
Beyond Anti-PD-1/PD-L1: Improving Immune Checkpoint Inhibitor Responses in Triple-Negative Breast Cancer


Abstract
:Simple Summary
Abstract
1. Introduction
2. TNBC Heterogeneity and Sub-Classifications
3. Immunotherapy and Immune Checkpoint Inhibition
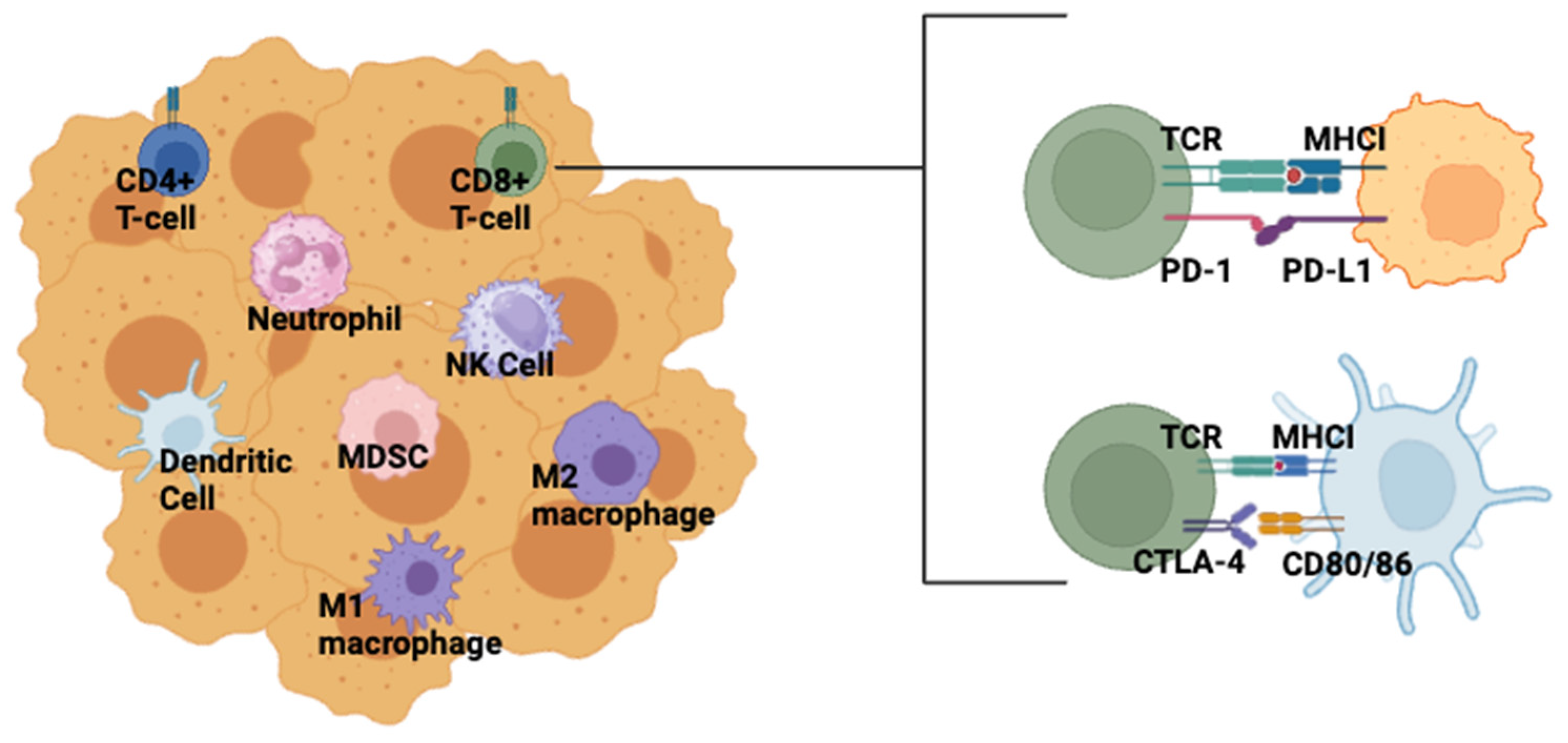
4. Clinical Status of ICI in TNBC
5. Improving Patient Selection
Immune Cell Signatures
6. Identifying Therapies That Will Sensitize Tumors to ICI
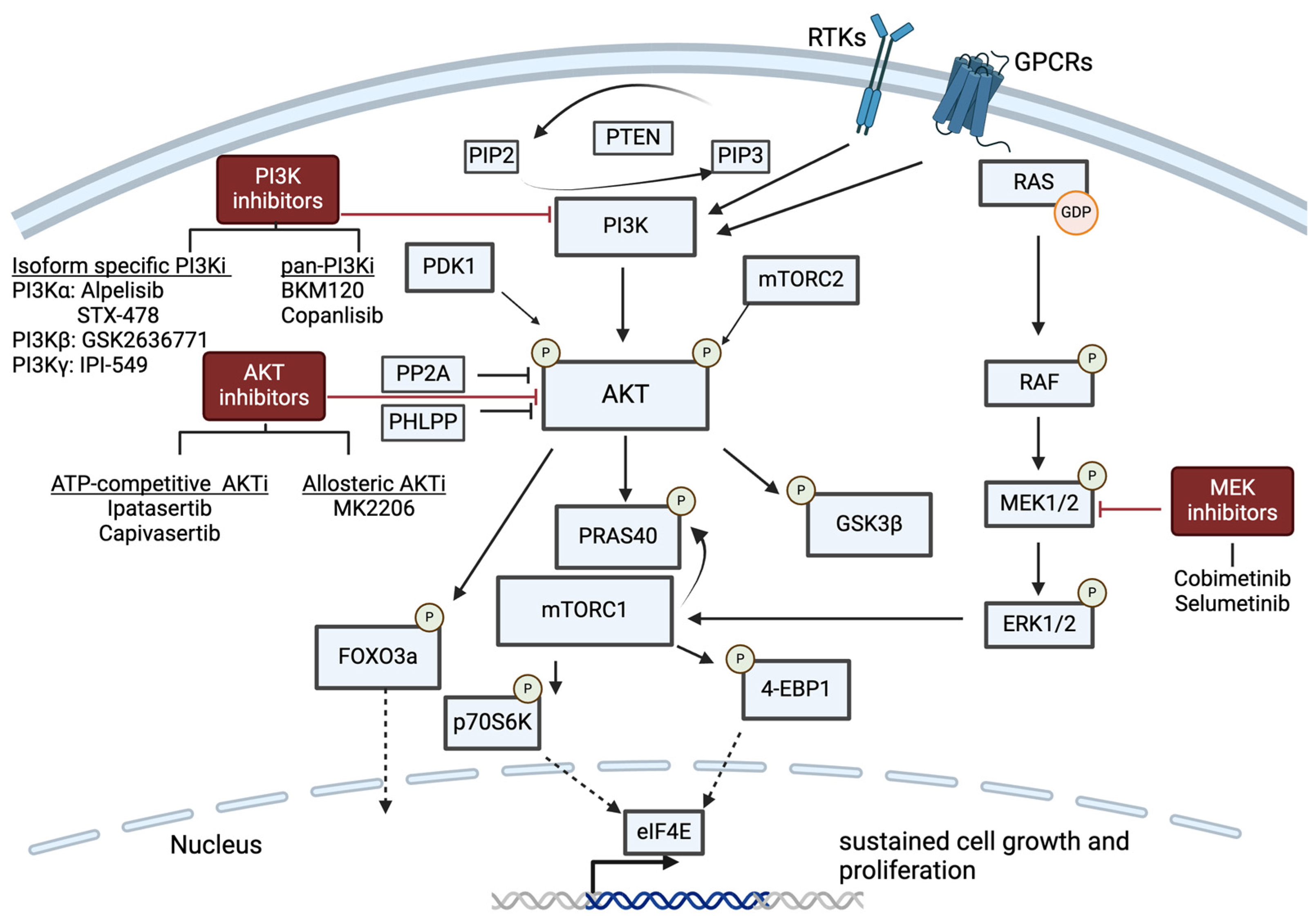
6.1. PI3K/AKT Pathway Inhibition
6.1.1. Isoform-Specific PI3K Inhibition

{kind=link}
{kind=link}
{kind=link}
| Identifier | Phase | Combination | Drug Names | Indications | Results | Ref |
|---|---|---|---|---|---|---|
| NCT04216472 | II | PI3Kαi + Chemo | Alpelisib + nab-PTX | Anthracycline-resistant TNBC with PIK3CA mutation or PTEN loss | Active, not recruiting | n/a |
| NCT04251533 | III | PI3Kαi + Chemo | Alpelisib + nab-PTX | Advanced stage TNBC with PIK3CA mutation or PTEN loss | Active, not recruiting | n/a |
| NCT05768139 | I/II | PI3Kαi (mutation specific) | STX-478 | Advanced solid tumors with PIK3CA mutation | Recruiting | n/a |
| NCT01458067 | I/IIa | PI3Kβi | GSK2636771 | Solid tumors with PTEN loss | Acceptable safety/toxicity profile | [73] |
| 9 + NCT04439188 (MATCH-Sub-protocol P) | II | PI3Kβi | GSK2636771 | Cancers with PTEN loss | Active, not recruiting | n/a |
| NCT02637531 (MARIO-1) | I/Ib | PI3Kγi +/− anti-PD1 | IPI-549 (eganelisib) +/− Nivolumab | TNBC | Acceptable safety/toxicity profile | [80] |
| NCT03961698 (MARIO-3) | II | PI3Kγi + anti-PD-L1 + Chemo | IPI-549 (eganelisib) + atezolizumab + nab-PTX | Locally advanced unresectable or metastatic TNBC | Interim analysis: ORR 55.3% | [81] |
6.1.2. Pan-PI3K Inhibition and AKT Inhibition
| Identifier | Phase | Combination | Drug Names | Indications | Results | Ref |
|---|---|---|---|---|---|---|
| NCT01790932 | II | pan-PI3Ki | BKM120 (buparlisib) | Metastatic TNBC | No OR | [82] |
| NCT04345913 | I/II | pan-PI3Ki + Chemo | Copanlisib + Eribulin | Advanced stage TNBC | Active, not recruiting | n/a |
| NCT01920061 | I/II | pan-PI3Ki + Chemo | Copanlisib+ Eribulin | Advanced-stage TNBC | Active, not recruiting | n/a |
| NCT01090960 | I | AKTi | IPAT | Metastatic TNBC | Acceptable safety/toxicity profile | [86] |
| LOTUS | II | AKTi + Chemo | IPAT + PTX | Metastatic TNBC | Trending increase OS for PTEN low and PIK3CA/AKT1/PTEN-altered subgroups (25.8 vs. 22.1 mo) | [88] |
| FAIRLANE | II | AKTi + Chemo | IPAT + PTX | Early-stage TNBC | No increase in pCR | [96] |
