Genome-Wide Methylation Patterns in Primary Uveal Melanoma: Development of MethylSig-UM, an Epigenomic Prognostic Signature to Improve Patient Stratification



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. pUM Samples
2.2. DNA Extraction, Bisulfite Conversion and Array Hybridization
2.3. Data Normalization
2.4. Data Analysis and Visualization
2.5. Differentially Methylated Probes (DMPs) and Univariate Prognosis
2.6. Cluster Analysis
2.7. MethylSig-UM Signature Development
2.8. Differentially Expressed Genes
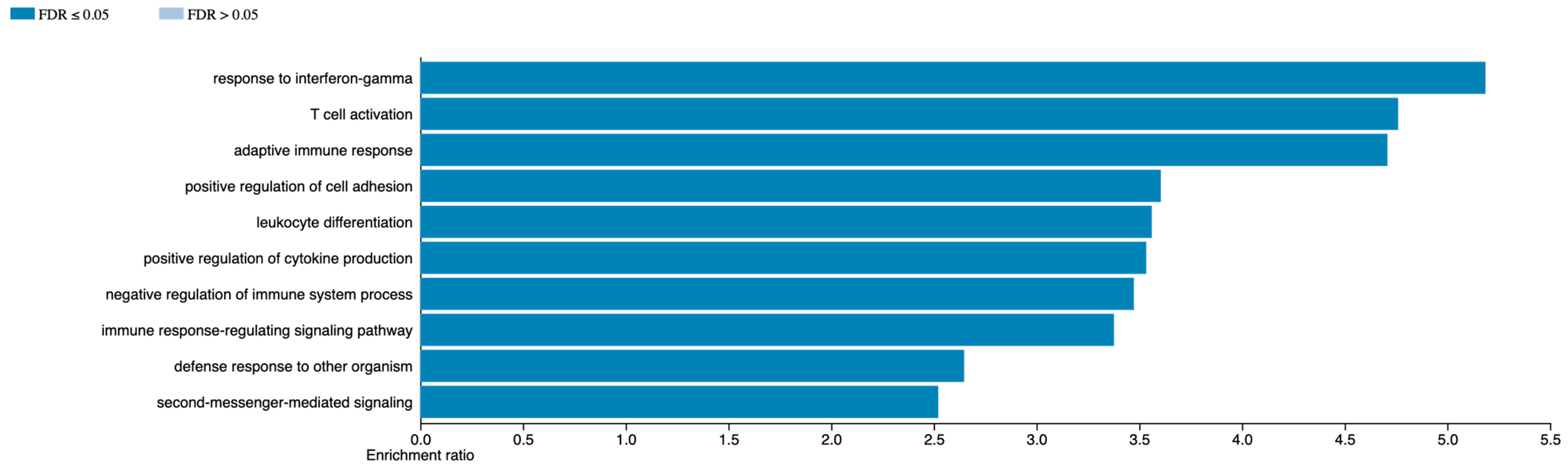
2.9. GO Term Analysis
3. Results
3.1. Patient Demographics
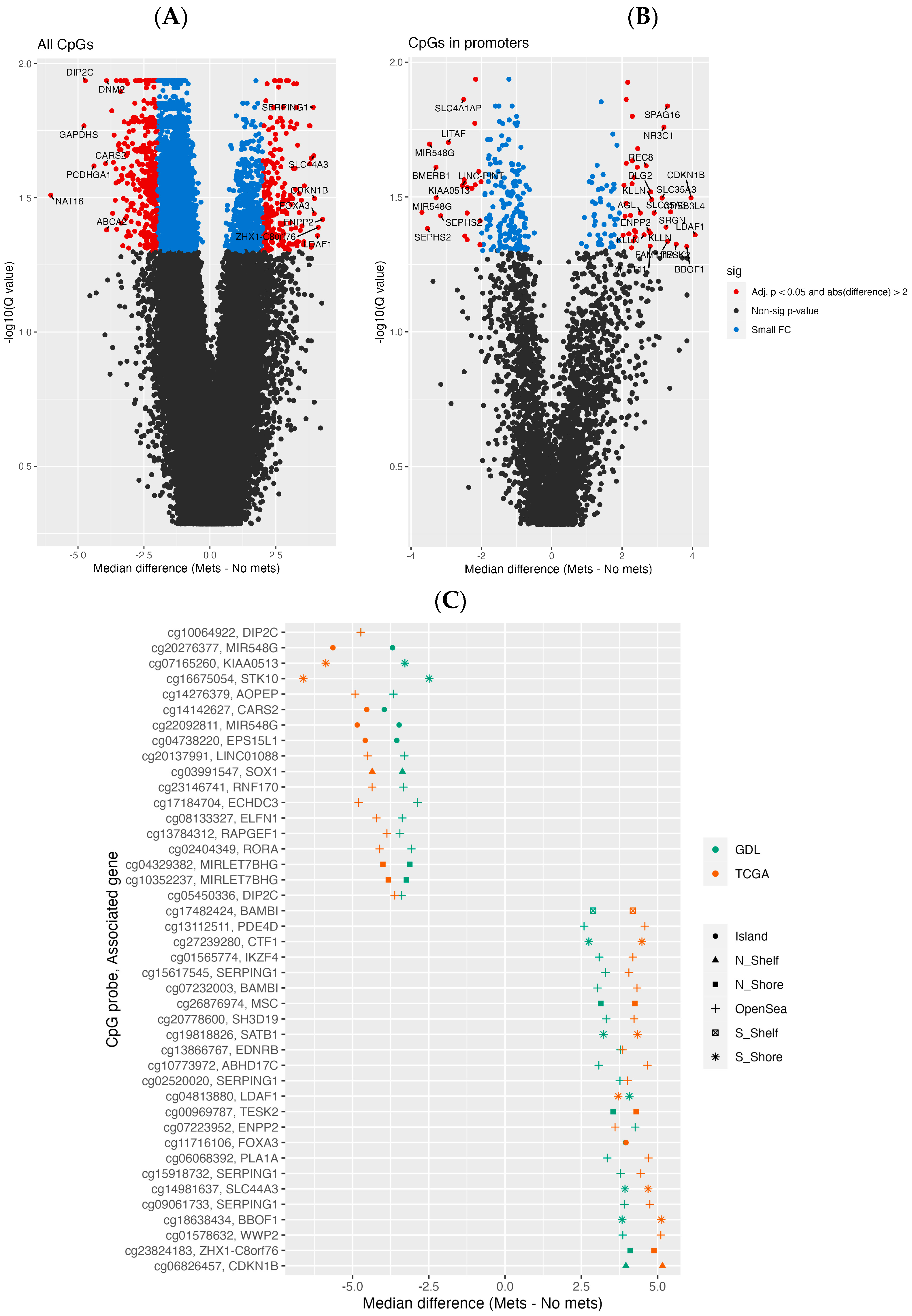
3.2. Differential Methylation Analysis
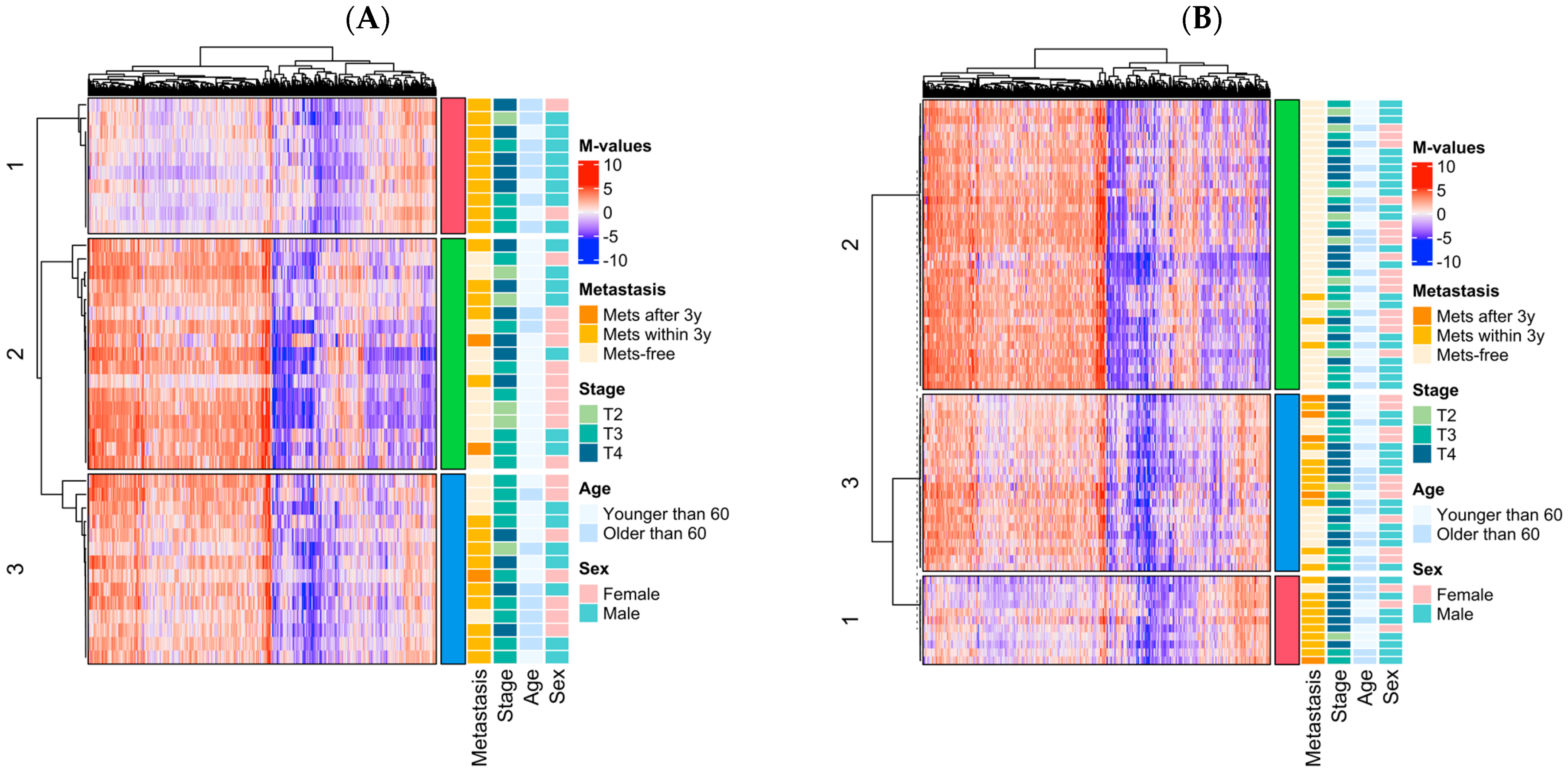
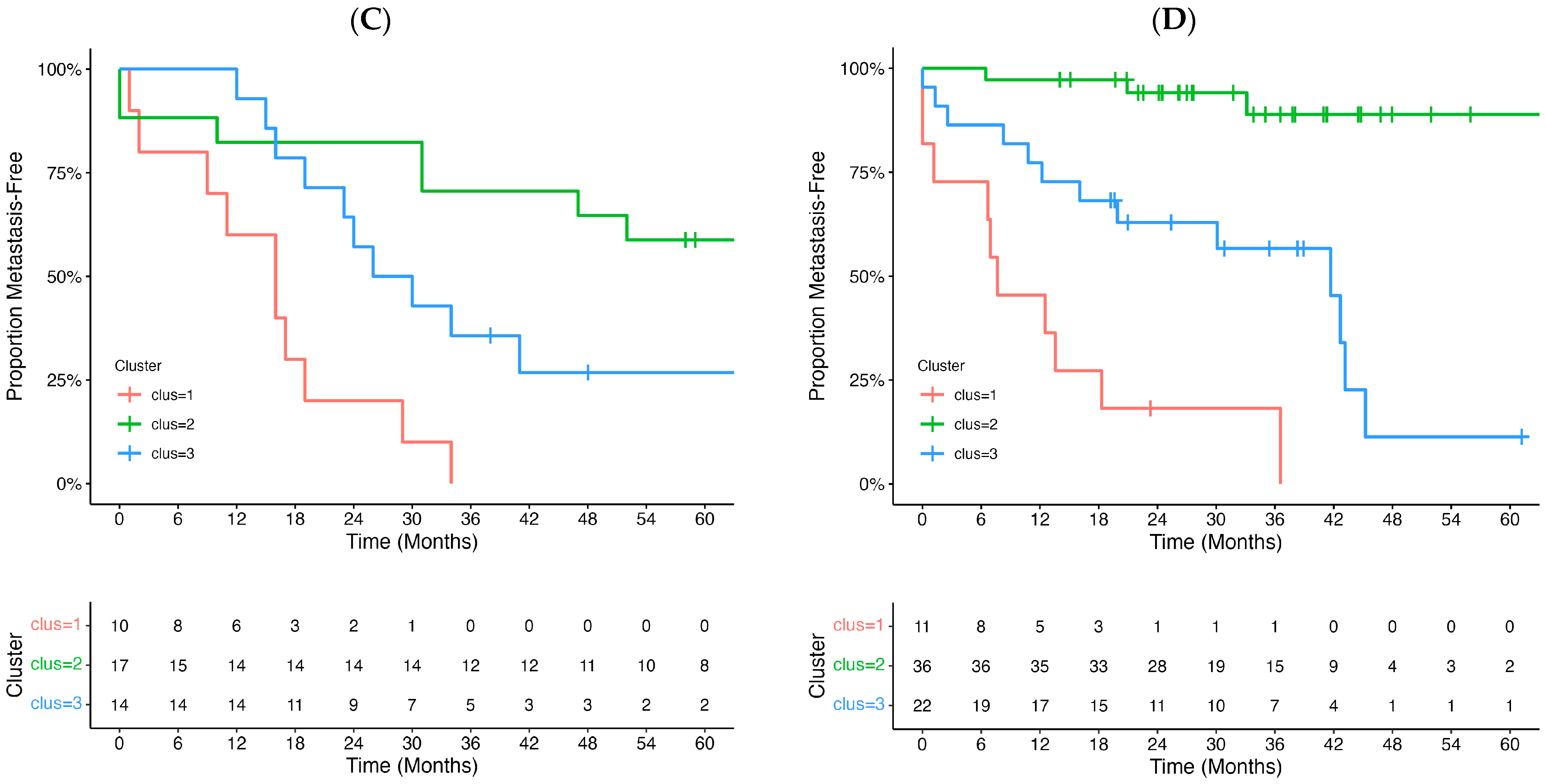
3.3. Epigenomic Subtype Analysis
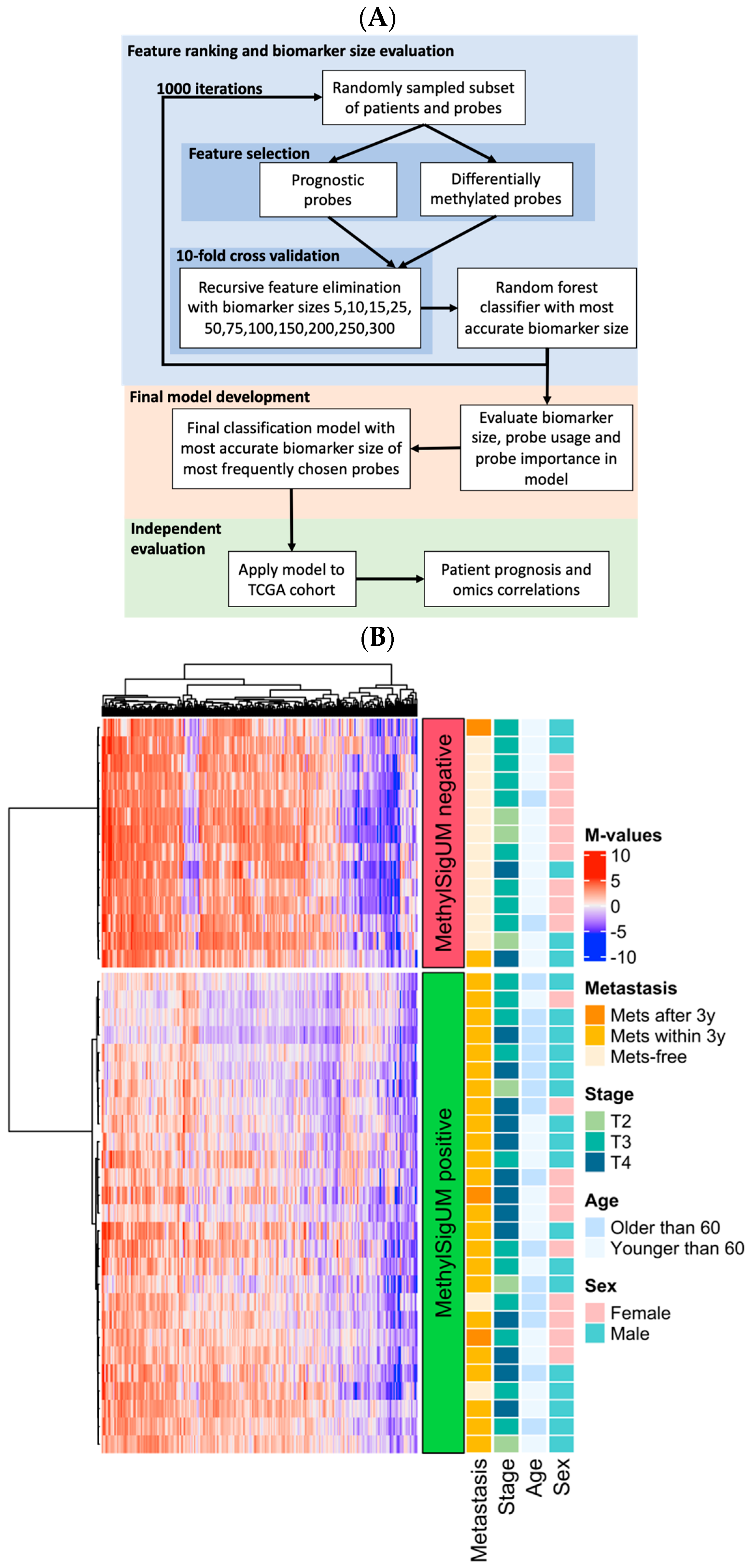
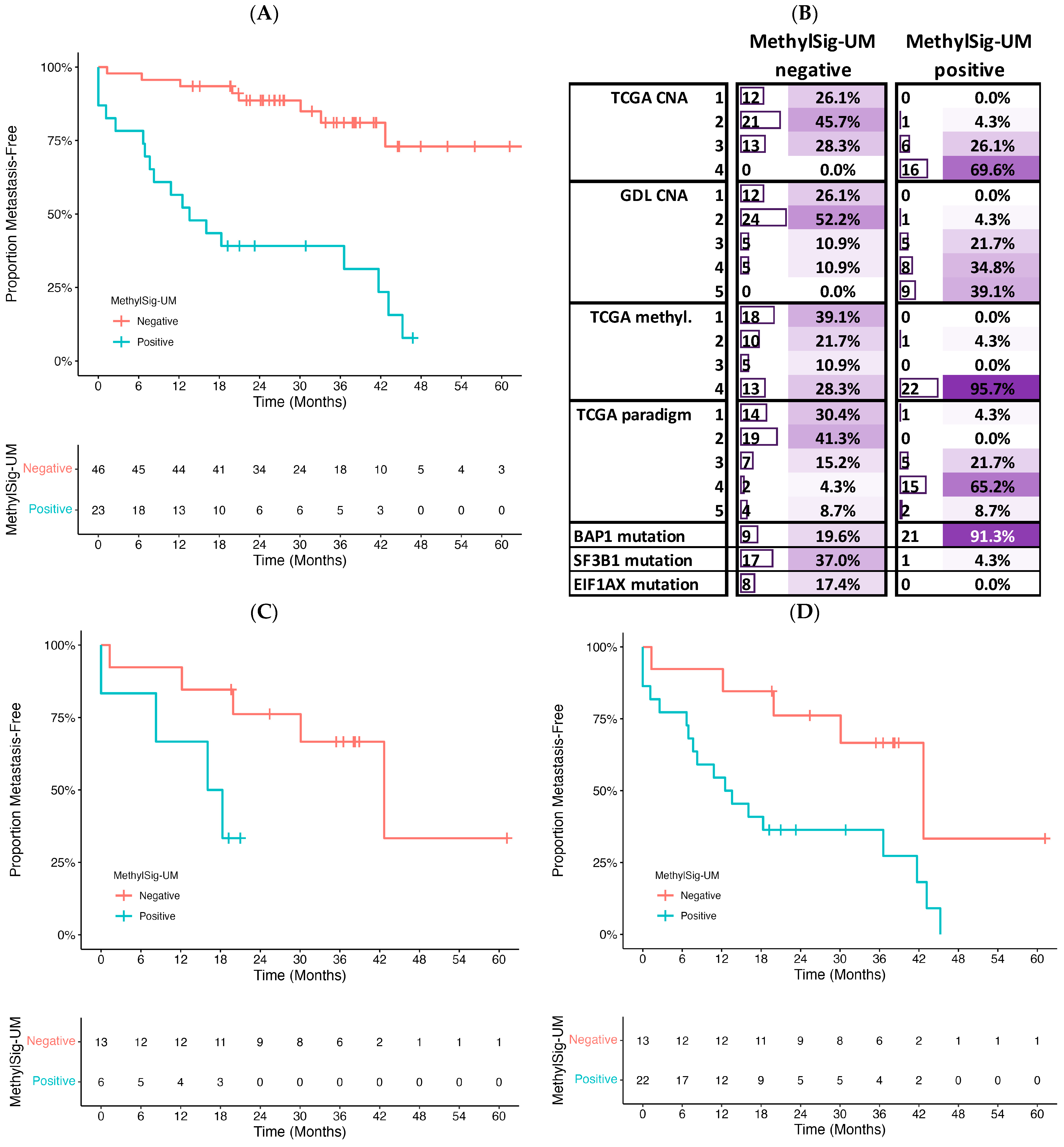
3.4. Prognostic Signature Development
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singh, A.D.; Turell, M.E.; Topham, A.K. Uveal melanoma: Trends in incidence, treatment, and survival. Ophthalmology 2011, 118, 1881–1885. [Google Scholar] [CrossRef] [PubMed]
- Ewens, K.G.; Kanetsky, P.A.; Richards-Yutz, J.; Al-Dahmash, S.; De Luca, M.C.; Bianciotto, C.G.; Shields, C.L.; Ganguly, A. Genomic profile of 320 uveal melanoma cases: Chromosome 8p-loss and metastatic outcome. Investig. Ophthalmol. Vis. Sci. 2013, 54, 5721–5729. [Google Scholar] [CrossRef] [PubMed]
- Vaquero-Garcia, J.; Lalonde, E.; Ewens, K.G.; Ebrahimzadeh, J.; Richard-Yutz, J.; Shields, C.L.; Barrera, A.; Green, C.J.; Barash, Y.; Ganguly, A. PRiMeUM: A Model for Predicting Risk of Metastasis in Uveal Melanoma. Investig. Ophthalmol. Vis. Sci. 2017, 58, 4096–4105. [Google Scholar] [CrossRef] [PubMed]
- Buzzacco, D.M.; Abdel-Rahman, M.H.; Park, S.; Davidorf, F.; Olencki, T.; Cebulla, C.M. Long-term survivors with metastatic uveal melanoma. Open Ophthalmol. J. 2012, 6, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Kaliki, S.; Shields, C.L.; Shields, J.A. Uveal melanoma: Estimating prognosis. Indian. J. Ophthalmol. 2015, 63, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Aalto, Y.; Eriksson, L.; Seregard, S.; Larsson, O.; Knuutila, S. Concomitant loss of chromosome 3 and whole arm losses and gains of chromosome 1, 6, or 8 in metastasizing primary uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2001, 42, 313–317. [Google Scholar]
