

Impact of Genomic Mutation on Melanoma Immune Microenvironment and IFN-1 Pathway-Driven Therapeutic Responses

 and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Database Analysis
2.3. Cell Treatments
2.3.1. Photodynamic Treatment:
2.3.2. Chemotherapeutic Treatments:
2.3.3. H151 (STING Inhibitor) Treatment and PDT:
2.4. Cell Viability Assay
2.5. Generation of IFN-1 Pathway Reporter Human Melanoma Cell Line
2.6. Flow Cytometry
2.7. Statistical Analysis
3. Results
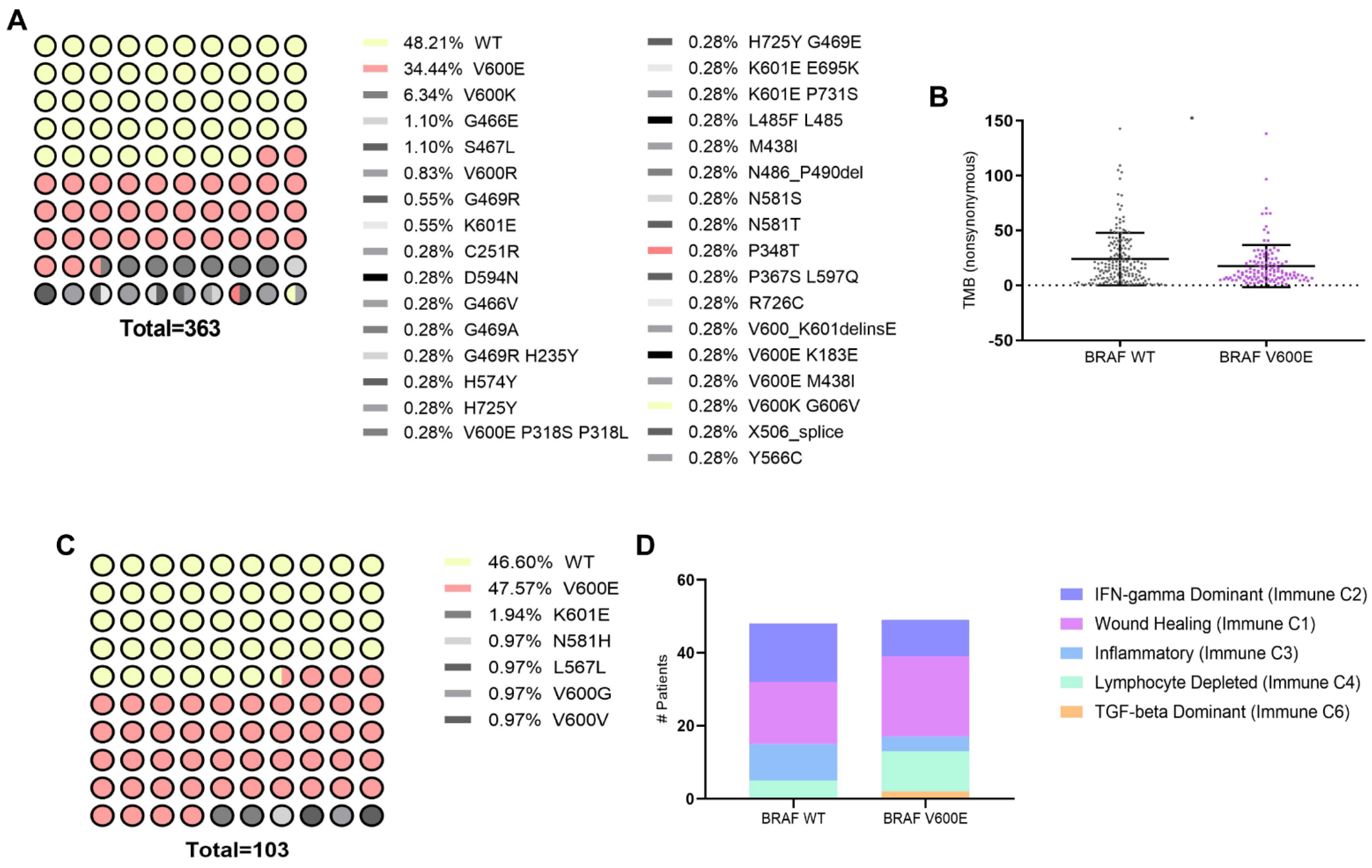
3.1. Association of BRAFV600E Mutation with Genomic and Immune Landscape Alterations in Melanoma Patients
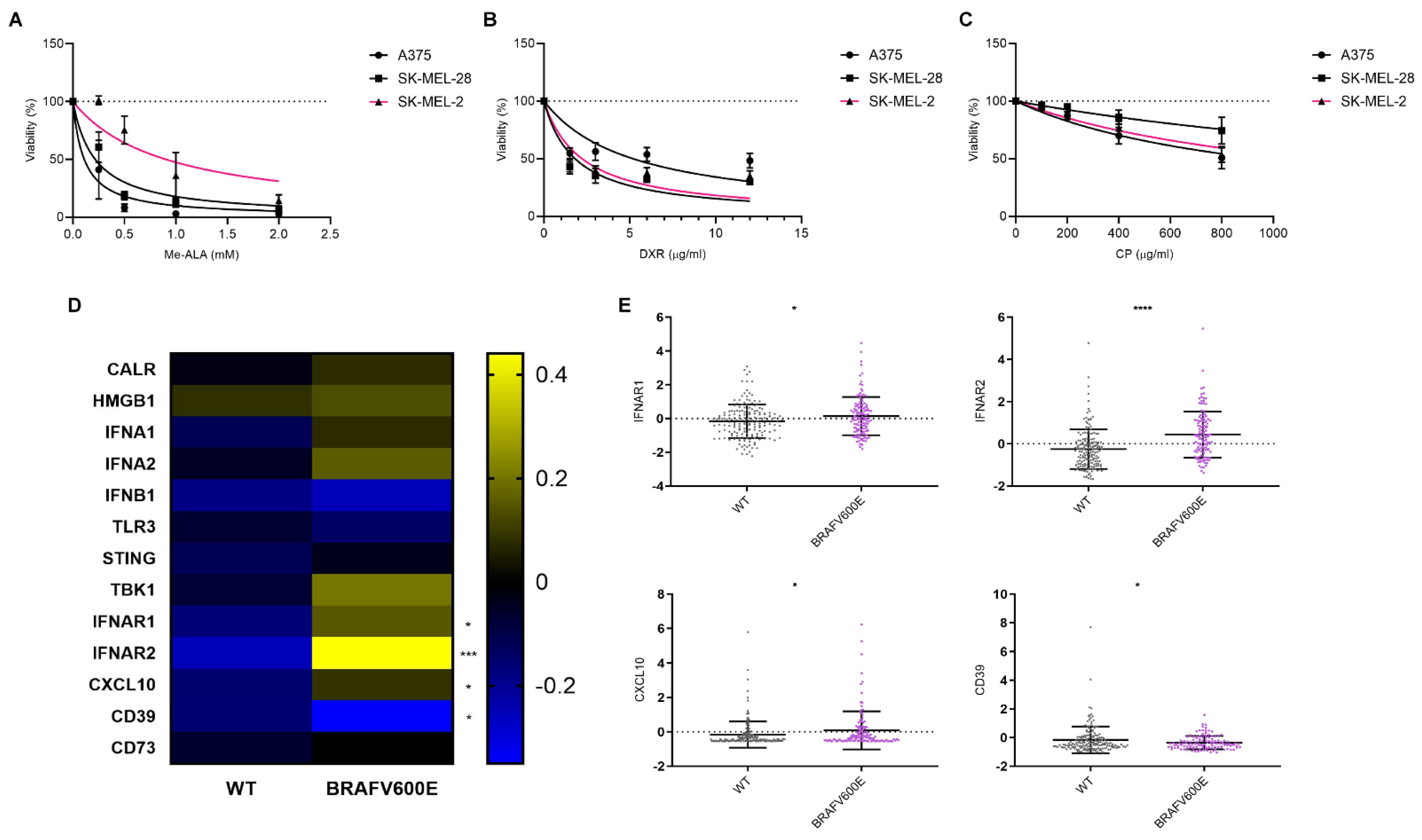
3.2. Sensitivity of Cell Lines Harboring Different Genomic Profiles to ICD and Non-ICD Inducer and Their Impact on ICD-Associated Gene Expression Profiles in Melanoma
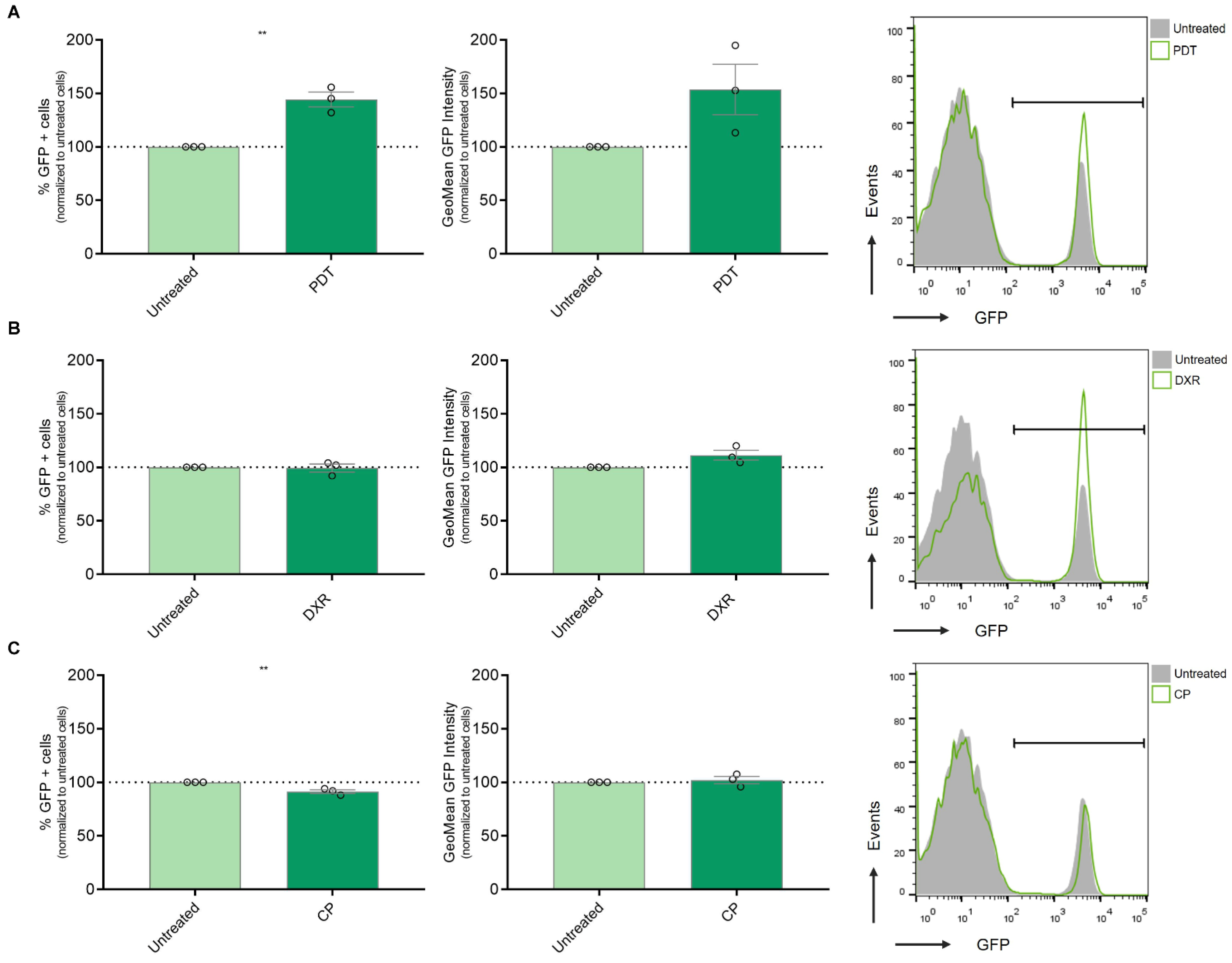
3.3. Modulation of IFN-1 Pathway Activity in Human Melanoma Cells by PDT-Induced ICD
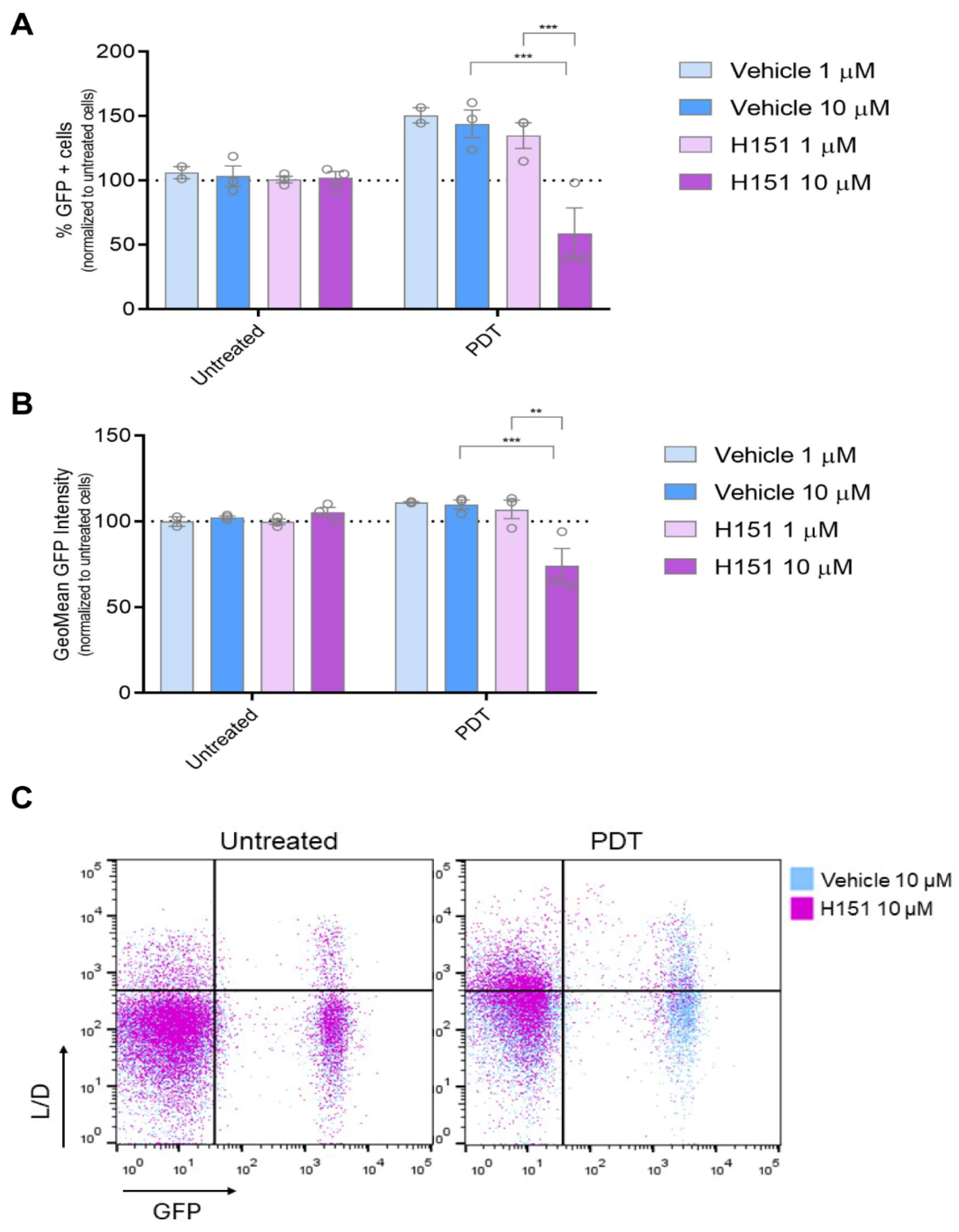
3.4. Inhibition of cGAS-STING Signaling Reverses PDT-Induced Upregulation of IFN-1 Pathway Activity Le Nano
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Long, G.V.; Swetter, S.M.; Menzies, A.M.; Gershenwald, J.E.; Scolyer, R.A. Cutaneous Melanoma. Lancet 2023, 402, 485–502. [Google Scholar] [CrossRef]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Palmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N. The Role of BRAF V600 Mutation in Melanoma. J. Transl. Med. 2012, 10, 85. [Google Scholar] [CrossRef]
- Mahumud, R.A.; Shahjalal, M. The Emerging Burden of Genetic Instability and Mutation in Melanoma: Role of Molecular Mechanisms. Cancers 2022, 14, 6202. [Google Scholar] [CrossRef]
- Kazandjian, S.; Rousselle, E.; Dankner, M.; Cescon, D.W.; Spreafico, A.; Ma, K.; Kavan, P.; Batist, G.; Rose, A.A.N. The Clinical, Genomic, and Transcriptomic Landscape of BRAF Mutant Cancers. Cancers 2024, 16, 445. [Google Scholar] [CrossRef]
- An, Q.; Liu, Z. Comparative Efficacy and Safety of Combination Therapies for Advanced Melanoma: A Network Meta-Analysis. BMC Cancer 2019, 19, 43. [Google Scholar] [CrossRef]
- Tong, J.; Kartolo, A.; Yeung, C.; Hopman, W.; Baetz, T. Long-Term Toxicities of Immune Checkpoint Inhibitor (ICI) in Melanoma Patients. Curr. Oncol. 2022, 29, 7953–7963. [Google Scholar] [CrossRef]
