Simple Summary
Our findings confirm the role of developmental aspects in extracranial germ cell tumors (eGCTs). Strong associations between syndromes or congenital anomalies and eGCTs may hint at the need for research to identify potential causes and mechanisms. Strong effects were observed for Swyer and Currarino syndrome. The strengths of the associations emphasize the need for further elaboration on presently discussed pathophysiological mechanisms. For other suggested risk factors such as Klinefelter, Turner, and Down syndrome, the strength of risk appears to be substantially low and may be missed because of diagnostic bias for the first two or lack of power for the latter. A number of further syndromes were identified in single cases. Most of these pertain to girls.
Abstract
GCTs are developmental tumors and are likely to reflect ontogenetic and teratogenetic determinants. The objective of this study was to identify syndromes with or without congenital anomalies and non-syndromic defects as potential risk factors. Patients with extracranial GCTs (eGCTs) registered in MAKEI 96/MAHO 98 between 1996 and 2017 were included. According to Teilum’s holistic concept, malignant and benign teratomas were registered. We used a case–control study design with Orphanet as a reference group for syndromic defects and the Mainz birth registry (EUROCAT) for congenital anomalies at birth. Co-occurring genetic syndromes and/or congenital anomalies were assessed accordingly. Odds ratios and 95% confidence intervals were calculated and p-values for Fisher’s exact test with Bonferroni correction if needed. A strong association was confirmed for Swyer (OR 338.6, 95% CI 43.7–2623.6) and Currarino syndrome (OR 34.2, 95% CI 13.2–88.6). We additionally found 16 isolated cases of eGCT with a wide range of syndromes. However, these were not found to be significantly associated following Bonferroni correction. Most of these cases pertained to girls. Regarding non-syndromic defects, no association with eGCTs could be identified. In our study, we confirmed a strong association for Swyer and Currarino syndromes with additional congenital anomalies.
1. Introduction
Extracranial pediatric germ cell tumors (eGCTs), a model of developmental tumorgenesis, are rare neoplasms occurring in different sites and ages [1]. According to data from the DKKR (Deutsches Kinderkrebsregister) from 1987 to 2011, the incidence rate is 4.8 per million (children aged <15 years). For girls, the incidence is 5.3, and for boys, it is 4.4 per million [2]. In a study from 2023, the 5-year survival rate (EFS) and overall survival were 82.33% (95% CI, 77.32% and 86.62%) and 94.13% (95% CI, 90.02% and 96.69%), respectively [3]. These tumors arise from primordial germ cells and derivatives and are localized in the gonads or in extragonadal tissues along the body midline, both extra- and intracranially [4,5]. The preferred regions of origin of eGCTs are the gonads (testis and ovary), sacrococcygeal region, and mediastinum [6]. eGCTs are classified according to originally Teilum with respect to their histological degree of embryonic differentiation (teratoma or embryonal carcinoma), extraembryonic differentiation (yolk sac tumor or choriocarcinoma), and germinal differentiation (seminoma in male patients, dysgerminoma in female patients, and germinoma when occurring in the CNS) [4]. Mixed forms with more than one histological subtype are possible, especially beyond childhood (ICD-11—Mortality and Morbidity Statistics (who.int (accessed on 18 April 2024)).
Some risk factors have been suggested, including genetic syndromes such as Swyer syndrome [7,8], Down syndrome [9,10,11], Currarino syndrome defined by the triad of a presacral mass, sacral and anorectal congenital anomalies [12], or Klinefelter syndrome [13,14]. Furthermore, the occurrence of eGCTs has been associated with non-syndromic defects as, e.g., cryptorchidism, inguinal hernias, congenital heart defects, and skeletal congenital anomalies [15,16]. A recent publication suggested that birth defects might only be related to eGCTs if related to syndromes (syndromic defects) but not for non-syndromic defects.
We analyzed cases of the German MAKEI 96/MAHO 98 registry providing data on eGCT. To identify potentially related risks in a broader range of syndromes, we identified all cases with any reported syndrome in MAKEI/MAHO and compared their prevalence to that reported in Orphanet. Additionally, we identified cases with reported non-syndromic defects and addressed their overall potential risk for any eGCT and by specific tumor site using a German birth cohort. We performed an aggregated data analysis. The objective was to confirm or refuse already known risk factors regarding syndromes with or without associated congenital anomalies, to identify potentially new syndromes associated with eGCTs, and to assess a potential role for non-syndromic defects.
2. Materials and Methods
The MAKEI 96 registry was conducted in accordance with the Declaration of Helsinki and was approved by the Ethics Committee of Heinrich-Heine-University, Düsseldorf (study number 837, decision of 22 August 1995). All participants provided their written informed consent. Informed consent was given by the respective legal guardians of the affected children. All patients had an eGCT. In the patient data evaluated here, cases with an additional non-oncologic diagnosis (genetic syndromes or congenital anomalies) were assessed in detail.
2.1. Study Population
The MAKEI 96/MAHO 98 registry comprises a large data collection of children and adolescents with an eGCT (n = 2610) [17,18]. Patients were prospectively enrolled in the registry over a 20-year period. Data were collected in collaboration with centers of the Society for Pediatric Oncology and Hematology (GPOH). Between January 1996 and October 2017, a total of 2610 patients with a newly diagnosed eGCT were prospectively registered by 62 pediatric oncology units in Germany, Austria, and Switzerland. Presacral, sacral, and coccygeal tumors were grouped together in the sacrococcygeal group. The presacral mass occurred as part of the Currarino syndrome.
In the current study, all patients with documented associated genetic syndromes and congenital anomalies of the genitourinary tract, the heart, the digestive system, the musculoskeletal system, and the central nervous system were identified. During the initial survey following the registration of patients, epidemiologic data were collected by the MAKEI 96/MAHO 98 registry. In case of incomplete reporting, the hospitals were reminded until presumed complete reporting was achieved. Basic demographic data were also documented. The CRF included information on anatomical site of origin, symptoms, diagnosis, tumor markers, and histology. Furthermore, non-oncologic diagnoses such as underlying genetic syndromes, chronic diseases, and congenital anomalies were documented.
2.2. Case Definitions
Syndromes categorized according to Orphanet in MAKEI/MAHO were identified. Verification of the diagnoses by molecular genetic analysis was beyond the scope of the MAKEI/MAHO registry. For analysis, we used any syndrome irrespective of any potential additional congenital anomalies.
