Radiosynthesis and Preclinical Evaluation of 18F-Labeled Estradiol Derivatives with Different Lipophilicity for PET Imaging of Breast Cancer


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. General
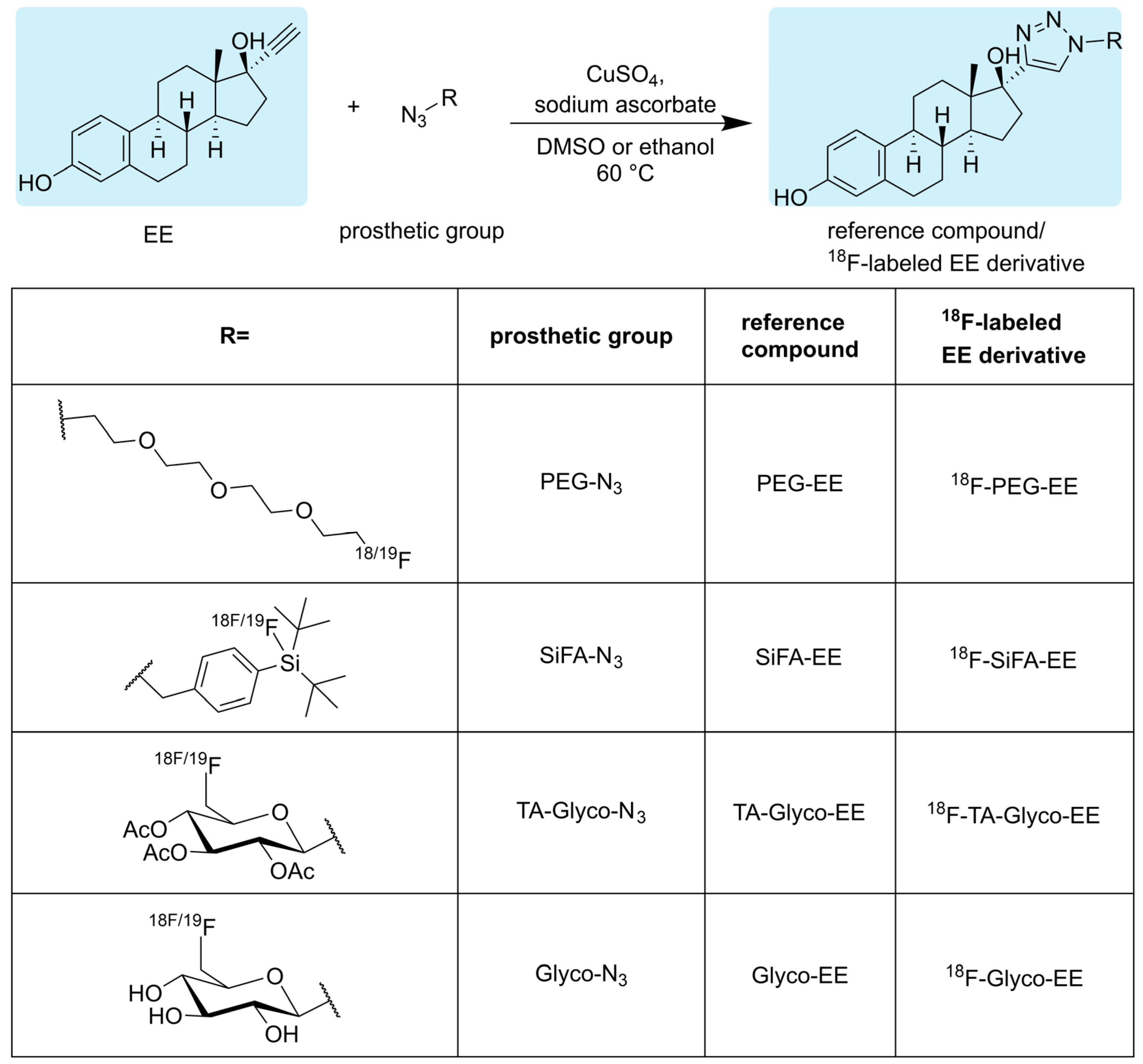
2.2. Syntheses
2.2.1. Synthesis of Glyco-EE