- Mahmoud, F.; Shields, B.; Makhoul, I.; Avaritt, N.; Wong, H.K.; Hutchins, L.F.; Shalin, S.; Tackett, A.J. Immune Surveillance in Melanoma: From Immune Attack to Melanoma Escape and Even Counterattack. Cancer. Biol. Ther. 2017, 18, 451–469. [Google Scholar] [CrossRef]
- Lee, J.H.; Shklovskaya, E.; Lim, S.Y.; Carlino, M.S.; Menzies, A.M.; Stewart, A.; Pedersen, B.; Irvine, M.; Alavi, S.; Yang, J.Y.H.; et al. Transcriptional Downregulation of MHC Class I and Melanoma De- Differentiation in Resistance to PD-1 Inhibition. Nat. Commun. 2020, 11, 1897. [Google Scholar] [CrossRef]
- Aiba, S.; Fujimura, T. Significance of Immunosuppressive Cells as a Target for Immunotherapies in Melanoma and Non-Melanoma Skin Cancers. Biomolecules 2020, 10, 1087. [Google Scholar] [CrossRef]
- Failmezger, H.; Muralidhar, S.; Rullan, A.; de Andrea, C.E.; Sahai, E.; Yuan, Y. Topological Tumor Graphs: A Graph-Based Spatial Model to Infer Stromal Recruitment for Immunosuppression in Melanoma Histology. Cancer Res. 2020, 256, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Agostinis, P.; Vitale, I.; Warren, S.; Adjemian, S.; Coukos, G.; Martinez, A.B.; Chan, T.A.; Edelson, R.L.; Demaria, S.; et al. Consensus Guidelines for the Definition, Detection and Interpretation of Immunogenic Cell Death. J. Immunother. Cancer 2020, 8, e000337. [Google Scholar] [CrossRef] [PubMed]
- Lamberti, M.; Mentucci, F.; Roselli, E.; Araya, P.; Rivarola, V.; Rumie Vittar, N.; Maccioni, M. Photodynamic Modulation of Type 1 Interferon Pathway on Melanoma Cells Promotes Dendritic Cell Activation. Front. Immunol. 2019, 10, 2614. [Google Scholar] [CrossRef] [PubMed]
- Borden, E.C. Interferons α and β in Cancer: Therapeutic Opportunities from New Insights. Nat. Rev. Drug Discov. 2019, 18, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Cheon, H.J.; Wang, Y.; Wightman, S.M.; Jackson, M.W.; Stark, G.R. How Cancer Cells Make and Respond to Interferon-I. Trends Cancer 2023, 9, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and Interpreting Cancer Genomics Data via the Xena Platform. Nat. Biotechnol. 2020, 38, 812–830. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [PubMed]
- Bürgi, M.; Prieto, C.; Oggero, M.; Bollati-Fogolín, M.; Etcheverrigaray, M.; Kratje, R. New Reporter Cell Clones to Determine the Biological Activity of Human Type I Interferons. BMC Proc. 2011, 5, 4. [Google Scholar] [CrossRef]
