
Optimizing Ex Vivo CAR-T Cell-Mediated Cytotoxicity Assay through Multimodality Imaging


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture, Lentiviral Transfection, and Single Cell Cloning
2.2. Co-Culture Setup for CAR-T Cytotoxicity Assays
2.3. Next Generation Sequencing and Expression Heat Map Generation
2.4. Luminescence Assay
2.5. Real-Time xCELLigence Live Cell Analysis
2.6. DiO Staining for Live Cell Imaging
2.7. Cytotox Red Dye Staining for Live Cell Imaging
2.8. NK Direct Killing and ADCC Assay
2.9. Flow Cytometry
3. Results
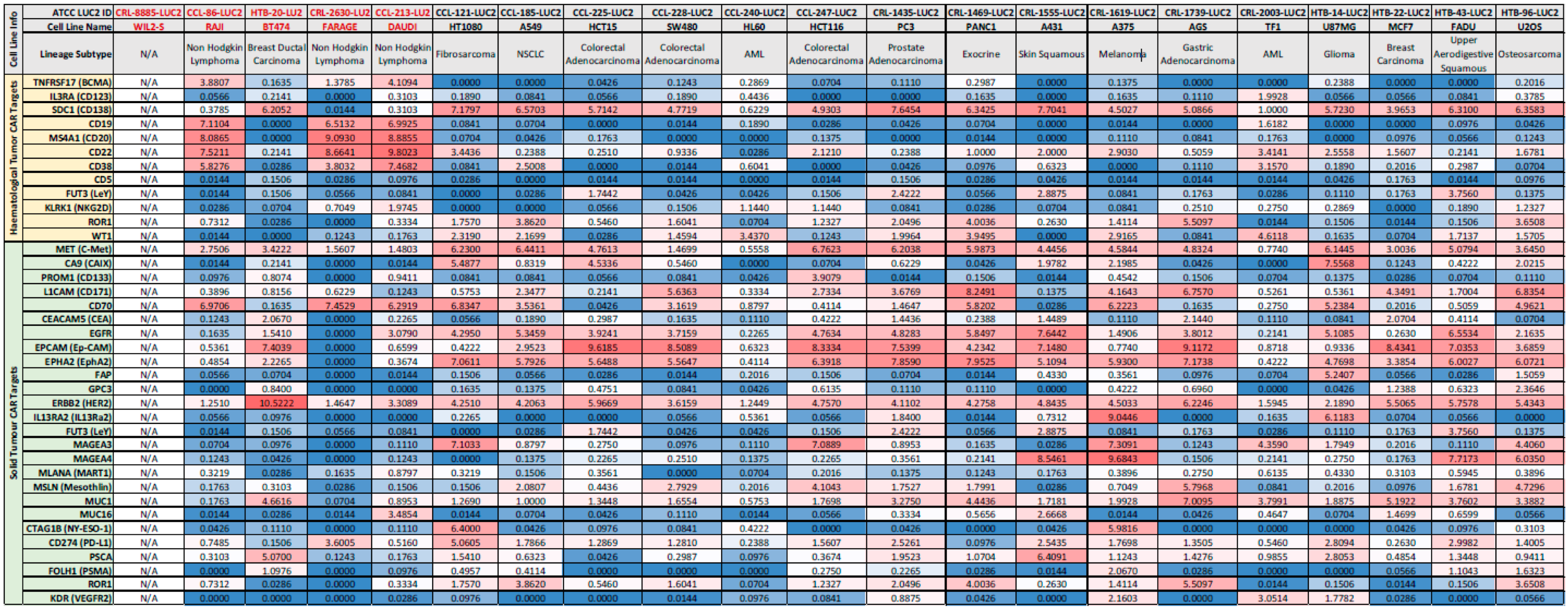
3.1. mRNA Profiling of Cancer Cell Lines for Common CAR-T Targets
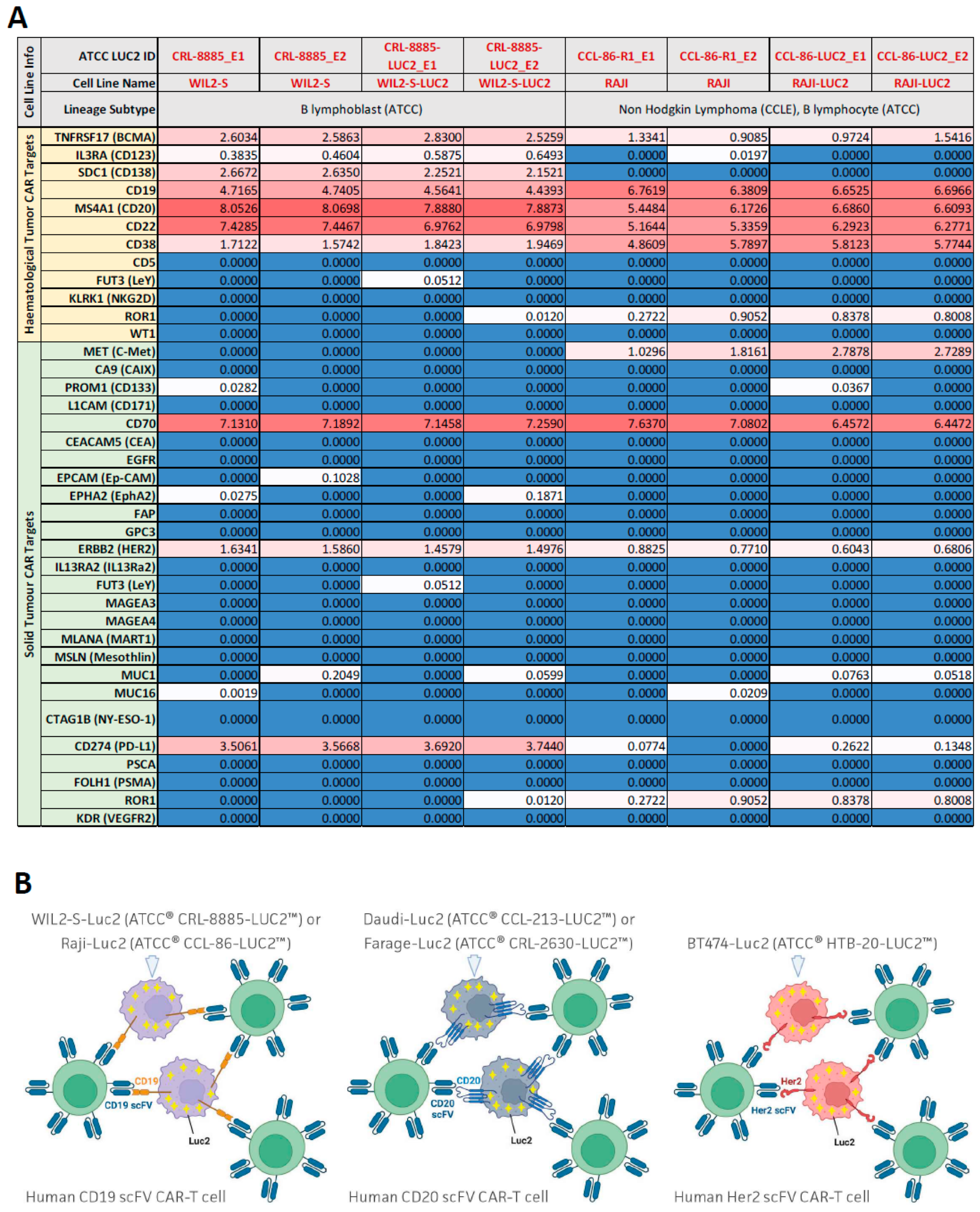
3.2. Optimizing Ex Vivo CD19 CAR-T Cell-Mediated Cytotoxicity Assay Using Luminescence and Live Cell Imaging
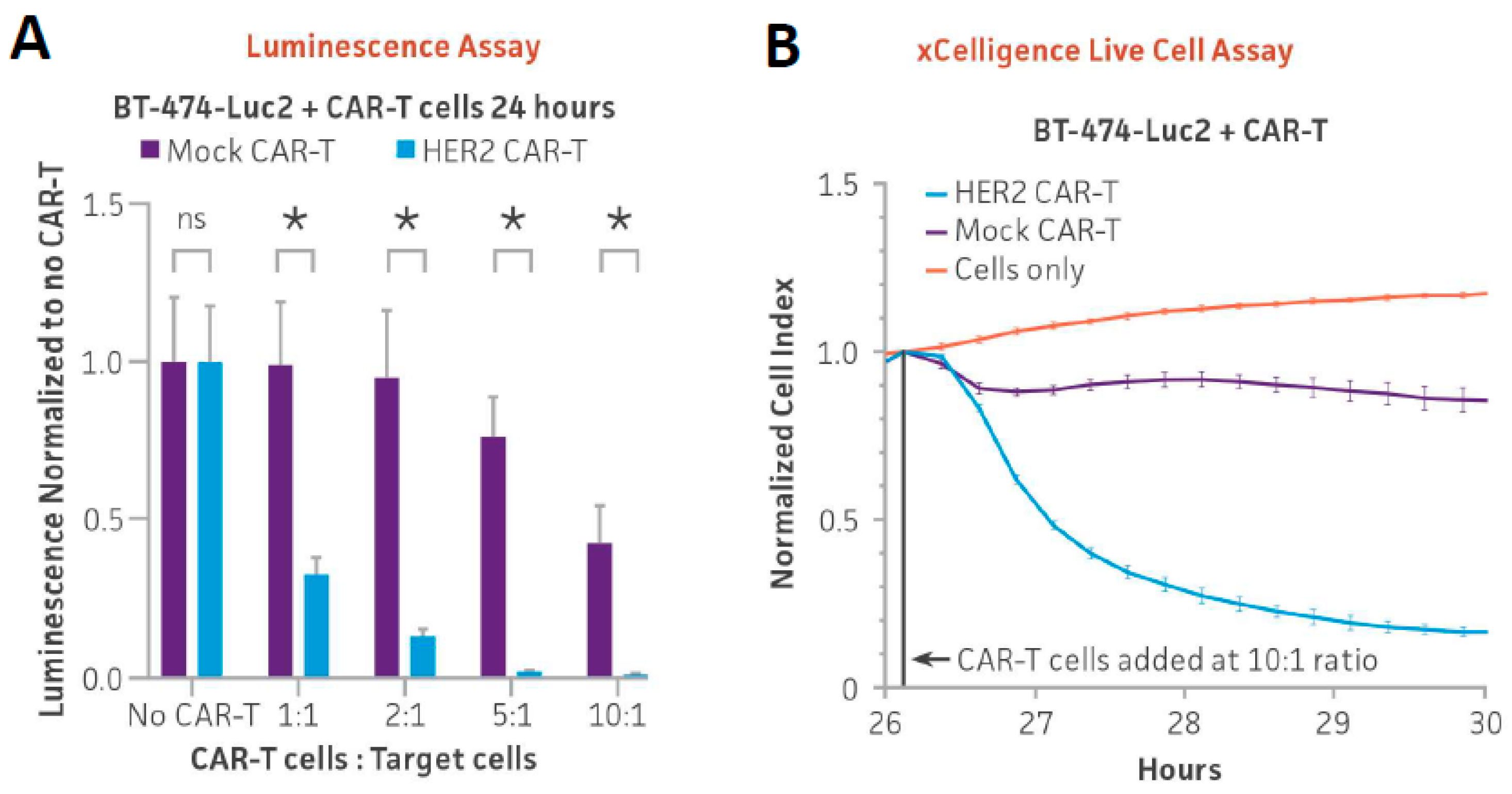
3.3. Optimizing Ex Vivo HER2 CAR-T Cell-Mediated Cytotoxicity Assay Using Luminescence and xCELLigence Live Cell Assay in Adherent Cells
3.4. Using Luminescence as a Readout for Ex Vivo NK Direct Killing and ADCC
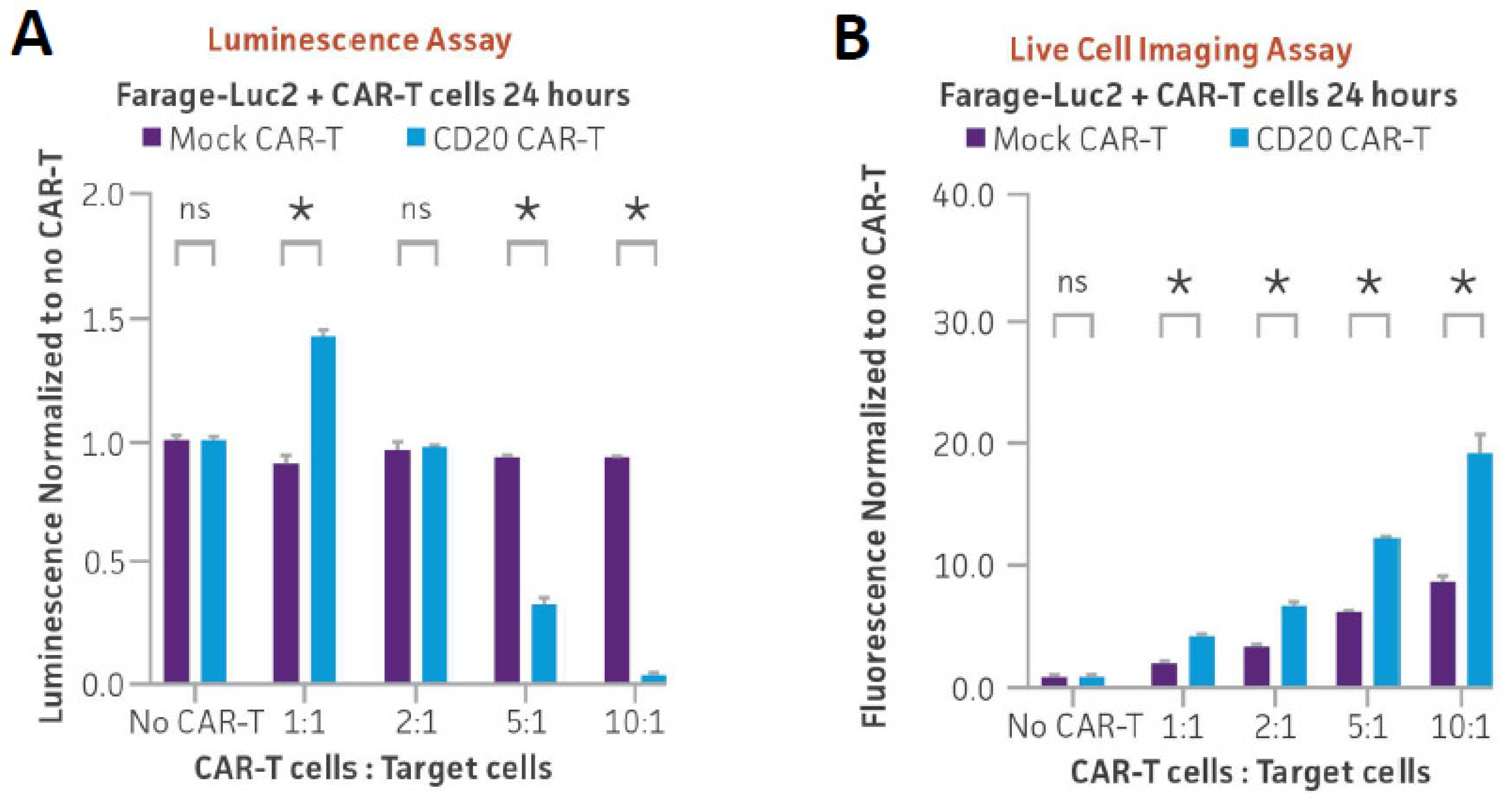
3.5. Optimizing Ex Vivo CD20 CAR-T Cell-Mediated Cytotoxicity Assay Using Luminescence and Cytotox Red Dye-Based Live Cell Imaging
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Sadelain, M.; Brentjens, R.; Riviere, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Wilson, W.H.; Janik, J.E.; Dudley, M.E.; Stetler-Stevenson, M.; Feldman, S.A.; Maric, I.; Raffeld, M.; Nathan, D.A.; Lanier, B.J.; et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010, 116, 4099–4102. [Google Scholar] [CrossRef]
