
Anticancer Activity of Delta-Tocotrienol in Human Hepatocarcinoma: Involvement of Autophagy Induction

 , ,
, ,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Antibodies and Reagents
2.2. Cell Culture
2.3. Proliferation Assays
2.4. MTT Viability Assay
2.5. Clonogenic Assay
2.6. Western Blot
2.7. Flow Cytometry Analysis (FACS)
2.8. Phase-Contrast Microscopy
2.9. Transmission Electron Microscopy (TEM)
2.10. Mitochondrial Staining
2.11. Mitochondrial Superoxide Analysis
2.12. TMRE-Mitochondrial Membrane Potential Assay
2.13. Immunofluorescence Analysis
2.14. Small Interfering RNA (siRNA)
2.15. Mitochondrial and Lysosomal Staining
2.16. Statistical Analysis
3. Results
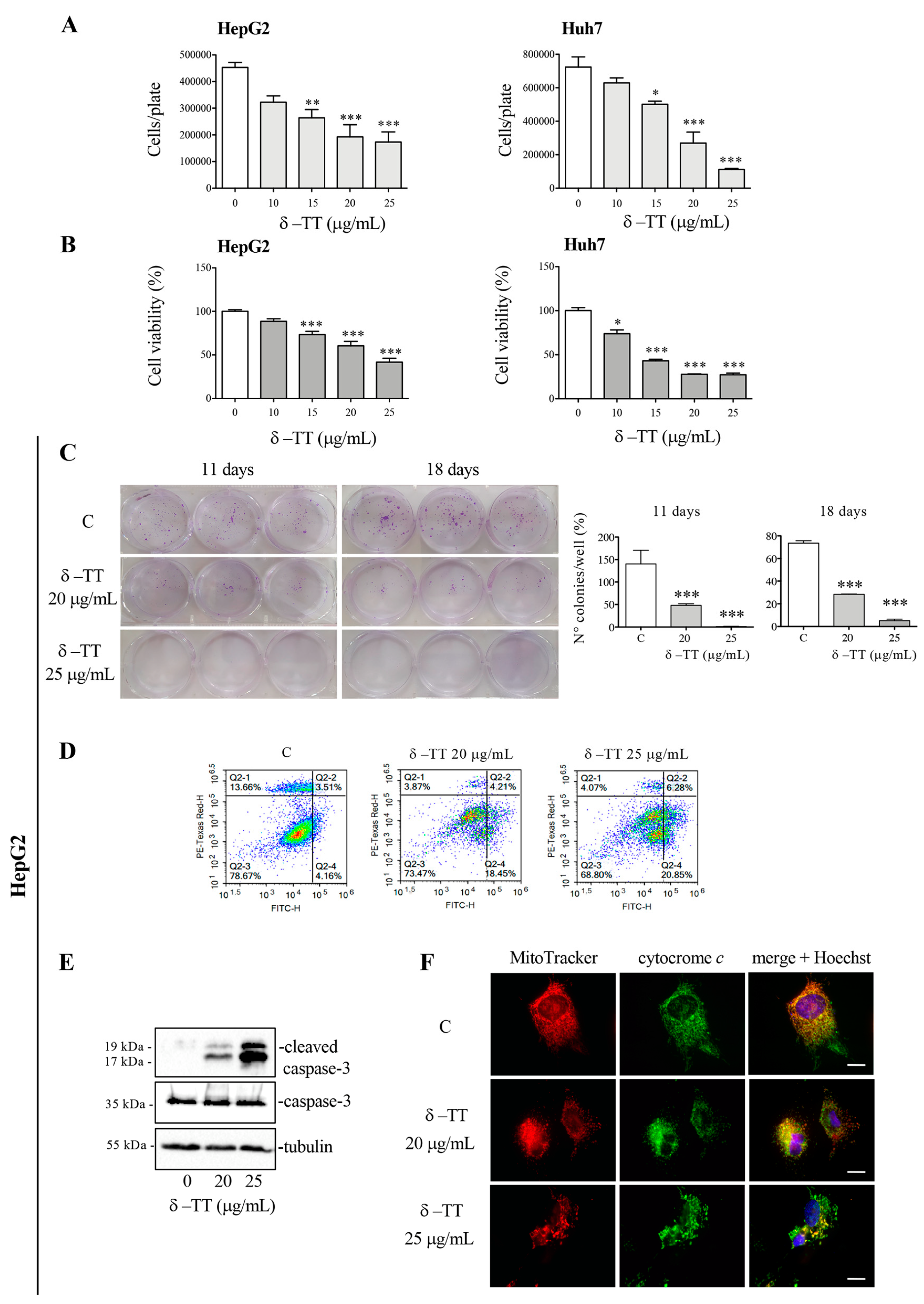
3.1. δ-TT Induces Antitumoral Effect in HCC Cells
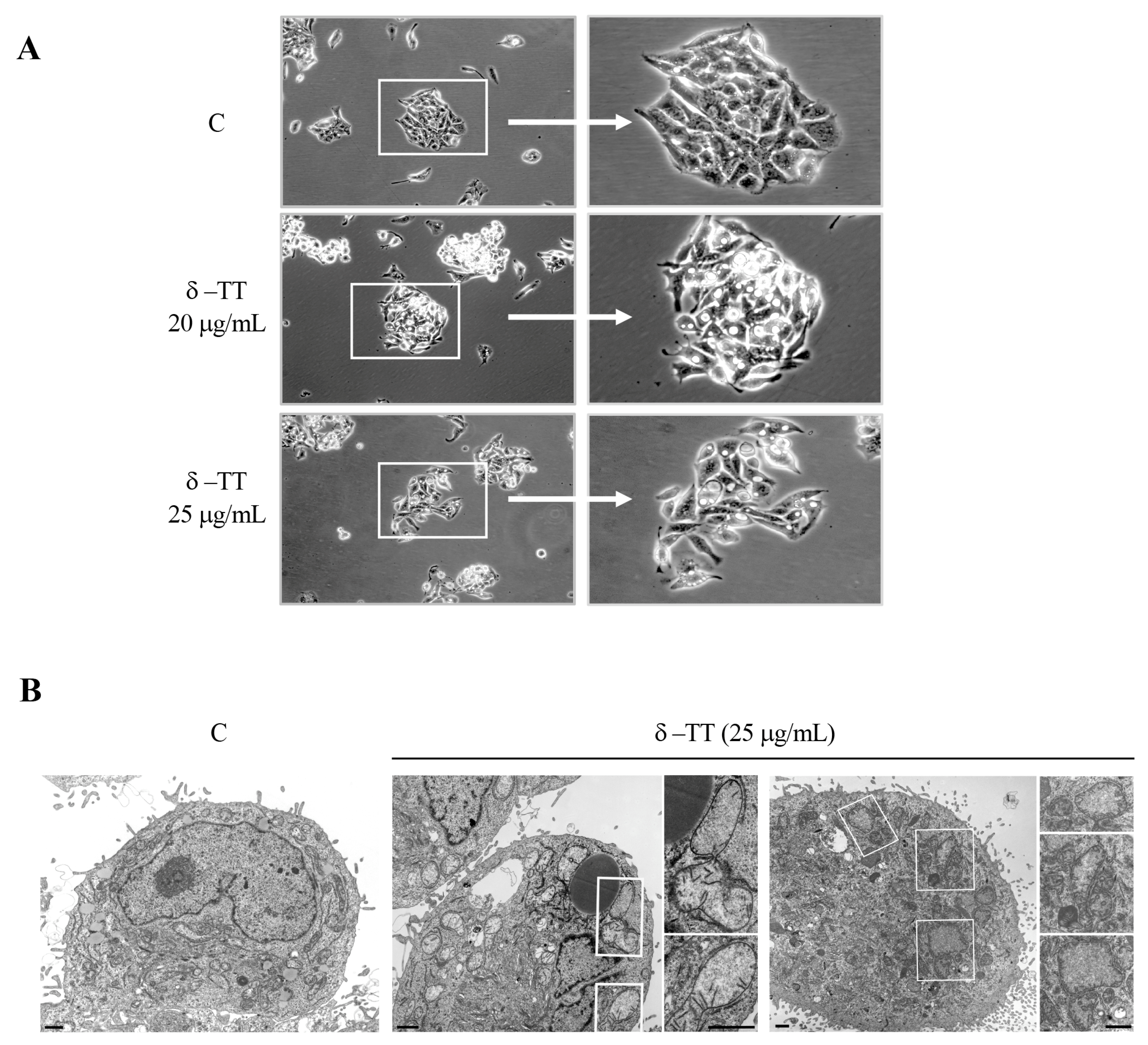
3.2. δ-TT Induces Mitochondrial Phenotypic Alteration in HCC Cells
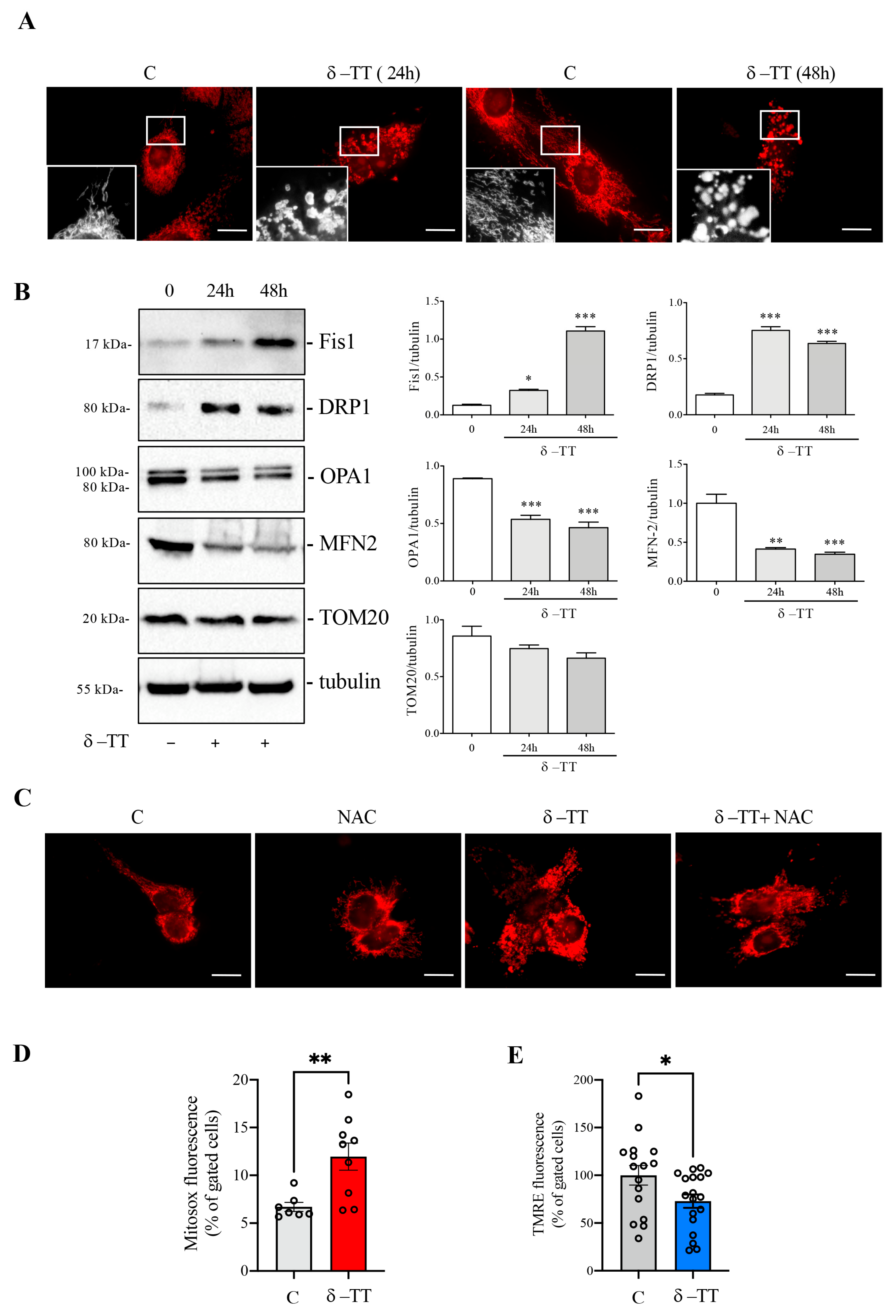
3.3. δ-TT Induces Mitochondrial Fission and ROS Release in HCC Cells
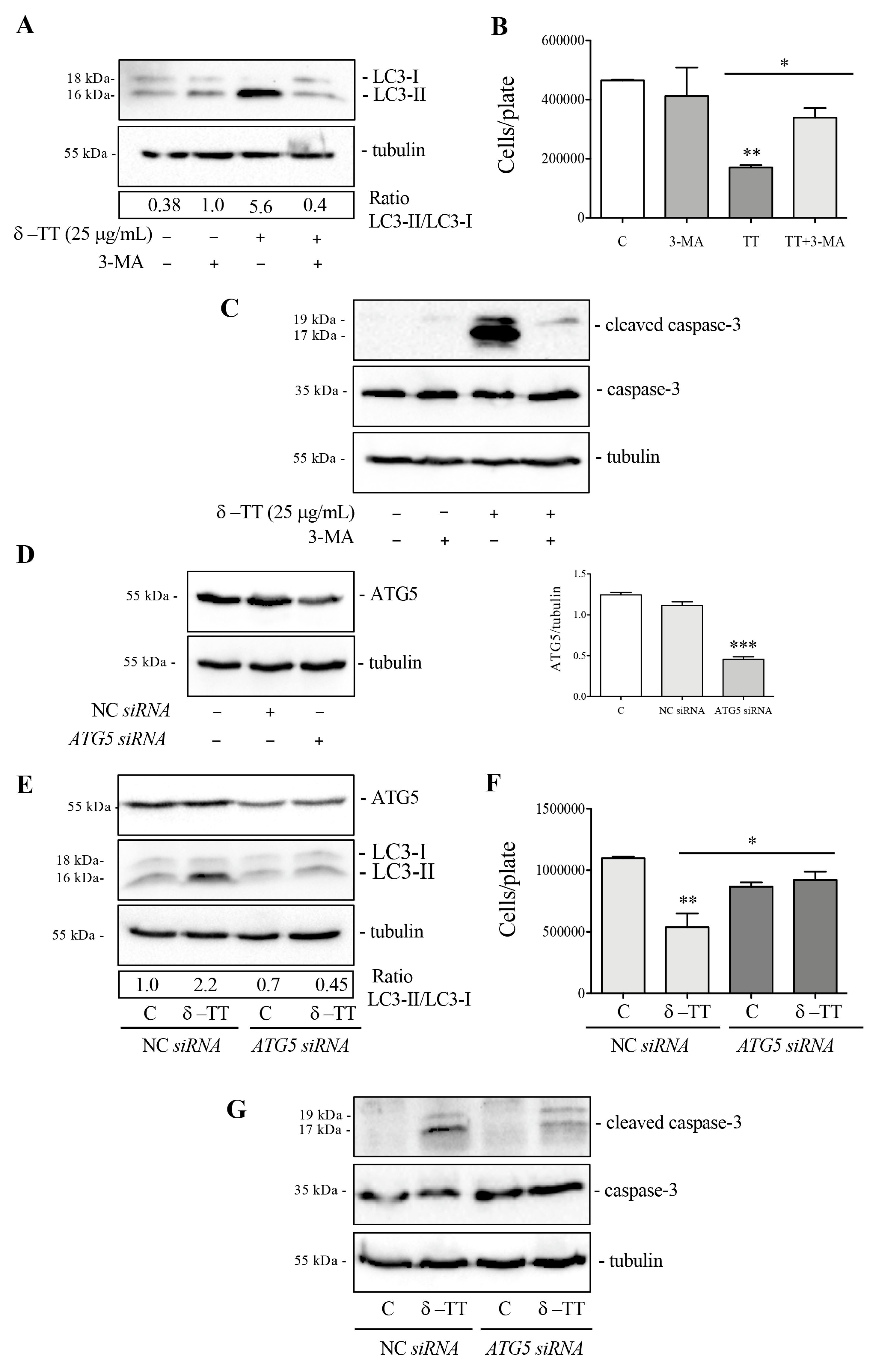
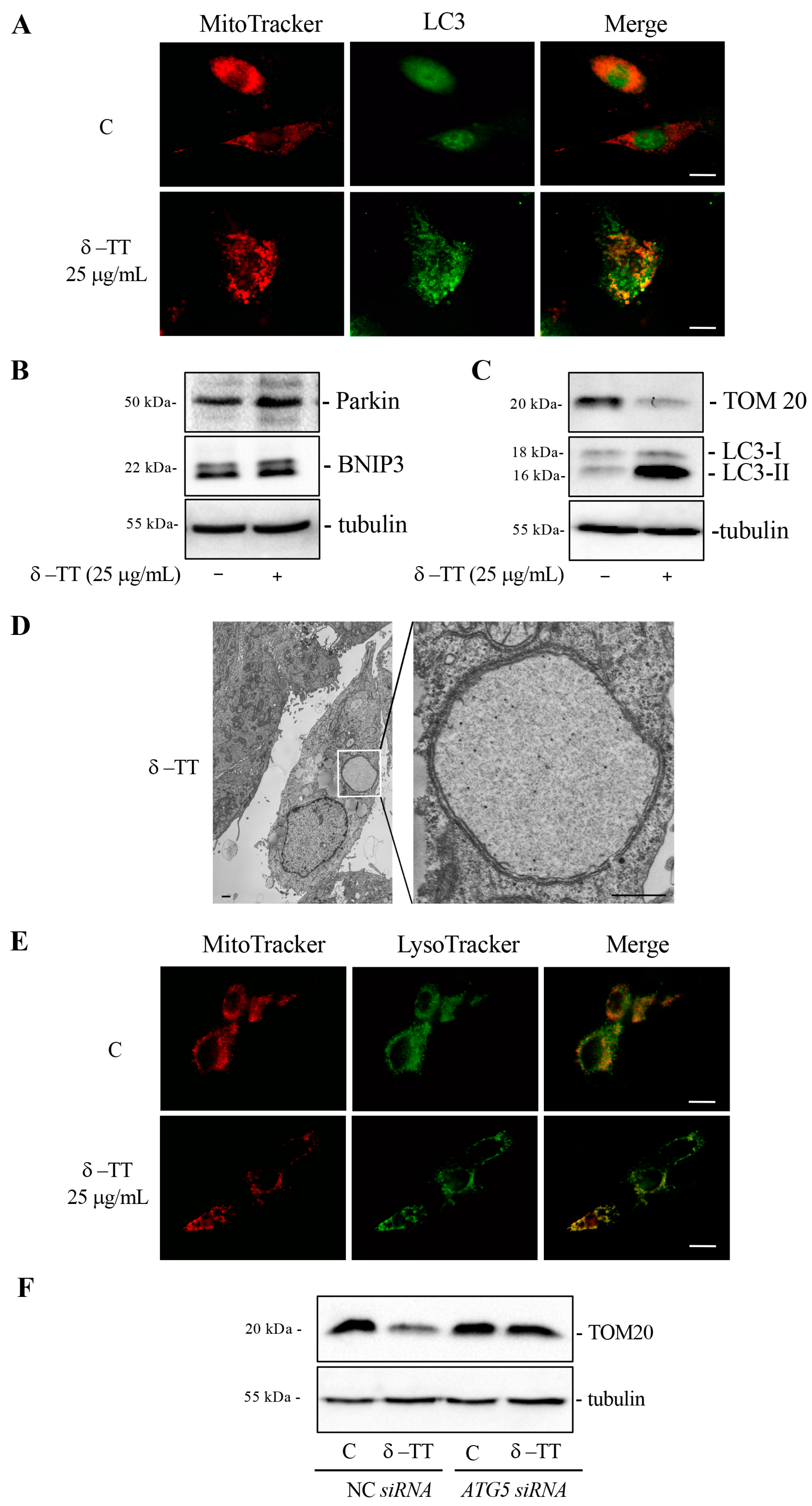
3.4. δ-TT Induces Autophagy in HCC Cells
3.5. Effect of Inhibition of Autophagy δ-TT-Induced on Survival of HCC Cells
3.6. δ-TT Induces Mitophagy in HCC Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M.; Takeda, H.; Takai, A.; Seno, H. Risk factors and diagnostic biomarkers for nonalcoholic fatty liver disease-associated hepatocellular carcinoma: Current evidence and future perspectives. World J. Gastroenterol. 2022, 28, 3410–3421. [Google Scholar] [CrossRef] [PubMed]
- Brown, Z.J.; Tsilimigras, D.I.; Ruff, S.M.; Mohseni, A.; Kamel, I.R.; Cloyd, J.M.; Pawlik, T.M. Management of Hepatocellular Carcinoma: A Review. JAMA Surg. 2023, 158, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Reig, M.; Forner, A.; Rimola, J.; Ferrer-Fàbrega, J.; Burrel, M.; Garcia-Criado, Á.; Kelley, R.K.; Galle, P.R.; Mazzaferro, V.; Salem, R.; et al. BCLC strategy for prognosis prediction and treatment recommendation: The 2022 update. J. Hepatol. 2022, 76, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Coffin, P.; He, A. Hepatocellular Carcinoma: Past and Present Challenges and Progress in Molecular Classification and Precision Oncology. Int. J. Mol. Sci. 2023, 24, 13274. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Klionsky, D.J. The emerging roles of autophagy in human diseases. Biomedicines 2021, 9, 1651. [Google Scholar] [CrossRef] [PubMed]