- Damato, B.; Dopierala, J.A.; Coupland, S.E. Genotypic profiling of 452 choroidal melanomas with multiplex ligation-dependent probe amplification. Clin. Cancer Res. 2010, 16, 6083–6092. [Google Scholar] [CrossRef] [PubMed]
- Kilic, E.; Naus, N.C.; van Gils, W.; Klaver, C.C.; van Til, M.E.; Verbiest, M.M.; Stijnen, T.; Mooy, C.M.; Paridaens, D.; Beverloo, H.B.; et al. Concurrent loss of chromosome arm 1p and chromosome 3 predicts a decreased disease-free survival in uveal melanoma patients. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2253–2257. [Google Scholar] [CrossRef] [PubMed]
- Onken, M.D.; Worley, L.A.; Harbour, J.W. A metastasis modifier locus on human chromosome 8p in uveal melanoma identified by integrative genomic analysis. Clin. Cancer Res. 2008, 14, 3737–3745. [Google Scholar] [CrossRef]
- van den Bosch, T.; van Beek, J.G.; Vaarwater, J.; Verdijk, R.M.; Naus, N.C.; Paridaens, D.; de Klein, A.; Kilic, E. Higher percentage of FISH-determined monosomy 3 and 8q amplification in uveal melanoma cells relate to poor patient prognosis. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2668–2674. [Google Scholar] [CrossRef]
- Damato, B.; Eleuteri, A.; Taktak, A.F.; Coupland, S.E. Estimating prognosis for survival after treatment of choroidal melanoma. Prog. Retin. Eye Res. 2011, 30, 285–295. [Google Scholar] [CrossRef] [PubMed]
- DeParis, S.W.; Taktak, A.; Eleuteri, A.; Enanoria, W.; Heimann, H.; Coupland, S.E.; Damato, B. External Validation of the Liverpool Uveal Melanoma Prognosticator Online. Investig. Ophthalmol. Vis. Sci. 2016, 57, 6116–6122. [Google Scholar] [CrossRef] [PubMed]
- Ewens, K.G.; Kanetsky, P.A.; Richards-Yutz, J.; Purrazzella, J.; Shields, C.L.; Ganguly, T.; Ganguly, A. Chromosome 3 status combined with BAP1 and EIF1AX mutation profiles are associated with metastasis in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5160–5167. [Google Scholar] [CrossRef] [PubMed]
