
Depicting Biomarkers for HER2-Inhibitor Resistance: Implication for Therapy in HER2-Positive Breast Cancer



Abstract
Simple Summary
Abstract
1. Introduction
2. Standard of Care for HER2+ BC
2.1. Monoclonal Antibodies (mAbs)
2.1.1. Trastuzumab
2.1.2. Pertuzumab
2.1.3. Margetuximab
2.2. Antibody–Drug Conjugates (ADCs)
2.2.1. T-DM1 (Trastuzumab Emtansine)
2.2.2. T-DXd (Trastuzumab Deruxtecan)
2.2.3. SYD985 (Trastuzumab Duocarmazine)
2.3. Tyrosine Kinase Inhibitors (TKIs)
2.4. Summary of Current HER2+ Therapies

{kind=link}
{kind=link}
{kind=link}
| Drug (Brand Name) | Type | Route | Approval Year | Treatment Setting | Practical Implications | |
|---|---|---|---|---|---|---|
| Clinical Benefits | Clinical Shortcomings * | |||||
| trastuzumab (Herceptin) | mAb | IV | 1998 | metastatic BC | fairly well tolerated gold standard first-line therapy: although HER2+ treatments have diversified greatly, trastuzumab remains integral to therapy, especially in combination with new generations of drugs | cardiotoxicity is the main ADR of interest, but is manageable with early detection and monitoring [24,25,26] susceptible to resistance limited CNS penetrance [90] |
| 2006 | adjuvant setting | |||||
| lapatinib (Tykerb) | TKI | oral | 2007 | metastatic BC | penetrates into CNS [76,77] in combination with trastuzumab, significantly increases pCR vs. trastuzumab alone [91] | has generally been overshadowed by newer TKIs ADRs include significant diarrhea and rash [85] |
| pertuzumab (Perjeta) | mAb | IV | 2012 | metastatic BC | highly synergistic with trastuzumab; significantly improves PFS and OS vs. trastuzumab alone with little difference in serious AEs [92,93] | may increase cardiac burden compared with trastuzumab [94] like trastuzumab, similarly susceptible to resistance and has limited CNS penetrance [90] |
| 2013 | neoadjuvant setting | |||||
| 2017 | adjuvant setting for early BC | |||||
| T-DM1 (Kadcyla) | ADC | IV | 2013 | metastatic BC | overcomes resistance to trastuzumab [95] and lapatinib [96] greatly outperforms trastuzumab in adjuvant setting (50% decrease in recurrence/death) [61] | may increase AE incidence in comparison to trastuzumab; severe thrombocytopenia is particularly noteworthy [65] |
| 2019 | adjuvant setting for early BC | |||||
| neratinib (Nerlynx) | TKI | oral | 2017 | adjuvant setting for early BC | only TKI for early BC outperforms lapatinib (with CHT), with improved PFS and time to CNS intervention [97] greater potency vs. other TKIs in biochemical assays due to irreversible binding [74] | more severe diarrhea than other TKIs [97] |
| 2020 | metastatic BC | |||||
| T-DXd (Enhertu) | ADC | IV | 2019 | metastatic BC | preferred second-line treatment in metastatic setting; outperforms T-DM1 [59,98] first targeted drug for HER2-low BC [98] | ILD (seen in 10%–15% patients) [68] |
| tucatinib (Tukysa) | TKI | oral | 2020 | metastatic BC | strongest activity for CNS metastases [99,100] in comparison with other TKIs, decreased GI and skin toxicities due to higher specificity for HER2 [101] | ADRs include diarrhea and rash [86] |
| margetuximab (Margenz) | mAb | IV | 2020 | metastatic BC | improved PFS vs. trastuzumab with chemotherapy [53,54] increased ADCC activity vs. trastuzumab [52] | no significant difference in OS vs. trastuzumab [54] similar cardiotoxicity issues as with trastuzumab [54] and pertuzumab |
3. Biomarkers for HER2 Therapy Resistance
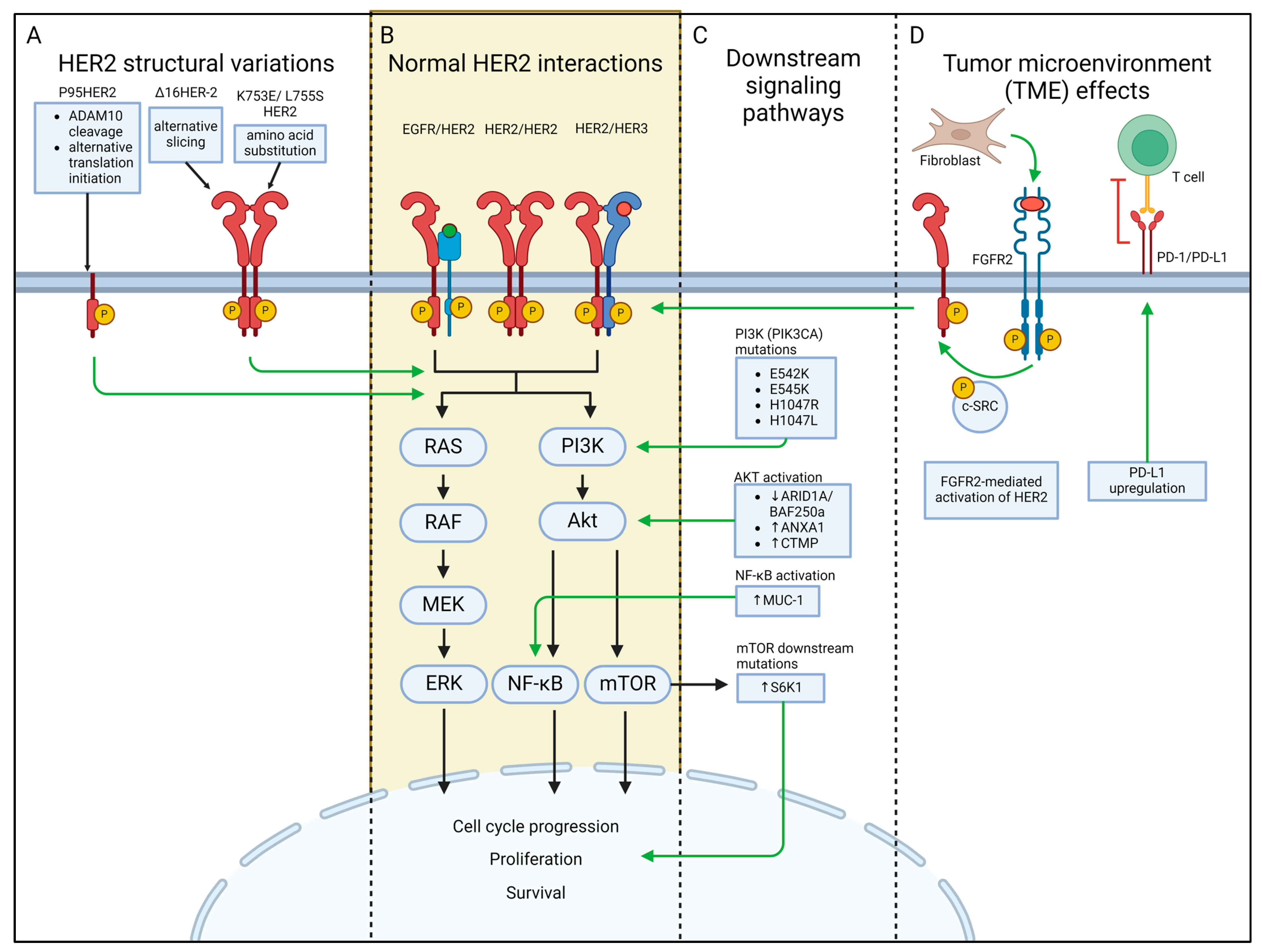
3.1. HER2 Structural Variations
3.1.1. P95HER2
3.1.2. Δ16HER-2
3.1.3. HER2 K753E and L755S
3.2. Downstream Signaling Proteins
3.2.1. PI3K/Akt/mTOR Pathway
PI3KCA H1047R and E545K
S6K1 Overexpression
ARID1A/BAF250a and ANXA1
CTMP Overexpression
3.2.2. The NF-κB Pathway
3.2.3. Other Pathways
3.3. Alterations of the Tumor Microenvironment (TME) as Biomarkers
3.4. Other Biomarkers
4. Strategies to Overcome Drug Resistance
4.1. Tyrosine Kinase Inhibitors (TKIs)
4.2. Monoclonal Antibodies (mAbs)
4.3. Bispecific Antibodies
4.4. Antibody–Drug Conjugates (ADC)
4.5. Combination Therapy
4.5.1. Combination with Oncogenic Pathway Inhibitors
4.5.2. Combination with Heat Shock Protein (HSP) Inhibitors
4.5.3. Combination with Immune Checkpoint Inhibitors (PD-1/PD-L1 Inhibitors)
4.5.4. Combination with Metabolic Inhibitors
4.5.5. Combination with CDK4/6 Inhibitors
4.5.6. Combination with Anti-Hormonal Therapy
5. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Giaquinto, A.N.; Sung, H.; Miller, K.D.; Kramer, J.L.; Newman, L.A.; Minihan, A.; Jemal, A.; Siegel, R.L. Breast Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 524–541. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Gnant, M. Breast cancer. Lancet 2017, 389, 1134–1150. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, D.; Pal, D.; Sharma, R.; Garg, V.K.; Goel, N.; Koundal, D.; Zaguia, A.; Koundal, S.; Belay, A. Global Increase in Breast Cancer Incidence: Risk Factors and Preventive Measures. Biomed. Res. Int. 2022, 2022, 9605439. [Google Scholar] [CrossRef] [PubMed]
- Tsang, J.Y.S.; Tse, G.M. Molecular Classification of Breast Cancer. Adv. Anat. Pathol. 2020, 27, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Henry, N.L.; Cannon-Albright, L.A. Breast cancer histologic subtypes show excess familial clustering. Cancer 2019, 125, 3131–3138. [Google Scholar] [CrossRef] [PubMed]
- Masood, S. Breast Cancer Subtypes: Morphologic and Biologic Characterization. Women’s Health 2016, 12, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Shastry, M.; Hamilton, E. Targeting HER2-positive breast cancer: Advances and future directions. Nat. Rev. Drug Discov. 2023, 22, 101–126. [Google Scholar] [CrossRef] [PubMed]
- Marra, A.; Chandarlapaty, S.; Modi, S. Management of patients with advanced-stage HER2-positive breast cancer: Current evidence and future perspectives. Nat. Rev. Clin. Oncol. 2024, 21, 185–202. [Google Scholar] [CrossRef]
- Wolff, A.C.; Hammond, M.E.H.; Hicks, D.G.; Dowsett, M.; McShane, L.M.; Allison, K.H.; Allred, D.C.; Bartlett, J.M.S.; Bilous, M.; Fitzgibbons, P.; et al. Recommendations for Human Epidermal Growth Factor Receptor 2 Testing in Breast Cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Update. J. Clin. Oncol. 2013, 31, 3997–4013. [Google Scholar] [CrossRef]
- Vicario, R.; Peg, V.; Morancho, B.; Zacarias-Fluck, M.; Zhang, J.; Martinez-Barriocanal, A.; Navarro Jimenez, A.; Aura, C.; Burgues, O.; Lluch, A.; et al. Patterns of HER2 Gene Amplification and Response to Anti-HER2 Therapies. PLoS ONE 2015, 10, e0129876. [Google Scholar] [CrossRef] [PubMed]
- Marotta, M.; Onodera, T.; Johnson, J.; Budd, G.T.; Watanabe, T.; Cui, X.; Giuliano, A.E.; Niida, A.; Tanaka, H. Palindromic amplification of the ERBB2 oncogene in primary HER2-positive breast tumors. Sci. Rep. 2017, 7, 41921. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of Chemotherapy plus a Monoclonal Antibody against HER2 for Metastatic Breast Cancer That Overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Han, F.; Zhu, L.; Ding, W.; Zhang, K.; Kan, C.; Hou, N.; Li, Q.; Sun, X. Advances in understanding the role and mechanisms of tumor stem cells in HER2-positive breast cancer treatment resistance (Review). Int. J. Oncol. 2023, 62, 1–11. [Google Scholar] [CrossRef]
- Carter, P.; Presta, L.; Gorman, C.M.; Ridgway, J.B.; Henner, D.; Wong, W.L.; Rowland, A.M.; Kotts, C.; Carver, M.E.; Shepard, H.M. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc. Natl. Acad. Sci. USA 1992, 89, 4285–4289. [Google Scholar] [CrossRef] [PubMed]