The diagnosis of congenital anomalies was based on clinical aspects, ultrasound, or CT/MRI imaging (in cases). In accordance with EUROCAT, non-syndromic defects were grouped according to their localization [19,20,21]. We additionally considered the associated type of tumor and potential syndrome.
2.3. Definition of Exposure
In accordance with Schraw et al. [22], we distinguished the effects of syndromes and isolated non-syndromic defects. In cases with an associated syndrome, we could not distinguish those potential additional non-syndromic defects because of the aggregated data structure regarding controls.
2.4. Reference Population
To assess the association with syndromes, we used the respective prevalence of genetic syndromes in the general population as documented in Orphanet. The prevalence of genetic syndromes in the general population was extracted from the Orphanet database (https://www.orpha.net/, accessed on 18 April 2024).
The principle of the case–control study is to compare the prevalence of cases (MAKEI 96/MAHO 98) with or without genetic syndromes to controls from the general population. The prevalence of non-syndromic defects in the general population was estimated from the Mainz malformation registry with a complete, extensive, and standardized assessment of birth defects in the first week of life, including ultrasound of the urogenital system at birth. The data were extracted from the EUROCAT registry for children born between 1996 and 2017 (http://www.eurocat-network.eu/accessprevalencedata/prevalencetables, accessed on 18 April 2024). In the Mainz registry, cases are highly likely to reflect the general population [23].
2.5. Statistical Analysis
Because the registries provided only aggregated data, case data were aggregated accordingly using Excel files (Version 2405). These aggregated data were entered into stat calc to calculate the odds ratios and 95% confidence intervals. Stat calc is a public domain app sponsored and provided by CDC (https://www.cdc.gov/epiinfo/user-guide/statcalc/statcalcintro.html, accessed on 18 April 2024). Stat calc also provides formal statistical testing using different methods. We used the p-values for Fisher’s exact test with additional Bonferroni correction for multiple testing regarding potential new syndromes associated with eGCTs. To assess whether the number of cases was sufficient to detect the observed effect size in previously reported associations with a power of at least 80%, we used the calculation of power with the power and sample size program (https://vbiostatps.app.vumc.org/ps/ accessed on 18 April 2024) in a post hoc analysis.
3. Results
3.1. Overall Frequency of Registered eGCTs
The most frequent eGCT in the total MAKEI 96/MAHO 98 registry sample of 2610 patients at the time of analysis were those of the ovaries (47%, n = 1232), followed by those of the sacrococcygeal region (21%, n = 540), the testis (17%, n = 447), and the mediastinum (4%, n = 114). A total of 277 tumors were found in other anatomical structures of the body midline (11%), neck, eye, or retroperitoneum. Of the 2610 patients with an eGCT, 754 were male (28.9%), and 1856 were female (71.1%). In comparison, in the group of patients with an eGCT and an additional non-oncologic diagnosis, 44 were male (45.9%), and 51 were female (54.1%).
3.2. Location and Histology of eGCTs in Patients with Genetic Syndromes
Gonadal GCTs: 34 patients with genetic syndromes were found in combination with a gonadal GCT (26 of 1232 ovarian GCTs and 7 of 447 testicular GCTs). A percentage of 62% of the 26 ovarian GCTs were dysgerminomas, followed by their precursor lesion gonadoblastomas (15%). When analyzing patients with a genetic syndrome and a testicular GCT, four (57%) presented with mixed eGCTs. In addition, one case each of seminoma, embryonal carcinoma, and embryonal carcinoma with a yolk sac tumor was detected.
Extragonadal GCTs: Nine patients with a tumor in the sacrococcygeal region (9 of 540 tumors of the sacrococcygeal region) were documented. In the group of sacrococcygeal tumors, only teratomas were found, and one of them was combined with a yolk sac tumor. Three patients with a genetic syndrome had a mediastinal GCT (3 of 114) comprising one choriocarcinoma and two mixed eGCTs (one composed of a yolk sac tumor and a teratoma, the other composed of a choriocarcinoma, a yolk sac tumor, and a teratoma). Among patients with an eGCT of the retroperitoneum, 3 patients (out of 277) had a genetic syndromic background comprising Down syndrome, neurofibromatosis type 1 and one patient with intellectual disability, thoraco-lumbar scoliosis, and tethered cord. Among these, two patients had teratomas, and one had a mixed eGCT (embryo carcinoma, choriocarcinoma, and yolk sac tumor).
3.3. eGCT and Associated Genetic Syndromes
Of the 95 patients with an additional non-oncologic diagnosis, 48 had a syndrome. Most syndromes recorded in the MAKEI/MAHO registry could be linked to a specific tumor (Table 1), except for Down syndrome, which was found in connection with eGCTs of different localizations. Most underlying genetic syndromes were found in the group of patients with an ovarian eGCT (n = 26). In terms of age, most eGCTs in patients with an additional syndrome were diagnosed in the prepubertal or pubertal period. Only in the group of sacrococcygeal GCTs was the peak incidence in the first year of life. The majority of eGCTs within this group were malignant; only in patients with Currarino syndrome were benign eGCT tumors predominated.

Table 1.
Germ cell tumors are divided by localization and co-occurring syndromes (number of cases, sex, age at diagnosis, and dignity).