- Karampa, A.D.; Goussia, A.C.; Glantzounis, G.K.; Mastoridou, E.M.; Anastasopoulos, N.A.T.; Charchanti, A.V. The Role of Macroautophagy and Chaperone-Mediated Autophagy in the Pathogenesis and Management of Hepatocellular Carcinoma. Cancers 2022, 14, 760. [Google Scholar] [CrossRef] [PubMed]
- Alim Al-Bari, A.; Ito, Y.; Thomes, P.G.; Menon, M.B.; García-Macia, M.; Fadel, R.; Stadlin, A.; Peake, N.; Faris, M.A.I.E.; Eid, N.; et al. Emerging mechanistic insights of selective autophagy in hepatic diseases. Front. Pharmacol. 2023, 14, 1149809. [Google Scholar] [CrossRef] [PubMed]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef]
- Cai, H.; Meng, Z.; Yu, F. The involvement of ROS-regulated programmed cell death in hepatocellular carcinoma. Crit. Rev. Oncol. Hematol. 2024, 197, 104361. [Google Scholar] [CrossRef]
- Li, Q.; Lin, Y.; Liang, G.; Xiao, N.; Zhang, H.; Yang, X.; Yang, J.; Liu, A. Autophagy and Senescence: The Molecular Mechanisms and Implications in Liver Diseases. Int. J. Mol. Sci. 2023, 24, 16880. [Google Scholar] [CrossRef] [PubMed]
- Kouroumalis, E.; Tsomidis, I.; Voumvouraki, A. Pathogenesis of Hepatocellular Carcinoma: The Interplay of Apoptosis and Autophagy. Biomedicines 2023, 11, 1166. [Google Scholar] [CrossRef]
- Yun, C.W.; Lee, S.H. The roles of autophagy in cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, J.; Li, Q. Autophagy, an accomplice or antagonist of drug resistance in HCC? Cell Death Dis. 2021, 12, 266. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Nguyen, T.M.; Ngoc, D.T.M.; You, T.; Park, M.K.; Lee, C.H. Unraveling the Janus-Faced Role of Autophagy in Hepatocellular Carcinoma: Implications for Therapeutic Interventions. Int. J. Mol. Sci. 2023, 24, 16255. [Google Scholar] [CrossRef]
- Eggersdorfer, M.; Schmidt, K.; Péter, S.; Richards, J.; Winklhofer-Roob, B.; Hahn, A.; Obermüller-Jevic, U. Vitamin E: Not only a single stereoisomer. Free Radic. Biol. Med. 2024, 215, 106–111. [Google Scholar] [CrossRef]