- Garfall, A.L.; Maus, M.V.; Hwang, W.T.; Lacey, S.F.; Mahnke, Y.D.; Melenhorst, J.J.; Zheng, Z.; Vogl, D.T.; Cohen, A.D.; Weiss, B.M.; et al. Chimeric Antigen Receptor T Cells against CD19 for Multiple Myeloma. N. Engl. J. Med. 2015, 373, 1040–1047. [Google Scholar] [CrossRef]
- Sengsayadeth, S.; Savani, B.N.; Oluwole, O.; Dholaria, B. Overview of approved CAR-T therapies, ongoing clinical trials, and its impact on clinical practice. EJHaem 2022, 3, 6–10. [Google Scholar] [CrossRef]
- Jackson, H.J.; Rafiq, S.; Brentjens, R.J. Driving CAR T-cells forward. Nat. Rev. Clin. Oncol. 2016, 13, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Kiesgen, S.; Messinger, J.C.; Chintala, N.K.; Tano, Z.; Adusumilli, P.S. Comparative analysis of assays to measure CAR T-cell-mediated cytotoxicity. Nat. Protoc. 2021, 16, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Brunner, K.T.; Mauel, J.; Cerottini, J.C.; Chapuis, B. Quantitative assay of the lytic action of immune lymphoid cells on 51-Cr-labelled allogeneic target cells in vitro; inhibition by isoantibody and by drugs. Immunology 1968, 14, 181–196. [Google Scholar] [PubMed]
- Erskine, C.L.; Henle, A.M.; Knutson, K.L. Determining optimal cytotoxic activity of human Her2neu specific CD8 T cells by comparing the Cr51 release assay to the xCELLigence system. J. Vis. Exp. 2012, e3683. [Google Scholar] [CrossRef]