2.2.2. Synthesis of TA-Glyco-EE
2.2.3. Synthesis of PEG-EE
2.2.4. Synthesis of SiFA-EE
2.3. Radiosyntheses
2.3.1. Radiosynthesis of 18F-TA-Glyco-EE and 18F-Glyco-EE
2.3.2. Radiosynthesis of 18F-PEG-EE
2.3.3. Radiosynthesis of 18F-SiFA-EE
2.4. In Vitro Studies
2.4.1. Determination of logD7.4
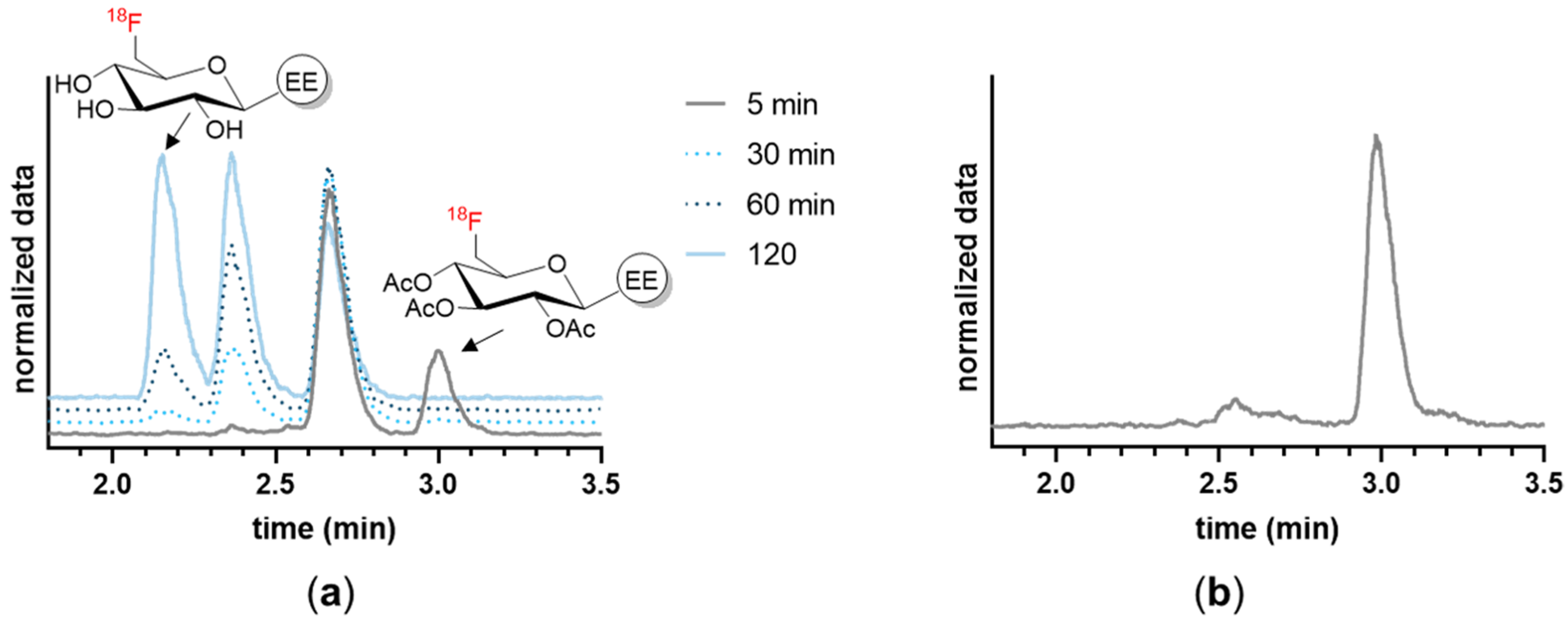
2.4.2. Stability in Serum and Plasma In Vitro
2.4.3. Plasma Protein Binding
2.4.4. Cell Lines
2.4.5. Stability in Cells
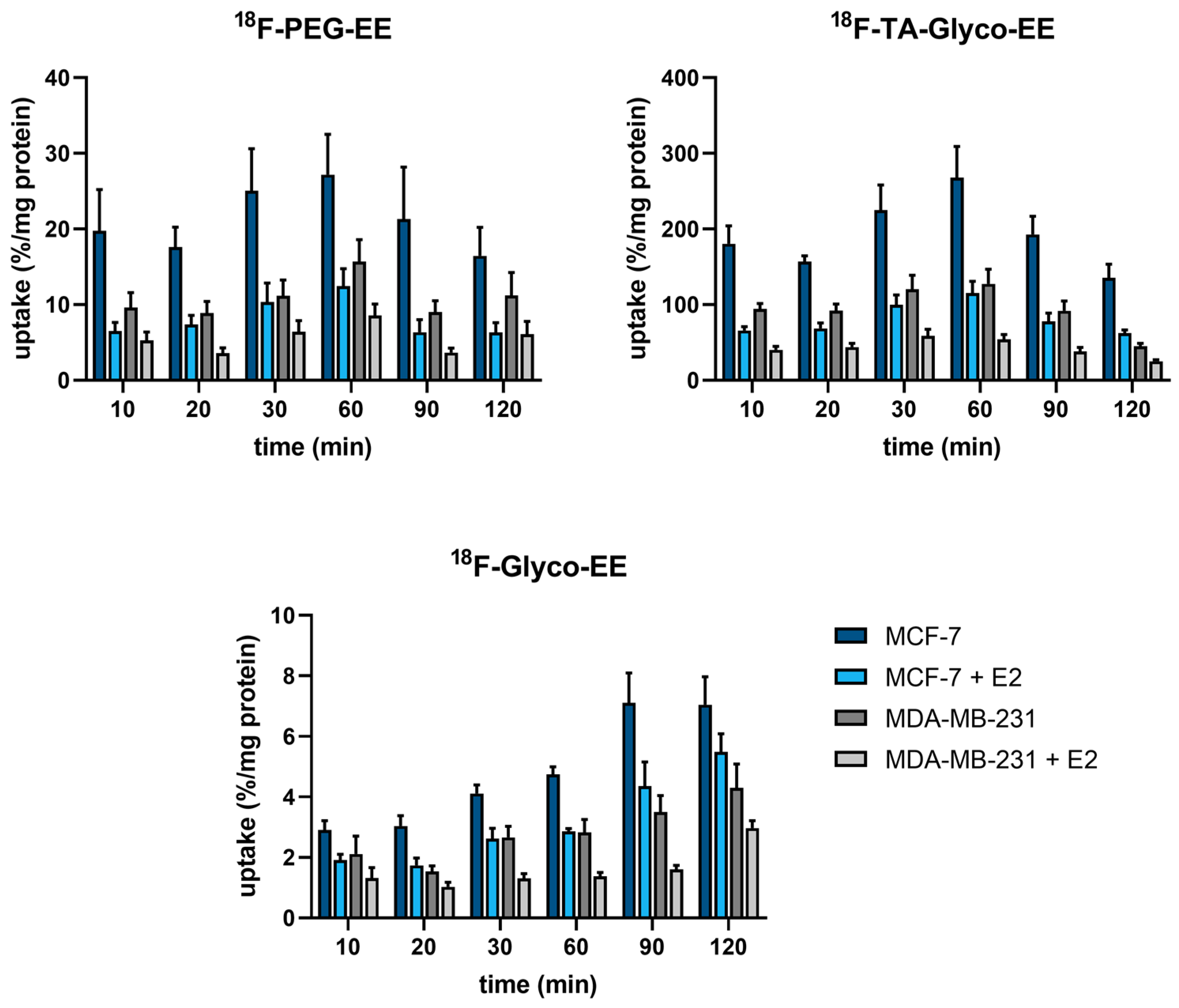
2.4.6. Cellular Uptake Assays
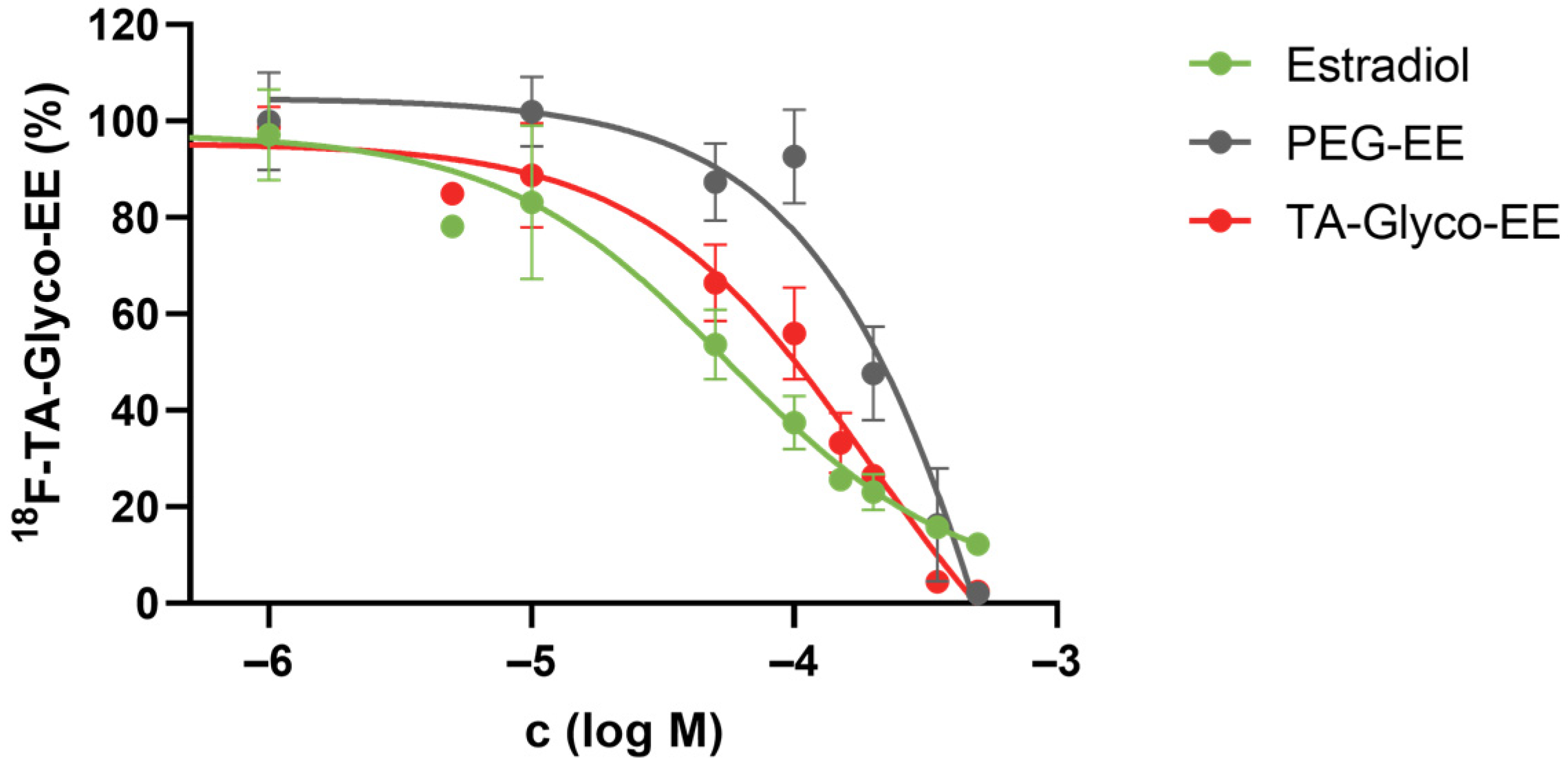