- Field, M.G.; Kuznetsov, J.N.; Bussies, P.L.; Cai, L.Z.; Alawa, K.A.; Decatur, C.L.; Kurtenbach, S.; Harbour, J.W. BAP1 Loss Is Associated with DNA Methylomic Repatterning in Highly Aggressive Class 2 Uveal Melanomas. Clin. Cancer Res. 2019, 25, 5663–5673. [Google Scholar] [CrossRef] [PubMed]
- Lamas, N.J.; Martel, A.; Nahon-Esteve, S.; Goffinet, S.; Macocco, A.; Bertolotto, C.; Lassalle, S.; Hofman, P. Prognostic Biomarkers in Uveal Melanoma: The Status Quo, Recent Advances and Future Directions. Cancers 2021, 14, 96. [Google Scholar] [CrossRef] [PubMed]
- Onken, M.D.; Worley, L.A.; Char, D.H.; Augsburger, J.J.; Correa, Z.M.; Nudleman, E.; Aaberg, T.M., Jr.; Altaweel, M.M.; Bardenstein, D.S.; Finger, P.T.; et al. Collaborative Ocular Oncology Group report number 1: Prospective validation of a multi-gene prognostic assay in uveal melanoma. Ophthalmology 2012, 119, 1596–1603. [Google Scholar] [CrossRef]
- Onken, M.D.; Worley, L.A.; Ehlers, J.P.; Harbour, J.W. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 2004, 64, 7205–7209. [Google Scholar] [CrossRef]
- Onken, M.D.; Worley, L.A.; Tuscan, M.D.; Harbour, J.W. An accurate, clinically feasible multi-gene expression assay for predicting metastasis in uveal melanoma. J. Mol. Diagn. 2010, 12, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Royer-Bertrand, B.; Torsello, M.; Rimoldi, D.; El Zaoui, I.; Cisarova, K.; Pescini-Gobert, R.; Raynaud, F.; Zografos, L.; Schalenbourg, A.; Speiser, D.; et al. Comprehensive Genetic Landscape of Uveal Melanoma by Whole-Genome Sequencing. Am. J. Hum. Genet. 2016, 99, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jia, R.; Ge, S. Role of Epigenetics in Uveal Melanoma. Int. J. Biol. Sci. 2017, 13, 426–433. [Google Scholar] [CrossRef]
- Sharma, A.; Stei, M.M.; Frohlich, H.; Holz, F.G.; Loeffler, K.U.; Herwig-Carl, M.C. Genetic and epigenetic insights into uveal melanoma. Clin. Genet. 2018, 93, 952–961. [Google Scholar] [CrossRef] [PubMed]
- van der Velden, P.A.; Metzelaar-Blok, J.A.; Bergman, W.; Monique, H.; Hurks, H.; Frants, R.R.; Gruis, N.A.; Jager, M.J. Promoter hypermethylation: A common cause of reduced p16(INK4a) expression in uveal melanoma. Cancer Res. 2001, 61, 5303–5306. [Google Scholar] [PubMed]