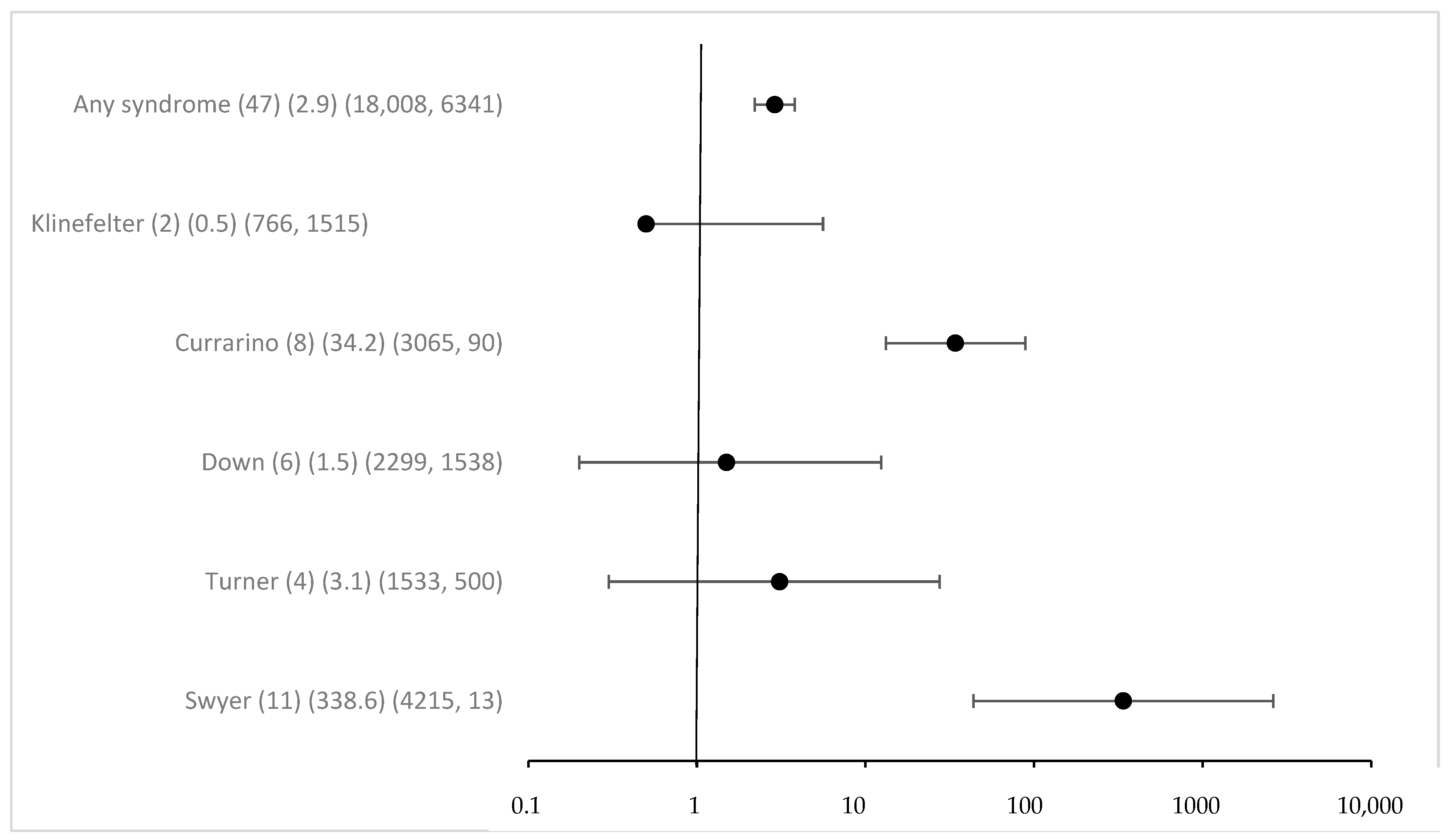
Overall associated syndromes were only rarely detected in the study group with eGCTs (1.8%). Regarding any syndrome registered in MAKEI 96/MAHO 98, the odds ratio was 2.9 (95% CI 2.2–3.8) (Figure 1). The previously described correlations between Swyer and Currarino syndromes and the occurrence of eGCTs were confirmed. The strength of the effect was substantial, with an OR for Swyer syndrome above 100 and 34 for Currarino syndrome, respectively, with 95% CI clearly excluding 1. For Down syndrome, the OR was 1.5 with a 95% CI including 1 (95% CI 0.2–12.4) despite a considerable number of cases. The power calculation in a post hoc analysis showed that the power to detect the observed effect with an alpha of <0.05 was only 0.08. For Klinefelter and Turner syndromes [41], we failed to identify a significant association [42]. The effect estimates for the known syndromes did not require Bonferroni correction because of a priori hypothesis (Figure 1).
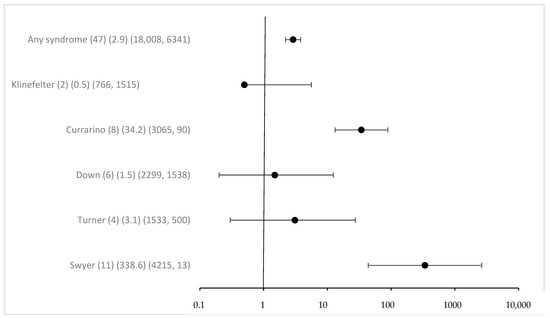
Figure 1.
Odds ratio and 95% confidence intervals for eGCT in patients with different syndromes. Provided data (n) (OR) (exposure per million in cases, controls).
Interestingly, 16 new syndromes were found in children with eGCTs, and most of them were female (75%, n = 12). However, these were only isolated cases, never more than one. Compared to the registry, these yielded low p-values for Fisher’s exact and for some high OR, which were driven by the extreme paucity of such syndromes in the general population. Following Bonferroni correction for multiple testing, the respective p-values were clearly above the Bonferroni adjusted p-value of 0.00238, however (see Figure S1). A remarkable finding, however, is the fact that most cases were tied to girls. While this is evident for the Triple X syndrome, the sex distribution of the other syndromes was either equal or slightly shifted to male or female patients. On average, the prevalence for girls in the general population was 42% for the syndromes mentioned (https://www.orpha.net/, accessed on 18 April 2024).
3.4. eGCT and Associated Malformations
In the MAKEI 96/MAHO 98 registry, 47 patients with a non-syndromic defect were recorded. Non-syndromic defects occurred in connection with eGCTs of all localizations. Most were described in the group of sacrococcygeal GCTs (n = 18) (Table 2). Urogenital malformations accounted for the largest proportion. The age distribution at the time of tumor diagnosis also showed a peak postnatally for the sacrococcygeal GCTs and prepubertal or pubertal for the other tumor localizations.
Table 2.
Germ cell tumors divided by co-occurring congenital anomalies and tumor location (number of cases, sex, age at diagnosis, and dignity (number of malignant tumors of all tumors belonging to a localization)).