2.4.7. Competitive Cellular Uptake
2.5. In Vivo Studies
2.5.1. Animal Models
2.5.2. Biodistribution
2.5.3. PET Imaging
2.6. In Vitro Autoradiography
3. Results
3.1. Chemistry and Radiochemistry
3.2. In Vitro Characterization
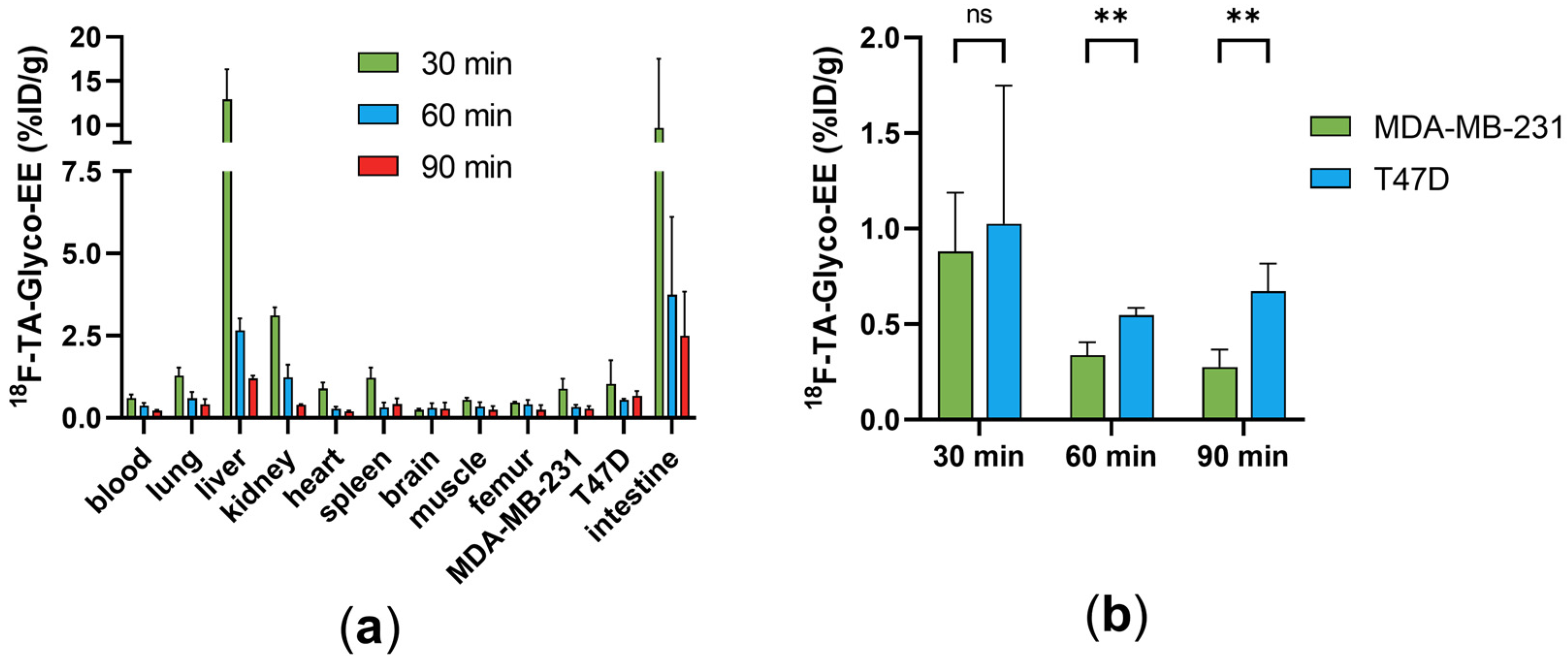
3.3. Biodistribution and Small Animal PET Imaging
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.E.M.; Lam, F.; Laversanne, M.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today. Available online: https://gco.iarc.who.int/today (accessed on 6 February 2024).