- Montagnani Marelli, M.; Marzagalli, M.; Fontana, F.; Raimondi, M.; Moretti, R.M.; Limonta, P. Anticancer properties of tocotrienols: A review of cellular mechanisms and molecular targets. J. Cell. Physiol. 2019, 234, 1147–1164. [Google Scholar] [CrossRef]
- Sailo, B.L.; Banik, K.; Padmavathi, G.; Javadi, M.; Bordoloi, D.; Kunnumakkara, A.B. Tocotrienols: The promising analogues of vitamin E for cancer therapeutics. Pharmacol. Res. 2018, 130, 259–272. [Google Scholar] [CrossRef]
- Tham, S.Y.; Loh, H.S.; Mai, C.W.; Fu, J.Y. Tocotrienols modulate a life or death decision in cancers. Int. J. Mol. Sci. 2019, 20, 372. [Google Scholar] [CrossRef]
- Zhang, J.S.; Li, D.M.; Ma, Y.; He, N.; Gu, Q.; Wang, F.S.; Jiang, S.Q.; Chen, B.Q.; Liu, J.R. γ-Tocotrienol Induces Paraptosis-Like Cell Death in Human Colon Carcinoma SW620 Cells. PLoS ONE 2013, 8, e57779. [Google Scholar] [CrossRef]
- Fontana, F.; Moretti, R.M.; Raimondi, M.; Marzagalli, M.; Beretta, G.; Procacci, P.; Sartori, P.; Montagnani Marelli, M.; Limonta, P. δ-Tocotrienol induces apoptosis, involving endoplasmic reticulum stress and autophagy, and paraptosis in prostate cancer cells. Cell Prolif. 2019, 52, e12576. [Google Scholar] [CrossRef]
- Montagnani Marelli, M.; Beretta, G.; Moretti, R.M. Necroptosis Induced by Delta-Tocotrienol Overcomes Docetaxel Chemoresistance in Prostate Cancer Cells. Int. J. Mol. Sci. 2023, 24, 4923. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, R.V.; Parajuli, P.; Sylvester, P.W. γ-Tocotrienol-induced autophagy in malignant mammary cancer cells. Exp. Biol. Med. 2014, 239, 33–44. [Google Scholar] [CrossRef]
- Montagnani Marelli, M.; Marzagalli, M.; Moretti, R.M.; Beretta, G.; Casati, L.; Comitato, R.; Gravina, G.L.; Festuccia, C.; Limonta, P. Vitamin E δ-tocotrienol triggers endoplasmic reticulum stress-mediated apoptosis in human melanoma cells. Sci. Rep. 2016, 6, 30502. [Google Scholar] [CrossRef] [PubMed]
- Pang, K.L.; Mai, C.W.; Chin, K.Y. Molecular Mechanism of Tocotrienol-Mediated Anticancer Properties: A Systematic Review of the Involvement of Endoplasmic Reticulum Stress and Unfolded Protein Response. Nutrients 2023, 15, 1854. [Google Scholar] [CrossRef]