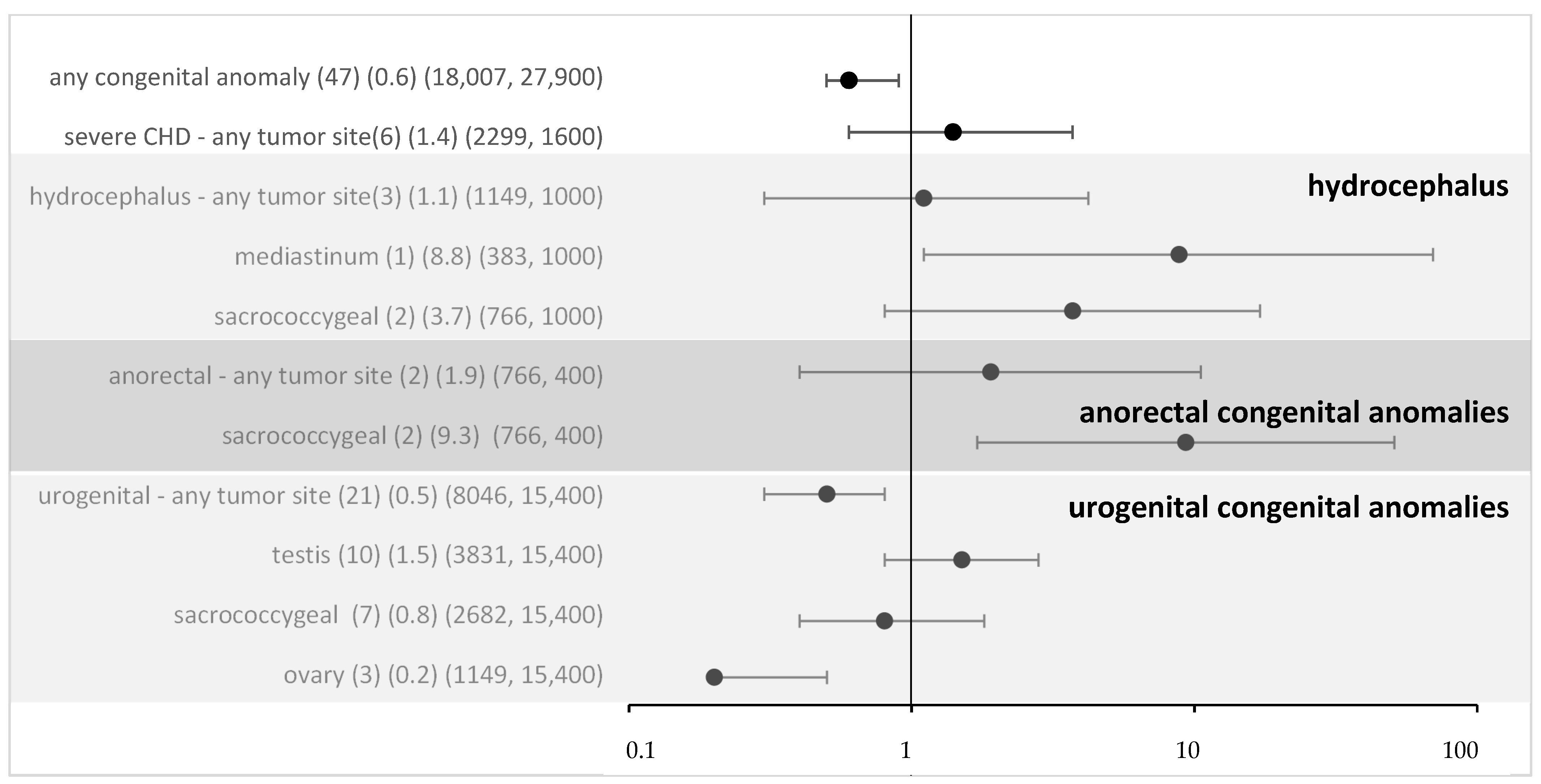
For all non-syndromic defects in the MAKEI/MAHO registry, compared to the data from the Mainz registry, the OR was 0.6 (95% CI 0.5–0.9) (Figure 2). Thus, an overall risk related to isolated congenital anomalies appears highly unlikely. To assess whether the local vicinity of tumors and congenital anomalies might be related to a higher risk, we performed a subgroup analysis. For most subgroups, there was no evidence, and following Bonferroni correction (Bonferroni corrected p-value 0.005), none of the associations reached significance. Overall, no strong effect sizes were found.
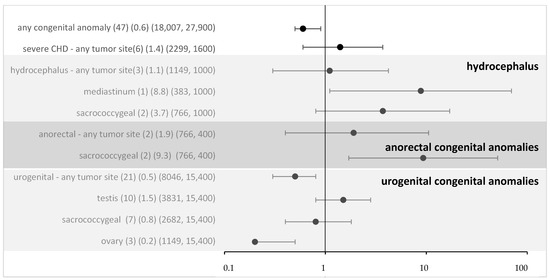
Figure 2.
Odds ratio and 95% confidence intervals for eGCT in patients with different congenital anomalies and by tumor sites (sensitivity analysis with data from Mainz). Provided data (n) (OR) (exposure per million in cases, controls).
4. Discussion
The main finding of our paper was the confirmation of the role of several syndromes in accounting for the risk of an eGCT. We observed high ORs for both Swyer and Currarino syndromes. Regarding non-syndromic defects, no overall association was found. Subgroup analysis might suggest a hint at a potential role for non-syndromic defects close to the tumor site.
The identification of risk factors requires a control population and cautious interpretation of statistical findings. In the literature, few papers used a control group describing potential associations of syndromes and the later emergence of eGCTs [43,44,45,46,47]. Whereas some previous papers only reported associations between tumor and syndrome in cases [48,49,50,51], a recent paper with a control group allowing for individual case–control analysis with adjustment confirmed a strong statistical association for Klinefelter with comparison to controls, but not for Turner and Down syndrome [22]. Our data confirmed the latter. Regarding our data, the estimate for Klinefelter syndrome was likely, however, to reflect diagnostic bias. Unlike in other registers [22] no systematic screening for Klinefelter was performed in our MAKEI/MAHO patients. Similarly, no systematic screening for Turner syndrome was performed in eGCT cases in MAKEI/MAHO. As many of the eGCTs occur prepubertal or in early puberty, such cases may well be missed because of the later occurrence of the lead symptoms. Down syndrome, in contrast, is most likely to be diagnosed early in life. Additionally, some biological probability for causality was described the in the literature. The result of a study by Cools et al. (2006) was a longer expression of OCT3/4, PLAP, and c-KIT, biomarkers for delayed primordial germ cell maturation, linking it to the development of the precursor of a malignant eGCT, via either GCNIS or gonadoblastioma, in male fetuses with trisomy 21 [52]. The estimate for the strength of the effect, however, was low (OR 1.5) with a wide 95% CI clearly including one. In a post hoc power analysis, we found that with the observed case numbers the power to detect such a small effect size was only very low (0.08).
