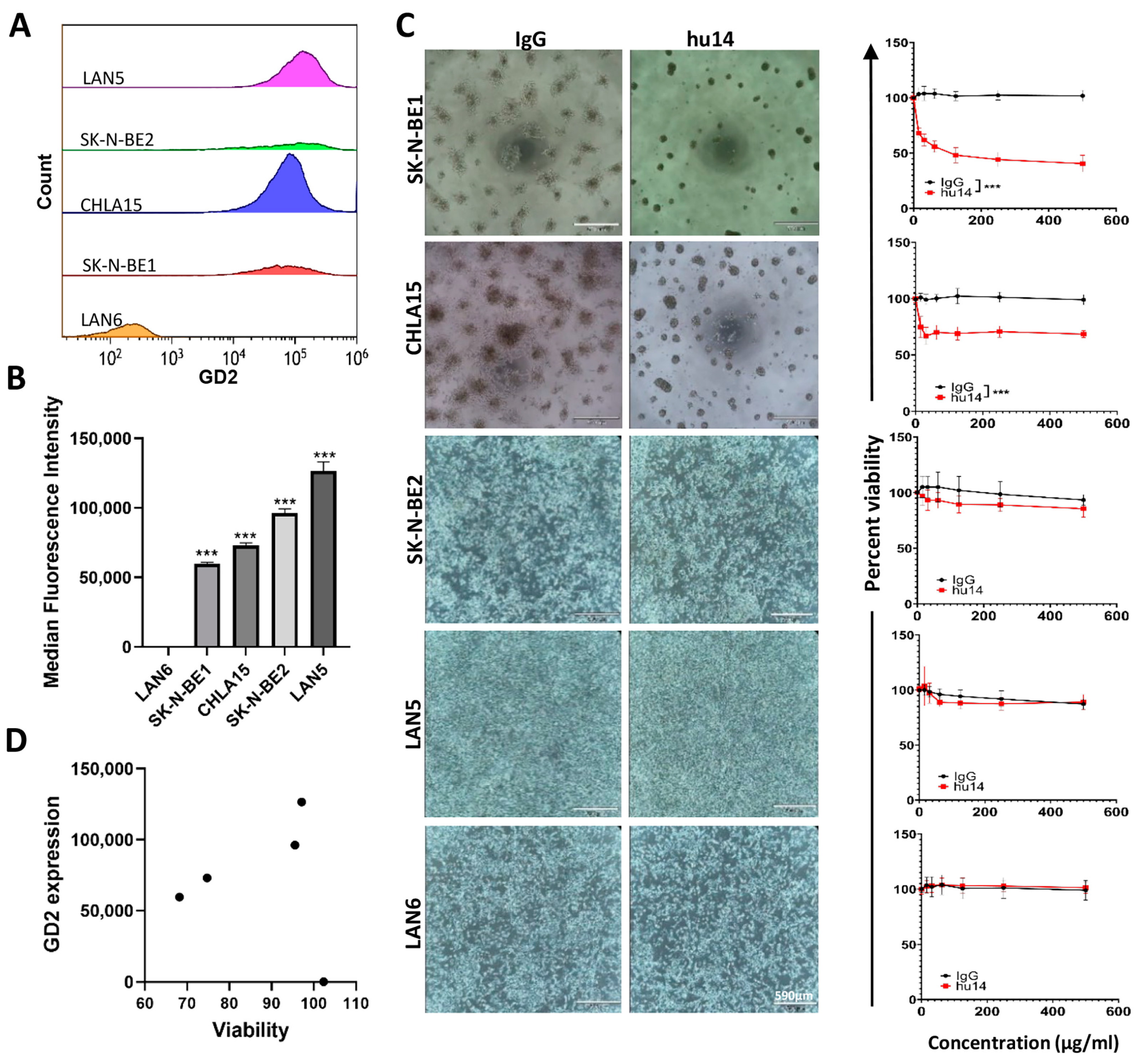
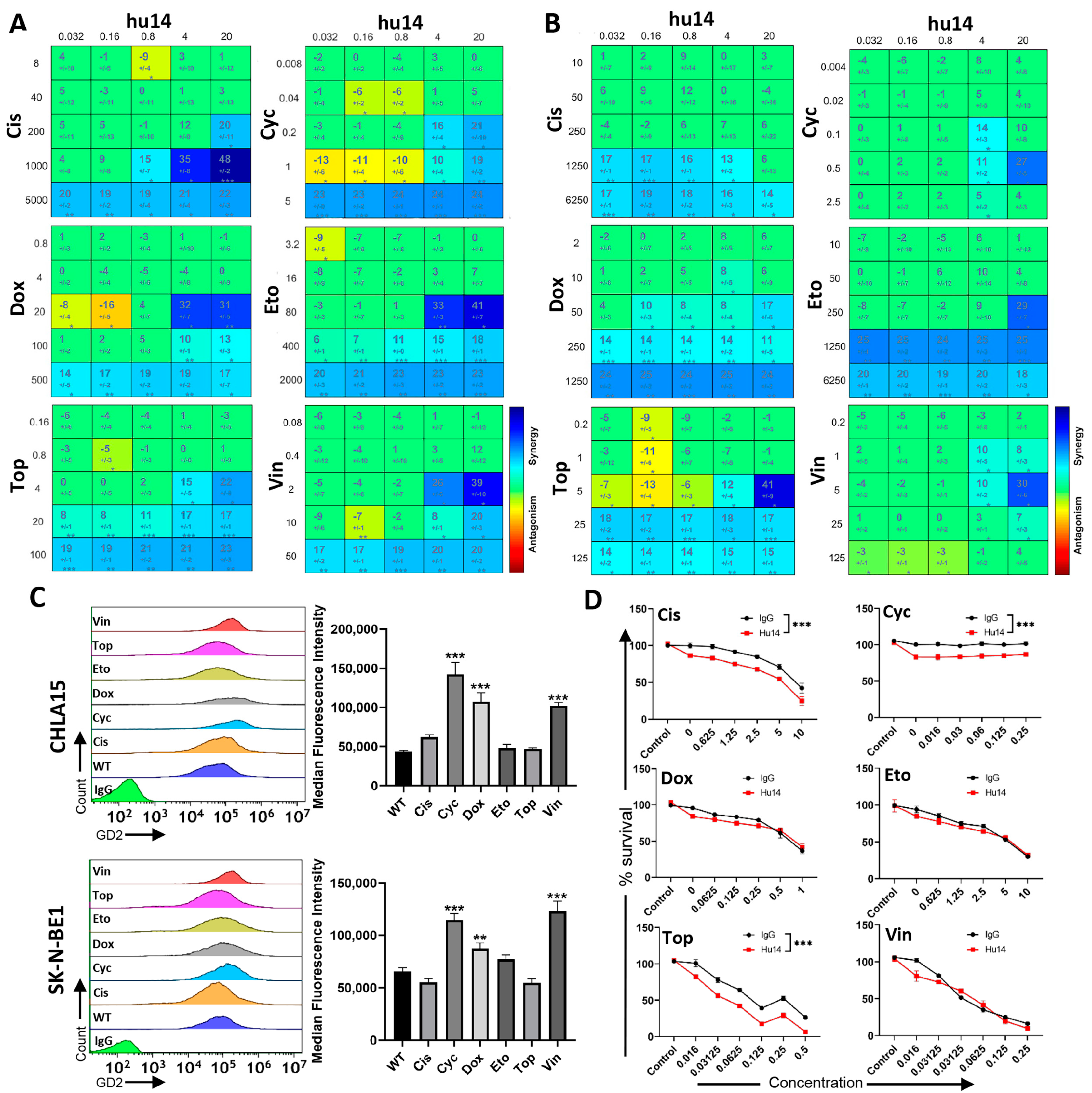
3.1. GD2 Expression Varies between NB Cell Lines, but Does Not Influence the Susceptibility of the Cells to Hu14
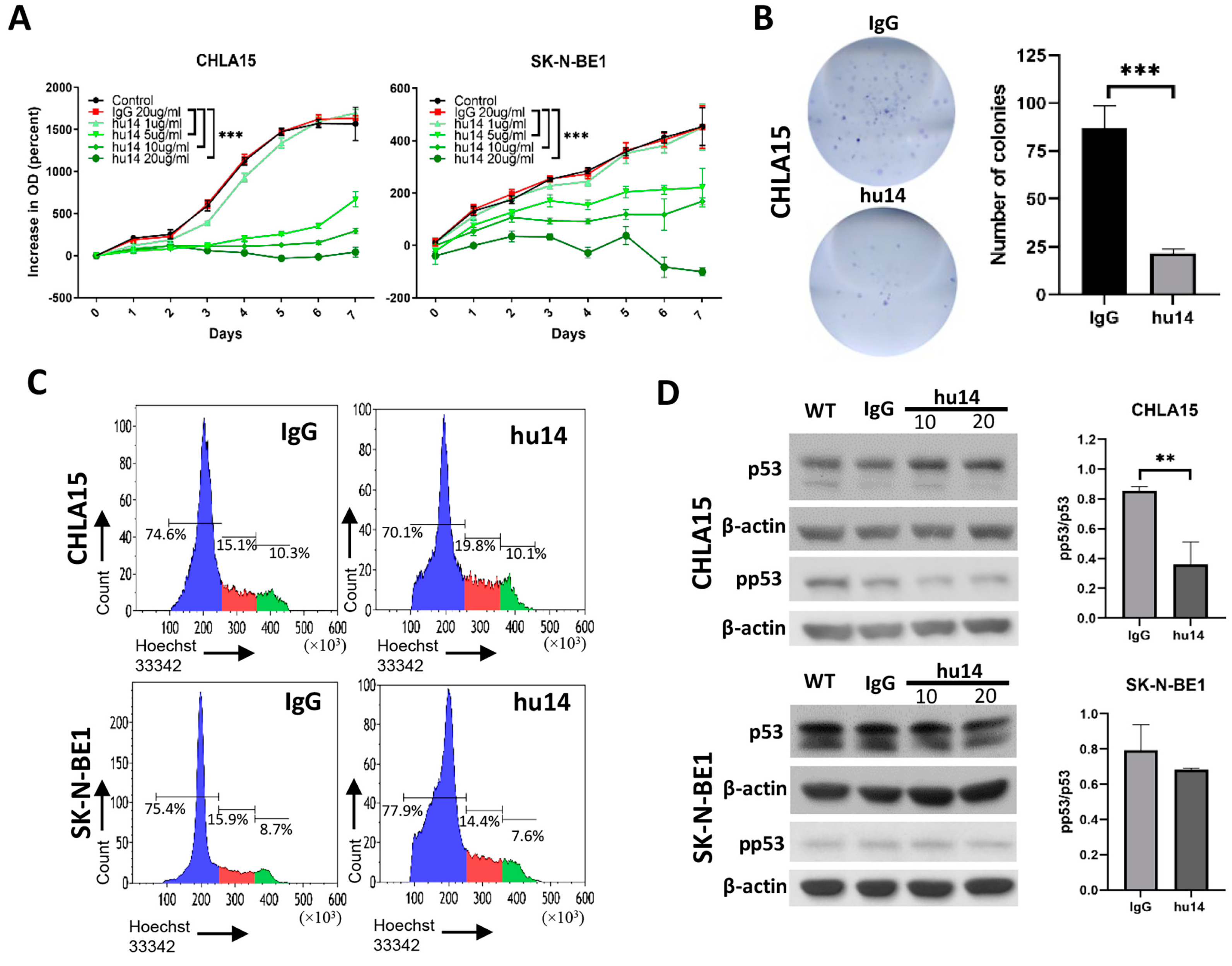
3.2. Hu14 Has an Anti-Proliferative Effect on Susceptible Cell Lines
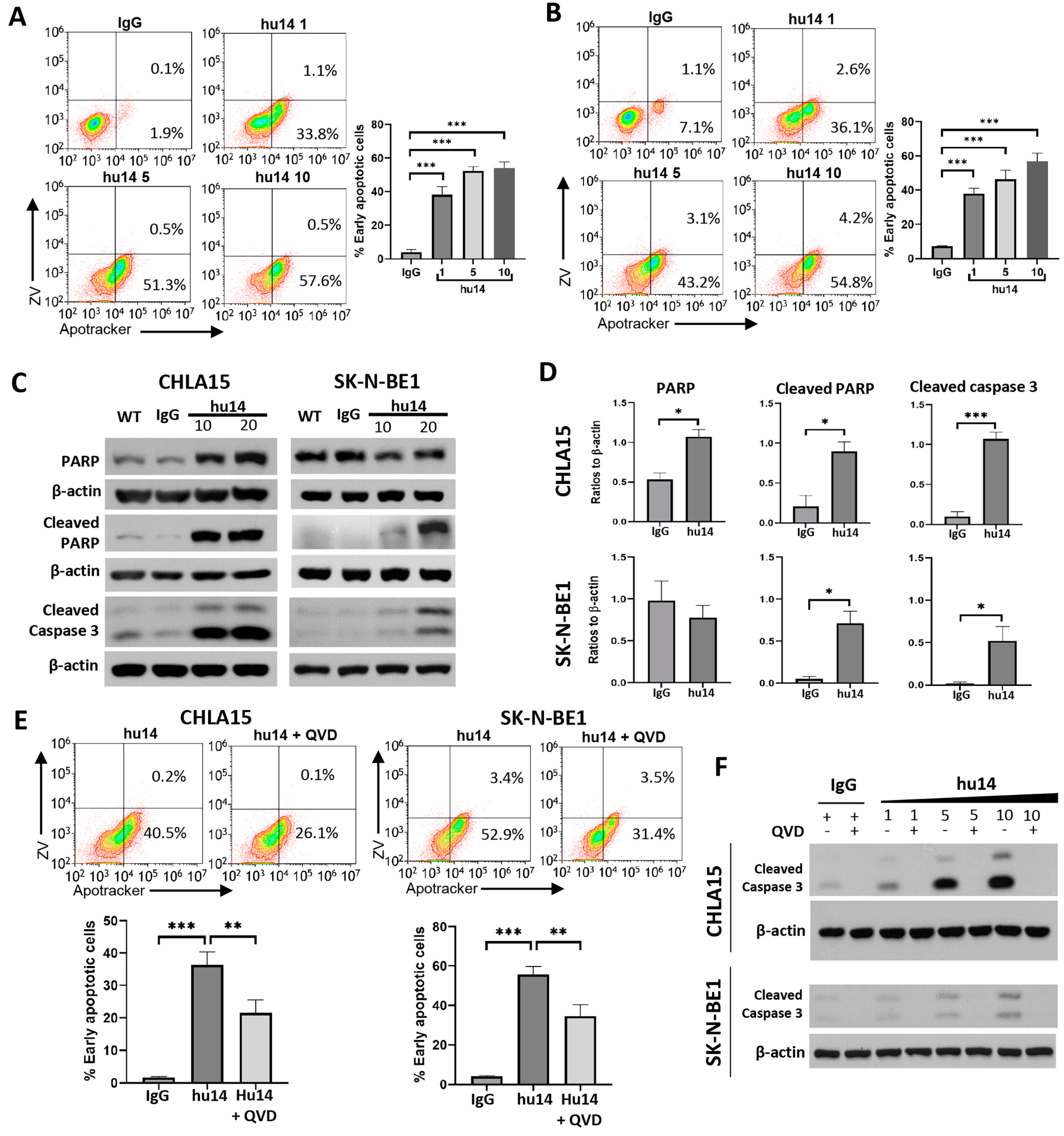
3.3. Apoptosis Is the Major Pathway Triggered by Hu14
3.4. Role of Other Cell-Death Pathways in NB Cells Treated with Hu14
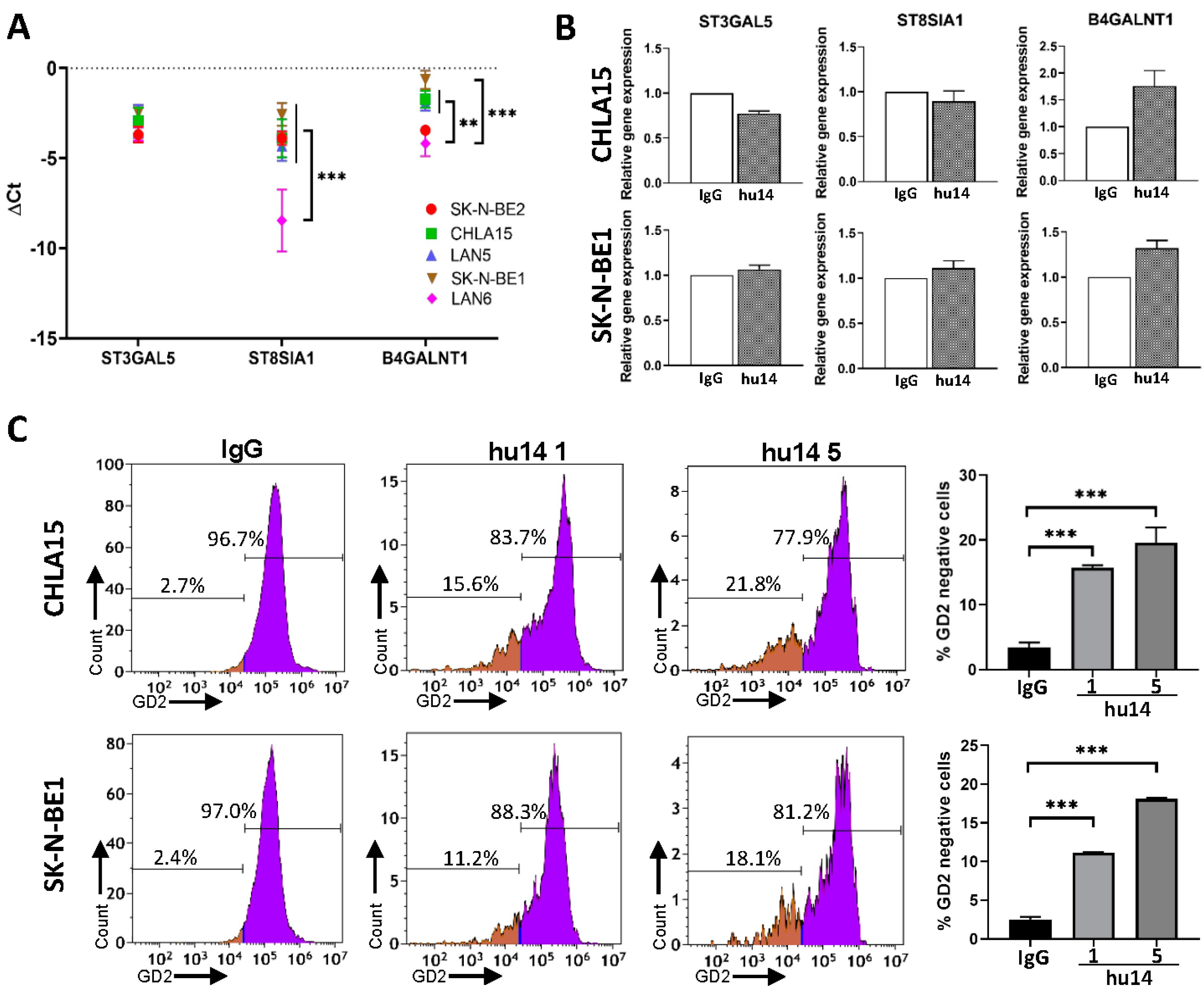
3.5. Effect of Hu14 on GD2 Biosynthesis
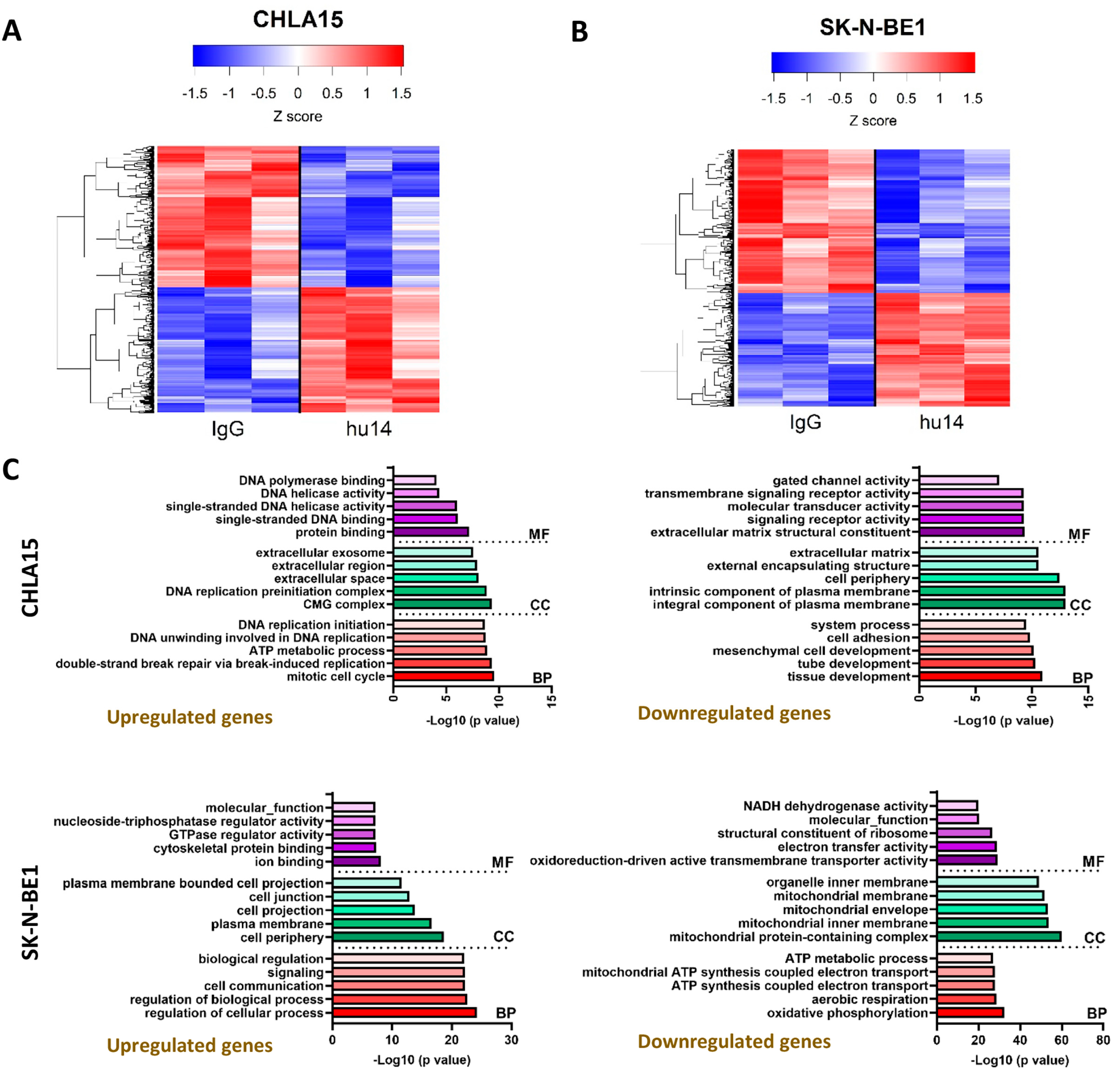
3.6. Transcriptional Changes Induced by Hu14 in CHLA15 and SK-N-BE1 Cells
3.7. Hu14 Synergizes with Induction Chemotherapy Drugs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schulz, G.; Cheresh, D.A.; Varki, N.M.; Yu, A.; Staffileno, L.K.; Reisfeld, R.A. Detection of Ganglioside GD2 in Tumor Tissues and Sera of Neuroblastoma Patients. Cancer Res. 1984, 44, 5914–5920. [Google Scholar] [PubMed]
- Yoshida, S.; Fukumoto, S.; Kawaguchi, H.; Sato, S.; Ueda, R.; Furukawa, K. Ganglioside GD2 in Small Cell Lung Cancer Cell Lines: Enhancement of Cell Proliferation and Mediation of Apoptosis. Cancer Res. 2001, 61, 4244–4252. [Google Scholar] [PubMed]
- Keyel, M.E.; Reynolds, C.P. Spotlight on Dinutuximab in the Treatment of High-Risk Neuroblastoma: Development and Place in Therapy. Biologics 2018, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Philippova, J.; Shevchenko, J.; Sennikov, S. GD2-Targeting Therapy: A Comparative Analysis of Approaches and Promising Directions. Front. Immunol. 2024, 15, 1371345. [Google Scholar] [CrossRef] [PubMed]
