
Unraveling Cancer’s Wnt Signaling: Dynamic Control through Protein Kinase Regulation
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Canonical and Non-Canonical Wnt Signaling: A Comprehensive Categorization
2.1. Canonical Wnt/β-Catenin Pathway
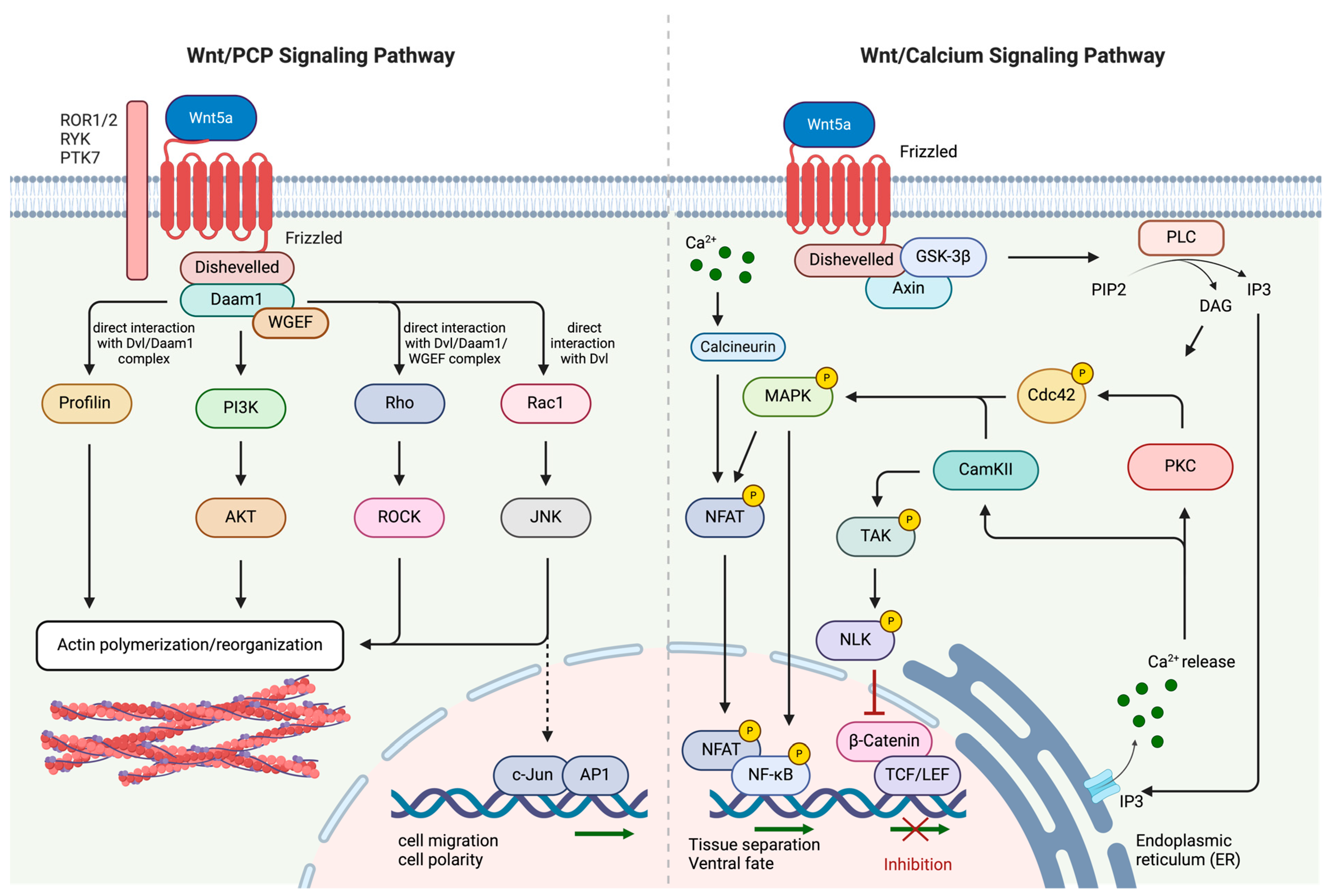
2.2. Non-Canonical Wnt/PCP and Wnt/Ca2+ Signaling Pathways
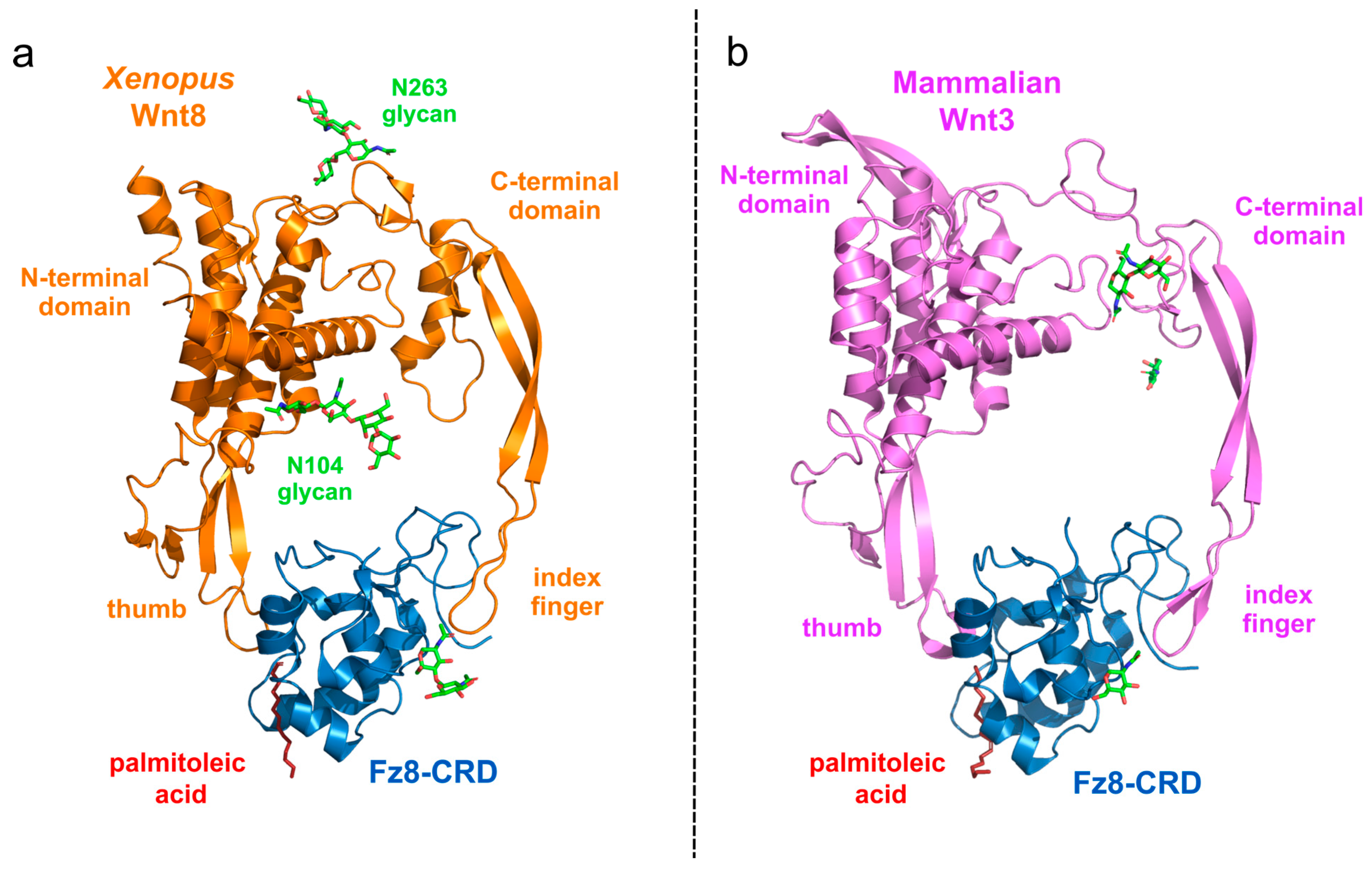
2.3. Protein Structure of Wnt Ligand and FZD Receptor Binding
3. Wnt Signaling in Cancer
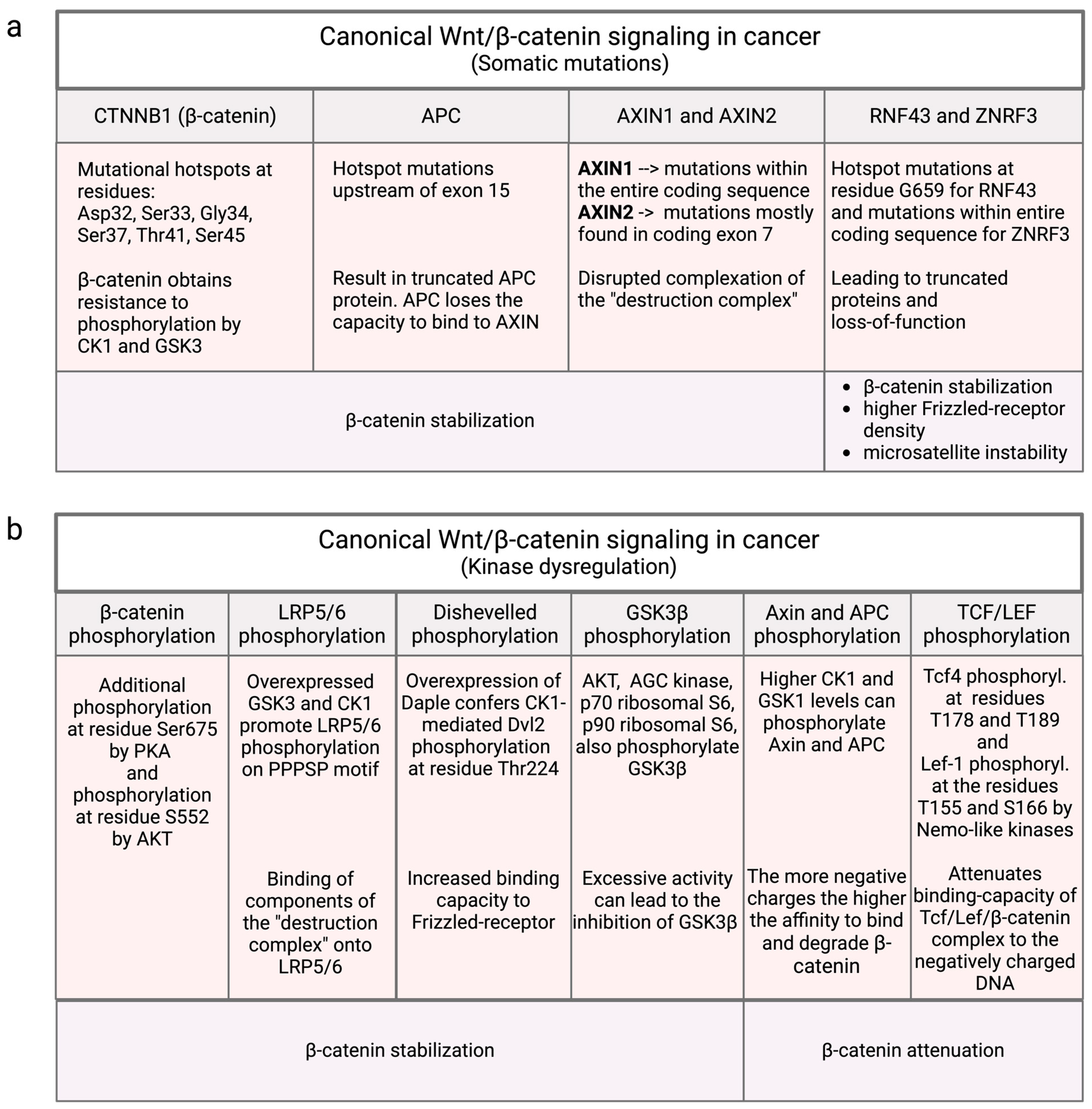
3.1. Mutational Influence on Canonical Wnt/β-Catenin Signaling
3.2. Non-Mutational Influences in Canonical Wnt/β-Catenin Signaling
3.2.1. β-Catenin Phosphorylation
3.2.2. LRP5/6 Phosphorylation
3.2.3. Dvl Phosphorylation
3.2.4. GSK3β Phosphorylation
3.2.5. Axin and APC Phosphorylation by CK1 and GSK3
3.2.6. Phosphorylation of TCF/LEF by Nlk and Casein Kinases
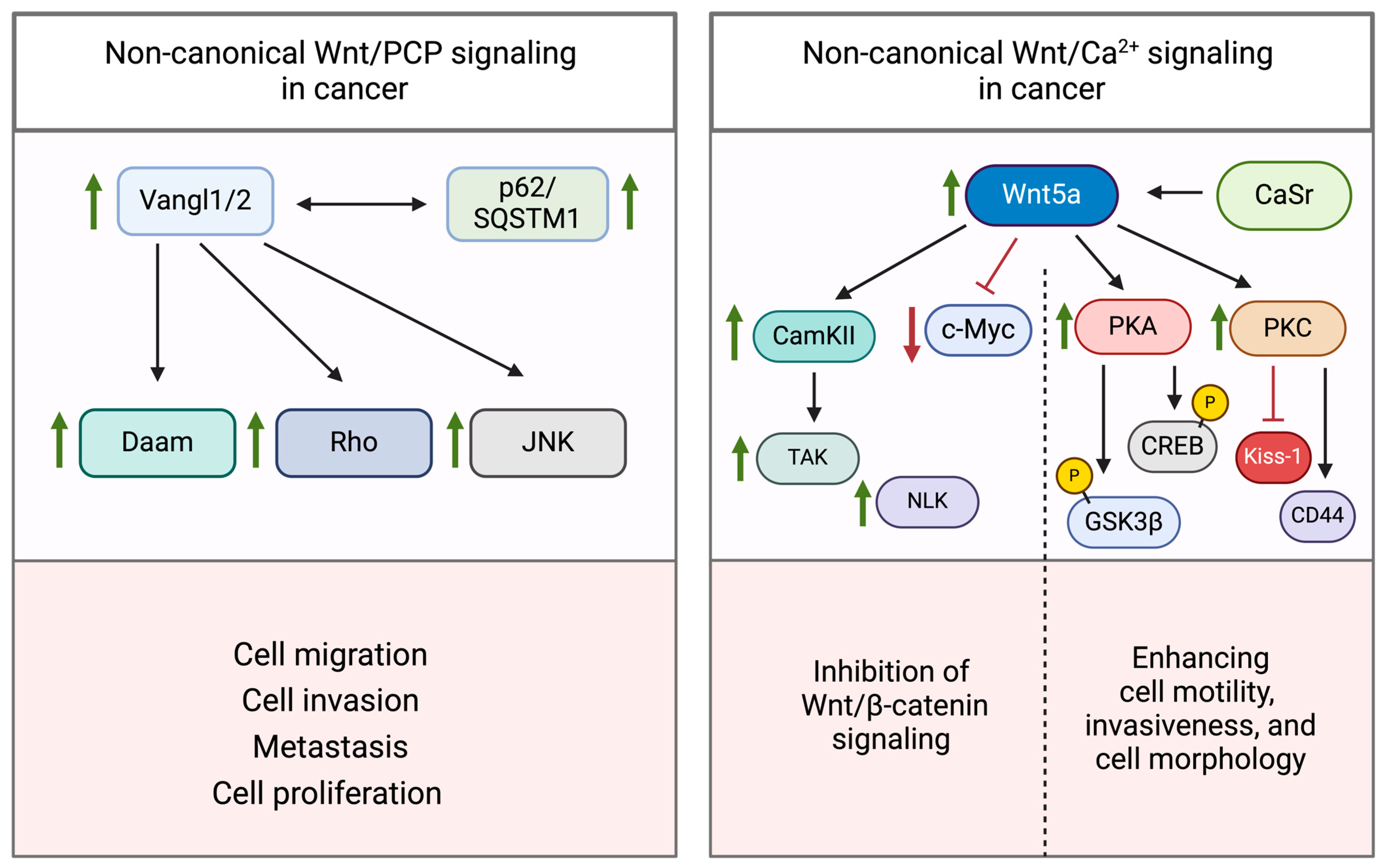
3.3. Dysregulation of Non-Canonical Wnt Signaling
3.3.1. Wnt/PCP Pathway
3.3.2. Wnt/Ca2+ Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Tumor Pro-Oncogene /Suppressor | Malignancy (Tissue) | References |
|---|---|---|---|