Regarding the presumed new syndromes, our data point to the need for a very careful interpretation of statistics in case of isolated findings of syndromes in eGCT cases. Although effect estimates based on comparison with control groups may appear huge, the risk is far from proven. In the case of many “new associations”, it is important to perform at least a Bonferroni correction. On the other hand, a causative association is difficult to rule out, especially when only isolated cases are identified. Although a role for the individual syndromes could not be established, confinement to girls is striking. Among patients with gonadal eGCT, associations with genetic syndromes based on disorders of sexual development have been described previously. About 1 in 4500 births (0.02%) is affected by a disorder of sexual development [20]. Several publications describe an increased risk of developing an eGCT in individuals with gonadal dysgenesis or in patients with Turner syndrome and Y chromosomal material [7,53,54,55,56,57,58]. The MAKEI 96/MAHO 98 registry comprised 18 patients with gonadal dysgenesis who had an underlying genetic syndrome (0.69%). Among patients with published results with disorders of sexual development, gonadoblastoma is diagnosed in 4.7–25% of cases. Of these, 50% develop a dysgerminoma [15,59,60,61]. These histology types were prominent in the 18 patients of the MAKEI 96/MAHO 98 registry (15 of 18 patients with pure gonadoblastoma and 11 of 18 patients with gonadoblastoma and dysgerminoma). eGCTs most commonly occur in gonadal dysgenesis in association with Swyer syndrome [62]. Various molecular biomarkers, such as miR-371a-3p, were discovered in patients with eGCT and disorders of sexual development [63].
Mutations of the MNX1 gene have been found in patients with Currarino syndrome, and further molecular genetic correlations are being sought [64].
For non-syndromic defects, we failed to identify an increased risk for eGCTs in accordance with Schraw et al. [22]. A subgroup analysis linking tumor site and congenital anomaly did not yield significant results either.
Confirmation of high risks for Currarino and Swyer syndrome might suggest a cause for screening in these syndromes because in children with these syndromes, the a priori probability of GCTs might be high enough to allow for screening with acceptable positive predictive values. Unfortunately, however, most cases of Currarino syndrome occur in the first year of life when the test is not specific. For Swyer syndrome with pure dysgerminomas, a screening is not useful because of the absence of known tumor markers such as AFP and beta-HCG.
Strengths and Limitations
The MAKEI 96/MAHO 98 registry is very complete regarding the recording of eGCT.
Beyond the presumed complete eGCT registration for Germany in MAKEI 96/MAHO 98 and comparison to a standard control group (Orphanet), the strength of our paper pertains to statistical analysis. We provide the mandatory Bonferroni correction if needed and show that an apparently strong effect suggested for one case only in extremely rare conditions may well be spurious. Such associations should only be discussed in case of possible causative pathways. Regarding congenital anomalies, the Mainz registry, like EUROCAT, aims to provide essential epidemiological information on the birth prevalence of congenital malformations and anomalies in Europe [18,19]. The strength of the Mainz registry, however, is a complete case ascertainment and a structured clinical assessment and ultrasound screening for the entire cohort. Therefore, the Mainz registry provides a unique, valid, comparator to presumed complete congenital anomaly assessment in the cases. The latter is likely because all the children with eGCT undergo extensive screening for metastasis. As with all registries, there is an issue about diagnostic and an ascertainment bias. Regarding the cases, this would account for underestimation of the effect sizes. As outlined, specific syndromes such as Turner and Klinefelter syndrome may be very plausible reasons for diagnostic bias. For other syndromes and non-syndromic defects, this may be less evident but cannot be excluded.
Previous articles on the association of an eGCT and an additional non-oncologic diagnosis were based on small data sets or case reports. As the proportion of diagnosed eGCTs increased over the observed period, we assume that this could be due to the improved diagnostic capabilities that developed over time. Still, our study has several limitations. Compared to today, fewer predispositions to tumors were known at the beginning of data collection. Not all co-occurring phenotypic features among other patients registered in the MAKEI 96/MAHO 98 registry have led to the diagnosis of a genetic syndrome. Any errors in diagnosis or coding may adversely affect the accuracy of the presented results. Furthermore, under-reporting may be an issue as patients with sacrococcygeal eGCTs; in particular, those who undergo postnatal surgery in smaller hospitals might not been registered in the MAKEI 96/MAHO 98 registry. In addition, potential candidates for the registry may have died from pathologies due to genetic syndrome or congenital anomalies before the eGCT could be diagnosed and registered. An evaluation of the success of the therapy in the reported cases was not part of this paper. In the eGCTs of the sacrococcygeal region, tumors with different relationships to the os sacrum and os coccyx were combined without considering the migration of the primordial germ cells. Further, only one leading syndromic abnormality has been documented per case. Regarding congenital anomalies, one case may be recorded for different anomalies, also affecting the statistical results. Patients who were diagnosed with eGCT and congenital anomalies shortly after birth were also included. No data was collected on premature births as a possible cause of the anomaly. Possible overlaps between the data sets of the MAKEI 96/MAHO 98 registry, EUROCAT, and the Mainz registry cannot be ruled out with certainty.
Unfortunately, we could not access the entire Mainz data to allow for individual case statistical analysis because the registration in the Mainz registry has been closed, and the involved persons have retired. This is why we could only use the Mainz registry as reported in EUROCAT.
5. Conclusions
In our study, we confirmed an association between Swyer and Currarino syndrome and eGCTs with high effect sizes. There is biological plausibility for these associations. Our failure to confirm an association between Klinefelter and Turner syndrome and eGCTs might be due to probable diagnostic bias of these conditions in cases. The presumed size of the association of Down syndrome with eGCT appeared to be small and thus could not be identified with the available case numbers. Case reports of associations of rare syndromes in children with eGCT are most likely to be caused by chance. Considering non-syndromic defects, no association was shown in relation to eGCTs.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/cancers16112157/s1, Figure S1: Odds ratio and 95% confidence intervals for eGCT in patients with syndromes.