- Calipel, A.; Abonnet, V.; Nicole, O.; Mascarelli, F.; Coupland, S.E.; Damato, B.; Mouriaux, F. Status of RASSF1A in uveal melanocytes and melanoma cells. Mol. Cancer Res. 2011, 9, 1187–1198. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.P.; Yoon, J.H.; Liu, L.; Tommasi, S.; Wilczynski, S.P.; Dammann, R. Methylation of the RASSF1A gene in human cancers. Biol. Chem. 2002, 383, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Maat, W.; van der Velden, P.A.; Out-Luiting, C.; Plug, M.; Dirks-Mulder, A.; Jager, M.J.; Gruis, N.A. Epigenetic inactivation of RASSF1a in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2007, 48, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Maat, W.; Beiboer, S.H.; Jager, M.J.; Luyten, G.P.; Gruis, N.A.; van der Velden, P.A. Epigenetic regulation identifies RASEF as a tumor-suppressor gene in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- Neumann, L.C.; Weinhausel, A.; Thomas, S.; Horsthemke, B.; Lohmann, D.R.; Zeschnigk, M. EFS shows biallelic methylation in uveal melanoma with poor prognosis as well as tissue-specific methylation. BMC Cancer 2011, 11, 380. [Google Scholar] [CrossRef] [PubMed]
- Moulin, A.P.; Clement, G.; Bosman, F.T.; Zografos, L.; Benhattar, J. Methylation of CpG island promoters in uveal melanoma. Br. J. Ophthalmol. 2008, 92, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, M.F.; Curtis, E.J.; Goldbaum, M.H.; Mischel, P.S. BAP1 methylation: A prognostic marker of uveal melanoma metastasis. NPJ Precis. Oncol. 2021, 5, 89. [Google Scholar] [CrossRef] [PubMed]
- Soltysova, A.; Dvorska, D.; Kajabova, V.H.; Pecimonova, M.; Cepcekova, K.; Ficek, A.; Demkova, L.; Buocikova, V.; Babal, P.; Juras, I.; et al. Uncovering accurate prognostic markers for high-risk uveal melanoma through DNA methylation profiling. Clin. Transl. Med. 2023, 13, e1317. [Google Scholar] [CrossRef]
- Ferrier, S.T.; Burnier, J.V. Novel Methylation Patterns Predict Outcome in Uveal Melanoma. Life 2020, 10, 248. [Google Scholar] [CrossRef]