- Wada, S.; Satomi, Y.; Murakoshi, M.; Noguchi, N.; Yoshikawa, T.; Nishino, H. Tumor suppressive effects of tocotrienol in vivo and in vitro. Cancer Lett. 2005, 229, 181–191. [Google Scholar] [CrossRef]
- Sakai, M.; Okabe, M.; Tachibana, H.; Yamada, K. Apoptosis induction by γ-tocotrienol in human hepatoma Hep3B cells. J. Nutr. Biochem. 2006, 17, 672–676. [Google Scholar] [CrossRef]
- Rajendran, P.; Li, F.; Manu, K.A.; Shanmugam, M.K.; Loo, S.Y.; Kumar, A.P.; Sethi, G. γ-Tocotrienol is a novel inhibitor of constitutive and inducible STAT3 signalling pathway in human hepatocellular carcinoma: Potential role as an antiproliferative, pro-apoptotic and chemosensitizing agent. Br. J. Pharmacol. 2011, 163, 283–298. [Google Scholar] [CrossRef]
- Burdeos, G.C.; Nakagawa, K.; Kimura, F.; Miyazawa, T. Tocotrienol attenuates triglyceride accumulation in HepG2 cells and F344 rats. Lipids 2012, 47, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Burdeos, G.C.; Ito, J.; Eitsuka, T.; Nakagawa, K.; Kimura, F.; Miyazawa, T. δ and γ tocotrienols suppress human hepatocellular carcinoma cell proliferation: Via regulation of Ras-Raf-MEK-ERK pathway-associated upstream signaling. Food Funct. 2016, 7, 4170–4174. [Google Scholar] [CrossRef]
- Sivaraman Siveen, K.; Seok Ahn, K.; Ong, T.H.; Shanmugam, M.K.; Li, F.; Ney Yap, W.; Prem Kumar, A.; Wai Fong, C.; Tergaonkar, V.; Hui, K.M.; et al. γ-tocotrienol inhibits angiogenesis-dependent growth of human hepatocellular carcinoma through abrogation of AKT/mTOR pathway in an orthotopic mouse model. Oncotarget 2014, 5, 1897–1911. [Google Scholar] [CrossRef] [PubMed]
- Beretta, G.; Gelmini, F.; Fontana, F.; Moretti, R.M.; Montagnani Marelli, M.; Limonta, P. Semi-preparative HPLC purification of δ-tocotrienol (δ-T3) from Elaeis guineensis Jacq. and Bixa orellana L. and evaluation of its in vitro anticancer activity in human A375 melanoma cells. Nat. Prod. Res. 2018, 32, 1130–1135. [Google Scholar] [CrossRef]
- Wang, S.; Tan, J.; Miao, Y.; Zhang, Q. Mitochondrial Dynamics, Mitophagy, and Mitochondria–Endoplasmic Reticulum Contact Sites Crosstalk Under Hypoxia. Front. Cell Dev. Biol. 2022, 10, 848214. [Google Scholar] [CrossRef]