- Peper, J.K.; Schuster, H.; Loffler, M.W.; Schmid-Horch, B.; Rammensee, H.G.; Stevanovic, S. An impedance-based cytotoxicity assay for real-time and label-free assessment of T-cell-mediated killing of adherent cells. J. Immunol. Methods 2014, 405, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Riss, T.; Niles, A.; Moravec, R.; Karassina, N.; Vidugiriene, J. Cytotoxicity Assays: In Vitro Methods to Measure Dead Cells. In Assay Guidance Manual; Markossian, S., Grossman, A., Brimacombe, K., Arkin, M., Auld, D., Austin, C., Baell, J., Chung, T.D.Y., Coussens, N.P., Dahlin, J.L., et al., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2019; pp. 379–394. [Google Scholar]
- Karimi, M.A.; Lee, E.; Bachmann, M.H.; Salicioni, A.M.; Behrens, E.M.; Kambayashi, T.; Baldwin, C.L. Measuring cytotoxicity by bioluminescence imaging outperforms the standard chromium-51 release assay. PLoS ONE 2014, 9, e89357. [Google Scholar] [CrossRef] [PubMed]
- Chu, F.; Cao, J.; Neelalpu, S.S. Versatile CAR T-cells for cancer immunotherapy. Contemp. Oncol. 2018, 22, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Cappell, K.M.; Kochenderfer, J.N. Long-term outcomes following CAR T cell therapy: What we know so far. Nat. Rev. Clin. Oncol. 2023, 20, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Budi, H.S.; Ahmad, F.N.; Achmad, H.; Ansari, M.J.; Mikhailova, M.V.; Suksatan, W.; Chupradit, S.; Shomali, N.; Marofi, F. Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor (CAR) for tumor immunotherapy; recent progress. Stem Cell Res. Ther. 2022, 13, 40. [Google Scholar] [CrossRef] [PubMed]