- Barker, E.; Mueller, B.M.; Handgretinger, R.; Herter, M.; Yu, A.L.; Reisfeld, R.A. Effect of a Chimeric Anti-Ganglioside GD2 Antibody on Cell-Mediated Lysis of Human Neuroblastoma Cells. Cancer Res. 1991, 51, 144–149. [Google Scholar] [PubMed]
- Voeller, J.; Sondel, P.M. Advances in Anti-GD2 Immunotherapy for Treatment of High-Risk Neuroblastoma. J. Pediatr. Hematol. Oncol. 2019, 41, 163–169. [Google Scholar] [CrossRef]
- Doronin, I.I.; Vishnyakova, P.A.; Kholodenko, I.V.; Ponomarev, E.D.; Ryazantsev, D.Y.; Molotkovskaya, I.M.; Kholodenko, R.V. Ganglioside GD2 in Reception and Transduction of Cell Death Signal in Tumor Cells. BMC Cancer 2014, 14, 295. [Google Scholar] [CrossRef]
- Aixinjueluo, W.; Furukawa, K.; Zhang, Q.; Hamamura, K.; Tokuda, N.; Yoshida, S.; Ueda, R.; Furukawa, K. Mechanisms for the Apoptosis of Small Cell Lung Cancer Cells Induced by Anti-GD2 Monoclonal Antibodies: Roles of Anoikis. J. Biol. Chem. 2005, 280, 29828–29836. [Google Scholar] [CrossRef] [PubMed]
- Sterner, E.; Peach, M.L.; Nicklaus, M.C.; Gildersleeve, J.C. Therapeutic Antibodies to Ganglioside GD2 Evolved from Highly Selective Germline Antibodies. Cell Rep. 2017, 20, 1681–1691. [Google Scholar] [CrossRef]
- Sorkin, L.S.; Otto, M.; Baldwin, W.M.; Vail, E.; Gillies, S.D.; Handgretinger, R.; Barfield, R.C.; Yu, H.M.; Yu, A.L. Anti-GD2 with an FC Point Mutation Reduces Complement Fixation and Decreases Antibody-Induced Allodynia. Pain 2010, 149, 135–142. [Google Scholar] [CrossRef]