- Moja, L.; Tagliabue, L.; Balduzzi, S.; Parmelli, E.; Pistotti, V.; Guarneri, V.; D’Amico, R. Trastuzumab containing regimens for early breast cancer. Cochrane Database Syst. Rev. 2012, 2021, CD006243. [Google Scholar] [CrossRef]
- Camejo, N.; Castillo, C.; Alonso, R.; Correa, F.; Rivero, E.; Mezquita, C.; Rosich, A.; Dellacasa, F.; Silveira, L.; Delgado, L. Effectiveness of Trastuzumab for Human Epidermal Growth Factor Receptor 2–Positive Breast Cancer in a Real-Life Setting: One Decade of Experience Under National Treatment Coverage Regulations. JCO Glob. Oncol. 2020, 6, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.; Eiermann, W.; Robert, N.; Pienkowski, T.; Martin, M.; Press, M.; Mackey, J.; Glaspy, J.; Chan, A.; Pawlicki, M.; et al. Adjuvant Trastuzumab in HER2-Positive Breast Cancer. N. Engl. J. Med. 2011, 365, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Erickson, A.W.; Ghodrati, F.; Habbous, S.; Jerzak, K.J.; Sahgal, A.; Ahluwalia, M.S.; Das, S. HER2-targeted therapy prolongs survival in patients with HER2-positive breast cancer and intracranial metastatic disease: A systematic review and meta-analysis. Neuro-Oncol. Adv. 2020, 2, vdaa136. [Google Scholar] [CrossRef]
- Mendes, D.; Alves, C.; Afonso, N.; Cardoso, F.; Passos-Coelho, J.L.; Costa, L.; Andrade, S.; Batel-Marques, F. The benefit of HER2-targeted therapies on overall survival of patients with metastatic HER2-positive breast cancer—A systematic review. Breast Cancer Res. 2015, 17, 140. [Google Scholar] [CrossRef]
- Mittal, A.; Tamimi, F.; Molto, C.; Meti, N.; Al-Showbaki, L.; Wilson, B.E.; Amir, E. Three-year disease-free survival in randomized trials of neoadjuvant chemotherapy and HER2-targeted therapy in breast cancer: A meta-analysis. Crit. Rev. Oncol./Hematol. 2023, 181, 103880. [Google Scholar] [CrossRef]
- Balduzzi, S.; Mantarro, S.; Guarneri, V.; Tagliabue, L.; Pistotti, V.; Moja, L.; D’Amico, R. Trastuzumab-containing regimens for metastatic breast cancer. Cochrane Database Syst. Rev. 2014, 2014, CD006242. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.L.; Barlow, M.; Barrett-Lee, P.J.; Canney, P.A.; Gilmour, I.M.; Robb, S.D.; Plummer, C.J.; Wardley, A.M.; Verrill, M.W. Management of cardiac health in trastuzumab-treated patients with breast cancer: Updated United Kingdom National Cancer Research Institute recommendations for monitoring. Br. J. Cancer 2009, 100, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Mazzotta, M.; Krasniqi, E.; Barchiesi, G.; Pizzuti, L.; Tomao, F.; Barba, M.; Vici, P. Long-Term Safety and Real-World Effectiveness of Trastuzumab in Breast Cancer. J. Clin. Med. 2019, 8, 254. [Google Scholar] [CrossRef] [PubMed]
- Barish, R.; Gates, E.; Barac, A. Trastuzumab-Induced Cardiomyopathy. Cardiol. Clin. 2019, 37, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Copeland-Halperin, R.S.; Liu, J.E.; Yu, A.F. Cardiotoxicity of HER2-targeted therapies. Curr. Opin. Cardiol. 2019, 34, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Greenblatt, K.; Khaddour, K. Trastuzumab. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Jackson, C.; Finikarides, L.; Freeman, A.L.J. The adverse effects of trastuzumab-containing regimes as a therapy in breast cancer: A piggy-back systematic review and meta-analysis. PLoS ONE 2022, 17, e0275321. [Google Scholar] [CrossRef] [PubMed]
- Molina, M.A.; Codony-Servat, J.; Albanell, J.; Rojo, F.; Arribas, J.; Baselga, J. Trastuzumab (herceptin), a humanized anti-Her2 receptor monoclonal antibody, inhibits basal and activated Her2 ectodomain cleavage in breast cancer cells. Cancer Res. 2001, 61, 4744–4749. [Google Scholar] [PubMed]
- Wang, J.; Xu, B. Targeted therapeutic options and future perspectives for HER2-positive breast cancer. Signal Transduct. Target. Ther. 2019, 4, 34. [Google Scholar] [CrossRef]
- Spector, N.L.; Blackwell, K.L. Understanding the mechanisms behind trastuzumab therapy for human epidermal growth factor receptor 2-positive breast cancer. J. Clin. Oncol. 2009, 27, 5838–5847. [Google Scholar] [CrossRef]
- Baselga, J.; Albanell, J.; Molina, M.A.; Arribas, J. Mechanism of action of trastuzumab and scientific update. Semin. Oncol. 2001, 28, 4–11. [Google Scholar] [CrossRef]
- Albanell, J.; Codony, J.; Rovira, A.; Mellado, B.; Gascón, P. Mechanism of Action of Anti-Her2 Monoclonal Antibodies: Scientific Update on Trastuzumab and 2c4; Springer: New York, NY, USA, 2003; pp. 253–268. [Google Scholar]
- Dubská, L.; Anděra, L.; Sheard, M.A. HER2 signaling downregulation by trastuzumab and suppression of the PI3K/Akt pathway: An unexpected effect on TRAIL-induced apoptosis. FEBS Lett. 2005, 579, 4149–4158. [Google Scholar] [CrossRef]
- Nagata, Y.; Lan, K.H.; Zhou, X.; Tan, M.; Esteva, F.J.; Sahin, A.A.; Klos, K.S.; Li, P.; Monia, B.P.; Nguyen, N.T.; et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 2004, 6, 117–127. [Google Scholar] [CrossRef]
- Yakes, F.M.; Chinratanalab, W.; Ritter, C.A.; King, W.; Seelig, S.; Arteaga, C.L. Herceptin-induced inhibition of phosphatidylinositol-3 kinase and Akt Is required for antibody-mediated effects on p27, cyclin D1, and antitumor action. Cancer Res. 2002, 62, 4132–4141. [Google Scholar]
- Maadi, H.; Soheilifar, M.H.; Choi, W.-S.; Moshtaghian, A.; Wang, Z. Trastuzumab Mechanism of Action; 20 Years of Research to Unravel a Dilemma. Cancers 2021, 13, 3540. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; Gianni, L. The immune system and response to HER2-targeted treatment in breast cancer. Lancet Oncol. 2014, 15, 58–68. [Google Scholar] [CrossRef]
- Costa, R.L.B.; Czerniecki, B.J. Clinical development of immunotherapies for HER2+ breast cancer: A review of HER2-directed monoclonal antibodies and beyond. npj Breast Cancer 2020, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Boone, J.J.M.; Bhosle, J.; Tilby, M.J.; Hartley, J.A.; Hochhauser, D. Involvement of the HER2 pathway in repair of DNA damage produced by chemotherapeutic agents. Mol. Cancer Ther. 2009, 8, 3015–3023. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Yarmand-Bagheri, R. The role of HER2 in angiogenesis. Semin. Oncol. 2001, 28, 27–32. [Google Scholar] [CrossRef]
- Petricevic, B.; Laengle, J.; Singer, J.; Sachet, M.; Fazekas, J.; Steger, G.; Bartsch, R.; Jensen-Jarolim, E.; Bergmann, M. Trastuzumab mediates antibody-dependent cell-mediated cytotoxicity and phagocytosis to the same extent in both adjuvant and metastatic HER2/neu breast cancer patients. J. Transl. Med. 2013, 11, 307. [Google Scholar] [CrossRef]
- Tsao, L.-C.; Crosby, E.J.; Trotter, T.N.; Wei, J.; Wang, T.; Yang, X.; Summers, A.N.; Lei, G.; Rabiola, C.A.; Chodosh, L.A.; et al. Trastuzumab/pertuzumab combination therapy stimulates antitumor responses through complement-dependent cytotoxicity and phagocytosis. JCI Insight 2022, 7, e155636. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, G.M.; Scher, N.S.; Cortazar, P.; Chattopadhyay, S.; Tang, S.; Song, P.; Liu, Q.; Ringgold, K.; Pilaro, A.M.; Tilley, A.; et al. First FDA Approval of Dual Anti-HER2 Regimen: Pertuzumab in Combination with Trastuzumab and Docetaxel for HER2-Positive Metastatic Breast Cancer. Clin. Cancer Res. 2013, 19, 4911–4916. [Google Scholar] [CrossRef] [PubMed]
- Barthélémy, P.; Leblanc, J.; Goldbarg, V.; Wendling, F.; Kurtz, J.E. Pertuzumab: Development beyond breast cancer. Anticancer Res. 2014, 34, 1483–1491. [Google Scholar] [PubMed]
- Sendur, M.A.; Aksoy, S.; Altundag, K.; Sendur, M.A.; Aksoy, S.; Altundag, K. Pertuzumab in HER2-positive breast cancer. Curr. Med Res. Opin. 2012, 28, 1709–1716. [Google Scholar] [CrossRef] [PubMed]
- Dean, L.; Kane, M. Trastuzumab Therapy and ERBB2 Genotype; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2012.