- Davis, E.J.; Johnson, D.B.; Sosman, J.A.; Chandra, S. Melanoma: What Do All the Mutations Mean? Cancer 2018, 124, 3490–3499. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 Human Cancer Genomes Reveals the Landscape of Tumor Mutational Burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Couselo, E.; Adelantado, E.Z.; Ortiz, C.; García, J.S.; Perez-Garcia, J. NRAS-Mutant Melanoma: Current Challenges and Future Prospect. Onco Targets Ther. 2017, 10, 3941–3947. [Google Scholar] [CrossRef] [PubMed]
- Acquaviva, J.; Smith, D.L.; Jimenez, J.P.; Zhang, C.; Sequeira, M.; He, S.; Sang, J.; Bates, R.C.; Proia, D.A. Overcoming Acquired BRAF Inhibitor Resistance in Melanoma via Targeted Inhibition of Hsp90 with Ganetespib. Mol. Cancer Ther. 2014, 13, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Egger, M.E.; McMasters, K.M.; Hao, H. Differential Expression of ABCB5 in BRAF Inhibitor-Resistant Melanoma Cell Lines. BMC Cancer 2018, 18, 675. [Google Scholar] [CrossRef] [PubMed]
- Morales, D.; Vigneron, P.; Ferreira, I.; Hamitou, W.; Magnano, M.; Mahenthiran, L.; Lok, C.; Vayssade, M. Fibroblasts Influence Metastatic Melanoma Cell Sensitivity to Combined BRAF and MEK Inhibition. Cancers 2021, 13, 4761. [Google Scholar] [CrossRef] [PubMed]
- Riverso, M.; Montagnani, V.; Stecca, B. KLF4 Is Regulated by RAS/RAF/MEK/ERK Signaling through E2F1 and Promotes Melanoma Cell Growth. Oncogene 2017, 36, 3322–3333. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; More, S.; Rufo, N.; Mece, O.; Sassano, M.L.; Agostinis, P.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial Watch: Immunogenic Cell Death Induction by Anticancer Chemotherapeutics. Oncoimmunology 2017, 6, e1386829. [Google Scholar] [CrossRef] [PubMed]
- Lamberti, M.J.; Montico, B.; Ravo, M.; Nigro, A.; Giurato, G.; Iorio, R.; Tarallo, R.; Weisz, A.; Stellato, C.; Steffan, A.; et al. Integration of MiRNA:MRNA Co-Expression Revealed Crucial Mechanisms Modulated in Immunogenic Cancer Cell Death. Biomedicines 2022, 10, 1896. [Google Scholar] [CrossRef] [PubMed]
- Casares, N.; Pequignot, M.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-Dependent Immunogenicity of Doxorubicin-Induced Tumor Cell Death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef]