| CTNNB1 (β-catenin) | Proto-oncogene | Hepatocellular carcinoma, pancreatic cancer, colorectal cancer, gastroesophageal/junction carcinoma, gastric adeno- carcinoma | [57,58] |
| APC | Suppressor | CRC, uterine endometrial cancer, stomach cancer, skin cutaneous melanoma | [61,62] |
| AXIN1 | Suppressor | HCC uterine endometrial cancer | [61,65] |
| AXIN2 | Suppressor | Colorectal cancer | [61,65] |
| RNF43 | Suppressor | Colorectal cancer, endometrial cancer | [67] |
| ZNRF3 | Suppressor | Uterine and skin cancer | [61] |
| Nlk | Suppressor | NSCLC (non-small-cell lung cancer) | [120] |
| Frizzled receptor | Proto-oncogene | Breast cancer | [125,128,129] |
| Prostate cancer | [130] | ||
| Gastric cancer | [131,132] | ||
| Colorectal cancer | [133,134] | ||
| Glioblastoma | [127] | ||
| Vangl2 | Proto-oncogene | Breast cancer, ovarian cancer, uterine corpus endometrial carcinoma | [137] |
| PRINCKLE1 | Suppressor | Breast cancer | [141] |
| MINK1 | Suppressor | Breast cancer | [141] |
| Wnt5a | Suppressor | Neuroblastoma | [142,143] |
| Esophageal squamous cell carcinoma | [144] | ||
| Acute myeloid lymphoma | [145,146,147] | ||
| Breast cancer | [148,149,150] | ||
| Thyroid carcinoma | [152] | ||