| IPATunity130 | III | AKTi + Chemo | IPAT + PTX | Locally advanced unresectable or metastatic TNBC | No improvement in PFS | [89] |
| PAKT | II | AKTi + Chemo | CAPI + PTX | Metastatic TNBC | PFS (5.9 vs. 4.2 months) OS (19.1 vs. 12.6 months) | [87] |
| CAPItello-290 | III | AKTi + Chemo | CAPI + PTX | Metastatic TNBC | Active, not recruiting | [97] |
| NCT03742102 (BEGONIA) | Ib/II | AKTi + anti-PD-L1 + Chemo | CAPI + Durvalumab+PTX | Metastatic PD-L1+ TNBC | Interim analysis: no change in ORR | [98] |
| NCT03800836 (CO40151) | Ib | AKTi + anti-PD-L1 + Chemo | IPAT + Atezolizumab + PTX or nab-PTX | Locally advanced or metastatic TNBC | Acceptable safety/toxicity profile: 73% ORR | [99] |
| NCT03424005 (Morpheus-panBC) | Ib/II | AKTi + anti-PD-L1 | IPAT + Atezolizumab | Locally advanced unresectable or metastatic TNBC | Recruiting | n/a |
| NCT04177108 (IPATunity170) | III | AKTi + anti-PD-L1 + Chemo | IPAT + Atezolizumab + PTX | Locally advanced unresectable or metastatic TNBC | No improvement in PFS or ORR | [67] |
| NCT01263145 | Ib | AKTi + Chemo | MK-2206 + PTX | Metastatic breast cancer | Acceptable safety/toxicity profile | [100] |
| NCT01277757 | II | AKTi | MK-2206 | Advanced breast cancer with PIK3CA mutation, AKT mutation, or PTEN loss | No improvement in PFS | [101] |
6.2. RAS/MAPK/ERK Pathway Inhibition
| Identifier | Phase | Combination | Drug Names | Indications | Results | Ref |
|---|---|---|---|---|---|---|
| NCT01562275 | Ib | AKTi + MEKi | Ipatasertib + Cobimetinib | Locally advanced or metastatic solid tumors | Limited tolerability and efficacy | [108] |
| NCT03202316 | II | Anti-PD-L1 + MEKi + Chemo | Atezolizumab + Cobimetinib + Eribulin | Chemotherapy-resistant metastatic inflammation breast cancer | Active, not recruiting | n/a |
| NCT03801369 | II | PARPi + anti-PD-LI or MEKi or AKTi | Olaparib + Durvalumab, or Selumetinib, or Capivasertib | Metastatic TNBC | Recruiting | n/a |
6.3. High Throughput Screening to Identify Targeted Therapies
6.4. Antibody–Drug Conjugates
6.5. Chemotherapies
7. Identifying New Immunotherapy Strategies for TNBC beyond Anti-PD-1/PD-L1
7.1. CTLA-4
7.2. Targeting Alternative Checkpoints
7.3. Novel Immunotherapy Targets
7.4. Myeloid Based Therapies
8. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- American Cancer Society Breast Cancer Facts & Figures 2022–2024; Atlanta. 2022. Available online: https://www.cancer.org/research/cancer-facts-statistics/breast-cancer-facts-figures.html (accessed on 19 February 2024).