- Chen, S.; Li, D.; Du, X.; He, X.; Huang, M.; Wang, Y.; Yang, X.; Wang, J. Carrier-Free Nanoassembly of Doxorubicin Prodrug and SiRNA for Combinationally Inducing Immunogenic Cell Death and Reversing Immunosuppression. Nano Today 2020, 35, 100924. [Google Scholar] [CrossRef]
- Martins, I.; Kepp, O.; Schlemmer, F.; Adjemian, S.; Tailler, M.; Shen, S.; Michaud, M.; Menger, L.; Gdoura, A.; Tajeddine, N.; et al. Restoration of the Immunogenicity of Cisplatin-Induced Cancer Cell Death by Endoplasmic Reticulum Stress. Oncogene 2011, 30, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Terenzi, A.; Pirker, C.; Keppler, B.K.; Berger, W. Anticancer Metal Drugs and Immunogenic Cell Death. J. Inorg. Biochem. 2016, 165, 71–79. [Google Scholar] [CrossRef]
- da Silva, D.B.; da Silva, C.L.; Davanzo, N.N.; da Silva Souza, R.; Correa, R.J.; Tedesco, A.C.; Riemma Pierre, M.B. Protoporphyrin IX (PpIX) Loaded PLGA Nanoparticles for Topical Photodynamic Therapy of Melanoma Cells. Photodiagnosis Photodyn. Ther. 2021, 35, 102317. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I Interferons in Anticancer Immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.S.; Chiang, S.F.; Chen, C.Y.; Hong, W.Z.; Chen, T.W.; Chen, W.T.L.; Ke, T.W.; Yang, P.C.; Liang, J.A.; Shiau, A.C.; et al. Targeting CD73 Increases Therapeutic Response to Immunogenic Chemotherapy by Promoting Dendritic Cell Maturation. Cancer Immunol. Immunother. 2023, 72, 2283–2297. [Google Scholar] [CrossRef]
- Vacchelli, E.; Sistigu, A.; Yamazaki, T.; Vitale, I.; Zitvogel, L.; Kroemer, G. Autocrine Signaling of Type 1 Interferons in Successful Anticancer Chemotherapy. Oncoimmunology 2015, 4, e988042. [Google Scholar]
- Obeid, M.; Panaretakis, T.; Joza, N.; Tufi, R.; Tesniere, A.; van Endert, P.; Zitvogel, L.; Kroemer, G. Calreticulin Exposure Is Required for the Immunogenicity of Gamma-Irradiation and UVC Light-Induced Apoptosis. Cell Death Differ. 2007, 14, 1848–1850. [Google Scholar] [CrossRef]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.; Remédios, C.; et al. Cancer Cell-Autonomous Contribution of Type I Interferon Signaling to the Efficacy of Chemotherapy. Nat. Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef]