| Colon carcinoma | [152,153] | ||
| Wnt5a | Proto-oncogene | Prostate cancer | [154,155] |
| Melanoma | [156,157] | ||
| Breast cancer | [158] | ||
| Pancreatic cancer | [159,160,161] |
4. Targeting the Wnt Signaling Pathway in Cancer Therapy
4.1. Targeting the Wnt Signaling Pathway on Distinct Cellular Levels
- (1)
- Extracellular and Membrane Levels: The activation of the Wnt pathway is influenced by the presence of Wnt ligands and the expression of receptors such as Frizzled (Fzd) and co-receptors LRP5/6. Preventing the interaction of Wnt ligands with their receptors effectively blocks the signal transduction and subsequently reduces tumor growth and metastasis. Niclosamide, for instance, is a pharmaceutical compound belonging to the class of anthelmintics. It is a salicylamide derivative that is normally used for the treatment of parasitic worm infestations and mollusks. Interestingly, it was found that niclosamide inhibits the Wnt/β-catenin signaling pathway through several mechanisms: It enhances the internalization of the Wnt receptor Frizzled 1 (Fzd1) [168], promotes the degradation of the Wnt co-receptor LRP6 [169], suppresses the expression of the Wnt signaling regulator Dishevelled 2 (Dvl2) [170] and inhibits the formation of the β-catenin/TCF complex [171].
- (2)
- Cytoplasmic Level: At the cytoplasmic level, the stability and concentration of beta-catenin are controlled by components like APC, Axin, and the phosphorylation status of COX-2. Therapeutics that stabilize Axin or other components of the destruction complex, promote the degradation of β-catenin. Inhibition of β-catenin effectively reduces Wnt signaling activity and thereby cell migration and proliferation. Such compounds include the small molecule pyrazole-4-carboxamide (YW2065) [172], which is listed and discussed below.
- (3)
- Nuclear Level: In the nucleus, Wnt signaling regulates gene transcription through factors such as LEF/TCFs, CBP, c-Myc, and cyclin D1, which are essential for cell proliferation and differentiation. Blocking the interaction between β-catenin and CBP leads to reduced β-catenin-mediated transcription. This approach has shown promise in preclinical studies, particularly in enhancing the efficacy of cytotoxic and targeted therapies. Such compounds include the small molecule Foscevivint (PRI-724), which is listed and discussed below [173].