Author Contributions
Conceptualization, J.H.S., R.v.K., H.R. (Heiko Reutter) and G.C.; Data curation, A.R., D.v.S., M.F., C.B., L.F., B.S.L., R.W., B.F., W.B., I.S., H.R. (Harald Reinhard), M.D., P.H., M.H., C.V., S.S., D.T.S., G.S., U.G. and G.C.; Formal analysis, J.H.S. and R.v.K.; Investigation, J.H.S.; Writing—original draft, J.H.S. and R.v.K.; Writing—review and editing, J.H.S., A.R., D.v.S., M.F., C.B., R.W., W.B., I.S., H.R. (Harald Reinhard), M.H., S.S., D.T.S., G.S., L.L., U.G., R.v.K., H.R. (Heiko Reutter) and G.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The MAKEI 96 registry was conducted in accordance with the Declaration of Helsinki and was approved by the Ethics Committee of Heinrich-Heine-University, Düsseldorf (study number 837, decision of 22 August 1995).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study. Written informed consent has been obtained from the patient(s) to publish this paper. Informed consent was given by the respective legal guardians of the affected children.
Data Availability Statement
Restrictions apply to the availability of these data. Data were obtained from EUROCAT (Mainz registry) and are available at https://eu-rd-platform.jrc.ec.europa.eu/eurocat/eurocat-data/prevalence_en (accessed on 18 April 2024) with the permission of EUROCAT (Mainz registry).
Acknowledgments
MAKEI 96 was supported by the German Cancer Aid. We acknowledge the Deutsche Forschungsgemeinschaft and the Friedrich-Alexander-Universität Erlangen-Nürnberg within the funding program “Open Access Publication Funding”. Open Access funding enabled and organized by Projekt DEAL. We thank all the patients and their families who participated in the study, as well as all teams of all participating centers, for their dedicated efforts. We would particularly like to thank A. Wiesel for providing the data of the Mainz registry and the data managers Carmen Teske and Jans-Enno Müller of the MAKEI group in Bonn.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Lobo, J.; Gillis, A.J.M.; Jerónimo, C.; Henrique, R.; Looijenga, L.H.J. Human Germ Cell Tumors are Developmental Cancers: Impact of Epigenetics on Pathobiology and Clinic. Int. J. Mol. Sci. 2019, 20, 258. [Google Scholar] [CrossRef] [PubMed]
- Kaatsch, P.; Häfner, C.; Calaminus, G.; Blettner, M.; Tulla, M. Pediatric germ cell tumors from 1987 to 2011: Incidence rates, time trends, and survival. Pediatrics 2015, 135, e136–e143. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Dong, K.; Li, K.; Liu, J.; Du, X.; Huang, C.; Jiao, Y.; Han, Y.; Yang, J.; Liao, X.; et al. Extracranial Germ Cell Tumors in Children: Ten Years of Experience in Three Children’s Medical Centers in Shanghai. Cancers 2023, 15, 5412. [Google Scholar] [CrossRef] [PubMed]
- Teilum, G. Classification of endodermal sinus tumour (mesoblastoma vitellinum) and so-called “embryonal carcinoma” of the ovary. Acta Pathol. Microbiol. Scand. 1965, 64, 407–429. [Google Scholar] [CrossRef] [PubMed]
- Oosterhuis, J.W.; Stoop, H.; Honecker, F.; Looijenga, L.H. Why human extragonadal germ cell tumours occur in the midline of the body: Old concepts, new perspectives. Int. J. Androl. 2007, 30, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.T.; Calaminus, G.; Koch, S.; Teske, C.; Schmidt, P.; Haas, R.J.; Harms, D.; Göbel, U. Epidemiologic analysis of 1442 children and adolescents registered in the German germ cell tumor protocols. Pediatr. Blood Cancer 2004, 42, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Swyer, G.I.M. Male Pseudohermaphroditism: A Hitherto Undescribed Form. Br. Med. J. 1955, 2, 709–712. [Google Scholar] [CrossRef]
- Matsumoto, F.; Shimada, K.; Ida, S. Tumors of Bilateral Streak Gonads in Patients with Disorders of Sex Development Containing Y Chromosome Material. Clin. Pediatr. Endocrinol. 2014, 23, 93–97. [Google Scholar] [CrossRef]
- Down, J.L. Observations on an ethnic classification of idiots. 1866. Ment. Retard. 1995, 33, 54–56. [Google Scholar]
- Hasle, H. Pattern of malignant disorders in individuals with Down’s syndrome. Lancet Oncol. 2001, 2, 429–436. [Google Scholar] [CrossRef]
- Hasle, H.; Clemmensen, I.H.; Mikkelsen, M. Risks of leukaemia and solid tumours in individuals with Down’s syndrome. Lancet 2000, 355, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Currarino, G.; Coln, D.; Votteler, T. Triad of anorectal, sacral, and presacral anomalies. AJR Am. J. Roentgenol. 1981, 137, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Klinefelter, H.F.; Reifenstein, E.C.; Albright, F. Syndrome characterized by gynecomastia, aspermatogenesis without a-Leydigism, and increased excretion of follicle-stimulating hormone. J. Clin. Endocrinol. Metab. 1942, 2, 615–627. [Google Scholar] [CrossRef]
- Sogge, M.R.; McDonald, S.D.; Cofold, B. The malignant potential of the dysgenetic germ cell in Klinefelter’s syndrome. Am. J. Med. 1979, 66, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Verp, M.S.; Simpson, J.L. Abnormal Sexual Differentiation and Neoplasia. Cancer Genet. Cytogenet. 1987, 25, 191–218. [Google Scholar] [CrossRef] [PubMed]
- Nishi, M.; Miyake, H.; Takeda, T.; Hatae, Y. Congenital Malformations and Childhood Cancer. Med. Pediatr. Oncol. 2000, 34, 250–254. [Google Scholar] [CrossRef]
- Calaminus, G.; Schneider, D.T.; von Schweinitz, D.; Jürgens, H.; Infed, N.; Schönberger, S.; Olson, T.A.; Albers, P.; Vokuhl, C.; Stein, R.; et al. Age-Dependent Presentation and Clinical Course of 1465 Patients Aged 0 to Less than 18 Years with Ovarian or Testicular Germ Cell Tumors; Data of the MAKEI 96 Protocol Revisited in the Light of Prenatal Germ Cell Biology. Cancers 2020, 12, 611. [Google Scholar] [CrossRef]
- Schneider, D.T.; Wessalowski, R.; Calaminus, G.; Pape, H.; Bamberg, M.; Engert, J.; Waag, K.; Gadner, H.; Göbel, U. Treatment of recurrent malignant sacrococcygeal germ cell tumors: Analysis of 22 patients registered in the German protocols MAKEI 83/86, 89, and 96. J. Clin. Oncol. 2001, 19, 1951–1960. [Google Scholar] [CrossRef] [PubMed]
- Greenlees, R.; Neville, A.; Addor, M.C.; Amar, E.; Arriola, L.; Bakker, M.; Barisic, I.; Boyd, P.A.; Calzolari, E.; Doray, B.; et al. Paper 6: EUROCAT Member Registries: Organization and Activities. Birth Defects Res. A Clin. Mol. Teratol. 2011, 91, S51–S100. [Google Scholar] [CrossRef]
- Tucker, F.D.; Morris, J.K.; on behalf of the JRC Management Committee; Neville, A.; Garne, E.; Kinsner-Ovaskainen, A.; Lanzoni, M.; Loane, M.A.; Martin, S.; Nicholl, C.; et al. EUROCAT: An update on its functions and activities. J. Community Genet. 2018, 9, 407–410. [Google Scholar] [CrossRef]
- EUROCAT. EUROCAT Guide 1.4: Instruction for the Registration of Congenital Anomalies. EUROCAT Central Registry, University of Ulster. 2013. Available online: https://eu-rd-platform.jrc.ec.europa.eu/eurocat/data-collection/guidelines-for-data-registration_en (accessed on 15 May 2024).