- Liu, Y.; Crowe, W.N.; Wang, L.; Lu, Y.; Petty, W.J.; Habib, A.A.; Zhao, D. An Inhalable Nanoparticulate STING Agonist Synergizes with Radiotherapy to Confer Long-Term Control of Lung Metastases. Nat. Commun 2019, 10, 5180. [Google Scholar] [CrossRef]
- Dasari, S.; Bernard Tchounwou, P. Cisplatin in Cancer Therapy: Molecular Mechanisms of Action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Li, T.; Cheng, H.; Yuan, H.; Xu, Q.; Shu, C.; Zhang, Y.; Xu, P.; Tan, J.; Rui, Y.; Li, P.; et al. Antitumor Activity of CGAMP via Stimulation of CGAS-CGAMP-STING-IRF3 Mediated Innate Immune Response. Sci. Rep. 2016, 6, 19049. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Liu, Z.; Zhang, X.; Han, C.; Samandi, L.Z.; Dong, C.; Sumer, B.D.; Lea, J.; Fu, Y.X.; Gao, J. Synergistic STING Activation by PC7A Nanovaccine and Ionizing Radiation Improves Cancer Immunotherapy. J. Control. Release 2019, 300, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Luo, K.; Li, N.; Ye, W.; Gao, H.; Luo, X.; Cheng, B. Activation of Stimulation of Interferon Genes (STING) Signal and Cancer Immunotherapy. Molecules 2022, 27, 4638. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liang, Z.; Guo, Y.; de Dieu Habimana, J.; Ren, Y.; Amissah, O.B.; Mukama, O.; Peng, S.; Ding, X.; Lv, L.; et al. STING Agonist DiABZI Enhances the Cytotoxicity of T Cell towards Cancer Cells. Cell Death Dis. 2024, 15, 265. [Google Scholar] [CrossRef] [PubMed]
- Sumner, R.P.; Blest, H.; Lin, M.; Maluquer de Motes, C.; Towers, G.J. HIV-1 with Gag Processing Defects Activates CGAS Sensing. Retrovirology 2024, 21, 10. [Google Scholar] [CrossRef] [PubMed]
- Bankell, E.; Liu, L.; van der Horst, J.; Rippe, C.; Jepps, T.A.; Nilsson, B.O.; Swärd, K. Suppression of Smooth Muscle Cell Inflammation by Myocardin-Related Transcription Factors Involves Inactivation of TANK-Binding Kinase 1. Sci. Rep. 2024, 14, 13321. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.C.; Zappasodi, R. A Decade of Checkpoint Blockade Immunotherapy in Melanoma: Understanding the Molecular Basis for Immune Sensitivity and Resistance. Nat. Immunol. 2022, 23, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Hauschild, A.; Larkin, J.; Ribas, A.; Dréno, B.; Flaherty, K.T.; Ascierto, P.A.; Lewis, K.D.; McKenna, E.; Zhu, Q.; Mun, Y.; et al. Modeled Prognostic Subgroups for Survival and Treatment Outcomes in BRAF V600-Mutated Metastatic Melanoma: Pooled Analysis of 4 Randomized Clinical Trials. JAMA Oncol. 2018, 4, 1382–1388. [Google Scholar] [CrossRef] [PubMed]