- Lee, Y.G.; Park, D.H.; Chae, Y.C. Role of Mitochondrial Stress Response in Cancer Progression. Cells 2022, 771. [Google Scholar] [CrossRef]
- Ciccarone, F.; Castelli, S.; Ciriolo, M.R. Oxidative stress-driven autophagy across onset and therapeutic outcome in hepatocellular carcinoma. Oxid. Med. Cell. Longev. 2019, 2019, 6050123. [Google Scholar] [CrossRef]
- Towers, C.G.; Wodetzki, D.; Thorburn, A. Autophagy and cancer: Modulation of cell death pathways and cancer cell adaptations. J. Cell Biol. 2020, 219, e201909033. [Google Scholar] [CrossRef]
- Chourasia, A.H.; Boland, M.L.; Macleod, K.F. Mitophagy and cancer. Cancer Metab. 2015, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- Ke, P.Y. Mitophagy in the Pathogenesis of Liver Diseases. Cells 2020, 9, 831. [Google Scholar] [CrossRef]
- Denisenko, T.V.; Gogvadze, V.; Zhivotovsky, B. Mitophagy in carcinogenesis and cancer treatment. Discov. Oncol. 2021, 12, 58. [Google Scholar] [CrossRef]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef]
- Lee, S.; Son, J.Y.; Lee, J.; Cheong, H. Unraveling the Intricacies of Autophagy and Mitophagy: Implications in Cancer Biology. Cells 2023, 12, 2742. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Pan, S.; Zhang, J.; Li, N.; Geng, Q. Mitophagy: A novel perspective for insighting into cancer and cancer treatment. Cell Prolif. 2022, 55, e13327. [Google Scholar] [CrossRef] [PubMed]
- Kiruthiga, C.; Devi, K.P.; Nabavi, S.M.; Bishayee, A. Autophagy: A potential therapeutic target of polyphenols in hepatocellular carcinoma. Cancers 2020, 12, 562. [Google Scholar] [CrossRef]
- Li, H.; Liu, Y.; Jiang, W.; Xue, J.; Cheng, Y.; Wang, J.; Yang, R.; Zhang, X. Icaritin promotes apoptosis and inhibits proliferation by down-regulating AFP gene expression in hepatocellular carcinoma. BMC Cancer 2021, 21, 318. [Google Scholar] [CrossRef]
- Chen, P.; Yao, L.; Yuan, M.; Wang, Z.; Zhang, Q.; Jiang, Y.; Li, L. Mitochondrial dysfunction: A promising therapeutic target for liver diseases. Genes Dis. 2024, 11, 101115. [Google Scholar] [CrossRef] [PubMed]