- Jagosky, M.; Tan, A.R. Combination of Pertuzumab and Trastuzumab in the Treatment of HER2-Positive Early Breast Cancer: A Review of the Emerging Clinical Data. Breast Cancer Targets Ther. 2021, 13, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Beckmann, M.W.; Rody, A.; Schneeweiss, A.; Müller, V.; Fehm, T.; Marschner, N.; Gluz, O.; Schrader, I.; Heinrich, G.; et al. HER2 Dimerization Inhibitor Pertuzumab—Mode of Action and Clinical Data in Breast Cancer. Breast Care 2013, 8, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Cruz, V.L.; Souza-Egipsy, V.; Gion, M.; Pérez-García, J.; Cortes, J.; Ramos, J.; Vega, J.F. Binding Affinity of Trastuzumab and Pertuzumab Monoclonal Antibodies to Extracellular HER2 Domain. Int. J. Mol. Sci. 2023, 24, 12031. [Google Scholar] [CrossRef] [PubMed]
- Royce, M.; Osgood, C.L.; Amatya, A.K.; Fiero, M.H.; Chang, C.J.G.; Ricks, T.K.; Shetty, K.A.; Kraft, J.; Qiu, J.; Song, P.; et al. FDA Approval Summary: Margetuximab plus Chemotherapy for Advanced or Metastatic HER2-Positive Breast Cancer. Clin. Cancer Res. 2022, 28, 1487–1492. [Google Scholar] [CrossRef] [PubMed]
- Mitani, S.; Kawakami, H. Emerging Targeted Therapies for HER2 Positive Gastric Cancer That Can Overcome Trastuzumab Resistance. Cancers 2020, 12, 400. [Google Scholar] [CrossRef]
- Rugo, H.S.; Im, S.-A.; Cardoso, F.; Cortés, J.; Curigliano, G.; Musolino, A.; Pegram, M.D.; Wright, G.S.; Saura, C.; Escrivá-De-Romaní, S.; et al. Efficacy of Margetuximab vs. Trastuzumab in Patients With Pretreated ERBB2-Positive Advanced Breast Cancer. JAMA Oncol. 2021, 7, 573. [Google Scholar] [CrossRef]
- Rugo, H.S.; Im, S.-A.; Cardoso, F.; Cortes, J.; Curigliano, G.; Musolino, A.; Pegram, M.D.; Bachelot, T.; Wright, G.S.; Saura, C.; et al. Margetuximab Versus Trastuzumab in Patients With Previously Treated HER2-Positive Advanced Breast Cancer (SOPHIA): Final Overall Survival Results From a Randomized Phase 3 Trial. J. Clin. Oncol. 2023, 41, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Catenacci, D.V.T.; Kang, Y.K.; Yoon, H.H.; Shim, B.Y.; Kim, S.T.; Oh, D.Y.; Spira, A.I.; Ulahannan, S.V.; Avery, E.J.; Boland, P.M.; et al. Margetuximab with retifanlimab as first-line therapy in HER2+/PD-L1+ unresectable or metastatic gastroesophageal adenocarcinoma: MAHOGANY cohort A. ESMO Open 2022, 7, 100563. [Google Scholar] [CrossRef] [PubMed]
- Pettinato, M.C. Introduction to Antibody-Drug Conjugates. Antibodies 2021, 10, 42. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Kreutzfeldt, J.; Rozeboom, B.; Dey, N.; De, P. The trastuzumab era: Current and upcoming targeted HER2+ breast cancer therapies. Am. J. Cancer Res. 2020, 10, 1045–1067. [Google Scholar]
- Hunter, F.W.; Barker, H.R.; Lipert, B.; Rothé, F.; Gebhart, G.; Piccart-Gebhart, M.J.; Sotiriou, C.; Jamieson, S.M.F. Mechanisms of resistance to trastuzumab emtansine (T-DM1) in HER2-positive breast cancer. Br. J. Cancer 2020, 122, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Diéras, V.; Miles, D.; Verma, S.; Pegram, M.; Welslau, M.; Baselga, J.; Krop, I.E.; Blackwell, K.; Hoersch, S.; Xu, J.; et al. Trastuzumab emtansine versus capecitabine plus lapatinib in patients with previously treated HER2-positive advanced breast cancer (EMILIA): A descriptive analysis of final overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Mamounas, E.P.; Untch, M.; Mano, M.S.; Huang, C.S.; Geyer, C.E., Jr.; Von Minckwitz, G.; Wolmark, N.; Pivot, X.; Kuemmel, S.; Digiovanna, M.P.; et al. Adjuvant T-DM1 versus trastuzumab in patients with residual invasive disease after neoadjuvant therapy for HER2-positive breast cancer: Subgroup analyses from KATHERINE. Ann. Oncol. 2021, 32, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Krop, I.E.; Kim, S.B.; Martin, A.G.; LoRusso, P.M.; Ferrero, J.M.; Badovinac-Crnjevic, T.; Hoersch, S.; Smitt, M.; Wildiers, H. Trastuzumab emtansine versus treatment of physician’s choice in patients with previously treated HER2-positive metastatic breast cancer (TH3RESA): Final overall survival results from a randomised open-label phase 3 trial. Lancet Oncol. 2017, 18, 743–754. [Google Scholar] [CrossRef]
- Perez, E.A.; Barrios, C.; Eiermann, W.; Toi, M.; Im, Y.H.; Conte, P.; Martin, M.; Pienkowski, T.; Pivot, X.; Burris, H.r.; et al. Trastuzumab Emtansine With or Without Pertuzumab Versus Trastuzumab Plus Taxane for Human Epidermal Growth Factor Receptor 2-Positive, Advanced Breast Cancer: Primary Results From the Phase III MARIANNE Study. J. Clin. Oncol. 2017, 35, 141–148. [Google Scholar] [CrossRef]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.Y.; Dieras, V.; Guardino, E.; et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Ke, J.; Song, Y. T-DM1-induced thrombocytopenia in breast cancer patients: New perspectives. Biomed. Pharmacother. 2020, 129, 110407. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Sun, J.; Liu, L.; Kong, X.; Lin, D.; Zhou, H.; Gao, J. Clinicopathological characteristics of HER2-low breast cancer: A retrospective study. Sci. Rep. 2023, 13, 12382. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, P.; Hamilton, E.; Tolaney, S.M.; Cortes, J.; Morganti, S.; Ferraro, E.; Marra, A.; Viale, G.; Trapani, D.; Cardoso, F.; et al. HER2-Low Breast Cancer: Pathological and Clinical Landscape. J. Clin. Oncol. 2020, 38, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Abuhelwa, Z.; Alloghbi, A.; Alqahtani, A.; Nagasaka, M. Trastuzumab Deruxtecan-Induced Interstitial Lung Disease/Pneumonitis in ERBB2-Positive Advanced Solid Malignancies: A Systematic Review. Drugs 2022, 82, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Yver, A.; Agatsuma, T.; Soria, J.C. The art of innovation: Clinical development of trastuzumab deruxtecan and redefining how antibody-drug conjugates target HER2-positive cancers. Ann. Oncol. 2020, 31, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Rassy, E.; Rached, L.; Pistilli, B. Antibody drug conjugates targeting HER2: Clinical development in metastatic breast cancer. Breast 2022, 66, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Dokter, W.; Ubink, R.; Van Der Lee, M.; Van Der Vleuten, M.; Van Achterberg, T.; Jacobs, D.; Loosveld, E.; Van Den Dobbelsteen, D.; Egging, D.; Mattaar, E.; et al. Preclinical Profile of the HER2-Targeting ADC SYD983/SYD985: Introduction of a New Duocarmycin-Based Linker-Drug Platform. Mol. Cancer Ther. 2014, 13, 2618–2629. [Google Scholar] [CrossRef] [PubMed]
- Nadal-Serrano, M.; Morancho, B.; Escriva-de-Romani, S.; Morales, C.B.; Luque, A.; Escorihuela, M.; Espinosa Bravo, M.; Peg, V.; Dijcks, F.A.; Dokter, W.H.A.; et al. The Second Generation Antibody-Drug Conjugate SYD985 Overcomes Resistances to T-DM1. Cancers 2020, 12, 670. [Google Scholar] [CrossRef]
- Escrivá-De-Romaní, S.; Saura, C. The change of paradigm in the treatment of HER2-positive breast cancer with the development of new generation antibody-drug conjugates. Cancer Drug Resist. 2023, 6, 45–58. [Google Scholar] [CrossRef]
- Collins, D.M.; Conlon, N.T.; Kannan, S.; Verma, C.S.; Eli, L.D.; Lalani, A.S.; Crown, J. Preclinical Characteristics of the Irreversible Pan-HER Kinase Inhibitor Neratinib Compared with Lapatinib: Implications for the Treatment of HER2-Positive and HER2-Mutated Breast Cancer. Cancers 2019, 11, 737. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Domchek, S.M.; Burstein, H.J.; Harris, L.; Younger, J.; Kuter, I.; Bunnell, C.; Rue, M.; Gelman, R.; Winer, E. Central nervous system metastases in women who receive trastuzumab-based therapy for metastatic breast carcinoma. Cancer 2003, 97, 2972–2977. [Google Scholar] [CrossRef] [PubMed]
- Montemurro, F.; Ellis, P.; Anton, A.; Wuerstlein, R.; Delaloge, S.; Bonneterre, J.; Quenel-Tueux, N.; Linn, S.C.; Irahara, N.; Donica, M.; et al. Safety of trastuzumab emtansine (T-DM1) in patients with HER2-positive advanced breast cancer: Primary results from the KAMILLA study cohort 1. Eur. J. Cancer 2019, 109, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Montemurro, F.; Delaloge, S.; Barrios, C.H.; Wuerstlein, R.; Anton, A.; Brain, E.; Hatschek, T.; Kelly, C.M.; Peña-Murillo, C.; Yilmaz, M.; et al. Trastuzumab emtansine (T-DM1) in patients with HER2-positive metastatic breast cancer and brain metastases: Exploratory final analysis of cohort 1 from KAMILLA, a single-arm phase IIIb clinical trial☆. Ann. Oncol. 2020, 31, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Bachelot, T.; Romieu, G.; Campone, M.; Diéras, V.; Cropet, C.; Dalenc, F.; Jimenez, M.; Le Rhun, E.; Pierga, J.Y.; Gonçalves, A.; et al. Lapatinib plus capecitabine in patients with previously untreated brain metastases from HER2-positive metastatic breast cancer (LANDSCAPE): A single-group phase 2 study. Lancet Oncol. 2013, 14, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Awada, A.; Colomer, R.; Inoue, K.; Bondarenko, I.; Badwe, R.A.; Demetriou, G.; Lee, S.-C.; Mehta, A.O.; Kim, S.-B.; Bachelot, T.; et al. Neratinib Plus Paclitaxel vs Trastuzumab Plus Paclitaxel in Previously Untreated Metastatic ERBB2-Positive Breast Cancer: The NEfERT-T Randomized Clinical Trial. JAMA Oncol. 2016, 2, 1557–1564. [Google Scholar] [CrossRef] [PubMed]
- Dinkel, V.; Anderson, D.; Winski, S.; Winkler, J.; Koch, K.; Lee, P.A. Abstract 852: ARRY-380, a potent, small molecule inhibitor of ErbB2, increases survival in intracranial ErbB2+ xenograft models in mice. Cancer Res. 2012, 72, 852. [Google Scholar] [CrossRef]