- Giugliano, F.; Crimini, E.; Tarantino, P.; Zagami, P.; Uliano, J.; Corti, C.; Trapani, D.; Curigliano, G.; Ascierto, P.A. First Line Treatment of BRAF Mutated Advanced Melanoma: Does One Size Fit All? Cancer Treat. Rev. 2021, 99, 102253. [Google Scholar] [CrossRef]
- Ziogas, D.C.; Theocharopoulos, C.; Lialios, P.P.; Foteinou, D.; Koumprentziotis, I.A.; Xynos, G.; Gogas, H. Beyond CTLA-4 and PD-1 Inhibition: Novel Immune Checkpoint Molecules for Melanoma Treatment. Cancers 2023, 15, 2718. [Google Scholar] [CrossRef]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martín-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949.e16. [Google Scholar] [CrossRef] [PubMed]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational Landscape Determines Sensitivity to PD-1 Blockade in Non–Small Cell Lung Cancer. Science (1979) 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Offin, M.; Rizvi, H.; Tenet, M.; Ni, A.; Sanchez-Vega, F.; Li, B.T.; Drilon, A.; Kris, M.G.; Rudin, C.M.; Schultz, N.; et al. Tumor Mutation Burden and Efficacy of EGFR-Tyrosine Kinase Inhibitors in Patients with EGFR-Mutant Lung Cancers. Clin. Cancer Res. 2019, 25, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.T.; Wu, Y.L. Combination Chemotherapy Alone Should Be Used in the Treatment of Patients With Stage IV EGFR-Mutant NSCLC Whose Disease Has Progressed on All Available Tyrosine Kinase Inhibitors. J. Thorac. Oncol. 2021, 16, 1627–1631. [Google Scholar] [CrossRef] [PubMed]
- Eckardt, J.; Schroeder, C.; Martus, P.; Armeanu-Ebinger, S.; Kelemen, O.; Gschwind, A.; Bonzheim, I.; Eigentler, T.; Amaral, T.; Ossowski, S.; et al. TMB and BRAF Mutation Status Are Independent Predictive Factors in High-Risk Melanoma Patients with Adjuvant Anti-PD-1 Therapy. J. Cancer Res. Clin. Oncol. 2023, 149, 833–840. [Google Scholar] [CrossRef]
- Patel, R.R.; Ramkissoon, S.H.; Ross, J.; Weintraub, L. Tumor Mutational Burden and Driver Mutations: Characterizing the Genomic Landscape of Pediatric Brain Tumors. Pediatr. Blood Cancer 2020, 67, e28338. [Google Scholar] [CrossRef] [PubMed]