- Viola, V.; Ciffolilli, S.; Legnaioli, S.; Piroddi, M.; Betti, M.; Mazzini, F.; Pierpaoli, E.; Provinciali, M.; Galli, F. Mitochondrial-dependent anticancer activity of δ-tocotrienol and its synthetic derivatives in HER-2/neu overexpressing breast adenocarcinoma cells. BioFactors 2013, 39, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Fontana, F.; Marzagalli, M.; Raimondi, M.; Zuco, V.; Zaffaroni, N.; Limonta, P. δ-Tocotrienol sensitizes and re-sensitizes ovarian cancer cells to cisplatin via induction of G1 phase cell cycle arrest and ROS/MAPK-mediated apoptosis. Cell Prolif. 2021, 54, e13111. [Google Scholar] [CrossRef] [PubMed]
- Gogvadze, V.; Norberg, E.; Orrenius, S.; Zhivotovsky, B. Involvement of Ca2+ and ROS in α-tocopheryl succinate-induced mitochondrial permeabilization. Int. J. Cancer 2010, 127, 1823–1832. [Google Scholar] [CrossRef]
- Raimondi, M.; Fontana, F.; Marzagalli, M.; Audano, M.; Beretta, G.; Procacci, P.; Sartori, P.; Mitro, N.; Limonta, P. Ca2+ overload- and ROS-associated mitochondrial dysfunction contributes to δ-tocotrienol-mediated paraptosis in melanoma cells. Apoptosis 2021, 26, 277–292. [Google Scholar] [CrossRef]
- Suman, S.; Datta, K.; Chakraborty, K.; Kulkarni, S.S.; Doiron, K.; Fornace, A.J.; Sree Kumar, K.; Hauer-Jensen, M.; Ghosh, S.P. Gamma tocotrienol, a potent radioprotector, preferentially upregulates expression of anti-apoptotic genes to promote intestinal cell survival. Food Chem. Toxicol. 2013, 60, 488–496. [Google Scholar] [CrossRef]
- Pathak, R.; Bachri, A.; Ghosh, S.P.; Koturbash, I.; Boerma, M.; Binz, R.K.; Sawyer, J.R.; Hauer-Jensen, M. The Vitamin E Analog Gamma-Tocotrienol (GT3) Suppresses Radiation-Induced Cytogenetic Damage. Pharm. Res. 2016, 33, 2117–2125. [Google Scholar] [CrossRef] [PubMed]
- Khor, S.C.; Wan Ngah, W.Z.; Mohd Yusof, Y.A.; Abdul Karim, N.; Makpol, S. Tocotrienol-Rich Fraction Ameliorates Antioxidant Defense Mechanisms and Improves Replicative Senescence-Associated Oxidative Stress in Human Myoblasts. Oxid. Med. Cell Longev. 2017, 2017, 3868305. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Casati, L.; Pagani, F.; Limonta, P.; Vanetti, C.; Stancari, G.; Sibilia, V. Beneficial effects of δ-tocotrienol against oxidative stress in osteoblastic cells: Studies on the mechanisms of action. Eur. J. Nutr. 2020, 59, 1975–1987. [Google Scholar] [CrossRef]