- Bianchini, G.; De Angelis, C.; Licata, L.; Gianni, L. Treatment landscape of triple-negative breast cancer—Expanded options, evolving needs. Nat. Rev. Clin. Oncol. 2022, 19, 91–113. [Google Scholar] [CrossRef] [PubMed]
- Schettini, F.; Chic, N.; Brasó-Maristany, F.; Paré, L.; Pascual, T.; Conte, B.; Martínez-Sáez, O.; Adamo, B.; Vidal, M.; Barnadas, E.; et al. Clinical, pathological, and PAM50 gene expression features of HER2-low breast cancer. NPJ Breast Cancer 2021, 7, 1. [Google Scholar] [CrossRef]
- Wolff, A.C.; Elizabeth, M.; Hammond, H.; Allison, K.H.; Harvey, B.E.; Mangu, P.B.; Bartlett, J.M.S.; Bilous, M.; Ellis, I.O.; Fitzgibbons, P.; et al. Human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Focused Update. J. Clin. Oncol. 2018, 36, 2105–2122. [Google Scholar]
- Modi, S.; Jacot, W.; Yamashita, T.; Sohn, J.; Vidal, M.; Tokunaga, E.; Tsurutani, J.; Ueno, N.T.; Prat, A.; Chae, Y.S.; et al. Trastuzumab deruxtecan in previously treated HER2-low advanced breast cancer. N. Engl. J. Med. 2022, 387, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of triple-negative breast cancer molecular subtypes: Implications for neoadjuvant chemotherapy selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef] [PubMed]
- Denkert, C.; Loibl, S.; Noske, A.; Roller, M.; Müller, B.M.; Komor, M.; Budczies, J.; Darb-Esfahani, S.; Kronenwett, R.; Hanusch, C.; et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J. Clin. Oncol. 2010, 28, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Denkert, C.; von Minckwitz, G.; Darb-Esfahani, S.; Lederer, B.; Heppner, B.I.; Weber, K.E.; Budczies, J.; Huober, J.; Klauschen, F.; Furlanetto, J.; et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: A pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol. 2018, 19, 40–50. [Google Scholar] [CrossRef]
- Loi, S.; Drubay, D.; Adams, S.; Pruneri, G.; Francis, P.A.; Lacroix-Triki, M.; Joensuu, H.; Maria, V.D.; Dieci, V.; Badve, S.; et al. Tumor-infiltrating lymphocytes and prognosis: A pooled individual patient analysis of early-stage triple-negative breast cancers. J. Clin. Oncol. 2019, 37, 559–569. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Teng, M.W.L.; Kershaw, M.H.; Smyth, M.J. Cancer immunoediting: From surveillance to escape. Nat. Immunol. 2013, 3, 85–99. [Google Scholar] [CrossRef]
- Smyth, M.J.; Godfrey, D.I.; Trapani, J.A. A fresh look at tumor immunosurveillance and immunotherapy. Nat. Immunol. 2001, 2, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Starnes, C.O. Coley’s toxins in perspective. Nature 1992, 357, 11–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Ballas, Z.K. The 2018 Nobel Prize in Physiology or Medicine: An exemplar of bench to bedside in immunology. J. Allergy Clin. Immunol. 2018, 142, 1752–1753. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Sobral-Leite, M.; Van de Vijver, K.; Michaut, M.; van der Linden, R.; Hooijer, G.K.J.; Horlings, H.M.; Severson, T.M.; Mulligan, A.M.; Weerasooriya, N.; Sanders, J.; et al. Assessment of PD-L1 expression across breast cancer molecular subtypes, in relation to mutation rate, BRCA1-like status, tumor-infiltrating immune cells and survival. Oncoimmunology 2018, 7, e1509820. [Google Scholar] [CrossRef]
- Lee, K.-M.; Chuang, E.; Griffin, M.; Khattri, R.; Hong, D.K.; Zhang, W.; Straus, D.; Samelson, L.E.; Thompson, C.B.; Bluestone, J.A. Molecular basis of T cell inactivation by CTLA-4. Science 1998, 282, 2263–2266. [Google Scholar]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1777. [Google Scholar] [CrossRef] [PubMed]
- Tivol, E.A.; Borriello, F.; Nicola Schweitzer, A.; Lynch, W.P.; Bluestone, J.A.; Sharpe, A.H. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 1995, 3, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Emens, L.A.; Adams, S.; Barrios, C.H.; Diéras, V.; Iwata, H.; Loi, S.; Rugo, H.S.; Schneeweiss, A.; Winer, E.P.; Patel, S.; et al. First-line atezolizumab plus nab-paclitaxel for unresectable, locally advanced, or metastatic triple-negative breast cancer: IMpassion130 final overall survival analysis. Ann. Oncol. 2021, 32, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.-A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.; Rugo, H.S.; Cescon, D.W.; Im, S.-A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Perez-Garcia, J.; Iwata, H.; et al. Pembrolizumab plus chemotherapy in advanced triple-negative breast cancer. N. Eng. J. Med. 2022, 387, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Ouyang, Q.; Sun, T.; Zhang, Q.; Teng, Y.; Cui, J.; Wang, H.; Yin, Y.; Wang, X.; Zhou, X.; et al. Toripalimab plus nab-paclitaxel in metastatic or recurrent triple-negative breast cancer: A randomized phase 3 trial. Nat. Med. 2024, 30, 249–256. [Google Scholar] [CrossRef]
- Loibl, S.; Untch, M.; Burchardi, N.; Huober, J.; Sinn, B.V.; Blohmer, J.U.; Grischke, E.M.; Furlanetto, J.; Tesch, H.; Hanusch, C.; et al. A randomised phase II study investigating durvalumab in addition to an anthracycline taxane-based neoadjuvant therapy in early triple-negative breast cancer: Clinical results and biomarker analysis of GeparNuevo study. Ann. Oncol. 2019, 30, 1279–1288. [Google Scholar] [CrossRef]
- Sharma, P.; Stecklein, S.R.; Yoder, R.; Staley, J.M.; Schwensen, K.; O’Dea, A.; Nye, L.; Satelli, D.; Crane, G.; Madan, R.; et al. Clinical and biomarker findings of neoadjuvant pembrolizumab and carboplatin plus docetaxel in triple-negative breast cancer. JAMA Oncol. 2023, 10, 227–235. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Zhang, H.; Barrios, C.H.; Saji, S.; Jung, K.H.; Hegg, R.; Koehler, A.; Sohn, J.; Iwata, H.; Telli, M.L.; et al. Neoadjuvant atezolizumab in combination with sequential nab-paclitaxel and anthracycline-based chemotherapy versus placebo and chemotherapy in patients with early-stage triple-negative breast cancer (IMpassion031): A randomised, double-blind, phase 3 trial. Lancet 2020, 396, 1090–1100. [Google Scholar] [CrossRef]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for early triple-negative breast cancer. N. Eng. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Cortes, J.; Dent, R.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; et al. Event-free survival with pembrolizumab in early triple-negative breast cancer. N. Eng. J. Med. 2022, 386, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Wood, S.J.; Gao, Y.; Lee, J.H.; Chen, J.; Wang, Q.; Meisel, J.L.; Li, X. High tumor infiltrating lymphocytes are significantly associated with pathological complete response in triple negative breast cancer treated with neoadjuvant KEYNOTE-522 chemoimmunotherapy. Breast Cancer Res. Treat. 