- Stringer-Reasor, E.M.; O’Brien, B.J.; Topletz-Erickson, A.; White, J.B.; Lobbous, M.; Riley, K.; Childress, J.; LaMaster, K.; Melisko, M.E.; Morikawa, A.; et al. Pharmacokinetic (PK) analyses in CSF and plasma from TBCRC049, an ongoing trial to assess the safety and efficacy of the combination of tucatinib, trastuzumab and capecitabine for the treatment of leptomeningeal metastasis (LM) in HER2 positive breast cancer. J. Clin. Oncol. 2021, 39, 1044. [Google Scholar] [CrossRef]
- Xu, B.; Yan, M.; Ma, F.; Hu, X.; Feng, J.; Ouyang, Q.; Tong, Z.; Li, H.; Zhang, Q.; Sun, T.; et al. Pyrotinib plus capecitabine versus lapatinib plus capecitabine for the treatment of HER2-positive metastatic breast cancer (PHOEBE): A multicentre, open-label, randomised, controlled, phase 3 trial. Lancet Oncol. 2021, 22, 351–360. [Google Scholar] [CrossRef]
- Zheng, Y.; Cao, W.-M.; Shao, X.; Shi, Y.; Cai, L.; Chen, W.; Liu, J.; Shen, P.; Chen, Y.; Wang, X.; et al. Pyrotinib plus docetaxel as first-line treatment for HER2-positive metastatic breast cancer: The PANDORA phase II trial. Nat. Commun. 2023, 14, 8314. [Google Scholar] [CrossRef]
- Ma, F.; Yan, M.; Li, W.; Ouyang, Q.; Tong, Z.; Teng, Y.; Wang, Y.; Wang, S.; Geng, C.; Luo, T.; et al. Pyrotinib versus placebo in combination with trastuzumab and docetaxel as first line treatment in patients with HER2 positive metastatic breast cancer (PHILA): Randomised, double blind, multicentre, phase 3 trial. BMJ 2023, 383, e076065. [Google Scholar] [CrossRef] [PubMed]
- Shyam Sunder, S.; Sharma, U.C.; Pokharel, S. Adverse effects of tyrosine kinase inhibitors in cancer therapy: Pathophysiology, mechanisms and clinical management. Signal Transduct. Target. Ther. 2023, 8, 262. [Google Scholar] [CrossRef] [PubMed]
- Murthy, R.K.; Loi, S.; Okines, A.; Paplomata, E.; Hamilton, E.; Hurvitz, S.A.; Lin, N.U.; Borges, V.; Abramson, V.; Anders, C.; et al. Tucatinib, Trastuzumab, and Capecitabine for HER2-Positive Metastatic Breast Cancer. N. Engl. J. Med. 2020, 382, 597–609. [Google Scholar] [CrossRef]
- Park, Y.H.; Lee, K.H.; Sohn, J.H.; Lee, K.S.; Jung, K.H.; Kim, J.H.; Lee, K.H.; Ahn, J.S.; Kim, T.Y.; Kim, G.M.; et al. A phase II trial of the pan-HER inhibitor poziotinib, in patients with HER2-positive metastatic breast cancer who had received at least two prior HER2-directed regimens: Results of the NOV120101-203 trial. Int. J. Cancer 2018, 143, 3240–3247. [Google Scholar] [CrossRef]
- Macpherson, I.R.; Spiliopoulou, P.; Rafii, S.; Saggese, M.; Baird, R.D.; Garcia-Corbacho, J.; Italiano, A.; Bonneterre, J.; Campone, M.; Cresti, N.; et al. A phase I/II study of epertinib plus trastuzumab with or without chemotherapy in patients with HER2-positive metastatic breast cancer. Breast Cancer Res. 2020, 22, 1. [Google Scholar] [CrossRef] [PubMed]
- Irie, H.; Kawabata, R.; Fujioka, Y.; Nakagawa, F.; Itadani, H.; Nagase, H.; Ito, K.; Uchida, J.; Ohkubo, S.; Matsuo, K. Acquired resistance to trastuzumab/pertuzumab or to T-DM1 in vivo can be overcome by HER2 kinase inhibition with TAS0728. Cancer Sci. 2020, 111, 2123–2131. [Google Scholar] [CrossRef]
- Wang, W.; He, H.; Marin-Ramos, N.I.; Zeng, S.; Swenson, S.D.; Cho, H.Y.; Fu, J.; Beringer, P.M.; Neman, J.; Chen, L.; et al. Enhanced brain delivery and therapeutic activity of trastuzumab after blood-brain barrier opening by NEO100 in mouse models of brain-metastatic breast cancer. Neuro Oncol. 2021, 23, 1656–1667. [Google Scholar] [CrossRef]
- Xin, Y.; Guo, W.W.; Huang, Q.; Zhang, P.; Zhang, L.Z.; Jiang, G.; Tian, Y. Effects of lapatinib or trastuzumab, alone and in combination, in human epidermal growth factor receptor 2-positive breast cancer: A meta-analysis of randomized controlled trials. Cancer Med. 2016, 5, 3454–3463. [Google Scholar] [CrossRef]
- Swain, S.M.; Baselga, J.; Kim, S.B.; Ro, J.; Semiglazov, V.; Campone, M.; Ciruelos, E.; Ferrero, J.M.; Schneeweiss, A.; Heeson, S.; et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N. Engl. J. Med. 2015, 372, 724–734. [Google Scholar] [CrossRef]
- Ishii, K.; Morii, N.; Yamashiro, H. Pertuzumab in the treatment of HER2-positive breast cancer: An evidence-based review of its safety, efficacy, and place in therapy. Core Evid. 2019, 14, 51–70. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Meng, W.; Zhao, W.; Tong, Z. Cardiac safety analysis of anti-HER2-targeted therapy in early breast cancer. Sci. Rep. 2022, 12, 14312. [Google Scholar] [CrossRef] [PubMed]
- Lewis Phillips, G.D.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blattler, W.A.; Lambert, J.M.; Chari, R.V.; Lutz, R.J.; et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef] [PubMed]
- Junttila, T.T.; Li, G.; Parsons, K.; Phillips, G.L.; Sliwkowski, M.X. Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Res. Treat. 2011, 128, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Saura, C.; Matito, J.; Oliveira, M.; Wildiers, H.; Brufksy, A.M.; Waters, S.H.; Hurvitz, S.A.; Moy, B.; Kim, S.B.; Gradishar, W.J.; et al. Biomarker Analysis of the Phase III NALA Study of Neratinib + Capecitabine versus Lapatinib + Capecitabine in Patients with Previously Treated Metastatic Breast Cancer. Clin. Cancer Res. 2021, 27, 5818–5827. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Jacot, W.; Yamashita, T.; Sohn, J.; Vidal, M.; Tokunaga, E.; Tsurutani, J.; Ueno, N.T.; Prat, A.; Chae, Y.S.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. N. Engl. J. Med. 2022, 387, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Wedam, S.; Cheng, J.; Fiero, M.H.; Xia, H.; Li, F.; Fan, J.; Zhang, X.; Yu, J.; Song, P.; et al. FDA Approval Summary: Tucatinib for the Treatment of Patients with Advanced or Metastatic HER2-positive Breast Cancer. Clin. Cancer Res. 2021, 27, 1220–1226. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jiang, J.; Bao, X.; Kumar, V.; Alley, S.C.; Peterson, S.; Lee, A.J. Mechanistic Modeling of Central Nervous System Pharmacokinetics and Target Engagement of HER2 Tyrosine Kinase Inhibitors to Inform Treatment of Breast Cancer Brain Metastases. Clin. Cancer Res. 2022, 28, 3329–3341. [Google Scholar] [CrossRef] [PubMed]
- Conlon, N.T.; Kooijman, J.J.; van Gerwen, S.J.C.; Mulder, W.R.; Zaman, G.J.R.; Diala, I.; Eli, L.D.; Lalani, A.S.; Crown, J.; Collins, D.M. Comparative analysis of drug response and gene profiling of HER2-targeted tyrosine kinase inhibitors. Br. J. Cancer 2021, 124, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Yeo, B.; Kotsori, K.; Mohammed, K.; Walsh, G.; Smith, I.E. Long-term outcome of HER2 positive metastatic breast cancer patients treated with first-line trastuzumab. Breast 2015, 24, 751–757. [Google Scholar] [CrossRef]
- Gajria, D.; Chandarlapaty, S. HER2-amplified breast cancer: Mechanisms of trastuzumab resistance and novel targeted therapies. Expert. Rev. Anticancer Ther. 2011, 11, 263–275. [Google Scholar] [CrossRef]
- Lv, L.; Yang, S.; Zhu, Y.; Zhai, X.; Li, S.; Tao, X.; Dong, D. Relationship between metabolic reprogramming and drug resistance in breast cancer. Front. Oncol. 2022, 12, 942064. [Google Scholar] [CrossRef]
- Pohlmann, P.R.; Mayer, I.A.; Mernaugh, R. Resistance to Trastuzumab in Breast Cancer. Clin. Cancer Res. 2009, 15, 7479–7491. [Google Scholar] [CrossRef]
- Anido, J.; Scaltriti, M.; Bech Serra, J.J.; Santiago Josefat, B.; Todo, F.R.; Baselga, J.; Arribas, J. Biosynthesis of tumorigenic HER2 C-terminal fragments by alternative initiation of translation. EMBO J. 2006, 25, 3234–3244. [Google Scholar] [CrossRef]
- Maria, A.M.; El-Shebiney, M.; El-Saka, A.M.; Zamzam, Y. Expression of truncated HER2 and its prognostic value in HER2-positive breast cancer patients. J. Egypt. Natl. Cancer Inst. 2018, 30, 49–55. [Google Scholar] [CrossRef]
- Scaltriti, M.; Rojo, F.; Ocana, A.; Anido, J.; Guzman, M.; Cortes, J.; Di Cosimo, S.; Matias-Guiu, X.; Ramon y Cajal, S.; Arribas, J.; et al. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J. Natl. Cancer Inst. 2007, 99, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Sperinde, J.; Huang, W.; Vehtari, A.; Chenna, A.; Kellokumpu-Lehtinen, P.L.; Winslow, J.; Bono, P.; Lie, Y.S.; Petropoulos, C.J.; Weidler, J.; et al. p95HER2 Methionine 611 Carboxy-Terminal Fragment Is Predictive of Trastuzumab Adjuvant Treatment Benefit in the FinHer Trial. Clin. Cancer Res. 2018, 24, 3046–3052. [Google Scholar] [CrossRef] [PubMed]
- Molina, M.A.; Saez, R.; Ramsey, E.E.; Garcia-Barchino, M.J.; Rojo, F.; Evans, A.J.; Albanell, J.; Keenan, E.J.; Lluch, A.; Garcia-Conde, J.; et al. NH(2)-terminal truncated HER-2 protein but not full-length receptor is associated with nodal metastasis in human breast cancer. Clin. Cancer Res. 2002, 8, 347–353. [Google Scholar] [PubMed]
- Yang, M.; Li, Y.; Kong, L.; Huang, S.; He, L.; Liu, P.; Mo, S.; Lu, X.; Lin, X.; Xiao, Y.; et al. Inhibition of DPAGT1 suppresses HER2 shedding and trastuzumab resistance in human breast cancer. J. Clin. Investig. 2023, 133, e164428. [Google Scholar] [CrossRef]
- Jackson, C.; Browell, D.; Gautrey, H.; Tyson-Capper, A. Clinical Significance of HER-2 Splice Variants in Breast Cancer Progression and Drug Resistance. Int. J. Cell Biol. 2013, 2013, 973584. [Google Scholar] [CrossRef]
- Pupa, S.M.; Ligorio, F.; Cancila, V.; Franceschini, A.; Tripodo, C.; Vernieri, C.; Castagnoli, L. HER2 Signaling and Breast Cancer Stem Cells: The Bridge behind HER2-Positive Breast Cancer Aggressiveness and Therapy Refractoriness. Cancers 2021, 13, 4778. [Google Scholar] [CrossRef]
- Mitra, D.; Brumlik, M.J.; Okamgba, S.U.; Zhu, Y.; Duplessis, T.T.; Parvani, J.G.; Lesko, S.M.; Brogi, E.; Jones, F.E. An oncogenic isoform of HER2 associated with locally disseminated breast cancer and trastuzumab resistance. Mol. Cancer Ther. 2009, 8, 2152–2162. [Google Scholar] [CrossRef] [PubMed]