4.2. Therapeutic Classes
4.2.1. Natural Compounds
4.2.2. Small Molecules
4.2.3. Therapeutic Peptides and Peptide Mimetics
4.2.4. Monoclonal Antibodies
4.2.5. Novel Emerging Strategies
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nüsslein-Volhard, C.; Wieschaus, E. Mutations Affecting Segment Number and Polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Varmus, H.E. Many Tumors Induced by the Mouse Mammary Tumor Virus Contain a Provirus Integrated in the Same Region of the Host Genome. Cell 1982, 31, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Wodarz, A.; Nusse, R. Mechanisms of Wnt Signaling in Development. Annu. Rev. Cell Dev. Biol. 1998, 14, 59–88. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.Y.; Nusse, R. The Wnt Signaling Pathway in Development and Disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef] [PubMed]
- Holstein, T.W. The Evolution of the Wnt Pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a007922. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Holzem, M.; Boutros, M.; Holstein, T.W. The Origin and Evolution of Wnt Signalling. Nat. Rev. Genet. 2024, 25, 500–512. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Wnt/β-Catenin Signaling in Development and Disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef]
- Kahn, M. Can We Safely Target the WNT Pathway? Nat. Rev. Drug Discov. 2014, 13, 513–532. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt Signaling in Cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Clevers, H.; Nusse, R. Wnt/β-Catenin Signaling and Disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef]
- Acebron, S.P.; Karaulanov, E.; Berger, B.S.; Huang, Y.-L.; Niehrs, C. Mitotic Wnt Signaling Promotes Protein Stabilization and Regulates Cell Size. Mol. Cell 2014, 54, 663–674. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Haas, A.; Bufe, A.; Parbin, S.; Hennecke, M.; Voloshanenko, O.; Gross, J.; Boutros, M.; Acebron, S.P.; Bastians, H. Wnt10b-GSK3β–Dependent Wnt/STOP Signaling Prevents Aneuploidy in Human Somatic Cells. Life Sci. Alliance 2021, 4. [Google Scholar] [CrossRef]
- Staal, F.J.T.; Luis, T.C.; Tiemessen, M.M. WNT Signalling in the Immune System: WNT Is Spreading Its Wings. Nat. Rev. Immunol. 2008, 8, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Verheyen, E.M.; Gottardi, C.J. Regulation of Wnt/β-Catenin Signaling by Protein Kinases. Dev. Dyn. 2010, 239, 34–44. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-Catenin Signaling: Components, Mechanisms, and Diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Voloshanenko, O.; Gmach, P.; Winter, J.; Kranz, D.; Boutros, M. Mapping of Wnt-Frizzled Interactions by Multiplex CRISPR Targeting of Receptor Gene Families. FASEB J. 2017, 31, 4832–4844. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G. Wnt/β-Catenin Signalling: Function, Biological Mechanisms, and Therapeutic Opportunities. Sig Transduct. Target. Ther. 2022, 7, 1–23. [Google Scholar] [CrossRef]
- Anthony, C.C.; Robbins, D.J.; Ahmed, Y.; Lee, E. Nuclear Regulation of Wnt/β-Catenin Signaling: It’s a Complex Situation. Genes 2020, 11, 886. [Google Scholar] [CrossRef]
- Sharma, M.; Jamieson, C.; Lui, C.; Henderson, B.R. Distinct Hydrophobic “Patches” in the N- and C-Tails of Beta-Catenin Contribute to Nuclear Transport. Exp. Cell Res. 2016, 348, 132–145. [Google Scholar] [CrossRef]
- Fagotto, F.; Glück, U.; Gumbiner, B.M. Nuclear Localization Signal-Independent and Importin/Karyopherin-Independent Nuclear Import of Beta-Catenin. Curr. Biol. 1998, 8, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.Y.; Kostiuk, V.; González, D.P.; Lusk, C.P.; Khokha, M.K. Kap-Β2/Transportin Mediates β-Catenin Nuclear Transport in Wnt Signaling. eLife 2022, 11, e70495. [Google Scholar] [CrossRef] [PubMed]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of C-MYC as a Target of the APC Pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Shtutman, M.; Zhurinsky, J.; Simcha, I.; Albanese, C.; D’Amico, M.; Pestell, R.; Ben-Ze’ev, A. The Cyclin D1 Gene Is a Target of the Beta-Catenin/LEF-1 Pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 5522–5527. [Google Scholar] [CrossRef] [PubMed]
- Angers, S.; Moon, R.T. Proximal Events in Wnt Signal Transduction. Nat. Rev. Mol. Cell Biol. 2009, 10, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Pate, K.T.; Stringari, C.; Sprowl-Tanio, S.; Wang, K.; TeSlaa, T.; Hoverter, N.P.; McQuade, M.M.; Garner, C.; Digman, M.A.; Teitell, M.A.; et al. Wnt Signaling Directs a Metabolic Program of Glycolysis and Angiogenesis in Colon Cancer. EMBO J. 2014, 33, 1454–1473. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-C.; Klein, P.S. Multiple Roles for Glycogen Synthase Kinase-3 as a Drug Target in Alzheimer’s Disease. Curr. Drug Targets 2006, 7, 1389–1397. [Google Scholar] [CrossRef] [PubMed]