2024, 205, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Eggermont, A.M.M.; Chiarion-Sileni, V.; Grob, J.-J.; Dummer, R.; Wolchok, J.D.; Schmidt, H.; Hamid, O.; Robert, C.; Ascierto, P.A.; Richards, J.M.; et al. Prolonged survival in stage III melanoma with ipilimumab adjuvant therapy. N. Eng. J. Med. 2016, 375, 1845–1855. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; Ascierto, P.A.; Middleton, M.R.; Hennicken, D.; Zoffoli, R.; Pieters, A.; Amadi, A.; Kupas, K.; Kotapati, S.; Moshyk, A.; et al. Indirect treatment comparison of nivolumab versus placebo as adjuvant treatment for resected melanoma. Eur. J. Cancer 2021, 158, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Ericsson, P.I.; Stovgaard, E.S.; Sua, L.F.; Reisenbichler, E.; Kos, Z.; Carter, J.M.; Michiels, S.; Le Quesne, J.; Nielsen, T.O.; Lænkholm, A.V.; et al. The path to a better biomarker: Application of a risk management framework for the implementation of PD-L1 and TILs as immuno-oncology biomarkers in breast cancer clinical trials and daily practice. J. Pathol. 2020, 250, 667–684. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Q.; Fan, J.; Lin, Q.; Liu, X.; Li, J.; Hong, R.; Wang, S. Tumor stromal type is associated with stromal PD-L1 expression and predicts outcomes in breast cancer. PLoS ONE 2019, 14, e0223325. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, F.S.; Gaule, P.; McGuire, J.; Patel, K.; Blenman, K.; Pusztai, L.; Rimm, D.L. PD-L1 protein expression on both tumor cells and macrophages are associated with response to neoadjuvant durvalumab with chemotherapy in triple-negative breast cancer. Clin. Cancer Res. 2020, 26, 5456–5461. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Geng, H.; Liu, Y.; Liu, L.; Chen, Y.; Wu, F.; Liu, Z.; Ling, S.; Wang, Y.; Zhou, L. Hot and cold tumors: Immunological features and the therapeutic strategies. MedComm 2023, 4, e343. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, K.E.; Yost, S.E.; Chang, C.W.; Johnson, R.M.; Carr, A.R.; McAdam, P.R.; Halligan, D.L.; Chang, C.C.; Schmolze, D.; Liang, J.; et al. Comprehensive profiling of poor-risk paired primary and recurrent triple-negative breast cancers reveals immune phenotype shifts. Clin. Cancer Res. 2020, 26, 657–668. [Google Scholar] [CrossRef]
- Iwase, T.; Blenman, K.R.M.; Li, X.; Reisenbichler, E.; Seitz, R.; Hout, D.; Nielsen, T.J.; Schweitzer, B.L.; Bailey, D.B.; Shen, Y.; et al. A novel immunomodulatory 27-gene signature to predict response to neoadjuvant immunochemotherapy for primary triple-negative breast cancer. Cancers 2021, 13, 4839. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef]
- Sreekumar, A.; Bado, I.; He, J.; Zong, C.; Westbrook, T.; Liu, J.; Lo, H.; Mo, Q.; Jebakumar, D.; Kim, I.; et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat. Cell Biol. 2019, 21, 1113–1126. [Google Scholar] [CrossRef]
- Xu, L.; Saunders, K.; Huang, S.-P.; Knutsdottir, H.; Martinez-Algarin, K.; Terrazas, I.; Chen, K.; McArthur, H.M.; Maués, J.; Hodgdon, C.; et al. A comprehensive single-cell breast tumor atlas defines epithelial and immune heterogeneity and interactions predicting anti-PD-1 therapy response. Cell Rep. Med. 2024, 5, 101511. [Google Scholar] [CrossRef] [PubMed]
- Bassez, A.; Vos, H.; Van Dyck, L.; Floris, G.; Arijs, I.; Desmedt, C.; Boeckx, B.; Vanden Bempt, M.; Nevelsteen, I.; Lambein, K.; et al. A single-cell map of intratumoral changes during Anti-PD1 treatment of patients with breast cancer. Nat. Med. 2021, 27, 820–832. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, H.; Mo, H.; Hu, X.; Gao, R.; Zhao, Y.; Liu, B.; Niu, L.; Sun, X.; Yu, X.; et al. Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative breast cancer. Cancer Cell 2021, 39, 1578–1593.e8. [Google Scholar] [CrossRef]
- Karn, T.; Denkert, C.; Weber, K.E.; Holtrich, U.; Hanusch, C.; Sinn, B.V.; Higgs, B.W.; Jank, P.; Sinn, H.P.; Huober, J.; et al. Tumor mutational burden and immune infiltration as independent predictors of response to neoadjuvant immune checkpoint inhibition in early TNBC in GeparNuevo. Ann. Oncol. 2020, 31, 1216–1222. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Amgad, M.; Stovgaard, E.S.; Balslev, E.; Thagaard, J.; Chen, W.; Dudgeon, S.; Sharma, A.; Kerner, J.K.; Denkert, C.; Yuan, Y.; et al. Report on computational assessment of tumor infiltrating lymphocytes from the International Immuno-Oncology Biomarker Working Group. NPJ Breast Cancer 2020, 6, 16. [Google Scholar] [CrossRef]
- Gruosso, T.; Gigoux, M.; Manem, V.S.K.; Bertos, N.; Zuo, D.; Perlitch, I.; Saleh, S.M.I.; Zhao, H.; Souleimanova, M.; Johnson, R.M.; et al. Spatially distinct tumor immune microenvironments stratify triple-negative breast cancers. J. Clin. Investig. 2019, 129, 1785–1800. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Q.; Danenberg, E.; Huang, C.S.; Egle, D.; Callari, M.; Bermejo, B.; Dugo, M.; Zamagni, C.; Thill, M.; Anton, A.; et al. Spatial predictors of immunotherapy response in triple-negative breast cancer. Nature 2023, 621, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K pathway in human disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [PubMed]
- Vasan, N.; Cantley, L.C. At a crossroads: How to translate the roles of PI3K in oncogenic and metabolic signaling into improvements in cancer therapy. Nat. Rev. Clin. Oncol. 2022, 19, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Wiechmann, S.; Ruprecht, B.; Siekmann, T.; Zheng, R.; Frejno, M.; Kunold, E.; Bajaj, T.; Zolg, D.P.; Sieber, S.A.; Gassen, N.C.; et al. Chemical phosphoproteomics sheds new light on the targets and modes of action of AKT inhibitors. ACS Chem. Biol. 2021, 16, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Lin, J.; Wu, W.-I.; Ballard, J.; Lee, B.B.; Gloor, S.L.; Vigers, G.P.; Morales, T.H.; Friedman, L.S.; Skelton, N.; et al. An ATP-site on-off switch that restricts phosphatase accessibility of Akt. Sci. Signal 2012, 5, ra37. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB signaling: Navigating the network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed]