- Blamey, R.W.; Hornmark-Stenstam, B.; Ball, G.; Blichert-Toft, M.; Cataliotti, L.; Fourquet, A.; Gee, J.; Holli, K.; Jakesz, R.; Kerin, M.; et al. ONCOPOOL—A European database for 16,944 cases of breast cancer. Eur. J. Cancer 2010, 46, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Zhang, H.; Kong, Q.; Jiang, Y. Mechanisms for estrogen receptor expression in human cancer. Exp. Hematol. Oncol. 2018, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, N.; Silveyra, P. Chapter Three—Estrogen receptor signaling mechanisms. In Advances in Protein Chemistry and Structural Biology; Donev, R., Ed.; Academic Press: Cambridge, MA, USA, 2019; Volume 116, pp. 135–170. [Google Scholar]
- van Kruchten, M.; de Vries, E.G.E.; Brown, M.; de Vries, E.F.J.; Glaudemans, A.W.J.M.; Dierckx, R.A.J.O.; Schröder, C.P.; Hospers, G.A.P. PET imaging of oestrogen receptors in patients with breast cancer. Lancet Oncol. 2013, 14, e465–e475. [Google Scholar] [CrossRef] [PubMed]
- Kiesewetter, D.O.; Kilbourn, M.R.; Landvatter, S.W.; Heiman, D.F.; Katzenellenbogen, J.A.; Welch, M.J. Preparation of four fluorine-18-labeled estrogens and their selective uptakes in target tissues of immature rats. J. Nucl. Med. 1984, 25, 1212–1221. [Google Scholar] [PubMed]
- Liu, G.; Wang, W.; Lin, J.; Li, K.; Lv, G.; Zhao, X.; Wang, S.; Luo, S.; Qiu, L. Kit-like 18F-labeling of an estradiol derivative as a potential PET imaging agent for estrogen receptor-positive breast cancer. J. Radioanal. Nucl. Chem. 2017, 312, 599–607. [Google Scholar] [CrossRef]
- Xu, D.; Zhuang, R.; You, L.; Guo, Z.; Wang, X.; Peng, C.; Zhang, D.; Zhang, P.; Wu, H.; Pan, W.; et al. 18F-labeled estradiol derivative for targeting estrogen receptor-expressing breast cancer. Nucl. Med. Biol. 2018, 59, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Seimbille, Y.; Rousseau, J.; Benard, F.; Morin, C.; Ali, H.; Avvakumov, G.; Hammond, G.L.; van Lier, J.E. 18F-labeled difluoroestradiols: Preparation and preclinical evaluation as estrogen receptor-binding radiopharmaceuticals. Steroids 2002, 67, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Nayak, T.K.; Hathaway, H.J.; Ramesh, C.; Arterburn, J.B.; Dai, D.; Sklar, L.A.; Norenberg, J.P.; Prossnitz, E.R. Preclinical Development of a Neutral, Estrogen Receptor–Targeted, Tridentate 99mTc(I)-Estradiol-Pyridin-2-yl Hydrazine Derivative for Imaging of Breast and Endometrial Cancers. J. Nucl. Med. 2008, 49, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, K.; Arun, A.; Singh, S.; Manohar, M.; Chuttani, K.; Konwar, R.; Dwivedi, A.; Soni, R.; Singh, A.K.; Mishra, A.K.; et al. Bivalent Approach for Homodimeric Estradiol Based Ligand: Synthesis and Evaluation for Targeted Theranosis of ER(+) Breast Carcinomas. Bioconjug Chem. 2016, 27, 961–972. [Google Scholar] [CrossRef]
- Xu, D.; Lin, X.; Zeng, X.; Wen, X.; Li, J.; Li, Y.; Huang, J.; Chen, X.; Guo, Z.; Zhang, X. Radioiodinated 4-(p-Iodophenyl) Butanoic Acid-Modified Estradiol Derivative for ER Targeting SPECT Imaging. Anal. Chem. 2021, 93, 13998–14006. [Google Scholar] [CrossRef]
- Xu, D.; Peng, C.; Gao, F.; Guo, Z.; Zhuang, R.; Su, X.; Zhang, X. Radioiodinated estradiol dimer for estrogen receptor targeted breast cancer imaging. Chem. Biol. Drug Des. 2020, 96, 1332–1340. [Google Scholar] [CrossRef]
- U.S. Food & Drug Administration. Drug Trial Snapshot: CERIANNA. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/drug-trial-snapshot-cerianna (accessed on 6 February 2024).