- Gazzano-Santoro, H.; Ralph, P.; Ryskamp, T.C.; Chen, A.B.; Mukku, V.R. A non-radioactive complement-dependent cytotoxicity assay for anti-CD20 monoclonal antibody. J. Immunol. Methods 1997, 202, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Tung, J.K.; Berglund, K.; Gutekunst, C.A.; Hochgeschwender, U.; Gross, R.E. Bioluminescence imaging in live cells and animals. Neurophotonics 2016, 3, 025001. [Google Scholar] [CrossRef] [PubMed]
- Dagar, G.; Gupta, A.; Masoodi, T.; Nisar, S.; Merhi, M.; Hashem, S.; Chauhan, R.; Dagar, M.; Mirza, S.; Bagga, P.; et al. Harnessing the potential of CAR-T cell therapy: Progress, challenges, and future directions in hematological and solid tumor treatments. J. Transl. Med. 2023, 21, 449. [Google Scholar] [CrossRef] [PubMed]
- Lisby, A.N.; Carlson, R.D.; Baybutt, T.R.; Weindorfer, M.; Snook, A.E. Evaluation of CAR-T cell cytotoxicity: Real-time impedance-based analysis. Methods Cell Biol. 2022, 167, 81–98. [Google Scholar] [CrossRef] [PubMed]
- Lo Nigro, C.; Macagno, M.; Sangiolo, D.; Bertolaccini, L.; Aglietta, M.; Merlano, M.C. NK-mediated antibody-dependent cell-mediated cytotoxicity in solid tumors: Biological evidence and clinical perspectives. Ann. Transl. Med. 2019, 7, 105. [Google Scholar] [CrossRef] [PubMed]
- Schwietzer, Y.A.; Susek, K.H.; Chen, Z.; Alici, E.; Wagner, A.K. A tractable microscopy- and flow cytometry-based method to measure natural killer cell-mediated killing and infiltration of tumor spheroids. Methods Cell Biol. 2023, 178, 43–61. [Google Scholar] [CrossRef] [PubMed]