- Vergadi, E.; Ieronymaki, E.; Lyroni, K.; Vaporidi, K.; Tsatsanis, C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J. Immunol. 2017, 198, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Suresh, M. Role of PI3K/Akt signaling in memory CD8 T cell differentiation. Front. Immunol. 2013, 4, 20. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the CBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell 2018, 34, 427–438.e6. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Saéz, O.; Chic, N.; Pascual, T.; Adamo, B.; Vidal, M.; González-Farré, B.; Sanfeliu, E.; Schettini, F.; Conte, B.; Brasó-Maristany, F.; et al. Frequency and spectrum of PIK3CA somatic mutations in breast cancer. Breast Cancer Res. 2020, 22, 45. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN promotes resistance to T cell–mediated immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, N.; Ivan, D.; Trinh, V.A.; Glitza, I.C.; Curry, J.L.; Torres-Cabala, C.; Tetzlaff, M.T.; Bassett, R.L.; Prieto, V.G.; Hwu, W.J. High cytotoxic T-lymphocyte-associated antigen 4 and phospho-Akt expression in tumor samples predicts poor clinical outcomes in ipilimumab-treated melanoma patients. Melanoma Res. 2017, 27, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Turner, N.C.; Barrios, C.H.; Isakoff, S.J.; Kim, S.-B.; Sablin, M.-P.; Saji, S.; Savas, P.; Vidal, G.A.; Oliveira, M.; et al. First-line ipatasertib, atezolizumab, and taxane triplet for metastatic triple-negative breast cancer: Clinical and biomarker results. Clin. Cancer Res. 2023, 30, 767–778. [Google Scholar] [CrossRef] [PubMed]
- André, F.; Ciruelos, E.M.; Juric, D.; Loibl, S.; Campone, M.; Mayer, I.A.; Rubovszky, G.; Yamashita, T.; Kaufman, B.; Lu, Y.S.; et al. Alpelisib plus fulvestrant for PIK3CA-mutated, hormone receptor-positive, human epidermal growth factor receptor-2–negative advanced breast cancer: Final overall survival results from SOLAR-1. Ann. Oncol. 2021, 32, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Hanker, A.B.; Kaklamani, V.; Arteaga, C.L. Challenges for the clinical development of PI3K inhibitors: Strategies to improve their impact in solid tumors. Cancer Discov. 2019, 9, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Buckbinder, L.; Jean, D.J.S.; Tieu, T.; Ladd, B.; Hilbert, B.; Wang, W.; Alltucker, J.T.; Manimala, S.; Kryukov, G.V.; Brooijmans, N.; et al. STX-478, a mutant-selective, allosteric PI3Kα inhibitor spares metabolic dysfunction and improves therapeutic response in PI3Kα-mutant xenografts. Cancer Discov. 2023, 13, 2432–2447. [Google Scholar] [CrossRef]
- Yan, C.; Yang, J.; Saleh, N.; Chen, S.C.; Ayers, G.D.; Abramson, V.G.; Mayer, I.A.; Richmond, A. Inhibition of the Pi3k/Mtor pathway in breast cancer to enhance response to immune checkpoint inhibitors in breast cancer. Int. J. Mol. Sci. 2021, 22, 5207. [Google Scholar] [CrossRef]
- Bergholz, J.S.; Wang, Q.; Wang, Q.; Ramseier, M.; Prakadan, S.; Wang, W.; Fang, R.; Kabraji, S.; Zhou, Q.; Gray, G.K.; et al. PI3Kβ controls immune evasion in PTEN-deficient breast tumors. Nature 2023, 617, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Ganji, G.; Lemech, C.; Burris, H.A.; Han, S.W.; Swales, K.; Decordova, S.; DeYoung, M.P.; Smith, D.A.; Kalyana-Sundaram, S.; et al. A first-time-in-human study of GSK2636771, a phosphoinositide 3 kinase beta-selective inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2017, 23, 5981–5992. [Google Scholar] [CrossRef] [PubMed]
- Ali, K.; Soond, D.R.; Piñeiro, R.; Hagemann, T.; Pearce, W.; Lim, E.L.; Bouabe, H.; Scudamore, C.L.; Hancox, T.; Maecker, H.; et al. Inactivation of PI(3)K P110δ breaks regulatory T-cell-mediated immune tolerance to cancer. Nature 2014, 510, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.L.; Cugliandolo, F.M.; Rosner, D.R.; Gyori, D.; Roychoudhuri, R.; Okkenhaug, K. Phosphoinositide 3-kinase δ inhibition promotes antitumor responses but antagonizes checkpoint inhibitors. JCI Insight 2018, 3, e120626. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, L.; Marchingo, J.M.; Nomura, A.; Damasio, M.P.; Cantrell, D.A. Phosphoinositide 3-kinase P110 delta differentially restrains and directs naïve versus effector CD8+ T cell transcriptional programs. Front. Immunol. 2021, 12, 691997. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kγ 3 is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [PubMed]
- De Henau, O.; Rausch, M.; Winkler, D.; Campesato, L.F.; Liu, C.; Cymerman, D.H.; Budhu, S.; Ghosh, A.; Pink, M.; Tchaicha, J.; et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature 2016, 539, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Sai, J.; Owens, P.; Novitskiy, S.V.; Hawkins, O.E.; Vilgelm, A.E.; Yang, J.; Sobolik, T.; Lavender, N.; Johnson, A.C.; McClain, C.; et al. PI3K inhibition reduces mammary tumor growth and facilitates antitumor immunity and anti-PD1 responses. Clin. Cancer Res. 2017, 23, 3371–3384. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Postow, M.; Chmielowski, B.; Sullivan, R.; Patnaik, A.; Cohen, E.E.W.; Shapiro, G.; Steuer, C.; Gutierrez, M.; Yeckes-Rodin, H.; et al. Eganelisib, a first-in-class PI3Kγ inhibitor, in patients with advanced solid tumors: Results of the phase 1/1b MARIO-1 trial. Clin. Cancer Res. 2023, 29, 2210–2219. [Google Scholar] [CrossRef]
- Hatem, S.; Hargis, J.; Elias, A.; Lee, A.; Swart, R.; Dahkil, S.; Drakaki, A.; Phan, V.; Kass, F.; Cobleigh, M.; et al. Abstract P5-16-02: Updated efficacy, safety and translational data from MARIO-3, a phase II open-label study evaluating a novel triplet combination of eganelisib (IPI-549), atezolizumab (Atezo), and nab-paclitaxel (Nab-Pac) as first-line (1L) therapy for locally advanced or metastatic triple-negative breast cancer (TNBC). Cancer Res. 2022, 82, P5-16-02. [Google Scholar] [CrossRef]
- Garrido-Castro, A.C.; Saura, C.; Barroso-Sousa, R.; Guo, H.; Ciruelos, E.; Bermejo, B.; Gavilá, J.; Serra, V.; Prat, A.; Paré, L.; et al. Phase 2 study of buparlisib (BKM120), a pan-class I PI3K inhibitor, in patients with metastatic triple-negative breast cancer. Breast Cancer Res. 2020, 22, 120. [Google Scholar] [CrossRef] [PubMed]
- Isoyama, S.; Mori, S.; Sugiyama, D.; Kojima, Y.; Tada, Y.; Shitara, K.; Hinohara, K.; Dan, S.; Nishikawa, H. Cancer Immunotherapy with PI3K and PD-1 dual-blockade via optimal modulation of T cell activation signal. J. Immunother. Cancer 2021, 9, e002279. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Huang, X.; Ben, L.T.; Jia, W.; Zhang, S.; Cohen, L.; Huang, S.; Fan, J.; Chen, X.; Liu, S.; et al. A novel pan-PI3K inhibitor KTC1101 synergizes with Anti-PD-1 therapy by targeting tumor suppression and immune activation. Mol. Cancer 2024, 23, 54. [Google Scholar] [CrossRef] [PubMed]
- Davies, B.R.; Greenwood, H.; Dudley, P.; Crafter, C.; Yu, D.H.; Zhang, J.; Li, J.; Gao, B.; Ji, Q.; Maynard, J.; et al. Preclinical pharmacology of AZD5363, an inhibitor of AKT: Pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol. Cancer Ther. 2012, 11, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Saura, C.; Roda, D.; Roselló, S.; Oliveira, M.; Macarulla, T.; Pérez-Fidalgo, J.A.; Morales-Barrera, R.; Sanchis-García, J.M.; Musib, L.; Budha, N.; et al. A first-in-human phase I study of the ATP-competitive AKT inhibitor ipatasertib demonstrates robust and safe targeting of AKT in patients with solid tumors. Cancer Discov. 2017, 7, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Abraham, J.; Chan, S.; Wheatley, D.; Murray Brunt, A.; Nemsadze, G.; Baird, R.D.; Hee Park, Y.; Hall, P.S.; Perren, T.; et al. Capivasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer: The PAKT Trial. J. Clin. Oncol. 2019, 38, 423–433. [Google Scholar]