- Katzenellenbogen, J.A. The quest for improving the management of breast cancer by functional imaging: The discovery and development of 16α-[18F]fluoroestradiol (FES), a PET radiotracer for the estrogen receptor, a historical review. Nucl. Med. Biol. 2021, 92, 24–37. [Google Scholar] [CrossRef]
- O’Brien, S.R.; Edmonds, C.E.; Katz, D.; Mankoff, D.A.; Pantel, A.R. 18F-Fluoroestradiol (FES) PET/CT: Review of current practice and future directions. Clin. Transl. Imaging 2022, 10, 331–341. [Google Scholar] [CrossRef]
- Oliveira, M.; Neto, C.; Ribeiro Morais, G.; Thiemann, T. Steroid receptor ligands for breast cancer targeting: An insight into their potential role as pet imaging agents. Curr. Med. Chem. 2013, 20, 222–245. [Google Scholar] [CrossRef] [PubMed]
- von Schoultz, B.; Carlström, K.; Collste, L.; Eriksson, A.; Henriksson, P.; Pousette, Å.; Stege, R. Estrogen therapy and liver function—Metabolic effects of oral and parenteral administration. Prostate 1989, 14, 389–395. [Google Scholar] [CrossRef]
- Wetzel, E.A.; Hanson, A.M.; Troutfetter, C.L.; Burkett, D.J.; Sem, D.S.; Donaldson, W.A. Synthesis and evaluation of 17α-triazolyl and 9α-cyano derivatives of estradiol. Bioorg. Med. Chem. 2020, 28, 115670. [Google Scholar] [CrossRef] [PubMed]
- Radford, L.L.; Lapi, S.E. Methods for the Production of Radionuclides for Medicine. In Radiopharmaceutical Chemistry; Lewis, J.S., Windhorst, A.D., Zeglis, B.M., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 63–83. [Google Scholar]
- Tietze, L.F.; Schmuck, K. SiFA Azide: A New Building Block for PET Imaging Using Click Chemistry. Synlett 2011, 2011, 1697–1700. [Google Scholar] [CrossRef]
- Schirrmacher, R.; Bradtmöller, G.; Schirrmacher, E.; Thews, O.; Tillmanns, J.; Siessmeier, T.; Buchholz, H.G.; Bartenstein, P.; Wängler, B.; Niemeyer, C.M.; et al. 18F-Labeling of Peptides by means of an Organosilicon-Based Fluoride Acceptor. Angewandte Chem. Int. Ed. 2006, 45, 6047–6050. [Google Scholar] [CrossRef]
- Maschauer, S.; Haubner, R.; Kuwert, T.; Prante, O. 18F-Glyco-RGD Peptides for PET Imaging of Integrin Expression: Efficient Radiosynthesis by Click Chemistry and Modulation of Biodistribution by Glycosylation. Mol. Pharm. 2014, 11, 505–515. [Google Scholar] [CrossRef]
- Breyholz, H.-J.; Wagner, S.; Faust, A.; Riemann, B.; Höltke, C.; Hermann, S.; Schober, O.; Schäfers, M.; Kopka, K. Radiofluorinated Pyrimidine-2,4,6-triones as Molecular Probes for Noninvasive MMP-Targeted Imaging. ChemMedChem 2010, 5, 777–789. [Google Scholar] [CrossRef]
- Witt, B.L.; Tollefsbol, T.O. Molecular, Cellular, and Technical Aspects of Breast Cancer Cell Lines as a Foundational Tool in Cancer Research. Life 2023, 13, 2311. [Google Scholar] [CrossRef]
- Schirrmacher, R.; Wängler, B.; Bailey, J.; Bernard-Gauthier, V.; Schirrmacher, E.; Wängler, C. Small Prosthetic Groups in 18F-Radiochemistry: Useful Auxiliaries for the Design of 18F-PET Tracers. Semin. Nucl. Med. 2017, 47, 474–492. [Google Scholar] [CrossRef]
- Hansch, C.L.A.; Hoekman, D. Exploring QSAR: Hydrophobic, Electronic, and Steric Constants; American Chemical Society: Washington, DC, USA, 1995; Volume 2. [Google Scholar]