- Schraw, J.M.; Sok, P.; Desrosiers, T.A.; Janitz, A.E.; Langlois, P.H.; Canfield, M.A.; Frazier, A.L.; Plon, S.E.; Lupo, P.J.; Poynter, J.N. Associations between birth defects and childhood and adolescent germ cell tumors according to sex, histologic subtype, and site. Cancer 2023, 129, 3300–3308. [Google Scholar] [CrossRef]
- Queißer-Luft, A.; Spranger, J. Fehlbildungen bei Neugeborenen. Dtsch. Arztebl. 2006, 103, A 2464–A 2471. [Google Scholar]
- Hirschhorn, K.; Cooper, H.L.; Firschein, I.L. Deletion of short arms of chromosome 4–5 in a child with defects of midline fusion. Humangenetik 1965, 1, 479–482. [Google Scholar] [CrossRef]
- Caffey, J. Prenatal bowing and thickening of tubular bones, with multiple cutaneous dimples in arms and legs; a congenital syndrome of mechanical origin. Am. J. Dis. Child. 1947, 74, 543–562. [Google Scholar] [CrossRef] [PubMed]
- Capito, C.; Leclair, M.D.; Arnaud, A.; David, A.; Baron, S.; Corradini, N.; Héloury, Y. 46, XY pure gonadal dysgenesis: Clinical presentations and management of the tumor risk. J. Pediatr. Urol. 2011, 7, 72–75. [Google Scholar] [CrossRef]
- Cost, N.G.; Ludwig, A.T.; Wilcox, D.T.; Rakheja, D.; Steinberg, S.J.; Baker, L.A. A novel SOX9 mutation, 972delC, causes 46, XY sex-reversed campomelic dysplasia with nephrocalcinosis, urolithiasis, and dysgerminoma. J. Pediatr. Surg. 2009, 44, 451–454. [Google Scholar] [CrossRef]
- Lloyd, K.M.; Dennis, M. Cowden’s disease. A possible new symptom complex with multiple system involvement. Ann. Int. Med. 1963, 58, 136–142. [Google Scholar] [CrossRef]
- Denys, P.; Malvaux, P.; Van Den Berghe, H.; Tanghe, W.; Proesmans, W. Association of an anatomo-pathological syndrome of male pseudohermaphroditism, Wilms’ tumor, parenchymatous nephropathy and XX/XY mosaicism. Arch. Fr. Pediatr. 1967, 24, 729–739. [Google Scholar] [PubMed]
- Frasier, S.D.; Bashore, R.A.; Mosier, H.D. Gonadoblastoma associated with pure gonadal dysgenesis in monozygous twins. J. Pediatr. 1964, 64, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Hersmus, R.; van Bever, Y.; Wolffenbuttel, K.P.; Biermann, K.; Cools, M.; Looijenga, L.H. The biology of germ cell tumors in disorders of sex development. Clin. Genet. 2017, 91, 292–301. [Google Scholar] [CrossRef]
- Imerslund, O. Idiopathic chronic megaloblastic anemia in children. Acta Paediatr. Suppl. 1960, 49, 1–115. [Google Scholar] [CrossRef]
- Lennox, W.G.; Davis, J.P. Clinical correlates of the fast and the slow spike-wave electroencephalogram. Pediatrics 1950, 5, 626–644. [Google Scholar] [CrossRef] [PubMed]
- Noonan, J.A.; Ehmke, D.A. Associated non-cardiac malformations in children with congenital heart disease. J. Pediatr. 1963, 63, 469. [Google Scholar]
- Poland, A. Deficiency of the pectoral muscles. Guy’s Hosp. Rep. 1841, 6, 191–193. [Google Scholar]
- Rett, A. Über ein eigenartiges hirnatrophisches Syndrom bei Hyperammonämie im Kindesalter. Wien. Med. Wochenschr. 1966, 116, 723–726. [Google Scholar] [PubMed]
- Sturge, W.A. A case of partial epilepsy apparently due to lesion of one of the vasomotor centres of the brain. Trans. Clin. Soc. Lond. 1879, 12, 162. [Google Scholar]
- West, W.J. On a peculiar form of infantile convulsions. Lancet 1841, 35, 724–725. [Google Scholar] [CrossRef]
- De Lange, C. Sur une type nouveau de dégénération (typus Amstelodamensis). Arch. Ned. Enfant. 1933, 36, 713–719. [Google Scholar]
- Cohen, M.M.; Hayden, P.W. A newly recognized hamartomatous syndrome. Birth Defects Orig. Artic. Ser. 1979, 15, 291–296. [Google Scholar]
- Turner, H.H. A syndrome of infantilism, congenital webbed neck and cubitus valgus. Endocrinology 1938, 23, 566–574. [Google Scholar] [CrossRef]
- Schneider, D.T.; Schuster, A.E.; Fritsch, M.K.; Calaminus, G.; Göbel, U.; Harms, D.; Lauer, S.; Olson, T.; Perlman, E.J. Genetic analysis of mediastinal nonseminomatous germ cell tumors in children and adolescents. Genes Chromosomes Cancer 2002, 34, 115–125. [Google Scholar] [CrossRef]
- Johnson, K.J.; Ross, J.A.; Poynter, J.N.; Linabery, A.M.; Robison, L.L.; Shu, X.O. Paediatric germ cell tumours and congenital abnormalities: A Children’s Oncology Group study. Br. J. Cancer 2009, 101, 518–521. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.J.; Lee, J.M.; Ahsan, K.; Padda, H.; Feng, Q.; Partap, S.; Fowler, S.A.; Druley, T.E. Pediatric cancer risk in association with birth defects: A systematic review. PLoS ONE 2017, 12, e0181246. [Google Scholar] [CrossRef]