- Ilieva, K.M.; Correa, I.; Josephs, D.H.; Karagiannis, P.; Egbuniwe, I.U.; Cafferkey, M.J.; Spicer, J.F.; Harries, M.; Nestle, F.O.; Lacy, K.E.; et al. Effects of BRAF Mutations and BRAF Inhibition on Immune Responses to Melanoma. Mol. Cancer Ther. 2014, 13, 2769–2783. [Google Scholar] [CrossRef] [PubMed]
- Lamberti, M.; Nigro, A.; Mentucci, F.; Rumie Vittar, N.; Casolaro, V.; Dal Col, J. Dendritic Cells and Immunogenic Cancer Cell Death: A Combination for Improving Antitumor Immunity. Pharmaceutics 2020, 3, 256. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Hett, E.; Kroemer, G.; Marincola, F.M. Immunogenic Cell Death in Cancer: Concept and Therapeutic Implications. J. Transl. Med. 2023, 21, 162. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic Cell Death in Cancer and Infectious Disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Z.; Zhao, X.; Xiao, J.; Bi, J.; Li, X.Y.; Chen, G.; Lu, L. Review Immune Response of Targeting CD39 in Cancer. Biomark. Res. 2023, 11, 63. [Google Scholar] [CrossRef]
- Pinto, S.; Martínez-Romero, A.; O’Connor, J.E.; Gil-Benso, R.; San-Miguel, T.; Terrádez, L.; Monteagudo, C.; Callaghan, R.C. Intracellular Coexpression of CXC- and CC- Chemokine Receptors and Their Ligands in Human Melanoma Cell Lines and Dynamic Variations after Xenotransplantation. BMC Cancer 2014, 14, 118. [Google Scholar] [CrossRef]
- Gatti, G.; Nuñez, N.; Nocera, D.; Dejager, L.; Libert, C.; Giraudo, C.; Maccioni, M. Direct Effect of DsRNA Mimetics on Cancer Cells Induces Endogenous IFN-β Production Capable of Improving Dendritic Cell Function. Eur. J. Immunol. 2013, 43, 1849–1861. [Google Scholar] [CrossRef]
- Núñez, N.; Andreani, V.; Crespo, M.; Nocera, D.; Breser, M.; Morón, G.; Dejager, L.; Libert, C.; Rivero, V.; Maccioni, M. IFNβ Produced by TLR4-Activated Tumor Cells Is Involved in Improving the Antitumoral Immune Response. Cancer Res. 2012, 72, 592–603. [Google Scholar] [CrossRef]
- Nocera, D.; Roselli, E.; Araya, P.; Nuñez, N.; Lienenklaus, S.; Jablonska, J.; Weiss, S.; Gatti, G.; Brinkmann, M.; Kröger, A.; et al. In Vivo Visualizing the IFN-β Response Required for Tumor Growth Control in a Therapeutic Model of Polyadenylic-Polyuridylic Acid Administration. J. Immunol. 2016, 196, 2860–2869. [Google Scholar] [CrossRef]
- Ernst, M.; Giubellino, A. The Current State of Treatment and Future Directions in Cutaneous Malignant Melanoma. Biomedicines 2022, 10, 822. [Google Scholar] [CrossRef]