- Yang, S.; Yang, J.; Zhao, H.; Deng, R.; Fan, H.; Zhang, J.; Yang, Z.; Zeng, H.; Kuang, B.; Shao, L. The Protective Effects of γ-Tocotrienol on Muscle Stem Cells Through Inhibiting Reactive Oxidative Stress Production. Front. Cell Dev. Biol. 2022, 10, 820520. [Google Scholar] [CrossRef]
- Tan, C.Y.; Saw, T.Y.; Fong, C.W.; Ho, H.K. Comparative hepatoprotective effects of tocotrienol analogs against drug-induced liver injury. Redox Biol. 2015, 4, 308–320. [Google Scholar] [CrossRef] [PubMed]
- Pervez, M.A.; Khan, D.A.; Ijaz, A.; Khan, S. Effects of delta-tocotrienol supplementation on liver enzymes, inflammation, oxidative stress and hepatic steatosis in patients with nonalcoholic fatty liver disease. Turkish J. Gastroenterol. 2018, 29, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Pervez, M.A.; Khan, D.A.; Gilani, S.T.A.; Fatima, S.; Ijaz, A.; Nida, S. Hepato-Protective Effects of Delta-Tocotrienol and Alpha-Tocopherol in Patients with Non-Alcoholic Fatty Liver Disease: Regulation of Circulating MicroRNA Expression. Int. J. Mol. Sci. 2023, 24, 79. [Google Scholar] [CrossRef] [PubMed]
- Magosso, E.; Ansari, M.A.; Gopalan, Y.; Shuaib, I.L.; Wong, J.W.; Khan, N.A.K.; Abu Bakar, M.R.; Ng, B.H.; Yuen, K.H. Tocotrienols for normalisation of hepatic echogenic response in nonalcoholic fatty liver: A randomised placebo-controlled clinical trial. Nutr. J. 2013, 12, 166. [Google Scholar] [CrossRef]
- Pervez, M.A.; Khan, D.A.; Slehria, A.U.R.; Ijaz, A. Delta-tocotrienol supplementation improves biochemical markers of hepatocellular injury and steatosis in patients with nonalcoholic fatty liver disease: A randomized, placebo-controlled trial. Complement. Ther. Med. 2020, 52, 102494. [Google Scholar] [CrossRef]