- Botto, L.D.; Flood, T.; Little, J.; Fluchel, M.N.; Krikov, S.; Feldkamp, M.L.; Wu, Y.; Goedken, R.; Puzhankara, S.; Romitti, P.A. Cancer risk in children and adolescents with birth defects: A population-based cohort study. PLoS ONE 2013, 8, e69077. [Google Scholar] [CrossRef]
- Altmann, A.E.; Halliday, J.L.; Giles, G.G. Associations between congenital malformations and childhood cancer. A register-based case-control study. Br. J. Cancer 1998, 78, 1244–1249. [Google Scholar] [CrossRef]
- Carozza, S.E.; Langlois, P.H.; Miller, E.A.; Canfield, M. Are children with birth defects at higher risk of childhood cancers? Am. J. Epidemiol. 2012, 175, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.L.; Bao, D.M.; Wang, Y.; Shen, D.H.; Li, Y.; Cui, H. Swyer’s Syndrome with Mixed Ovarian Malignant Germ Cell Tumor and Ovarian Gonadoblastoma. Chin. Med. J. 2016, 129, 1752–1754. [Google Scholar] [CrossRef]
- Satgé, D.; Sasco, A.J.; Curé, H.; Leduc, B.; Sommelet, D.; Vekemans, M.J. An excess of testicular germ cell tumors in Down’s syndrome: Three case reports and a review of the literature. Cancer 1997, 80, 929–935. [Google Scholar] [CrossRef]
- Rod, J.; Cretolle, C.; Faivre, L.; Jacquot, C.; Yacoub, O.; Ravasse, P.; Cheynel, N.; Sarnacki, S. Malignant transformation of presacral mass in Currarino syndrome. Pediatr. Blood Cancer 2019, 66, e27659. [Google Scholar] [CrossRef]
- Bonouvrie, K.; van der Werff Ten Bosch, J.; van den Akker, M. Klinefelter syndrome and germ cell tumors: Review of the literature. Int. J. Pediatr. Endocrinol. 2020, 2020, 18. [Google Scholar] [CrossRef]
- Cools, M.; Honecker, F.; Stoop, H.; Veltman, J.D.; de Krijger, R.R.; Steyerberg, E.; Wolffenbuttel, K.P.; Bokemeyer, C.; Lau, Y.F.C.; Drop, S.L.; et al. Maturation delay of germ cells in fetuses with trisomy 21 results in increased risk for the development of testicular germ cell tumors. Hum. Pathol. 2006, 37, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Hughes, I.A.; Houk, C.; Ahmed, S.F.; Lee, P.A. Consensus statement on management of intersex disorders. Arch. Dis. Child. 2006, 91, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Abel, R. Ein Fall von Pseudohermaphroditismus masculinus mit sarcomatöser Cryptorchis sinistra. Arch. Pathol. Anat. Physiol. Für Klin. Med. 1891, 126, 420–437. [Google Scholar] [CrossRef]
- Bispo, A.V.S.; Burégio-Frota, P.; dos Santos, L.O.; Leal, G.F.; Duarte, A.R.; Araújo, J.; da Silva, V.C.; Muniz, M.T.C.; Liehr, T.; Santos, N. Y chromosome in Turner syndrome: Detection of hidden mosaicism and the report of a rare X; Y translocation case. Reprod. Fertil. Dev. 2014, 26, 1176–1182. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, F.; Matsuyama, S.; Matsui, F.; Yazawa, K.; Matsuoka, K. Variation of Gonadal Dysgenesis and Tumor Risk in Patients with 45,X/46, XY Mosaicism. Urology 2020, 137, 157–160. [Google Scholar] [CrossRef]
- Piazza, M.J.; Urbanetz, A.A. Germ Cell Tumors in Dysgenetic Gonads. Clinics 2019, 74, e408. [Google Scholar] [CrossRef] [PubMed]
- Hennes, E.; Zahn, S.; Lopes, L.F.; Schönberger, S.; Leuschner, I.; Göbel, U.; Calaminus, G.; Schneider, D.T. Molecular genetic analysis of bilateral ovarian germ cell tumors. Klin. Padiatr. 2012, 224, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Kathrins, M.; Kolon, T.F. Malignancy in disorders of sex development. Transl. Androl. Urol. 2016, 5, 794–798. [Google Scholar] [CrossRef] [PubMed]
- Pleskacova, J.; Hersmus, R.; Oosterhuis, J.W.; Setyawati, B.A.; Faradz, S.M.; Cools, M.; Wolffenbuttel, K.P.; Lebl, J.; Drop, S.L.; Looijenga, L.H. Tumor risk in disorders of sex development. Sex. Dev. 2010, 4, 259–269. [Google Scholar] [CrossRef]
- Cools, M.; Drop, S.L.; Wolffenbuttel, K.P.; Oosterhuis, J.W.; Looijenga, L.H. Germ Cell Tumors in the Intersex Gonad: Old Paths, New Directions, Moving Frontiers. Endocr. Rev. 2006, 27, 468–484. [Google Scholar] [CrossRef]
- Pyle, L.C.; Nathanson, K.L. A practical guide for evaluating gonadal germ cell tumor predisposition in differences of sex development. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Looijenga, L.H.J.; Kao, C.-S.; Idrees, M.T. Predicting gonadal germ cell cancer in people with disorders of sex development: Insights from developmental biology. Int. J. Mol. Sci. 2019, 20, 5017. [Google Scholar] [CrossRef] [PubMed]
- Dworschak, G.C.; Reutter, H.M.; Ludwig, M. Currarino syndrome: A comprehensive genetic review of a rare congenital disorder. Orphanet J. Rare Dis. 2021, 16, 167. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).