- Wei, A.Z.; Maniar, A.B.; Carvajal, R.D. New targeted and epigenetic therapeutic strategies for the treatment of uveal melanoma. Cancer Gene Ther. 2022, 29, 1819–1826. [Google Scholar] [CrossRef] [PubMed]
- Ness, C.; Katta, K.; Garred, O.; Kumar, T.; Olstad, O.K.; Petrovski, G.; Moe, M.C.; Noer, A. Integrated differential DNA methylation and gene expression of formalin-fixed paraffin-embedded uveal melanoma specimens identifies genes associated with early metastasis and poor prognosis. Exp. Eye Res. 2021, 203, 108426. [Google Scholar] [CrossRef] [PubMed]
- Smit, K.N.; Boers, R.; Vaarwater, J.; Boers, J.; Brands, T.; Mensink, H.; Verdijk, R.M.; van IJcken, W.F.J.; Gribnau, J.; de Klein, A.; et al. Genome-wide aberrant methylation in primary metastatic UM and their matched metastases. Sci. Rep. 2022, 12, 42. [Google Scholar] [CrossRef]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell 2017, 32, 204–220.e215. [Google Scholar] [CrossRef] [PubMed]
- Fortin, J.P.; Triche, T.J., Jr.; Hansen, K.D. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics 2017, 33, 558–560. [Google Scholar] [CrossRef] [PubMed]
- Naeem, H.; Wong, N.C.; Chatterton, Z.; Hong, M.K.; Pedersen, J.S.; Corcoran, N.M.; Hovens, C.M.; Macintyre, G. Reducing the risk of false discovery enabling identification of biologically significant genome-wide methylation status using the HumanMethylation450 array. BMC Genom. 2014, 15, 51. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.A.; Lemire, M.; Choufani, S.; Butcher, D.T.; Grafodatskaya, D.; Zanke, B.W.; Gallinger, S.; Hudson, T.J.; Weksberg, R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013, 8, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- P’ng, C.; Green, J.; Chong, L.C.; Waggott, D.; Prokopec, S.D.; Shamsi, M.; Nguyen, F.; Mak, D.Y.F.; Lam, F.; Albuquerque, M.A.; et al. BPG: Seamless, automated and interactive visualization of scientific data. BMC Bioinform. 2019, 20, 42. [Google Scholar] [CrossRef] [PubMed]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M. Building predictive models in r using the caret package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Vasaikar, S.; Shi, Z.; Greer, M.; Zhang, B. WebGestalt 2017: A more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017, 45, W130–W137. [Google Scholar] [CrossRef]