- Dent, R.; Oliveira, M.; Isakoff, S.J.; Im, S.A.; Espié, M.; Blau, S.; Tan, A.R.; Saura, C.; Wongchenko, M.J.; Xu, N.; et al. Final results of the double-blind placebo-controlled randomized phase 2 LOTUS trial of first-line ipatasertib plus paclitaxel for inoperable locally advanced/metastatic triple-negative breast cancer. Breast Cancer Res. Treat. 2021, 189, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Dent, R.A.; O’Shaughnessy, J.; Kim, S.B.; Isakoff, S.J.; Barrios, C.; Saji, S.; Bondarenko, I.; Nowecki, Z.; Lian, Q.; et al. Ipatasertib plus paclitaxel for PIK3CA/AKT1/PTEN-altered hormone receptor-positive HER2-negative advanced breast cancer: Primary results from Cohort B of the IPATunity130 randomized phase 3 trial. Breast Cancer Res. Treat. 2022, 191, 565–576. [Google Scholar] [CrossRef]
- Wu, W.I.; Voegtli, W.C.; Sturgis, H.L.; Dizon, F.P.; Vigers, G.P.A.; Brandhuber, B.J. Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS ONE 2010, 5, e12913. [Google Scholar] [CrossRef]
- Marks, D.K.; Gartrell, R.D.; El Asmar, M.; Boboila, S.; Hart, T.; Lu, Y.; Pan, Q.; Yu, J.; Hibshoosh, H.; Guo, H.; et al. Akt inhibition is associated with favorable immune profile changes within the tumor microenvironment of hormone receptor positive, HER2 negative breast cancer. Front. Oncol. 2020, 10, 968. [Google Scholar] [CrossRef]
- Wang, H.; Yee, D. I-SPY 2: A neoadjuvant adaptive clinical trial designed to improve outcomes in high-risk breast cancer. Curr. Breast Cancer Rep. 2019, 11, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.M.; Yau, C.; Wulfkuhle, J.; Brown-Swigart, L.; Gallagher, R.I.; Jesus, M.; Magbanua, M.; O’Grady, N.; Hirst, G.; Asare, S.; et al. Mechanism of action biomarkers predicting response to AKT inhibition in the I-SPY 2 breast cancer trial. NPJ Breast Cancer 2020, 6, 48. [Google Scholar] [CrossRef] [PubMed]
- Bullock, K.K.; Shattuck-Brandt, R.; Scalise, C.; Luo, W.; Chen, S.C.; Saleh, N.; Gonzalez-Ericsson, P.I.; Garcia, G.; Sanders, M.E.; Ayers, G.D.; et al. Endogenous PAKT activity is associated with response to AKT inhibition alone and in combination with immune checkpoint inhibition in murine models of TNBC. Cancer Lett. 2024, 586, 216681. [Google Scholar] [CrossRef] [PubMed]
- Savill, K.M.Z.; Lee, B.B.; Oeh, J.; Lin, J.; Lin, E.; Chung, W.J.; Young, A.; Chen, W.; Miś, M.; Mesh, K.; et al. Distinct resistance mechanisms arise to allosteric vs. ATP-competitive AKT inhibitors. Nat. Commun. 2022, 13, 2057. [Google Scholar] [CrossRef]
- Oliveira, M.; Saura, C.; Nuciforo, P.; Calvo, I.; Andersen, J.; Passos-Coelho, J.L.; Gil Gil, M.; Bermejo, B.; Patt, D.A.; Ciruelos, E.; et al. FAIRLANE, a double-blind placebo-controlled randomized phase II trial of neoadjuvant ipatasertib plus paclitaxel for early triple-negative breast cancer. Ann. Oncol. 2019, 30, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Cortes, J.; Robson, M.E.; Iwata, H.; Hegg, R.; Nechaeva, M.; Xu, B.; Verma, S.; Haddad, V.; Rodrigo Imedio, E.; et al. A phase III trial of capivasertib and paclitaxel in first-line treatment of patients with metastatic triple-negative breast cancer (CAPItello290). J. Clin. Oncol. 2020, 38. [Google Scholar] [CrossRef]
- Schmid, P.; Nowecki, Z.; Im, S.-A.; Chung, W.-P.; Lord, S.; Armstrong, A.; Ma, C.X.; Huisden, R.; Stewart, R.; Kumar, R.; et al. Abstract PD10-03: BEGONIA: Phase 1b/2 study of Durvalumab (D) combinations in locally advanced/metastatic triple-negative breast cancer (TNBC): Results from Arm 1 D + Paclitaxel (P), Arm 2 D+P + Capivasertib (C), and Arm 5 D+P + Oleclumab (O). Cancer Res. 2022, 82, PD10-03. [Google Scholar] [CrossRef]
- Schmid, P.; Loirat, D.; Savas, P.; Espinosa, E.; Boni, V.; Italiano, A.; White, S.; Singel, S.M.; Withana, N.; Mani, A.; et al. Abstract CT049: Phase Ib study evaluating a triplet combination of ipatasertib (IPAT), atezolizumab (Atezo), and paclitaxel (PAC) or nab-PAC as first-line (1L) therapy for locally advanced/metastatic triple negative breast cancer (TNBC). Cancer Res. 2019, 79, CT049. [Google Scholar] [CrossRef]
- Gonzalez-Angulo, A.M.; Krop, I.; Akcakanat, A.; Chen, H.; Liu, S.; Li, Y.; Culotta, K.S.; Tarco, E.; Piha-Paul, S.; Moulder-Thompson, S.; et al. SU2C Phase Ib study of paclitaxel and MK-2206 in advanced solid tumors and metastatic breast cancer. J. Natl. Cancer Inst. 2015, 107, dju493. [Google Scholar] [CrossRef]
- Xing, Y.; Lin, N.U.; Maurer, M.A.; Chen, H.; Mahvash, A.; Sahin, A.; Akcakanat, A.; Li, Y.; Abramson, V.; Litton, J.; et al. Phase II trial of AKT inhibitor MK-2206 in patients with advanced breast cancer who have tumors with PIK3CA or AKT mutations, and/or PTEN Loss/PTEN mutation. Breast Cancer Res. 2019, 21, 78. [Google Scholar] [CrossRef]
- Loi, S.; Dushyanthen, S.; Beavis, P.A.; Salgado, R.; Denkert, C.; Savas, P.; Combs, S.; Rimm, D.L.; Giltnane, J.M.; Estrada, M.V.; et al. RAS/MAPK activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: Therapeutic cooperation between MEK and PD-1/PD-L1 immune checkpoint inhibitors. Clin. Cancer Res. 2016, 22, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- Marín-Ramos, N.I.; Ortega-Gutiérrez, S.; López-Rodríguez, M.L. Blocking Ras inhibition as an antitumor strategy. Semin. Cancer Biol. 2018, 54, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-MTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Galiè, M. RAS as supporting actor in breast cancer. Front. Oncol. 2019, 9, 1199. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Liu, Y.; Yang, S.; Wu, X.; Li, H.; Wang, Q. MEK inhibitors for the treatment of non-small cell lung cancer. J. Hematol. Oncol. 2021, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Bartholomeusz, C.; Xie, X.; Pitner, M.K.; Kondo, K.; Dadbin, A.; Lee, J.; Saso, H.; Smith, P.D.; Dalby, K.N.; Ueno, N.T. MEK inhibitor selumetinib (AZD6244; ARRY-142886) prevents lung metastasis in a triple-negative breast cancer xenograft model. Mol. Cancer Ther. 2015, 14, 2773–2781. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, G.I.; Lorusso, P.; Cho, D.C.; Musib, L.; Yan, Y.; Wongchenko, M.; Chang, I.; Patel, P.; Chan, I.T.; Sanabria-Bohorquez, S.; et al. PHASE I STUDIES A Phase Ib open-label dose escalation study of the safety, pharmacokinetics, and pharmacodynamics of cobimetinib (GDC-0973) and ipatasertib (GDC-0068) in patients with locally advanced or metastatic solid tumors. Investig. New Drugs 2021, 47, 163–174. [Google Scholar]