- Van de Wiele, C.; De Vos, F.; Slegers, G.; Van Belle, S.; Dierckx, R.A. Radiolabeled estradiol derivatives to predict response to hormonal treatment in breast cancer: A review. Eur. J. Nucl. Med. 2000, 27, 1421–1433. [Google Scholar] [CrossRef] [PubMed]
- Paquette, M.; Phoenix, S.; Ouellet, R.; Langlois, R.; van Lier, J.E.; Turcotte, É.E.; Bénard, F.; Lecomte, R. Assessment of the Novel Estrogen Receptor PET Tracer 4-Fluoro-11β-methoxy-16α-[18F]fluoroestradiol (4FMFES) by PET Imaging in a Breast Cancer Murine Model. Mol. Imaging Biol. 2013, 15, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Antunes, I.F.; van Waarde, A.; Dierckx, R.A.J.O.; de Vries, E.G.E.; Hospers, G.A.P.; de Vries, E.F.J. Synthesis and Evaluation of the Estrogen Receptor β–Selective Radioligand 2-18F-Fluoro-6-(6-Hydroxynaphthalen-2-yl)Pyridin-3-ol: Comparison with 16α-18F-Fluoro-17β-Estradiol. J. Nucl. Med. 2017, 58, 554–559. [Google Scholar] [CrossRef]
- Tejería, M.E.; Pereira, M.P.; Gambini, J.P.; Duarte, P.; Giglio, J.G.; Rey, A.M. Synthesis of a [18F]F Estradiol Derivative via Click Chemistry Using an Automated Synthesis Module: In Vitro Evaluation as Potential Radiopharmaceutical for Breast Cancer Imaging. Pharmaceuticals 2024, 17, 388. [Google Scholar] [CrossRef] [PubMed]
- Holen, I.; Walker, M.; Nutter, F.; Fowles, A.; Evans, C.A.; Eaton, C.L.; Ottewell, P.D. Oestrogen receptor positive breast cancer metastasis to bone: Inhibition by targeting the bone microenvironment in vivo. Clin. Exp. Metastasis 2016, 33, 211–224. [Google Scholar] [CrossRef]
- Carlson, R.W. The History and Mechanism of Action of Fulvestrant. Clin. Breast Cancer 2005, 6, S5–S8. [Google Scholar] [CrossRef]
- Aliaga, A.; Rousseau, J.A.; Ouellette, R.; Cadorette, J.; van Lier, J.E.; Lecomte, R.; Bénard, F. Breast cancer models to study the expression of estrogen receptors with small animal PET imaging. Nucl. Med. Biol. 2004, 31, 761–770. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AY 1 (Total Synthesis Time) | logD7.4 | Plasma Protein Binding | Stability in Human Serum or Plasma | |
|---|---|---|---|---|
| 18F-PEG-EE | 19% (81 min) | 2.2 ± 0.1 | 71 ± 4% | >99% over 2 h 2 |
| 18F-SiFA-EE | 44% (27 min) | n.d. 3,4 | n.d. 3,4 | <50% after 10 min 2 |
| 18F-TA-Glyco-EE | 25% (77 min) | 2.3 ± 0.1 | 79 ± 1% | 0% after 30 min 2,5 |
| 18F-Glyco-EE | 22% (90 min) | 1.7 ± 0.1 | n.d. 3 | >99% over 2 h 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Friedel, A.; Prante, O.; Maschauer, S. Radiosynthesis and Preclinical Evaluation of 18F-Labeled Estradiol Derivatives with Different Lipophilicity for PET Imaging of Breast Cancer. Cancers 2024, 16, 2639. https://doi.org/10.3390/cancers16152639
Friedel A, Prante O, Maschauer S. Radiosynthesis and Preclinical Evaluation of 18F-Labeled Estradiol Derivatives with Different Lipophilicity for PET Imaging of Breast Cancer. Cancers. 2024; 16(15):2639. https://doi.org/10.3390/cancers16152639
Chicago/Turabian StyleFriedel, Anna, Olaf Prante, and Simone Maschauer. 2024. "Radiosynthesis and Preclinical Evaluation of 18F-Labeled Estradiol Derivatives with Different Lipophilicity for PET Imaging of Breast Cancer" Cancers 16, no. 15: 2639. https://doi.org/10.3390/cancers16152639