- Shang, S.; Hua, F.; Hu, Z.-W. The Regulation of β-Catenin Activity and Function in Cancer: Therapeutic Opportunities. Oncotarget 2017, 8, 33972–33989. [Google Scholar] [CrossRef]
- Haseeb, M.; Pirzada, R.H.; Ain, Q.U.; Choi, S. Wnt Signaling in the Regulation of Immune Cell and Cancer Therapeutics. Cells 2019, 8, 1380. [Google Scholar] [CrossRef]
- Liu, C.; Li, Y.; Semenov, M.; Han, C.; Baeg, G.H.; Tan, Y.; Zhang, Z.; Lin, X.; He, X. Control of Beta-Catenin Phosphorylation/Degradation by a Dual-Kinase Mechanism. Cell 2002, 108, 837–847. [Google Scholar] [CrossRef]
- Daniels, D.L.; Weis, W.I. Beta-Catenin Directly Displaces Groucho/TLE Repressors from Tcf/Lef in Wnt-Mediated Transcription Activation. Nat. Struct. Mol. Biol. 2005, 12, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Yang, Y.; Li, Q.; He, X.; Zhu, W.; Wang, J.; Gan, X. Oligomerization of Frizzled and LRP5/6 Protein Initiates Intracellular Signaling for the Canonical WNT/β-Catenin Pathway. J. Biol. Chem. 2018, 293, 19710–19724. [Google Scholar] [CrossRef] [PubMed]
- Graham, T.A.; Weaver, C.; Mao, F.; Kimelman, D.; Xu, W. Crystal Structure of a β-Catenin/Tcf Complex. Cell 2000, 103, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Boonekamp, K.E.; Heo, I.; Artegiani, B.; Asra, P.; van Son, G.; de Ligt, J.; Clevers, H. Identification of Novel Human Wnt Target Genes Using Adult Endodermal Tissue-Derived Organoids. Dev. Biol. 2021, 474, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Qin, K.; Yu, M.; Fan, J.; Wang, H.; Zhao, P.; Zhao, G.; Zeng, W.; Chen, C.; Wang, Y.; Wang, A.; et al. Canonical and Noncanonical Wnt Signaling: Multilayered Mediators, Signaling Mechanisms and Major Signaling Crosstalk. Genes Dis. 2024, 11, 103–134. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Mlodzik, M. Wnt-Frizzled/Planar Cell Polarity Signaling: Cellular Orientation by Facing the Wind (Wnt). Annu. Rev. Cell Dev. Biol. 2015, 31, 623–646. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Borchers, A.G.M.; Jolicoeur, C.; Rayburn, H.; Baker, J.C.; Tessier-Lavigne, M. PTK7/CCK-4 Is a Novel Regulator of Planar Cell Polarity in Vertebrates. Nature 2004, 430, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Grumolato, L.; Liu, G.; Mong, P.; Mudbhary, R.; Biswas, R.; Arroyave, R.; Vijayakumar, S.; Economides, A.N.; Aaronson, S.A. Canonical and Noncanonical Wnts Use a Common Mechanism to Activate Completely Unrelated Coreceptors. Genes. Dev. 2010, 24, 2517–2530. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, K.; Hall, A.; Tolwinski, N. Wnt Signaling Pathways Meet Rho GTPases. Genes. Dev. 2009, 23, 265–277. [Google Scholar] [CrossRef]
- Boutros, M.; Paricio, N.; Strutt, D.I.; Mlodzik, M. Dishevelled Activates JNK and Discriminates between JNK Pathways in Planar Polarity and Wingless Signaling. Cell 1998, 94, 109–118. [Google Scholar] [CrossRef]
- Barkó, S.; Bugyi, B.; Carlier, M.-F.; Gombos, R.; Matusek, T.; Mihály, J.; Nyitrai, M. Characterization of the Biochemical Properties and Biological Function of the Formin Homology Domains of Drosophila DAAM. J. Biol. Chem. 2010, 285, 13154–13169. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, M.; Xu, F.; Jiang, S. Wnt Signaling in Breast Cancer: Biological Mechanisms, Challenges and Opportunities. Mol. Cancer 2020, 19, 165. [Google Scholar] [CrossRef] [PubMed]
- Komiya, Y.; Habas, R. Wnt Signal Transduction Pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Xie, Z.; Wang, J.; Li, M.; Jing, N.; Li, L. Nuclear Factor of Activated T Cells (NFAT) Proteins Repress Canonical Wnt Signaling via Its Interaction with Dishevelled (Dvl) Protein and Participate in Regulating Neural Progenitor Cell Proliferation and Differentiation. J. Biol. Chem. 2011, 286, 37399–37405. [Google Scholar] [CrossRef]
- De, A. Wnt/Ca2+ Signaling Pathway: A Brief Overview. Acta Biochimica et Biophysica Sinica 2011, 43, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Kohn, A.D.; Moon, R.T. Wnt and Calcium Signaling: β-Catenin-Independent Pathways. Cell Calcium 2005, 38, 439–446. [Google Scholar] [CrossRef]
- Berridge, M.J. The Inositol Trisphosphate/Calcium Signaling Pathway in Health and Disease. Physiol. Rev. 2016, 96, 1261–1296. [Google Scholar] [CrossRef]
- Kühl, M.; Sheldahl, L.C.; Malbon, C.C.; Moon, R.T. Ca2+/Calmodulin-Dependent Protein Kinase II Is Stimulated by Wnt and Frizzled Homologs and Promotes Ventral Cell Fates in Xenopus. J. Biol. Chem. 2000, 275, 12701–12711. [Google Scholar] [CrossRef]