- Ebert, P.J.R.; Cheung, J.; Yang, Y.; McNamara, E.; Hong, R.; Moskalenko, M.; Gould, S.E.; Maecker, H.; Irving, B.A.; Kim, J.M.; et al. MAP kinase inhibition promotes T cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity 2016, 44, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Dushyanthen, S.; Teo, Z.L.; Caramia, F.; Savas, P.; Mintoff, C.P.; Virassamy, B.; Henderson, M.A.; Luen, S.J.; Mansour, M.; Kershaw, M.H.; et al. Agonist immunotherapy restores T cell function following MEK inhibition improving efficacy in breast cancer. Nat. Commun. 2017, 8, 606. [Google Scholar] [CrossRef]
- Dennison, L.; Ruggieri, A.; Mohan, A.; Leatherman, J.; Cruz, K.; Woolman, S.; Azad, N.; Lesinski, G.B.; Jaffee, E.M.; Yarchoan, M. Context-dependent immunomodulatory effects of MEK inhibition are enhanced with T-cell agonist therapy. Cancer Immunol. Res. 2021, 9, 1187–1201. [Google Scholar] [CrossRef]
- Franklin, D.A.; James, J.L.; Axelrod, M.L.; Balko, J.M. MEK inhibition activates STAT signaling to increase breast cancer immunogenicity via MHC-I expression. Cancer Drug Resist. 2020, 3, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Wabitsch, S.; Tandon, M.; Ruf, B.; Zhang, Q.; McCallen, J.D.; McVey, J.C.; Ma, C.; Green, B.L.; Diggs, L.P.; Heinrich, B.; et al. Anti–PD-1 in combination with trametinib suppresses tumor growth and improves survival of intrahepatic cholangiocarcinoma in mice. Cell Mol. Gastroenterol. Hepatol. 2021, 12, 1166–1178. [Google Scholar] [CrossRef]
- Lizotte, P.H.; Hong, R.L.; Luster, T.A.; Cavanaugh, M.E.; Taus, L.J.; Wang, S.; Dhaneshwar, A.; Mayman, N.; Yang, A.; Kulkarni, M.; et al. A high-throughput immune-oncology screen identifies EGFR inhibitors as potent enhancers of antigen-specific cytotoxic T-lymphocyte tumor cell killing. Cancer Immunol. Res. 2018, 6, 1511–1523. [Google Scholar] [CrossRef] [PubMed]
- Mbofung, R.M.; McKenzie, J.A.; Malu, S.; Zhang, M.; Peng, W.; Liu, C.; Kuiatse, I.; Tieu, T.; Williams, L.; Devi, S.; et al. HSP90 inhibition enhances cancer immunotherapy by upregulating interferon response genes. Nat. Commun. 2017, 8, 451. [Google Scholar] [CrossRef] [PubMed]
- Olivo Pimentel, V.; Yaromina, A.; Marcus, D.; Dubois, L.J.; Lambin, P. A novel co-culture assay to assess anti-tumor CD8+ T cell cytotoxicity via luminescence and multicolor flow cytometry. J. Immunol. Methods 2020, 487, 112899. [Google Scholar] [CrossRef]
- Zhou, Z.; Van der Jeught, K.; Fang, Y.; Yu, T.; Li, Y.; Ao, Z.; Liu, S.; Zhang, L.; Yang, Y.; Eyvani, H.; et al. An organoid-based screen for epigenetic inhibitors that stimulate antigen presentation and potentiate T-cell-mediated cytotoxicity. Nat. Biomed. Eng. 2021, 5, 1320–1335. [Google Scholar] [CrossRef]
- Xu, H.; Van der Jeught, K.; Zhou, Z.; Zhang, L.; Yu, T.; Sun, Y.; Li, Y.; Wan, C.; So, K.; Liu, D.; et al. Atractylenolide I enhances responsiveness to immune checkpoint blockade therapy by activating tumor antigen presentation. J. Clin. Investig. 2021, 131, e146832. [Google Scholar] [CrossRef]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody drug conjugate: The “biological missile” for targeted cancer therapy. Signal Transduct. Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef]
- Saini, K.S.; Punie, K.; Twelves, C.; Bortini, S.; de Azambuja, E.; Anderson, S.; Criscitiello, C.; Awada, A.; Loi, S. Antibody-drug conjugates, immune-checkpoint inhibitors, and their combination in breast cancer therapeutics. Expert. Opin. Biol. Ther. 2021, 21, 945–962. [Google Scholar] [CrossRef]
- Schmid, P.; Im, S.-A.; Armstrong, A.; Park, Y.H.; Chung, W.-P.; Nowecki, Z.; Lord, S.; Wysocki, P.J.; Lu, Y.-S.; Dry, H.; et al. BEGONIA: Phase 1b/2 study of Durvalumab (D) combinations in locally advanced/metastatic triple-negative breast cancer (TNBC)—Initial results from Arm 1, D+paclitaxel (P), and Arm 6, D+trastuzumab Deruxtecan (T-DXd). J. Clin. Oncol. 2021, 39. [Google Scholar] [CrossRef]
- Bardia, A.; Hurvitz, S.A.; Tolaney, S.M.; Loirat, D.; Punie, K.; Oliveira, M.; Brufsky, A.; Sardesai, S.D.; Kalinsky, K.; Zelnak, A.B.; et al. Sacituzumab govitecan in metastatic triple-negative breast cancer. N. Eng. J. Med. 2021, 384, 1529–1541. [Google Scholar] [CrossRef]
- Galmarini, D.; Galmarini, C.M.; Galmarini, F.C. Cancer Chemotherapy: A critical analysis of its 60 years of history. Crit. Rev. Oncol. Hematol. 2012, 84, 181–199. [Google Scholar] [CrossRef] [PubMed]
- Schiff, P.B.; Fant, J.; Horwitz, S.B. Promotion of microtubule assembly in vitro by taxol. Nature 1979, 277, 665–667. [Google Scholar] [CrossRef]
- Montecucco, A.; Zanetta, F.; Biamonti, G. Molecular mechanisms of etoposide. EXCLI J. 2015, 14, 95–108. [Google Scholar] [CrossRef]
- Tchounwou, P.B.; Dasari, S.; Noubissi, F.K.; Ray, P.; Kumar, S. Advances in our understanding of the molecular mechanisms of action of cisplatin in cancer therapy. J. Exp. Pharmacol. 2021, 13, 303–328. [Google Scholar] [CrossRef] [PubMed]
- Fucikova, J.; Kepp, O.; Kasikova, L.; Petroni, G.; Yamazaki, T.; Liu, P.; Zhao, L.; Spisek, R.; Kroemer, G.; Galluzzi, L. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 2020, 11, 1013. [Google Scholar] [CrossRef]
- Galluzzi, L.; Humeau, J.; Buqué, A.; Zitvogel, L.; Kroemer, G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2020, 17, 725–741. [Google Scholar] [CrossRef]
- Gradishar, W.J.; Tjulandin, S.; Davidson, N.; Shaw, H.; Desai, N.; Bhar, P.; Hawkins, M.; O’Shaughnessy, J. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J. Clin. Oncol. 2005, 23, 7794–7803. [Google Scholar] [CrossRef]
- Untch, M.; Jackisch, C.; Schneeweiss, A.; Conrad, B.; Aktas, B.; Denkert, C.; Eidtmann, H.; Wiebringhaus, H.; Kümmel, S.; Hilfrich, J.; et al. Nab-paclitaxel versus solvent-based paclitaxel in neoadjuvant chemotherapy for early breast cancer (GeparSepto-GBG 69): A randomised, phase 3 trial. Lancet Oncol. 2016, 17, 345–356. [Google Scholar] [CrossRef]
- Luhn, P.; Chui, S.Y.; Hsieh, A.F.C.; Yi, J.; Mecke, A.; Bajaj, P.S.; Hasnain, W.; Falgas, A.; Ton, T.G.N.; Kurian, A.W. Comparative effectiveness of first-line nab-paclitaxel versus paclitaxel monotherapy in triple-negative breast cancer. J. Comp. Eff. Res. 2019, 8, 1173–1185. [Google Scholar] [CrossRef]
- Jacob, S.L.; Huppert, L.A.; Rugo, H.S. Role of immunotherapy in breast cancer. JCO Oncol. Pract. 2023, 19, 167–179. [Google Scholar]