- Matta, H.; Gopalakrishnan, R.; Choi, S.; Prakash, R.; Natarajan, V.; Prins, R.; Gong, S.; Chitnis, S.D.; Kahn, M.; Han, X.; et al. Development and characterization of a novel luciferase based cytotoxicity assay. Sci. Rep. 2018, 8, 199. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, H.; Schäfer, A.; Kiderlen, A.F.; Masihi, K.N.; Burger, R. A highly sensitive cytotoxicity assay based on the release of reporter enzymes, from stably transfected cell lines. J. Immunol. Methods 1997, 204, 89–98. [Google Scholar] [CrossRef]
- Witzel, F.; Fritsche-Guenther, R.; Lehmann, N.; Sieber, A.; Bluthgen, N. Analysis of impedance-based cellular growth assays. Bioinformatics 2015, 31, 2705–2712. [Google Scholar] [CrossRef] [PubMed]
- Giaever, I.; Keese, C.R. A morphological biosensor for mammalian cells. Nature 1993, 366, 591–592. [Google Scholar] [CrossRef] [PubMed]
- Langhans, B.; Ahrendt, M.; Nattermann, J.; Sauerbruch, T.; Spengler, U. Comparative study of NK cell-mediated cytotoxicity using radioactive and flow cytometric cytotoxicity assays. J. Immunol. Methods 2005, 306, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Carannante, V.; Wiklund, M.; Onfelt, B. In vitro models to study natural killer cell dynamics in the tumor microenvironment. Front. Immunol. 2023, 14, 1135148. [Google Scholar] [CrossRef]
- Flores-Montero, J.; Kalina, T.; Corral-Mateos, A.; Sanoja-Flores, L.; Perez-Andres, M.; Martin-Ayuso, M.; Sedek, L.; Rejlova, K.; Mayado, A.; Fernandez, P.; et al. Fluorochrome choices for multi-color flow cytometry. J. Immunol. Methods 2019, 475, 112618. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromium Release | Bioluminescence Imaging | Impedance | Flow Cytometry | Fluorescence Imaging–Cell Labeling | Fluorescence Imaging–Cytotox Dye in Media | |
|---|---|---|---|---|---|---|