- Kim, H.R.; Park, J.S.; Karabulut, H.; Yasmin, F.; Jun, C.D. Transgelin-2: A Double-Edged Sword in Immunity and Cancer Metastasis. Front. Cell Dev. Biol. 2021, 9, 606149. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Wang, S.; Wang, Z. An Integrated Analysis Identified TAGLN2 As an Oncogene Indicator Related to Prognosis and Immunity in Pan-Cancer. J. Cancer 2023, 14, 1809–1836. [Google Scholar] [CrossRef]
- Lalonde, E.; Ewens, K.; Richards-Yutz, J.; Ebrahimzedeh, J.; Terai, M.; Gonsalves, C.F.; Sato, T.; Shields, C.L.; Ganguly, A. Improved Uveal Melanoma Copy Number Subtypes Including an Ultra-High-Risk Group. Ophthalmol. Sci. 2022, 2, 100121. [Google Scholar] [CrossRef]
- Estecio, M.R.; Issa, J.P. Dissecting DNA hypermethylation in cancer. FEBS Lett. 2011, 585, 2078–2086. [Google Scholar] [CrossRef]
- Lim, D.H.K.; Maher, E.R. DNA Methylation: A form of epigenetic control of gene expression. Obstet. Gynaecol. 2010, 12, 37–42. [Google Scholar] [CrossRef]
- Huang, H.; Qiu, D.; Zhou, Z.; Wu, B.; Shao, L.; Pu, Y.; He, T.; Wu, Y.; Cui, D.; Zhong, F. A pan-cancer analysis for the oncogenic role of cyclin-dependent kinase inhibitor 1B in human cancers. Discov. Oncol. 2023, 14, 126. [Google Scholar] [CrossRef]
- Matas-Rico, E.; Frijlink, E.; van der Haar Avila, I.; Menegakis, A.; van Zon, M.; Morris, A.J.; Koster, J.; Salgado-Polo, F.; de Kivit, S.; Lanca, T.; et al. Autotaxin impedes anti-tumor immunity by suppressing chemotaxis and tumor infiltration of CD8(+) T cells. Cell Rep. 2021, 37, 110013. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulou, M.; Fanidis, D.; Aidinis, V.; Chatzaki, E. ENPP2 Methylation in Health and Cancer. Int. J. Mol. Sci. 2021, 22, 11958. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Hoefsmit, E.P.; Rozeman, E.A.; Van, T.M.; Dimitriadis, P.; Krijgsman, O.; Conway, J.W.; Pires da Silva, I.; van der Wal, J.E.; Ketelaars, S.L.C.; Bresser, K.; et al. Comprehensive analysis of cutaneous and uveal melanoma liver metastases. J. Immunother. Cancer 2020, 8, e001501. [Google Scholar] [CrossRef]