- Karasarides, M.; Cogdill, A.P.; Robbins, P.B.; Bowden, M.; Burton, E.M.; Butterfield, L.H.; Cesano, A.; Hammer, C.; Haymaker, C.L.; Horak, C.E.; et al. Hallmarks of resistance to immune-checkpoint inhibitors. Cancer Immunol. Res. 2022, 10, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Santa-Maria, C.A.; Kato, T.; Park, J.-H.; Kiyotani, K.; Rademaker, A.; Shah, A.N.; Gross, L.; Blanco, L.Z.; Jain, S.; Flaum, L.; et al. A Pilot study of durvalumab and tremelimumab and immunogenomic dynamics in metastatic breast cancer. Oncotarget 2018, 9, 18985–18996. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; Othus, M.; Patel, S.P.; Miller, K.D.; Chugh, R.; Schuetze, S.M.; Chamberlin, M.D.; Haley, B.J.; Storniolo, A.M.V.; Reddy, M.P.; et al. A multicenter phase II trial of ipilimumab and nivolumab in unresectable or metastatic metaplastic breast cancer: Cohort 36 of Dual Anti–CTLA-4 and Anti–PD-1 Blockade in Rare Tumors (DART, SWOG S1609). Clin. Cancer Res. 2022, 28, 271–278. [Google Scholar] [CrossRef]
- Das, R.; Verma, R.; Sznol, M.; Boddupalli, C.S.; Gettinger, S.N.; Kluger, H.; Callahan, M.; Wolchok, J.D.; Halaban, R.; Dhodapkar, M.V.; et al. Combination therapy with anti–CTLA-4 and anti–PD-1 leads to distinct immunologic changes in vivo. J. Immunol. 2015, 194, 950–959. [Google Scholar] [CrossRef]
- Wei, S.C.; Levine, J.H.; Cogdill, A.P.; Zhao, Y.; Anang, N.A.A.S.; Andrews, M.C.; Sharma, P.; Wang, J.; Wargo, J.A.; Pe’er, D.; et al. Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell 2017, 170, 1120–1133.e17. [Google Scholar] [CrossRef]
- Wei, S.C.; Anang, N.A.A.S.; Sharma, R.; Andrews, M.C.; Reuben, A.; Levine, J.H.; Cogdill, A.P.; Mancuso, J.J.; Wargo, J.A.; Pe’er, D.; et al. Combination anti–CTLA-4 plus anti–PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies. Proc. Natl. Acad. Sci. USA 2019, 116, 22699–22709. [Google Scholar] [CrossRef] [PubMed]
- Dovedi, S.J.; Elder, M.J.; Yang, C.; Sitnikova, S.I.; Irving, L.; Hansen, A.; Hair, J.; Jones, D.C.; Hasani, S.; Wang, B.; et al. Design and efficacy of a monovalent bispecific PD-1/CTLA4 antibody that enhances CTLA4 blockade on PD-1 + activated T cells. Cancer Discov. 2021, 11, 1100–1117. [Google Scholar] [CrossRef]
- Fang, J.; Chen, F.; Liu, D.; Gu, F.; Chen, Z.; Wang, Y. Prognostic value of immune checkpoint molecules in breast cancer. Biosci. Rep. 2020, 40, BSR20201054. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Castillo Gutiérrez, E.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef]
- Frentzas, S.; Kao, S.; Gao, R.; Zheng, H.; Rizwan, A.; Budha, N.; De La Hoz Pedroza, L.; Tan, W.; Meniawy, T. AdvanTIG-105: A phase I dose escalation etudy of the anti-TIGIT monoclonal antibody ociperlimab in combination with tislelizumab in patients with advanced solid tumors. J. Immunother. Cancer 2023, 11, e005829. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Deng, F.; Jia, W. Inhibition of indoleamine 2,3-dioxygenase enhances the therapeutic efficacy of immunogenic chemotherapeutics in breast cancer. J. Breast Cancer 2019, 22, 196–209. [Google Scholar] [CrossRef] [PubMed]
- Le Naour, J.; Galluzzi, L.; Zitvogel, L.; Kroemer, G.; Vacchelli, E. Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology 2020, 9, 1777625. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C.; Malachowski, W.P.; DuHadaway, J.B.; Muller, A.J. Discovery of IDO1 inhibitors: From bench to bedside. Cancer Res. 2017, 77, 6795–6811. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.B.; Wang, G.; Chow, R.D.; Ye, L.; Zhu, L.; Dai, X.; Park, J.J.; Kim, H.R.; Errami, Y.; Guzman, C.D.; et al. Systematic immunotherapy target discovery using genome-scale in vivo CRISPR screens in CD8 T cells. Cell 2019, 178, 1189–1204.e23. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Gong, Y.; Jin, M.; Wu, H.; Guo, L.-W.; Pei, Y.-C.; Chai, W.-J.; Jiang, Y.-Z.; Liu, Y.; Ma, X.-Y.; et al. In vivo multidimensional CRISPR screens identify Lgals2 as an immunotherapy target in triple-negative breast cancer. Sci. Adv. 2022, 8, eabl8247. [Google Scholar] [CrossRef] [PubMed]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; La Fleur, M.W.; Juneja, V.R.; et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017, 547, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Li, B.; Chen, J.; Dang, J.; Chen, S.; Gunes, E.G.; Xu, B.; Tian, L.; Muend, S.; Raoof, M.; et al. Effect of cabazitaxel on macrophages improves CD47-targeted immunotherapy for triple-negative breast cancer. J. Immunother. Cancer 2021, 9, e002022. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, B.; Coussens, L.M. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Abdin, S.M.; Paasch, D.; Lachmann, N. CAR macrophages on a fast track to solid tumor therapy. Nat. Immunol. 2024, 25, 11–12. [Google Scholar] [CrossRef] [PubMed]
- Glass, E.B.; Hoover, A.A.; Bullock, K.K.; Madden, M.Z.; Reinfeld, B.I.; Harris, W.; Parker, D.; Hufnagel, D.H.; Crispens, M.A.; Khabele, D.; et al. Stimulating TAM-mediated anti-tumor immunity with mannose-decorated nanoparticles in ovarian cancer. BMC Cancer 2022, 22, 497. [Google Scholar] [CrossRef] [PubMed]
- Wróblewska, A.; Szczygieł, A.; Szermer-Olearnik, B.; Pajtasz-Piasecka, E. Macrophages as promising carriers for nanoparticle delivery in anticancer therapy. Int. J. Nanomedicine 2023, 18, 4521–4539. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Li, Z.; Wang, Z.; Chen, C.; Luo, Y. Chimeric antigen receptor macrophages activated through TLR4 or IFN-γ receptors suppress breast cancer growth by targeting VEGFR2. Cancer Immunol. Immunother. 2023, 72, 3243–3257. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, L.; Su, H.F.; Liu, Q.; Shen, J.; Dai, H.; Zheng, W.; Lu, Y.; Zhang, W.; Bei, Y.; et al. Chimeric antigen receptor macrophage therapy for breast tumours mediated by targeting the tumour extracellular matrix. Br. J. Cancer 2019, 121, 837–845. [Google Scholar] [CrossRef]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M.; et al. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell 2018, 174, 1293–1308.e36. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bullock, K.K.; Richmond, A. Beyond Anti-PD-1/PD-L1: Improving Immune Checkpoint Inhibitor Responses in Triple-Negative Breast Cancer. Cancers 2024, 16, 2189. https://doi.org/10.3390/cancers16122189
Bullock KK, Richmond A. Beyond Anti-PD-1/PD-L1: Improving Immune Checkpoint Inhibitor Responses in Triple-Negative Breast Cancer. Cancers. 2024; 16(12):2189. https://doi.org/10.3390/cancers16122189
Chicago/Turabian StyleBullock, Kennady K., and Ann Richmond. 2024. "Beyond Anti-PD-1/PD-L1: Improving Immune Checkpoint Inhibitor Responses in Triple-Negative Breast Cancer" Cancers 16, no. 12: 2189. https://doi.org/10.3390/cancers16122189