- Sheldahl, L.C.; Park, M.; Malbon, C.C.; Moon, R.T. Protein Kinase C Is Differentially Stimulated by Wnt and Frizzled Homologs in aG-Protein-Dependent Manner. Curr. Biol. 1999, 9, S1. [Google Scholar] [CrossRef]
- Mezzacappa, C.; Komiya, Y.; Habas, R. Activation and Function of Small GTPases Rho, Rac, and Cdc42 During Gastrulation. Methods Mol. Biol. 2012, 839, 119–131. [Google Scholar] [CrossRef]
- Zhang, Q.; Yu, J.; Chen, Q.; Yan, H.; Du, H.; Luo, W. Regulation of Pathophysiological and Tissue Regenerative Functions of MSCs Mediated via the WNT Signaling Pathway (Review). Mol. Med. Rep. 2021, 24, 1–14. [Google Scholar] [CrossRef]
- Ishitani, T.; Kishida, S.; Hyodo-Miura, J.; Ueno, N.; Yasuda, J.; Waterman, M.; Shibuya, H.; Moon, R.T.; Ninomiya-Tsuji, J.; Matsumoto, K. The TAK1-NLK Mitogen-Activated Protein Kinase Cascade Functions in the Wnt-5a/Ca2+ Pathway To Antagonize Wnt/β-Catenin Signaling. Mol. Cell Biol. 2003, 23, 131–139. [Google Scholar] [CrossRef]
- Hayat, R.; Manzoor, M.; Hussain, A. Wnt Signaling Pathway: A Comprehensive Review. Cell Biol. Int. 2022, 46, 863–877. [Google Scholar] [CrossRef] [PubMed]
- Janda, C.Y.; Waghray, D.; Levin, A.M.; Thomas, C.; Garcia, K.C. Structural Basis of Wnt Recognition by Frizzled. Science 2012, 337, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Hirai, H.; Matoba, K.; Mihara, E.; Arimori, T.; Takagi, J. Crystal Structure of a Mammalian Wnt–Frizzled Complex. Nat. Struct. Mol. Biol. 2019, 26, 372–379. [Google Scholar] [CrossRef]
- Nie, X.; Liu, H.; Liu, L.; Wang, Y.-D.; Chen, W.-D. Emerging Roles of Wnt Ligands in Human Colorectal Cancer. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Tümen, D.; Heumann, P.; Gülow, K.; Demirci, C.-N.; Cosma, L.-S.; Müller, M.; Kandulski, A. Pathogenesis and Current Treatment Strategies of Hepatocellular Carcinoma. Biomedicines 2022, 10, 3202. [Google Scholar] [CrossRef]
- Kim, S.; Jeong, S. Mutation Hotspots in the β-Catenin Gene: Lessons from the Human Cancer Genome Databases. Mol. Cells 2019, 42, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhao, T.; Zhang, S.; Wang, J.; Chen, Y.; Zhao, H.; Yang, Y.; Shi, S.; Chen, Q.; Liu, K. The Wnt Signaling Pathway in Tumorigenesis, Pharmacological Targets, and Drug Development for Cancer Therapy. Biomark. Res. 2021, 9, 68. [Google Scholar] [CrossRef]
- Birkeland, A.C.; Burgin, S.J.; Yanik, M.; Scott, M.V.; Bradford, C.R.; McHugh, J.B.; McLean, S.A.; Sullivan, S.E.; Nor, J.E.; McKean, E.L.; et al. Pathogenetic Analysis of Sinonasal Teratocarcinosarcomas Reveal Actionable β-Catenin Overexpression and a β-Catenin Mutation. J. Neurol. Surg. B Skull Base 2017, 78, 346–352. [Google Scholar] [CrossRef]
- Groenewald, W.; Lund, A.H.; Gay, D.M. The Role of WNT Pathway Mutations in Cancer Development and an Overview of Therapeutic Options. Cells 2023, 12, 990. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Song, I.H.; Lee, A.; Kang, J.; Lee, Y.S.; Lee, I.K.; Song, Y.S.; Lee, S.H. Enhancing the Landscape of Colorectal Cancer Using Targeted Deep Sequencing. Sci. Rep. 2021, 11, 8154. [Google Scholar] [CrossRef] [PubMed]
- Christie, M.; Jorissen, R.N.; Mouradov, D.; Sakthianandeswaren, A.; Li, S.; Day, F.; Tsui, C.; Lipton, L.; Desai, J.; Jones, I.T.; et al. Different APC Genotypes in Proximal and Distal Sporadic Colorectal Cancers Suggest Distinct WNT/β-Catenin Signalling Thresholds for Tumourigenesis. Oncogene 2013, 32, 4675–4682. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, C.; Breukel, C.; van der Luijt, R.; Fidalgo, P.; Lage, P.; Slors, F.J.M.; Leitão, C.N.; Fodde, R.; Smits, R. The ‘Just-Right’ Signaling Model: APC Somatic Mutations Are Selected Based on a Specific Level of Activation of the β-Catenin Signaling Cascade. Hum. Mol. Genet. 2002, 11, 1549–1560. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, P.; Lavrijsen, M.; Li, S.; Zhang, R.; Li, S.; van de Geer, W.S.; van de Werken, H.J.G.; Peppelenbosch, M.P.; Smits, R. Evaluation of AXIN1 and AXIN2 as Targets of Tankyrase Inhibition in Hepatocellular Carcinoma Cell Lines. Sci. Rep. 2021, 11, 7470. [Google Scholar] [CrossRef] [PubMed]