- Yatim, N.; Cullen, S.; Albert, M. Dying Cells Actively Regulate Adaptive Immune Responses. Nat. Rev. Immunol. 2017, 17, 262–275. [Google Scholar] [CrossRef]
- Musella, M.; Manic, G.; De Maria, R.; Vitale, I.; Sistigu, A. Type-I-Interferons in Infection and Cancer: Unanticipated Dynamics with Therapeutic Implications. Oncoimmunology 2017, 6, e1314424. [Google Scholar] [CrossRef]
- Ivashkiv, L.; Donlin, L. Regulation of Type I Interferon Responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef]
- Won, J.K.; Bakhoum, S.F. The Cytosolic DNA-Sensing CGAS-STING Pathway in Cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDT | |||
|---|---|---|---|
| SK-MEL-2 | A375 | SK-MEL-28 | |
| SK-MEL-2 | 0.0001 | 0.0001 | |
| A375 | 0.0001 | 0.87 | |
| SK-MEL-28 | 0.0001 | 0.87 | |
| DXR | |||
| SK-MEL-2 | A375 | SK-MEL-28 | |
| SK-MEL-2 | 0.0072 | 0.4364 | |
| A375 | 0.0072 | 0.005 | |
| SK-MEL-28 | 0.4364 | 0.005 | |
| CP | |||
| SK-MEL-2 | A375 | SK-MEL-28 | |
| SK-MEL-2 | 0.2866 | 0.003 | |
| A375 | 0.2866 | 0.0004 | |
| SK-MEL-28 | 0.003 | 0.0004 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mentucci, F.M.; Romero Nuñez, E.A.; Ercole, A.; Silvetti, V.; Dal Col, J.; Lamberti, M.J. Impact of Genomic Mutation on Melanoma Immune Microenvironment and IFN-1 Pathway-Driven Therapeutic Responses. Cancers 2024, 16, 2568. https://doi.org/10.3390/cancers16142568
Mentucci FM, Romero Nuñez EA, Ercole A, Silvetti V, Dal Col J, Lamberti MJ. Impact of Genomic Mutation on Melanoma Immune Microenvironment and IFN-1 Pathway-Driven Therapeutic Responses. Cancers. 2024; 16(14):2568. https://doi.org/10.3390/cancers16142568
Chicago/Turabian StyleMentucci, Fátima María, Elisa Ayelén Romero Nuñez, Agustina Ercole, Valentina Silvetti, Jessica Dal Col, and María Julia Lamberti. 2024. "Impact of Genomic Mutation on Melanoma Immune Microenvironment and IFN-1 Pathway-Driven Therapeutic Responses" Cancers 16, no. 14: 2568. https://doi.org/10.3390/cancers16142568
APA StyleMentucci, F. M., Romero Nuñez, E. A., Ercole, A., Silvetti, V., Dal Col, J., & Lamberti, M. J. (2024). Impact of Genomic Mutation on Melanoma Immune Microenvironment and IFN-1 Pathway-Driven Therapeutic Responses. Cancers, 16(14), 2568. https://doi.org/10.3390/cancers16142568