- Castagnoli, L.; Iorio, E.; Dugo, M.; Koschorke, A.; Faraci, S.; Canese, R.; Casalini, P.; Nanni, P.; Vernieri, C.; Di Nicola, M.; et al. Intratumor lactate levels reflect HER2 addiction status in HER2-positive breast cancer. J. Cell. Physiol. 2019, 234, 1768–1779. [Google Scholar] [CrossRef] [PubMed]
- Turpin, J.; Ling, C.; Crosby, E.J.; Hartman, Z.C.; Simond, A.M.; Chodosh, L.A.; Rennhack, J.P.; Andrechek, E.R.; Ozcelik, J.; Hallett, M.; et al. The ErbB2DeltaEx16 splice variant is a major oncogenic driver in breast cancer that promotes a pro-metastatic tumor microenvironment. Oncogene 2016, 35, 6053–6064. [Google Scholar] [CrossRef] [PubMed]
- Castiglioni, F.; Tagliabue, E.; Campiglio, M.; Pupa, S.M.; Balsari, A.; Menard, S. Role of exon-16-deleted HER2 in breast carcinomas. Endocr. Relat. Cancer 2006, 13, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Alajati, A.; Sausgruber, N.; Aceto, N.; Duss, S.; Sarret, S.; Voshol, H.; Bonenfant, D.; Bentires-Alj, M. Mammary tumor formation and metastasis evoked by a HER2 splice variant. Cancer Res. 2013, 73, 5320–5327. [Google Scholar] [CrossRef] [PubMed]
- Castagnoli, L.; Iezzi, M.; Ghedini, G.C.; Ciravolo, V.; Marzano, G.; Lamolinara, A.; Zappasodi, R.; Gasparini, P.; Campiglio, M.; Amici, A.; et al. Activated d16HER2 homodimers and SRC kinase mediate optimal efficacy for trastuzumab. Cancer Res. 2014, 74, 6248–6259. [Google Scholar] [CrossRef]
- Volpi, C.C.; Pietrantonio, F.; Gloghini, A.; Fuca, G.; Giordano, S.; Corso, S.; Pruneri, G.; Antista, M.; Cremolini, C.; Fasano, E.; et al. The landscape of d16HER2 splice variant expression across HER2-positive cancers. Sci. Rep. 2019, 9, 3545. [Google Scholar] [CrossRef]
- Verma, S.; Goyal, S.; Kumari, A.; Singh, A.; Jamal, S.; Grover, A. Structural investigations on mechanism of lapatinib resistance caused by HER-2 mutants. PLoS ONE 2018, 13, e0190942. [Google Scholar] [CrossRef]
- Kancha, R.K.; von Bubnoff, N.; Bartosch, N.; Peschel, C.; Engh, R.A.; Duyster, J. Differential sensitivity of ERBB2 kinase domain mutations towards lapatinib. PLoS ONE 2011, 6, e26760. [Google Scholar] [CrossRef]
- Trowe, T.; Boukouvala, S.; Calkins, K.; Cutler, R.E., Jr.; Fong, R.; Funke, R.; Gendreau, S.B.; Kim, Y.D.; Miller, N.; Woolfrey, J.R.; et al. EXEL-7647 inhibits mutant forms of ErbB2 associated with lapatinib resistance and neoplastic transformation. Clin. Cancer Res. 2008, 14, 2465–2475. [Google Scholar] [CrossRef]
- Zuo, W.J.; Jiang, Y.Z.; Wang, Y.J.; Xu, X.E.; Hu, X.; Liu, G.Y.; Wu, J.; Di, G.H.; Yu, K.D.; Shao, Z.M. Dual Characteristics of Novel HER2 Kinase Domain Mutations in Response to HER2-Targeted Therapies in Human Breast Cancer. Clin. Cancer Res. 2016, 22, 4859–4869. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xiao, Q.; Bao, Y.; Wang, W.; Goh, J.; Wang, P.; Yu, Q. HER2-L755S mutation induces hyperactive MAPK and PI3K-mTOR signaling, leading to resistance to HER2 tyrosine kinase inhibitor treatment. Cell Cycle 2019, 18, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.Y.; Qi, S.; Wu, H.; Wang, A.L.; Liu, Q.W.; Li, X.X.; Wang, B.L.; Ge, J.; Zou, F.M.; Chen, C.; et al. CHMFL-26 is a highly potent irreversible HER2 inhibitor for use in the treatment of HER2-positive and HER2-mutant cancers. Acta Pharmacol. Sin. 2022, 43, 2678–2686. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Saez, O.; Chic, N.; Pascual, T.; Adamo, B.; Vidal, M.; Gonzalez-Farre, B.; Sanfeliu, E.; Schettini, F.; Conte, B.; Braso-Maristany, F.; et al. Frequency and spectrum of PIK3CA somatic mutations in breast cancer. Breast Cancer Res. 2020, 22, 45. [Google Scholar] [CrossRef]
- Koren, S.; Reavie, L.; Couto, J.P.; De Silva, D.; Stadler, M.B.; Roloff, T.; Britschgi, A.; Eichlisberger, T.; Kohler, H.; Aina, O.; et al. PIK3CA(H1047R) induces multipotency and multi-lineage mammary tumours. Nature 2015, 525, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Hanker, A.B.; Pfefferle, A.D.; Balko, J.M.; Kuba, M.G.; Young, C.D.; Sanchez, V.; Sutton, C.R.; Cheng, H.; Perou, C.M.; Zhao, J.J.; et al. Mutant PIK3CA accelerates HER2-driven transgenic mammary tumors and induces resistance to combinations of anti-HER2 therapies. Proc. Natl. Acad. Sci. USA 2013, 110, 14372–14377. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Liu, P.; Ohlson, C.; Xu, E.; Symonds, L.; Isabella, A.; Muller, W.J.; Lin, N.U.; Krop, I.E.; Roberts, T.M.; et al. PIK3CA(H1047R)- and Her2-initiated mammary tumors escape PI3K dependency by compensatory activation of MEK-ERK signaling. Oncogene 2016, 35, 2961–2970. [Google Scholar] [CrossRef]
- Eichhorn, P.J.; Gili, M.; Scaltriti, M.; Serra, V.; Guzman, M.; Nijkamp, W.; Beijersbergen, R.L.; Valero, V.; Seoane, J.; Bernards, R.; et al. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res. 2008, 68, 9221–9230. [Google Scholar] [CrossRef]
- Gustin, J.P.; Cosgrove, D.P.; Park, B.H. The PIK3CA gene as a mutated target for cancer therapy. Curr. Cancer Drug Targets 2008, 8, 733–740. [Google Scholar] [CrossRef]
- Leontiadou, H.; Galdadas, I.; Athanasiou, C.; Cournia, Z. Insights into the mechanism of the PIK3CA E545K activating mutation using MD simulations. Sci. Rep. 2018, 8, 15544. [Google Scholar] [CrossRef]
- Sridhar, J.; Komati, R.; Kumar, S. Targeting RPS6K1 for Refractory Breast Cancer Therapy. In Breast Cancer; Mayrovitz, H.N., Ed.; Exon Publications: Brisbane, Australia, 2022. [Google Scholar]
- Huynh, F.C.; Nguyen, D.; Jones, F.E. Trastuzumab stimulation of ribosomal protein S6 kinase 1 (S6K1) predicts de novo trastuzumab resistance. Biochem. Biophys. Res. Commun. 2017, 483, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Zhao, J.X.; Dong, F.; Cao, X.C. ARID1A Mutation in Metastatic Breast Cancer: A Potential Therapeutic Target. Front. Oncol. 2021, 11, 759577. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.M.; Qu, X.; Lin, C.-F.; Huw, L.-Y.; Venkatanarayan, A.; Sokol, E.; Ou, F.-S.; Ihuegbu, N.; Zill, O.A.; Kabbarah, O.; et al. ARID1A mutations confer intrinsic and acquired resistance to cetuximab treatment in colorectal cancer. Nat. Commun. 2022, 13, 5478. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, C.; Zhao, Z. ARID1A: A potential prognostic factor for breast cancer. Tumour Biol. 2014, 35, 4813–4819. [Google Scholar] [CrossRef] [PubMed]
- Berns, K.; Sonnenblick, A.; Gennissen, A.; Brohee, S.; Hijmans, E.M.; Evers, B.; Fumagalli, D.; Desmedt, C.; Loibl, S.; Denkert, C.; et al. Loss of ARID1A Activates ANXA1, which Serves as a Predictive Biomarker for Trastuzumab Resistance. Clin. Cancer Res. 2016, 22, 5238–5248. [Google Scholar] [CrossRef] [PubMed]
- Silva-Oliveira, R.; Pereira, F.F.; Petronilho, S.; Martins, A.T.; Lameirinhas, A.; Constancio, V.; Caldas-Ribeiro, I.; Salta, S.; Lopes, P.; Antunes, L.; et al. Clinical Significance of ARID1A and ANXA1 in HER-2 Positive Breast Cancer. J. Clin. Med. 2020, 9, 3911. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Gu, Y.; Guo, S.; Dai, Q.; Zhang, W. Expressing Status and Correlation of ARID1A and Histone H2B on Breast Cancer. Biomed. Res. Int. 2016, 2016, 7593787. [Google Scholar] [CrossRef]
- Cho, H.D.; Lee, J.E.; Jung, H.Y.; Oh, M.H.; Lee, J.H.; Jang, S.H.; Kim, K.J.; Han, S.W.; Kim, S.Y.; Kim, H.J.; et al. Loss of Tumor Suppressor ARID1A Protein Expression Correlates with Poor Prognosis in Patients with Primary Breast Cancer. J. Breast Cancer 2015, 18, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hou, S.; You, Z.; Li, G.; Xu, S.; Li, X.; Zhang, X.; Lei, B.; Pang, D. Expression and prognostic values of ARID family members in breast cancer. Aging 2021, 13, 5621–5637. [Google Scholar] [CrossRef]
- Chen, Y.C.; Li, H.Y.; Liang, J.L.; Ger, L.P.; Chang, H.T.; Hsiao, M.; Calkins, M.J.; Cheng, H.C.; Chuang, J.H.; Lu, P.J. CTMP, a predictive biomarker for trastuzumab resistance in HER2-enriched breast cancer patient. Oncotarget 2017, 8, 29699–29710. [Google Scholar] [CrossRef]
- Lin, C.H.; Lin, W.D.; Huang, Y.C.; Chen, Y.C.; Loh, Z.J.; Ger, L.P.; Lin, F.C.; Li, H.Y.; Cheng, H.C.; Lee, K.H.; et al. Carboxyl-terminal modulator protein facilitates tumor metastasis in triple-negative breast cancer. Cancer Gene Ther. 2023, 30, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Maira, S.M.; Galetic, I.; Brazil, D.P.; Kaech, S.; Ingley, E.; Thelen, M.; Hemmings, B.A. Carboxyl-terminal modulator protein (CTMP), a negative regulator of PKB/Akt and v-Akt at the plasma membrane. Science 2001, 294, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; Liao, W.C.; Ger, L.P.; Chen, J.C.; Hsu, T.I.; Lee, Y.C.; Chang, H.T.; Chen, Y.C.; Jan, Y.H.; Lee, K.H.; et al. Carboxyl-terminal modulator protein positively regulates Akt phosphorylation and acts as an oncogenic driver in breast cancer. Cancer Res. 2013, 73, 6194–6205. [Google Scholar] [CrossRef] [PubMed]
- Ono, H.; Sakoda, H.; Fujishiro, M.; Anai, M.; Kushiyama, A.; Fukushima, Y.; Katagiri, H.; Ogihara, T.; Oka, Y.; Kamata, H.; et al. Carboxy-terminal modulator protein induces Akt phosphorylation and activation, thereby enhancing antiapoptotic, glycogen synthetic, and glucose uptake pathways. Am. J. Physiol. Cell Physiol. 2007, 293, C1576–C1585. [Google Scholar] [CrossRef] [PubMed]
- Bafna, S.; Kaur, S.; Batra, S.K. Membrane-bound mucins: The mechanistic basis for alterations in the growth and survival of cancer cells. Oncogene 2010, 29, 2893–2904. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Raina, D.; Joshi, M.D.; Kawano, T.; Ren, J.; Kharbanda, S.; Kufe, D. MUC1-C oncoprotein functions as a direct activator of the nuclear factor-kappaB p65 transcription factor. Cancer Res. 2009, 69, 7013–7021. [Google Scholar] [CrossRef] [PubMed]