| Principal measure of cytotoxicity | 51Cr release | Luciferase activity | Cell detachment | Live/dead staining phenotype | Decrease in fluorescent signal | Dye infiltration into dead cell |
| Radioactive materials needed | Yes | No | No | No | No | No |
| Target cell labeling required | Yes | No | No | Yes | Yes | No |
| Genetic modification of target cells | No | Yes (reporter gene) | No | No | No | No |
| Endpoint/kinetic | Endpoint | Temporal & Spatial | Temporal | Endpoint | Temporal & Spatial | Temporal & Spatial |
| Real-time measurement | No | Yes | Yes | No | Yes | Yes |
| Maximum time point measured | 18–24 h | Days | Days | Days | Days | Days |
| Ability to measure different cytotoxicity heterogenous targets | No | No | No | Yes | Yes | Yes |
| Throughput and automatability | Low | High | High | High | High | High |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Foulke, J.G.; Chen, L.; Chang, H.; McManus, C.E.; Tian, F.; Gu, Z. Optimizing Ex Vivo CAR-T Cell-Mediated Cytotoxicity Assay through Multimodality Imaging. Cancers 2024, 16, 2497. https://doi.org/10.3390/cancers16142497
Foulke JG, Chen L, Chang H, McManus CE, Tian F, Gu Z. Optimizing Ex Vivo CAR-T Cell-Mediated Cytotoxicity Assay through Multimodality Imaging. Cancers. 2024; 16(14):2497. https://doi.org/10.3390/cancers16142497
Chicago/Turabian StyleFoulke, John G., Luping Chen, Hyeyoun Chang, Catherine E. McManus, Fang Tian, and Zhizhan Gu. 2024. "Optimizing Ex Vivo CAR-T Cell-Mediated Cytotoxicity Assay through Multimodality Imaging" Cancers 16, no. 14: 2497. https://doi.org/10.3390/cancers16142497