- Furman, W.L.; McCarville, B.; Shulkin, B.L.; Davidoff, A.; Krasin, M.; Hsu, C.; Pan, H.; Wu, J.; Brennan, R.; Bishop, M.W.; et al. Improved Outcome in Children with Newly Diagnosed High-Risk Neuroblastoma Treated with Chemoimmunotherapy: Updated Results of a Phase II Study using hu14.18K322A. J. Clin. Oncol. 2022, 40, 335–344. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell Death: A Review of the Major Forms of Apoptosis, Necrosis and Autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar]
- Pang, Q.; Tang, Z.; Luo, L. The Crosstalk between Oncogenic Signaling and Ferroptosis in Cancer. Crit. Rev. Oncol. Hematol. 2024, 197, 104349. [Google Scholar] [CrossRef]
- Gao, J.; Xiong, A.; Liu, J.; Li, X.; Wang, J.; Zhang, L.; Liu, Y.; Xiong, Y.; Li, G.; He, X. PANoptosis: Bridging Apoptosis, Pyroptosis, and Necroptosis in Cancer Progression and Treatment. Cancer Gene Ther. 2024. [Google Scholar] [CrossRef]
- Radosevich, J. (Ed.) Apoptosis and Beyond: The Many Ways Cells Die; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2018. [Google Scholar]
- Kowalczyk, A.; Gil, M.; Horwacik, I.; Odrowaz, Z.; Kozbor, D.; Rokita, H. The GD2-Specific 14G2a Monoclonal Antibody Induces Apoptosis and Enhances Cytotoxicity of Chemotherapeutic Drugs in IMR-32 Human Neuroblastoma Cells. Cancer Lett. 2009, 281, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Skowron, M.; Berthold, F. Reliable Transcript Quantification by Real-Time Reverse Transcriptase-Polymerase Chain Reaction in Primary Neuroblastoma using Normalization to Averaged Expression Levels of the Control Genes HPRT1 and SDHA. J. Mol. Diagn. 2005, 7, 89–96. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential Analyses for RNA-Seq: Transcript-Level Estimates Improve Gene-Level Inferences. F1000Research 2015, 4, 1521. [Google Scholar] [CrossRef]