- Gezgin, G.; Dogrusoz, M.; van Essen, T.H.; Kroes, W.G.M.; Luyten, G.P.M.; van der Velden, P.A.; Walter, V.; Verdijk, R.M.; van Hall, T.; van der Burg, S.H.; et al. Genetic evolution of uveal melanoma guides the development of an inflammatory microenvironment. Cancer Immunol. Immunother. 2017, 66, 903–912. [Google Scholar] [CrossRef]
- Ringel, A.E.; Drijvers, J.M.; Baker, G.J.; Catozzi, A.; Garcia-Canaveras, J.C.; Gassaway, B.M.; Miller, B.C.; Juneja, V.R.; Nguyen, T.H.; Joshi, S.; et al. Obesity Shapes Metabolism in the Tumor Microenvironment to Suppress Anti-Tumor Immunity. Cell 2020, 183, 1848–1866.e1826. [Google Scholar] [CrossRef]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef]
- Lietz, C.E.; Newman, E.T.; Kelly, A.D.; Xiang, D.H.; Zhang, Z.; Luscko, C.A.; Lozano-Calderon, S.A.; Ebb, D.H.; Raskin, K.A.; Cote, G.M.; et al. Genome-wide DNA methylation patterns reveal clinically relevant predictive and prognostic subtypes in human osteosarcoma. Commun. Biol. 2022, 5, 213. [Google Scholar] [CrossRef]
- Xia, D.; Leon, A.J.; Yan, J.; Silva, A.; Bakhtiari, M.; Tremblay-LeMay, R.; Selvarajah, S.; Sabatini, P.; Diamandis, P.; Pugh, T.; et al. DNA Methylation-Based Classification of Small B-Cell Lymphomas: A Proof-of-Principle Study. J. Mol. Diagn. 2021, 23, 1774–1786. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, L.C.; Mathew, J.; Hirte, H.; Provias, J.; Pare, G.; Chong, M.; Grafodatskaya, D.; McCready, E. Evaluation of DNA Methylation Array for Glioma Tumor Profiling and Description of a Novel Epi-Signature to Distinguish IDH1/IDH2 Mutant and Wild-Type Tumors. Genes 2022, 13, 2075. [Google Scholar] [CrossRef] [PubMed]
- Sadikovic, B.; Levy, M.A.; Kerkhof, J.; Aref-Eshghi, E.; Schenkel, L.; Stuart, A.; McConkey, H.; Henneman, P.; Venema, A.; Schwartz, C.E.; et al. Clinical epigenomics: Genome-wide DNA methylation analysis for the diagnosis of Mendelian disorders. Genet. Med. 2021, 23, 1065–1074. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GDL | TCGA | ||
|---|---|---|---|
| Cohort size | 41 | 69 1 | |
| Sex | Male | 22 | 41 |
| Female | 19 | 28 | |
| Age at diagnosis | Median | 57 | 60 |
| Range | (19–77) | (22–86) | |
| AJCC tumor stage | T2 | 6 | 10 |