- Atta Manu, E.; Bedu-Addo, K.; Titiloye, N.A.; Ameh-Mensah, C.; Opoku, F.; Duduyemi, B.M. Expression of Tumour-Associated MUC1 Is a Poor Prognostic Marker in Breast Cancer in Kumasi, Ghana. J. Oncol. 2020, 2020, 9752952. [Google Scholar] [CrossRef] [PubMed]
- Fessler, S.P.; Wotkowicz, M.T.; Mahanta, S.K.; Bamdad, C. MUC1* is a determinant of trastuzumab (Herceptin) resistance in breast cancer cells. Breast Cancer Res. Treat. 2009, 118, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Liang, H.; Hao, C.; Yang, X.; Cui, X. Overexpression of MUC1 predicts poor prognosis in patients with breast cancer. Oncol. Rep. 2019, 41, 801–810. [Google Scholar] [CrossRef]
- Raina, D.; Uchida, Y.; Kharbanda, A.; Rajabi, H.; Panchamoorthy, G.; Jin, C.; Kharbanda, S.; Scaltriti, M.; Baselga, J.; Kufe, D. Targeting the MUC1-C oncoprotein downregulates HER2 activation and abrogates trastuzumab resistance in breast cancer cells. Oncogene 2014, 33, 3422–3431. [Google Scholar] [CrossRef]
- Sand, A.; Piacsek, M.; Donohoe, D.L.; Duffin, A.T.; Riddell, G.T.; Sun, C.; Tang, M.; Rovin, R.A.; Tjoe, J.A.; Yin, J. WEE1 inhibitor, AZD1775, overcomes trastuzumab resistance by targeting cancer stem-like properties in HER2-positive breast cancer. Cancer Lett. 2020, 472, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.; Friedlander, E.; Tanner, M.; Kapanen, A.I.; Carraway, K.L.; Isola, J.; Jovin, T.M. Decreased accessibility and lack of activation of ErbB2 in JIMT-1, a herceptin-resistant, MUC4-expressing breast cancer cell line. Cancer Res. 2005, 65, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Price-Schiavi, S.A.; Jepson, S.; Li, P.; Arango, M.; Rudland, P.S.; Yee, L.; Carraway, K.L. Rat Muc4 (sialomucin complex) reduces binding of anti-ErbB2 antibodies to tumor cell surfaces, a potential mechanism for herceptin resistance. Int. J. Cancer 2002, 99, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Mercogliano, M.F.; De Martino, M.; Venturutti, L.; Rivas, M.A.; Proietti, C.J.; Inurrigarro, G.; Frahm, I.; Allemand, D.H.; Deza, E.G.; Ares, S.; et al. TNFalpha-Induced Mucin 4 Expression Elicits Trastuzumab Resistance in HER2-Positive Breast Cancer. Clin. Cancer Res. 2017, 23, 636–648. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.H.; Zheng, Z.Q.; Jia, S.C.; Liu, S.N.; Xiao, X.F.; Chen, G.Y.; Liang, W.Q.; Lu, X.F. Trastuzumab resistance in HER2-positive breast cancer: Mechanisms, emerging biomarkers and targeting agents. Front. Oncol. 2022, 12, 1006429. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Fernández-Nogueira, P.; Mancino, M.; Fuster, G.; López-Plana, A.; Jauregui, P.; Almendro, V.; Enreig, E.; Menéndez, S.; Rojo, F.; Noguera-Castells, A.; et al. Tumor-Associated Fibroblasts Promote HER2-Targeted Therapy Resistance through FGFR2 Activation. Clin. Cancer Res. 2020, 26, 1432–1448. [Google Scholar] [CrossRef] [PubMed]
- Watson, S.S.; Dane, M.; Chin, K.; Tatarova, Z.; Liu, M.; Liby, T.; Thompson, W.; Smith, R.; Nederlof, M.; Bucher, E.; et al. Microenvironment-Mediated Mechanisms of Resistance to HER2 Inhibitors Differ between HER2+ Breast Cancer Subtypes. Cell Syst. 2018, 6, 329–342.e326. [Google Scholar] [CrossRef]
- Vathiotis, I.A.; Moutafi, M.K.; Divakar, P.; Aung, T.N.; Qing, T.; Fernandez, A.; Yaghoobi, V.; El-Abed, S.; Wang, Y.; Guillaume, S.; et al. Alpha-smooth Muscle Actin Expression in the Stroma Predicts Resistance to Trastuzumab in Patients with Early-stage HER2-positive Breast Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 6156–6163. [Google Scholar] [CrossRef]
- Duro-Sánchez, S.; Alonso, M.R.; Arribas, J. Immunotherapies against HER2-Positive Breast Cancer. Cancers 2023, 15, 1069. [Google Scholar] [CrossRef]
- Kurozumi, S.; Inoue, K.; Matsumoto, H.; Fujii, T.; Horiguchi, J.; Oyama, T.; Kurosumi, M.; Shirabe, K. Clinicopathological values of PD-L1 expression in HER2-positive breast cancer. Sci. Rep. 2019, 9, 16662. [Google Scholar] [CrossRef] [PubMed]
- Chaganty, B.K.R.; Qiu, S.; Gest, A.; Lu, Y.; Ivan, C.; Calin, G.A.; Weiner, L.M.; Fan, Z. Trastuzumab upregulates PD-L1 as a potential mechanism of trastuzumab resistance through engagement of immune effector cells and stimulation of IFNγ secretion. Cancer Lett. 2018, 430, 47–56. [Google Scholar] [CrossRef]
- Vo, T.H.; El-Sherbieny Abdelaal, E.; Jordan, E.; O’Donovan, O.; McNeela, E.A.; Mehta, J.P.; Rani, S. miRNAs as biomarkers of therapeutic response to HER2-targeted treatment in breast cancer: A systematic review. Biochem. Biophys. Rep. 2024, 37, 101588. [Google Scholar] [CrossRef]
- Luo, L.; Zhang, Z.; Qiu, N.; Ling, L.; Jia, X.; Song, Y.; Li, H.; Li, J.; Lyu, H.; Liu, H.; et al. Disruption of FOXO3a-miRNA feedback inhibition of IGF2/IGF-1R/IRS1 signaling confers Herceptin resistance in HER2-positive breast cancer. Nat. Commun. 2021, 12, 2699. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Bai, W.; Zhu, H.; Zhang, X.; Chen, Y.; Wang, L.; Yang, A.; Zhao, J.; Jia, L. MiR-221 promotes trastuzumab-resistance and metastasis in HER2-positive breast cancers by targeting PTEN. BMB Rep. 2014, 47, 268–273. [Google Scholar] [CrossRef]
- Wynn, C.S.; Tang, S.C. Anti-HER2 therapy in metastatic breast cancer: Many choices and future directions. Cancer Metastasis Rev. 2022, 41, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Collins, D.M.; Madden, S.F.; Gaynor, N.; AlSultan, D.; Le Gal, M.; Eustace, A.J.; Gately, K.A.; Hughes, C.; Davies, A.M.; Mahgoub, T.; et al. Effects of HER Family-targeting Tyrosine Kinase Inhibitors on Antibody-dependent Cell-mediated Cytotoxicity in HER2-expressing Breast Cancer. Clin. Cancer Res. 2021, 27, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Li, W.; Jin, N.; Cai, D.; Sun, J.; Sun, C.; Yang, F.; Wu, X.; Huang, X.; Wang, B.; et al. Treatment with pyrotinib-based therapy in lapatinib-resistant HER2-positive metastatic breast cancer: A multicenter real-world study. Ther. Adv. Med. Oncol. 2022, 14, 17588359221085232. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, K.L.; Bedard, P.L.; Lee, S.-H.; Lin, C.-C.; Tabernero, J.; Alsina, M.; Cohen, E.; Baselga, J.; Blumenschein, G.; Graham, D.M.; et al. A phase I open-label dose-escalation study of the anti-HER3 monoclonal antibody LJM716 in patients with advanced squamous cell carcinoma of the esophagus or head and neck and HER2-overexpressing breast or gastric cancer. BMC Cancer 2017, 17, 646. [Google Scholar] [CrossRef]
- Holbro, T.; Beerli, R.R.; Maurer, F.; Koziczak, M.; Barbas, C.F., 3rd; Hynes, N.E. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc. Natl. Acad. Sci. USA 2003, 100, 8933–8938. [Google Scholar] [CrossRef]
- Dorraji, E.; Borgen, E.; Segura-Pena, D.; Rawat, P.; Smorodina, E.; Dunn, C.; Greiff, V.; Sekulic, N.; Russnes, H.; Kyte, J.A. Development of a High-Affinity Antibody against the Tumor-Specific and Hyperactive 611-p95HER2 Isoform. Cancers 2022, 14, 4859. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.; Angelini, P.D.; Laos, S.; Bach-Faig, A.; Cunningham, M.P.; Ferrer-Ramon, C.; Luque-Garcia, A.; Garcia-Castillo, J.; Parra-Palau, J.L.; Scaltriti, M.; et al. A naturally occurring HER2 carboxy-terminal fragment promotes mammary tumor growth and metastasis. Mol. Cell. Biol. 2009, 29, 3319–3331. [Google Scholar] [CrossRef]
- Ma, J.; Mo, Y.; Tang, M.; Shen, J.; Qi, Y.; Zhao, W.; Huang, Y.; Xu, Y.; Qian, C. Bispecific Antibodies: From Research to Clinical Application. Front. Immunol. 2021, 12, 626616. [Google Scholar] [CrossRef] [PubMed]
- Baeuerle, P.A.; Wesche, H. T-cell-engaging antibodies for the treatment of solid tumors: Challenges and opportunities. Curr. Opin. Oncol. 2022, 34, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Rius Ruiz, I.; Vicario, R.; Morancho, B.; Morales, C.B.; Arenas, E.J.; Herter, S.; Freimoser-Grundschober, A.; Somandin, J.; Sam, J.; Ast, O.; et al. p95HER2-T cell bispecific antibody for breast cancer treatment. Sci. Transl. Med. 2018, 10, eaat1445. [Google Scholar] [CrossRef] [PubMed]
- Kodack, D.P.; Askoxylakis, V.; Ferraro, G.B.; Fukumura, D.; Jain, R.K. Emerging strategies for treating brain metastases from breast cancer. Cancer Cell 2015, 27, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Li, L.; Qiu, Y.; Sun, W.; Ren, T.; Lv, Y.; Liu, M.; Wang, X.; Tao, H.; Zhao, L.; et al. A novel humanized MUC1 antibody-drug conjugate for the treatment of trastuzumab-resistant breast cancer. Acta Biochim. Biophys. Sin. 2021, 53, 1625–1639. [Google Scholar] [CrossRef]
- Chang, H.L.; Schwettmann, B.; McArthur, H.L.; Chan, I.S. Antibody-drug conjugates in breast cancer: Overcoming resistance and boosting immune response. J. Clin. Investig. 2023, 133, e172156. [Google Scholar] [CrossRef]
- Iwata, T.N.; Ishii, C.; Ishida, S.; Ogitani, Y.; Wada, T.; Agatsuma, T. A HER2-Targeting Antibody-Drug Conjugate, Trastuzumab Deruxtecan (DS-8201a), Enhances Antitumor Immunity in a Mouse Model. Mol. Cancer Ther. 2018, 17, 1494–1503. [Google Scholar] [CrossRef]
- Muller, P.; Kreuzaler, M.; Khan, T.; Thommen, D.S.; Martin, K.; Glatz, K.; Savic, S.; Harbeck, N.; Nitz, U.; Gluz, O.; et al. Trastuzumab emtansine (T-DM1) renders HER2+ breast cancer highly susceptible to CTLA-4/PD-1 blockade. Sci. Transl. Med. 2015, 7, 315ra188. [Google Scholar] [CrossRef]
- Barok, M.; Joensuu, H.; Isola, J. Trastuzumab emtansine: Mechanisms of action and drug resistance. Breast Cancer Res. 2014, 16, 209. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.P.; Huang, W.L.; Lee, C.H.; Hsu, H.P.; Huang, W.L.; Liu, Y.Y.; Su, W.C. PI3K inhibitors in trastuzumab-resistant HER2-positive breast cancer cells with PI3K pathway alterations. Am. J. Cancer Res. 2022, 12, 3067–3082. [Google Scholar] [PubMed]