- Di Veroli, G.Y.; Fornari, C.; Wang, D.; Mollard, S.; Bramhall, J.L.; Richards, F.M.; Jodrell, D.I. Combenefit: An Interactive Platform for the Analysis and Visualization of Drug Combinations. Bioinformatics 2016, 32, 2866–2868. [Google Scholar] [CrossRef]
- Martens, S.; Bridelance, J.; Roelandt, R.; Vandenabeele, P.; Takahashi, N. MLKL in Cancer: More than a Necroptosis Regulator. Cell Death Differ. 2021, 28, 1757–1772. [Google Scholar] [CrossRef] [PubMed]
- Liebl, M.P.; Meister, S.C.; Frey, L.; Hendrich, K.; Klemmer, A.; Wohlfart, B.; Untucht, C.; Nuber, J.; Pohl, C.; Lakics, V. Robust LC3B Lipidation Analysis by Precisely Adjusting Autophagic Flux. Sci. Rep. 2022, 12, 79. [Google Scholar] [CrossRef] [PubMed]
- Tibbetts, R.; Yeo, K.K.; Muthugounder, S.; Lee, M.; Jung, C.; Porras-Corredor, T.; Sheard, M.A.; Asgharzadeh, S. Anti-Disialoganglioside Antibody Internalization by Neuroblastoma Cells as a Mechanism of Immunotherapy Resistance. Cancer Immunol. Immunother. 2022, 71, 153–164. [Google Scholar] [CrossRef]
- Horwacik, I.; Rokita, H. Modulation of Interactions of Neuroblastoma Cell Lines with Extracellular Matrix Proteins Affects their Sensitivity to Treatment with the Anti-GD2 Ganglioside Antibody 14G2a. Int. J. Oncol. 2017, 50, 1899–1914. [Google Scholar] [CrossRef] [PubMed]