| T3 | 20 | 27 | |
| T4 | 15 | 32 | |
| Metastasis | No | 14 | 43 |
| Yes | 27 | 26 | |
| Average follow-up time (time to metastasis) | Years | 7.2 | 2.6 |
| (A) | |||
| HR | 95% CI (Low, High) | p-Value | |
| Cluster 3 (vs. 2) | 2.14 | 0.73, 6.27 | 0.165 |
| Cluster 1 (vs. 2) | 4.57 | 1.38, 15.2 | 0.013 |
| T4 vs. T2–T3 | 3.39 | 1.44, 7.97 | 0.0051 |
| Age (≥60 vs. <60 years) | 2.11 | 0.87, 5.11 | 0.097 |
| Model Wald p-value | 0.00022 | ||
| Model logrank p-value | 8.0 × 10−5 | ||
| (B) | |||
| HR | 95% CI (Low, High) | p-Value | |
| Cluster 3 (vs. 2) | 9.90 | 2.79, 35.2 | 0.000405 |
| Cluster 1 (vs. 2) | 31.6 | 8.00, 12.4 | 8.2 × 10−7 |
| T4 vs. T2–T3 | 1.92 | 0.82, 4.50 | 0.136 |
| Model Wald p-value | 5.0 × 10−6 | ||
| Model logrank p-value | 2.0 × 10−10 | ||
| (A) | |||
| HR | 95% CI (Low, High) | p-Value | |
| MethylSig-UM | 7.20 | 3.08, 16.8 | 5.0 × 10−6 |
| T4 vs. T2–T3 | 2.35 | 1.04, 5.33 | 0.0408 |
| Model Wald p-value | 7.0 × 10−6 | ||
| Model logrank p-value | 2.0 × 10−7 | ||
| (B) | |||
| HR | 95% CI (Low, High) | p-Value | |
| MethylSig-UM | 4.51 | 1.77, 11.5 | 0.00162 |
| PRiMeUM | 2.56 | 0.9, 17.2 | 0.0747 |
| Model Wald p-value | 0.00001 | ||
| Model logrank p-value | 3.0 × 10−7 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lalonde, E.; Li, D.; Ewens, K.; Shields, C.L.; Ganguly, A. Genome-Wide Methylation Patterns in Primary Uveal Melanoma: Development of MethylSig-UM, an Epigenomic Prognostic Signature to Improve Patient Stratification. Cancers 2024, 16, 2650. https://doi.org/10.3390/cancers16152650
Lalonde E, Li D, Ewens K, Shields CL, Ganguly A. Genome-Wide Methylation Patterns in Primary Uveal Melanoma: Development of MethylSig-UM, an Epigenomic Prognostic Signature to Improve Patient Stratification. Cancers. 2024; 16(15):2650. https://doi.org/10.3390/cancers16152650
Chicago/Turabian StyleLalonde, Emilie, Dong Li, Kathryn Ewens, Carol L. Shields, and Arupa Ganguly. 2024. "Genome-Wide Methylation Patterns in Primary Uveal Melanoma: Development of MethylSig-UM, an Epigenomic Prognostic Signature to Improve Patient Stratification" Cancers 16, no. 15: 2650. https://doi.org/10.3390/cancers16152650