- Fujimoto, Y.; Morita, T.Y.; Ohashi, A.; Haeno, H.; Hakozaki, Y.; Fujii, M.; Kashima, Y.; Kobayashi, S.S.; Mukohara, T. Combination treatment with a PI3K/Akt/mTOR pathway inhibitor overcomes resistance to anti-HER2 therapy in PIK3CA-mutant HER2-positive breast cancer cells. Sci. Rep. 2020, 10, 21762. [Google Scholar] [CrossRef] [PubMed]
- André, F.; O’Regan, R.; Ozguroglu, M.; Toi, M.; Xu, B.; Jerusalem, G.; Masuda, N.; Wilks, S.; Arena, F.; Isaacs, C.; et al. Everolimus for women with trastuzumab-resistant, HER2-positive, advanced breast cancer (BOLERO-3): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2014, 15, 580–591. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Santagata, S.; Lin, N.U. Inhibiting HSP90 to treat cancer: A strategy in evolution. Curr. Mol. Med. 2012, 12, 1108–1124. [Google Scholar] [CrossRef] [PubMed]
- Powers, M.V.; Workman, P. Targeting of multiple signalling pathways by heat shock protein 90 molecular chaperone inhibitors. Endocr. Relat. Cancer 2006, 13 (Suppl. 1), S125–S135. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Bagatell, R.; Falsey, R. The stress response: Implications for the clinical development of hsp90 inhibitors. Curr. Cancer Drug Targets 2003, 3, 349–358. [Google Scholar] [CrossRef]
- Whitesell, L.; Santagata, S.; Mendillo, M.L.; Lin, N.U.; Proia, D.A.; Lindquist, S. HSP90 empowers evolution of resistance to hormonal therapy in human breast cancer models. Proc. Natl. Acad. Sci. USA 2014, 111, 18297–18302. [Google Scholar] [CrossRef] [PubMed]
- Chandarlapaty, S.; Scaltriti, M.; Angelini, P.; Ye, Q.; Guzman, M.; Hudis, C.A.; Norton, L.; Solit, D.B.; Arribas, J.; Baselga, J.; et al. Inhibitors of HSP90 block p95-HER2 signaling in Trastuzumab-resistant tumors and suppress their growth. Oncogene 2010, 29, 325–334. [Google Scholar] [CrossRef]
- O’Sullivan, C.C.; Smith, K.L. Therapeutic Considerations in Treating HER2-Positive Metastatic Breast Cancer. Curr. Breast Cancer Rep. 2014, 6, 169–182. [Google Scholar] [CrossRef]
- Jhaveri, K.; Wang, R.; Teplinsky, E.; Chandarlapaty, S.; Solit, D.; Cadoo, K.; Speyer, J.; D’Andrea, G.; Adams, S.; Patil, S.; et al. A phase I trial of ganetespib in combination with paclitaxel and trastuzumab in patients with human epidermal growth factor receptor-2 (HER2)-positive metastatic breast cancer. Breast Cancer Res. 2017, 19, 89. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Huang, W.; Liu, R.; Kong, Y.; Liu, Y.; Chen, X.; Xu, J. Synergistic Activity of the HSP90 Inhibitor Ganetespib With Lapatinib Reverses Acquired Lapatinib Resistance in HER2-Positive Breast Cancer Cells. Front. Pharmacol. 2021, 12, 651516. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Kim, Y.J.; Park, S.; Park, M.; Farrand, L.; Nguyen, C.T.; Ann, J.; Nam, G.; Park, H.J.; Lee, J.; et al. A novel HSP90 inhibitor targeting the C-terminal domain attenuates trastuzumab resistance in HER2-positive breast cancer. Mol. Cancer 2020, 19, 161. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Lee, H.J.; Lee, J.; Hong, J.; Hong Kim, Y.; Disis, M.L.; Gim, J.A.; Park, K.H. Novel peptide-based vaccine targeting heat shock protein 90 induces effective antitumor immunity in a HER2+ breast cancer murine model. J. Immunother. Cancer 2022, 10. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, R.; Kheraldine, H.S.; Meskin, N.; Vranic, S.; Al Moustafa, A.E. Crosstalk between HER2 and PD-1/PD-L1 in Breast Cancer: From Clinical Applications to Mathematical Models. Cancers 2020, 12, 636. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Lee, S.J.; Kim, Y.K.; Park, W.Y.; Park, D.Y.; Kim, J.Y.; Lee, C.H.; Gong, G.; Huh, G.Y.; Choi, K.U. Programmed death-ligand 1 (PD-L1) expression in tumour cell and tumour infiltrating lymphocytes of HER2-positive breast cancer and its prognostic value. Sci. Rep. 2017, 7, 11671. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Dong, P.; Ren, M.; Song, Y.; Qian, X.; Yang, Y.; Li, S.; Zhang, X.; Liu, F. PD-L1 Expression Is Associated with Tumor FOXP3(+) Regulatory T-Cell Infiltration of Breast Cancer and Poor Prognosis of Patient. J. Cancer 2016, 7, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Ubago, J.M.; Blanco, L.Z.; Shen, T.; Siziopikou, K.P. The PD-1/PD-L1 Axis in HER2+ Ductal Carcinoma In Situ (DCIS) of the Breast. Am. J. Clin. Pathol. 2019, 152, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Muenst, S.; Schaerli, A.R.; Gao, F.; Däster, S.; Trella, E.; Droeser, R.A.; Muraro, M.G.; Zajac, P.; Zanetti, R.; Gillanders, W.E.; et al. Expression of programmed death ligand 1 (PD-L1) is associated with poor prognosis in human breast cancer. Breast Cancer Res. Treat. 2014, 146, 15–24. [Google Scholar] [CrossRef]
- Stagg, J.; Loi, S.; Divisekera, U.; Ngiow, S.F.; Duret, H.; Yagita, H.; Teng, M.W.; Smyth, M.J. Anti-ErbB-2 mAb therapy requires type I and II interferons and synergizes with anti-PD-1 or anti-CD137 mAb therapy. Proc. Natl. Acad. Sci. USA 2011, 108, 7142–7147. [Google Scholar] [CrossRef]
- Loi, S.; Giobbie-Hurder, A.; Gombos, A.; Bachelot, T.; Hui, R.; Curigliano, G.; Campone, M.; Biganzoli, L.; Bonnefoi, H.; Jerusalem, G.; et al. Pembrolizumab plus trastuzumab in trastuzumab-resistant, advanced, HER2-positive breast cancer (PANACEA): A single-arm, multicentre, phase 1b-2 trial. Lancet Oncol. 2019, 20, 371–382. [Google Scholar] [CrossRef]
- Emens, L.A.; Esteva, F.J.; Beresford, M.; Saura, C.; De Laurentiis, M.; Kim, S.-B.; Im, S.-A.; Wang, Y.; Salgado, R.; Mani, A.; et al. Trastuzumab emtansine plus atezolizumab versus trastuzumab emtansine plus placebo in previously treated, HER2-positive advanced breast cancer (KATE2): A phase 2, multicentre, randomised, double-blind trial. Lancet Oncol. 2020, 21, 1283–1295. [Google Scholar] [CrossRef]
- Cortes, J.; Rugo, H.S.; Cescon, D.W.; Im, S.A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Perez-Garcia, J.; Iwata, H.; et al. Pembrolizumab plus Chemotherapy in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2022, 387, 217–226. [Google Scholar] [CrossRef]
- Hurvitz, S.A.; Bachelot, T.; Bianchini, G.; Harbeck, N.; Loi, S.; Park, Y.H.; Prat, A.; Gilham, L.; Boulet, T.; Gochitashvili, N.; et al. ASTEFANIA: Adjuvant ado-trastuzumab emtansine and atezolizumab for high-risk, HER2-positive breast cancer. Future Oncol. 2022, 18, 3563–3572. [Google Scholar] [CrossRef]
- Wu, X.; Huang, S.; He, W.; Song, M. Emerging insights into mechanisms of trastuzumab resistance in HER2-positive cancers. Int. Immunopharmacol. 2023, 122, 110602. [Google Scholar] [CrossRef]
- Vazquez-Martin, A.; Colomer, R.; Brunet, J.; Lupu, R.; Menendez, J.A. Overexpression of fatty acid synthase gene activates HER1/HER2 tyrosine kinase receptors in human breast epithelial cells. Cell Prolif. 2008, 41, 59–85. [Google Scholar] [CrossRef]
- Corominas-Faja, B.; Vellon, L.; Cuyàs, E.; Buxó, M.; Martin-Castillo, B.; Serra, D.; García, J.; Lupu, R.; Menendez, J.A. Clinical and therapeutic relevance of the metabolic oncogene fatty acid synthase in HER2+ breast cancer. Histol. Histopathol. 2017, 32, 687–698. [Google Scholar] [CrossRef]
- Feng, W.W.; Wilkins, O.; Bang, S.; Ung, M.; Li, J.; An, J.; Del Genio, C.; Canfield, K.; DiRenzo, J.; Wells, W.; et al. CD36-Mediated Metabolic Rewiring of Breast Cancer Cells Promotes Resistance to HER2-Targeted Therapies. Cell Rep. 2019, 29, 3405–3420.e3405. [Google Scholar] [CrossRef]
- Ligorio, F.; Di Cosimo, S.; Verderio, P.; Ciniselli, C.M.; Pizzamiglio, S.; Castagnoli, L.; Dugo, M.; Galbardi, B.; Salgado, R.; Loi, S.; et al. Predictive Role of CD36 Expression in HER2-Positive Breast Cancer Patients Receiving Neoadjuvant Trastuzumab. JNCI J. Natl. Cancer Inst. 2022, 114, 1720–1727. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R.; Colomer, R. Targeting fatty acid synthase: Potential for therapeutic intervention in her-2/neu-overexpressing breast cancer. Drug News Perspect. 2005, 18, 375–385. [Google Scholar] [CrossRef]
- Wang, X.; Tian, W. Green tea epigallocatechin gallate: A natural inhibitor of fatty-acid synthase. Biochem. Biophys. Res. Commun. 2001, 288, 1200–1206. [Google Scholar] [CrossRef]
- Eddy, S.F.; Kane, S.E.; Sonenshein, G.E. Trastuzumab-resistant HER2-driven breast cancer cells are sensitive to epigallocatechin-3 gallate. Cancer Res. 2007, 67, 9018–9023. [Google Scholar] [CrossRef]
- Blancafort, A.; Giró-Perafita, A.; Oliveras, G.; Palomeras, S.; Turrado, C.; Campuzano, Ò.; Carrión-Salip, D.; Massaguer, A.; Brugada, R.; Palafox, M.; et al. Dual fatty acid synthase and HER2 signaling blockade shows marked antitumor activity against breast cancer models resistant to anti-HER2 drugs. PLoS ONE 2015, 10, e0131241. [Google Scholar] [CrossRef]
- Corona, S.P.; Ravelli, A.; Cretella, D.; Cappelletti, M.R.; Zanotti, L.; Dester, M.; Gobbi, A.; Petronini, P.G.; Generali, D. CDK4/6 inhibitors in HER2-positive breast cancer. Crit. Rev. Oncol. Hematol. 2017, 112, 208–214. [Google Scholar] [CrossRef]
- Goel, S.; Wang, Q.; Watt, A.C.; Tolaney, S.M.; Dillon, D.A.; Li, W.; Ramm, S.; Palmer, A.C.; Yuzugullu, H.; Varadan, V.; et al. Overcoming Therapeutic Resistance in HER2-Positive Breast Cancers with CDK4/6 Inhibitors. Cancer Cell 2016, 29, 255–269. [Google Scholar] [CrossRef]
- Ciruelos, E.; Villagrasa, P.; Pascual, T.; Oliveira, M.; Pernas, S.; Paré, L.; Escrivá-de-Romaní, S.; Manso, L.; Adamo, B.; Martínez, E.; et al. Palbociclib and Trastuzumab in HER2-Positive Advanced Breast Cancer: Results from the Phase II SOLTI-1303 PATRICIA Trial. Clin. Cancer Res. 2020, 26, 5820–5829. [Google Scholar] [CrossRef]