- Horwacik, I.; Durbas, M.; Boraatyn, E.; Wegrzyn, P.; Rokita, H. Targeting GD2 Ganglioside and Aurora A Kinase as a Dual Strategy Leading to Cell Death in Cultures of Human Neuroblastoma Cells. Cancer Lett. 2013, 341, 248–264. [Google Scholar] [CrossRef]
- Cochonneau, D.; Terme, M.; Michaud, A.; Dorvillius, M.; Gautier, N.; Frikeche, J.; Alvarez-Rueda, N.; Bougras, G.; Aubry, J.; Paris, F.; et al. Cell Cycle Arrest and Apoptosis Induced by O-Acetyl-GD2-Specific Monoclonal Antibody 8B6 Inhibits Tumor Growth in Vitro and in Vivo. Cancer Lett. 2013, 333, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef]
- Minamoto, T.; Buschmann, T.; Habelhah, H.; Matusevich, E.; Tahara, H.; Boerresen-Dale, A.L.; Harris, C.; Sidransky, D.; Ronai, Z. Distinct Pattern of p53 Phosphorylation in Human Tumors. Oncogene 2001, 20, 3341–3347. [Google Scholar] [CrossRef]
- Higashimoto, Y.; Saito, S.; Tong, X.H.; Hong, A.; Sakaguchi, K.; Appella, E.; Anderson, C.W. Human p53 is Phosphorylated on Serines 6 and 9 in Response to DNA Damage-Inducing Agents. J. Biol. Chem. 2000, 275, 23199–23203. [Google Scholar] [CrossRef]
- Choi, M.E.; Price, D.R.; Ryter, S.W.; Choi, A.M.K. Necroptosis: A Crucial Pathogenic Mediator of Human Disease. JCI Insight 2019, 4, e128834. [Google Scholar] [CrossRef]
- Seong, D.; Jeong, M.; Seo, J.; Lee, J.; Hwang, C.H.; Shin, H.; Shin, J.Y.; Nam, Y.W.; Jo, J.Y.; Lee, H.; et al. Identification of MYC as an Antinecroptotic Protein that Stifles RIPK1-RIPK3 Complex Formation. Proc. Natl. Acad. Sci. USA 2020, 117, 19982–19993. [Google Scholar] [CrossRef] [PubMed]