- Pervez, M.A.; Khan, D.A.; Mirza, S.A.; Slehria, A.U.R.; Nisar, U.; Aamir, M. Comparison of delta-tocotrienol and alpha-tocopherol effects on hepatic steatosis and inflammatory biomarkers in patients with non-alcoholic fatty liver disease: A randomized double-blind active-controlled trial. Complement. Ther. Med. 2022, 70, 102866. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Green, D.R.; Zou, W. Autophagy in tumour immunity and therapy. Nat. Rev. Cancer 2021, 21, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Ariosa, A.R.; Lahiri, V.; Lei, Y.; Yang, Y.; Yin, Z.; Zhang, Z.; Klionsky, D.J. A perspective on the role of autophagy in cancer. Biochim. Biophys. Acta-Mol. Basis Dis. 2021, 1867, 166262. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Klionsky, D.J. Life and Death Decisions—The Many Faces of Autophagy in Cell Survival and Cell Death. Biomolecules 2022, 12, 866. [Google Scholar] [CrossRef]
- Wang, H.X.; Luo, J.T.; Tian, W.X.; Yan, W.Q.; Ge, S.; Zhang, Y.H.; Sun, W.G. γ-Tocotrienol inhibits oxidative phosphorylation and triggers apoptosis by inhibiting mitochondrial complex I subunit NDUFB8 and complex II subunit SDHB. Toxicology 2019, 417, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Ferro, F.; Servais, S.; Besson, P.; Roger, S.; Dumas, J.F.; Brisson, L. Autophagy and mitophagy in cancer metabolic remodelling. Semin. Cell Dev. Biol. 2020, 98, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, W.; Bian, Y.; Li, Y.; Cong, L. Multifaceted roles of aerobic glycolysis and oxidative phosphorylation in hepatocellular carcinoma. PeerJ 2023, 11, e14797. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montagnani Marelli, M.; Macchi, C.; Ruscica, M.; Sartori, P.; Moretti, R.M. Anticancer Activity of Delta-Tocotrienol in Human Hepatocarcinoma: Involvement of Autophagy Induction. Cancers 2024, 16, 2654. https://doi.org/10.3390/cancers16152654
Montagnani Marelli M, Macchi C, Ruscica M, Sartori P, Moretti RM. Anticancer Activity of Delta-Tocotrienol in Human Hepatocarcinoma: Involvement of Autophagy Induction. Cancers. 2024; 16(15):2654. https://doi.org/10.3390/cancers16152654
Chicago/Turabian StyleMontagnani Marelli, Marina, Chiara Macchi, Massimiliano Ruscica, Patrizia Sartori, and Roberta Manuela Moretti. 2024. "Anticancer Activity of Delta-Tocotrienol in Human Hepatocarcinoma: Involvement of Autophagy Induction" Cancers 16, no. 15: 2654. https://doi.org/10.3390/cancers16152654