- Tolaney, S.M.; Wardley, A.M.; Zambelli, S.; Hilton, J.F.; Troso-Sandoval, T.A.; Ricci, F.; Im, S.A.; Kim, S.B.; Johnston, S.R.; Chan, A.; et al. Abemaciclib plus trastuzumab with or without fulvestrant versus trastuzumab plus standard-of-care chemotherapy in women with hormone receptor-positive, HER2-positive advanced breast cancer (monarcHER): A randomised, open-label, phase 2 trial. Lancet Oncol. 2020, 21, 763–775. [Google Scholar] [CrossRef]
- Yan, M.; Niu, L.; Lv, H.; Zhang, M.; Wang, J.; Liu, Z.; Chen, X.; Lu, Z.; Zhang, C.; Zeng, H.; et al. Dalpiciclib and pyrotinib in women with HER2-positive advanced breast cancer: A single-arm phase II trial. Nat. Commun. 2023, 14, 6272. [Google Scholar] [CrossRef]
- Arpino, G.; Wiechmann, L.; Osborne, C.K.; Schiff, R. Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: Molecular mechanism and clinical implications for endocrine therapy resistance. Endocr. Rev. 2008, 29, 217–233. [Google Scholar] [CrossRef]
- Cotrim, C.Z.; Fabris, V.; Doria, M.L.; Lindberg, K.; Gustafsson, J.; Amado, F.; Lanari, C.; Helguero, L.A. Estrogen receptor beta growth-inhibitory effects are repressed through activation of MAPK and PI3K signalling in mammary epithelial and breast cancer cells. Oncogene 2013, 32, 2390–2402. [Google Scholar] [CrossRef]
- Rimawi, M.F.; Wiechmann, L.S.; Wang, Y.C.; Huang, C.; Migliaccio, I.; Wu, M.F.; Gutierrez, C.; Hilsenbeck, S.G.; Arpino, G.; Massarweh, S.; et al. Reduced dose and intermittent treatment with lapatinib and trastuzumab for potent blockade of the HER pathway in HER2/neu-overexpressing breast tumor xenografts. Clin. Cancer Res. 2011, 17, 1351–1361. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-C.; Morrison, G.; Gillihan, R.; Guo, J.; Ward, R.M.; Fu, X.; Botero, M.F.; Healy, N.A.; Hilsenbeck, S.G.; Phillips, G.L.; et al. Different mechanisms for resistance to trastuzumab versus lapatinib in HER2- positive breast cancers—Role of estrogen receptor and HER2 reactivation. Breast Cancer Res. 2011, 13, R121. [Google Scholar] [CrossRef] [PubMed]
- Pegram, M.; Jackisch, C.; Johnston, S.R.D. Estrogen/HER2 receptor crosstalk in breast cancer: Combination therapies to improve outcomes for patients with hormone receptor-positive/HER2-positive breast cancer. npj Breast Cancer 2023, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Prat Aparicio, A.; Cortes Castan, J.; Pare, L.; Galvan, P.; Bermejo, B.; Martínez, N.; Vidal, M.; Pernas, S.; López, R.; Muñoz, M.; et al. Abstract S3-03: PAM50 intrinsic subtype as a predictor of pathological complete response following neoadjuvant dual HER2 blockade without chemotherapy in HER2-positive breast cancer: First results of the PAMELA clinical trial. Cancer Res. 2017, 77, S3-03. [Google Scholar] [CrossRef]
- Pinhel, I.; Hills, M.; Drury, S.; Salter, J.; Sumo, G.; A’Hern, R.; Bliss, J.M.; Sestak, I.; Cuzick, J.; Barrett-Lee, P.; et al. ER and HER2 expression are positively correlated in HER2 non-overexpressing breast cancer. Breast Cancer Res. 2012, 14, R46. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, S.; Ye, C.; Liu, Q.; Cheng, Y.; Ye, J.; Liu, Y.; Duan, X.; Xin, L.; Zhang, H.; et al. Androgen Receptor: A New Marker to Predict Pathological Complete Response in HER2-Positive Breast Cancer Patients Treated with Trastuzumab Plus Pertuzumab Neoadjuvant Therapy. J. Pers. Med. 2022, 12, 261. [Google Scholar] [CrossRef]
- Gordon, M.A.; D’Amato, N.C.; Gu, H.; Babbs, B.; Wulfkuhle, J.; Petricoin, E.F.; Gallagher, I.; Dong, T.; Torkko, K.; Liu, B.; et al. Synergy between Androgen Receptor Antagonism and Inhibition of mTOR and HER2 in Breast Cancer. Mol. Cancer Ther. 2017, 16, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Wardley, A.; Cortes, J.; Provencher, L.; Miller, K.; Chien, A.J.; Rugo, H.S.; Steinberg, J.; Sugg, J.; Tudor, I.C.; Huizing, M.; et al. The efficacy and safety of enzalutamide with trastuzumab in patients with HER2+ and androgen receptor-positive metastatic or locally advanced breast cancer. Breast Cancer Res. Treat. 2021, 187, 155–165. [Google Scholar] [CrossRef]
- Lammers, P.; Criscitiello, C.; Curigliano, G.; Jacobs, I. Barriers to the Use of Trastuzumab for HER2+ Breast Cancer and the Potential Impact of Biosimilars: A Physician Survey in the United States and Emerging Markets. Pharmaceuticals 2014, 7, 943–953. [Google Scholar] [CrossRef]


| Anti-HER2 Regimen | Mechanism of Action | Mechanism For/Against Resistance |
|---|---|---|
| monoclonal antibodies (mAbs) | binds to ECD of HER2 receptor, inhibiting downstream signaling pathways (PI3K, MAPK) and inducing immune responses (ADCC) | challenged by various mechanisms of resistance, including receptor structural mutations and mutations in downstream signaling pathways |
| trastuzumab | binds to ECD IV more strongly inhibits downstream signaling pathways (PI3K/AKT) inhibits ligand-independent HER2/HER3 dimerization; inhibits HER2/HER2 homodimerization | structural mutations prevent binding; mutations in downstream signaling pathways bypass trastuzumab binding altogether |
| pertuzumab | binds to ECD II inhibits ligand-dependent HER2 dimerization | designed to delay trastuzumab resistance, potentially by enhancing trastuzumab-induced ADCC; however, similarly susceptible to resistance |
| margetuximab | binds to ECD IV structurally similar to trastuzumab; Fc domain of antibody optimized to enhance ADCC | designed to delay trastuzumab resistance by enhancing ADCC, but has not proven very effective |
| antibody–drug conjugates (ADCs) | deliver potent cytotoxic drugs specifically to antibody targets (HER2+ cells) | can overcome trastuzumab resistance via increased anti-tumor immunity |
| T-DM1 | includes the anti-tumor activities of trastuzumab and DM1, which inhibits microtubule polymerization once the HER2-T-DM1 complex is internalized via receptor-mediated endocytosis and lysed | resistance can arise from mechanisms of resistance to trastuzumab, dysfunctional intracellular trafficking (which reverses internalization of HER2-T-DM1 complexes), and impairment of DM1-mediated toxicity |
| T-DXd | includes anti-tumor activities of trastuzumab and DXd, a topoisomerase inhibitor in contrast to T-DM1, antibody–drug linker is cleavable, allowing DXd action in nearby non-HER2 expressing cells in contrast to T-DM1, higher drug-to-antibody ratio | decrease in HER2 expression and payload resistance confers resistance to T-DXd |
| tyrosine kinase inhibitors (TKIs) | block intracellular TK domain of HER receptors, preventing phosphorylation and inhibiting downstream signaling pathways; can cross BBB | mutations to TK domain of HER2 receptor may confer resistance; however, are effective against many mechanisms of resistance to mAbs and ADCs |
| lapatinib | reversibly binds to TK domain of HER1/HER2 receptors | |
| neratinib | irreversibly binds to TK domain of HER1/HER2/HER4 receptors | |
| tucatinib | reversible; binds specifically to HER2 receptor |
| Anti-HER2 Regimen | In Combination With | Clinical Trial | Treatment Setting |
|---|---|---|---|
| P13K/Akt/mTOR inhibitors | |||
| trastuzumab (mAb) | everolimus (mTOR inhibitor) + vinorelbine (chemo): improved PFS | NCT01007942 | trastuzumab-resistant and taxane-pretreated, HER2+ mBC |
| trastuzumab (mAb) | alpelisib (PI3K inhibitor) | NCT05063786 * | HER2+ BC bearing PIK3CA mutations, previously treated with HER2 inhibitors |
| HSP inhibitors | |||
| trasuzumab (mAb) | ganetespib + paclitaxel (chemo): clinical benefit rate of 44% | NCT02060253 | Trastuzumab-refractory HER2+ mBC |
| PD-1/PD-L1 inhibitors | |||
| trasuzumab (mAb) | pembrolizumab: some degree of clinical benefit; ORR of 15% | NCT02129556 | Trastuzumab-resistant mBC, PD-L1+ |
| T-DM1 (ADC) | atezolizumab: no obvious improvement of PFS | NCT02924883 | HER2+ mBC, PD-L1+, previously treated |
| metabolic inhibitors | |||
| trasuzumab (mAb) | TVB-2640 (FASN inhibitor) + paclitaxel or endocrine therapy | NCT03179904 * | HER2+ mBC |
| CDK4/6 inhibitors | |||
| trasuzumab (mAb) | palbociclib: promising survival outcomes | NCT02448420 | ER+/HER2+ mBC, resistant to HER2 inhibitor |
| trasuzumab (mAb) | abemaciclib: improvement of PFS | NCT02675231 | HR+/HER2+ advanced BC |
| pryotinib (TKI) | dalpiciclib: promising activity, manageable toxicity | NCT05328440 | HER2+ advanced BC |
| hormonal therapy | |||
| lapatinib (TKI) + trasuzumab (mAb) | letrozole or tamoxifen: increased pCR + pCRB rates in HER2+/HR+ group | NCT01973660 | stage I-IIIA HER2+ BC |
| trasuzumab (mAb) | enzalutamide (androgen receptor inhibitor): CBR24 of 24%, median PFS of 3.4 months | NCT02091960 | HER2+ BC, previously received ≥ 1 anti-HER2 regimen |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, A.; Chen, Y.; Wang, L.S.; Cusick, J.K.; Shi, Y. Depicting Biomarkers for HER2-Inhibitor Resistance: Implication for Therapy in HER2-Positive Breast Cancer. Cancers 2024, 16, 2635. https://doi.org/10.3390/cancers16152635
Cai A, Chen Y, Wang LS, Cusick JK, Shi Y. Depicting Biomarkers for HER2-Inhibitor Resistance: Implication for Therapy in HER2-Positive Breast Cancer. Cancers. 2024; 16(15):2635. https://doi.org/10.3390/cancers16152635
Chicago/Turabian StyleCai, Alvan, Yuan Chen, Lily S. Wang, John K. Cusick, and Yihui Shi. 2024. "Depicting Biomarkers for HER2-Inhibitor Resistance: Implication for Therapy in HER2-Positive Breast Cancer" Cancers 16, no. 15: 2635. https://doi.org/10.3390/cancers16152635
APA StyleCai, A., Chen, Y., Wang, L. S., Cusick, J. K., & Shi, Y. (2024). Depicting Biomarkers for HER2-Inhibitor Resistance: Implication for Therapy in HER2-Positive Breast Cancer. Cancers, 16(15), 2635. https://doi.org/10.3390/cancers16152635