- Nicolai, S.; Pieraccioli, M.; Peschiaroli, A.; Melino, G.; Raschella, G. Neuroblastoma: Oncogenic Mechanisms and Therapeutic Exploitation of Necroptosis. Cell Death Dis. 2015, 6, e2010. [Google Scholar] [CrossRef] [PubMed]
- Horne, C.R.; Samson, A.L.; Murphy, J.M. The Web of Death: The Expanding Complexity of Necroptotic Signaling. Trends Cell Biol. 2023, 33, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Zhang, C.; Guo, L.; He, H.; Jiang, K.; Huang, Y.; Zhang, X.; Zhang, H.; Wei, W.; Zhang, Y.; et al. A Necroptotic-Independent Function of MLKL in Regulating Endothelial Cell Adhesion Molecule Expression. Cell Death Dis. 2020, 11, 282–283. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Kovalenko, A.; Bogdanov, K.; Wallach, D. MLKL, the Protein that Mediates Necroptosis, also Regulates Endosomal Trafficking and Extracellular Vesicle Generation. Immunity 2017, 47, 51–65.e7. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Alborzinia, H.; Florez, A.F.; Kreth, S.; Bruckner, L.M.; Yildiz, U.; Gartlgruber, M.; Odoni, D.I.; Poschet, G.; Garbowicz, K.; Shao, C.; et al. MYCN Mediates Cysteine Addiction and Sensitizes Neuroblastoma to Ferroptosis. Nat. Cancer 2022, 3, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Floros, K.V.; Chawla, A.T.; Johnson-Berro, M.O.; Khatri, R.; Stamatouli, A.M.; Boikos, S.A.; Dozmorov, M.G.; Cowart, L.A.; Faber, A.C. MYCN Upregulates the Transsulfuration Pathway to Suppress the Ferroptotic Vulnerability in MYCN-Amplified Neuroblastoma. Cell Stress 2022, 6, 21–29. [Google Scholar] [CrossRef]
- Durbas, M.; Pabisz, P.; Wawak, K.; Wisniewska, A.; Boratyn, E.; Nowak, I.; Horwacik, I.; Woznicka, O.; Rokita, H. GD2 Ganglioside-Binding Antibody 14G2a and Specific Aurora A Kinase Inhibitor MK-5108 Induce Autophagy in IMR-32 Neuroblastoma Cells. Apoptosis 2018, 23, 492–511. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. P62 Links the Autophagy Pathway and the Ubiqutin-Proteasome System upon Ubiquitinated Protein Degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef]
- Julien, S.; Bobowski, M.; Steenackers, A.; Le Bourhis, X.; Delannoy, P. How do Gangliosides Regulate RTKs Signaling? Cells 2013, 2, 751–767. [Google Scholar] [CrossRef] [PubMed]
- Kroesen, M.; Bull, C.; Gielen, P.R.; Brok, I.C.; Armandari, I.; Wassink, M.; Looman, M.W.G.; Boon, L.; den Brok, M.H.; Hoogerbrugge, P.M.; et al. Anti-GD2 mAb and Vorinostat Synergize in the Treatment of Neuroblastoma. Oncoimmunology 2016, 5, e1164919. [Google Scholar] [CrossRef] [PubMed]
- Troschke-Meurer, S.; Zumpe, M.; Meissner, L.; Siebert, N.; Grabarczyk, P.; Forkel, H.; Maletzki, C.; Bekeschus, S.; Lode, H.N. Chemotherapeutics used for High-Risk Neuroblastoma Therapy Improve the Efficacy of Anti-GD2 Antibody Dinutuximab Beta in Preclinical Spheroid Models. Cancers 2023, 15, 904. [Google Scholar] [CrossRef] [PubMed]
- Yeh, S.; Wang, P.; Lou, Y.; Khoo, K.; Hsiao, M.; Hsu, T.; Wong, C. Glycolipid GD3 and GD3 Synthase are Key Drivers for Glioblastoma Stem Cells and Tumorigenicity. Proc. Natl. Acad. Sci. USA 2016, 113, 5592–5597. [Google Scholar] [CrossRef]
- Cazet, A.; Lefebvre, J.; Adriaenssens, E.; Julien, S.; Bobowski, M.; Grigoriadis, A.; Tutt, A.; Tulasne, D.; Le Bourhis, X.; Delannoy, P. GD3 Synthase Expression Enhances Proliferation and Tumor Growth of MDA-MB-231 Breast Cancer Cells through C-Met Activation. Mol. Cancer Res. 2010, 8, 1526–1535. [Google Scholar] [CrossRef] [PubMed]
- Cazet, A.; Groux-Degroote, S.; Teylaert, B.; Kwon, K.; Lehoux, S.; Slomianny, C.; Kim, C.; Le Bourhis, X.; Delannoy, P. GD3 Synthase Overexpression Enhances Proliferation and Migration of MDA-MB-231 Breast Cancer Cells. Biol. Chem. 2009, 390, 601–609. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomas, M.; Nguyen, T.H.; Drnevich, J.; D’Souza, A.M.; de Alarcon, P.A.; Gnanamony, M. Hu14.18K.322A Causes Direct Cell Cytotoxicity and Synergizes with Induction Chemotherapy in High-Risk Neuroblastoma. Cancers 2024, 16, 2064. https://doi.org/10.3390/cancers16112064
Thomas M, Nguyen TH, Drnevich J, D’Souza AM, de Alarcon PA, Gnanamony M. Hu14.18K.322A Causes Direct Cell Cytotoxicity and Synergizes with Induction Chemotherapy in High-Risk Neuroblastoma. Cancers. 2024; 16(11):2064. https://doi.org/10.3390/cancers16112064
Chicago/Turabian StyleThomas, Maria, Thu Hien Nguyen, Jenny Drnevich, Amber M. D’Souza, Pedro A. de Alarcon, and Manu Gnanamony. 2024. "Hu14.18K.322A Causes Direct Cell Cytotoxicity and Synergizes with Induction Chemotherapy in High-Risk Neuroblastoma" Cancers 16, no. 11: 2064. https://doi.org/10.3390/cancers16112064
APA StyleThomas, M., Nguyen, T. H., Drnevich, J., D’Souza, A. M., de Alarcon, P. A., & Gnanamony, M. (2024). Hu14.18K.322A Causes Direct Cell Cytotoxicity and Synergizes with Induction Chemotherapy in High-Risk Neuroblastoma. Cancers, 16(11), 2064. https://doi.org/10.3390/cancers16112064