- Salahshor, S.; Woodgett, J.R. The Links between Axin and Carcinogenesis. J. Clin. Pathol. 2005, 58, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Giannakis, M.; Hodis, E.; Jasmine Mu, X.; Yamauchi, M.; Rosenbluh, J.; Cibulskis, K.; Saksena, G.; Lawrence, M.S.; Qian, Z.R.; Nishihara, R.; et al. RNF43 Is Frequently Mutated in Colorectal and Endometrial Cancers. Nat. Genet. 2014, 46, 1264–1266. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Ford-Roshon, D.; Russo, M.; O’Brien, C.; Xiong, X.; Gurjao, C.; Grandclaudon, M.; Raghavan, S.; Corsello, S.M.; Carr, S.A.; et al. RNF43 G659fs Is an Oncogenic Colorectal Cancer Mutation and Sensitizes Tumor Cells to PI3K/mTOR Inhibition. Nat. Commun. 2022, 13, 3181. [Google Scholar] [CrossRef]
- Elez, E.; Ros, J.; Fernández, J.; Villacampa, G.; Moreno-Cárdenas, A.B.; Arenillas, C.; Bernatowicz, K.; Comas, R.; Li, S.; Kodack, D.P.; et al. RNF43 Mutations Predict Response to Anti-BRAF/EGFR Combinatory Therapies in BRAFV600E Metastatic Colorectal Cancer. Nat. Med. 2022, 28, 2162–2170. [Google Scholar] [CrossRef]
- Yamamoto, D.; Oshima, H.; Wang, D.; Takeda, H.; Kita, K.; Lei, X.; Nakayama, M.; Murakami, K.; Ohama, T.; Takemura, H.; et al. Characterization of RNF43 Frameshift Mutations That Drive Wnt Ligand- and R-Spondin-Dependent Colon Cancer. J. Pathol. 2022, 257, 39–52. [Google Scholar] [CrossRef]
- Gavagan, M.; Fagnan, E.; Speltz, E.B.; Zalatan, J.G. The Scaffold Protein Axin Promotes Signaling Specificity within the Wnt Pathway by Suppressing Competing Kinase Reactions. Cell Syst. 2020, 10, 515–525.e5. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Xiao, G.; Hu, J. Regulation of Wnt/β-Catenin Signaling by Posttranslational Modifications. Cell Biosci. 2014, 4, 13. [Google Scholar] [CrossRef]
- Liu, C.; Kato, Y.; Zhang, Z.; Do, V.M.; Yankner, B.A.; He, X. β-Trcp Couples β-Catenin Phosphorylation-Degradation and Regulates Xenopus Axis Formation. Proc. Natl. Acad. Sci. USA 1999, 96, 6273–6278. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, T.; Zhu, H.; Yan, R.; Li, X.; Zhang, C.; Tao, W.; Ke, X.; Hao, P.; Qu, Y. Neddylation Is Essential for β-Catenin Degradation in Wnt Signaling Pathway. Cell Rep. 2022, 38, 110538. [Google Scholar] [CrossRef]
- Korinek, V.; Barker, N.; Morin, P.J.; van Wichen, D.; de Weger, R.; Kinzler, K.W.; Vogelstein, B.; Clevers, H. Constitutive Transcriptional Activation by a β-Catenin-Tcf Complex in APC−/− Colon Carcinoma. Science 1997, 275, 1784–1787. [Google Scholar] [CrossRef]
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of β-Catenin-Tcf Signaling in Colon Cancer by Mutations in β-Catenin or APC. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef]
- Rubinfeld, B.; Robbins, P.; El-Gamil, M.; Albert, I.; Porfiri, E.; Polakis, P. Stabilization of β-Catenin by Genetic Defects in Melanoma Cell Lines. Science 1997, 275, 1790–1792. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Mahoney, E.; Zuo, T.; Manchanda, P.K.; Davuluri, R.V.; Kirschner, L.S. Protein Kinase A Activation Enhances β-Catenin Transcriptional Activity through Nuclear Localization to PML Bodies. PLoS ONE 2014, 9, e109523. [Google Scholar] [CrossRef]
- Fang, D.; Hawke, D.; Zheng, Y.; Xia, Y.; Meisenhelder, J.; Nika, H.; Mills, G.B.; Kobayashi, R.; Hunter, T.; Lu, Z. Phosphorylation of β-Catenin by Akt Promotes β-Catenin Transcriptional Activity. J. Biol. Chem. 2007, 282, 11221–11229. [Google Scholar] [CrossRef]
- Ren, Q.; Chen, J.; Liu, Y. LRP5 and LRP6 in Wnt Signaling: Similarity and Divergence. Front. Cell Dev. Biol. 2021, 9, 670960. [Google Scholar] [CrossRef]
- Kim, S.-E.; Huang, H.; Zhao, M.; Zhang, X.; Zhang, A.; Semonov, M.V.; MacDonald, B.T.; Zhang, X.; Abreu, J.G.; Peng, L.; et al. Wnt Stabilization of β-Catenin Reveals Principles for Morphogen Receptor-Scaffold Assemblies. Science 2013, 340, 867–870. [Google Scholar] [CrossRef] [PubMed]
- Beagle, B.; Mi, K.; Johnson, G.V.W. Phosphorylation of PPP(S/T)P Motif of the Free LRP6 Intracellular Domain Is Not Required to Activate the Wnt/Beta-Catenin Pathway and Attenuate GSK3beta Activity. J. Cell Biochem. 2009, 108, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Tamai, K.; Zeng, X.; Liu, C.; Zhang, X.; Harada, Y.; Chang, Z.; He, X. A Mechanism for Wnt Coreceptor Activation. Mol. Cell 2004, 13, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Tamai, K.; Doble, B.; Li, S.; Huang, H.; Habas, R.; Okamura, H.; Woodgett, J.; He, X. A Dual-Kinase Mechanism for Wnt Co-Receptor Phosphorylation and Activation. Nature 2005, 438, 873–877. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; Yokota, C.; Tamai, K.; Zeng, X.; He, X. Wnt Signal Amplification via Activity, Cooperativity, and Regulation of Multiple Intracellular PPPSP Motifs in the Wnt Co-Receptor LRP6. J. Biol. Chem. 2008, 283, 16115–16123. [Google Scholar] [CrossRef] [PubMed]
- González-Sancho, J.M.; Greer, Y.E.; Abrahams, C.L.; Takigawa, Y.; Baljinnyam, B.; Lee, K.H.; Lee, K.S.; Rubin, J.S.; Brown, A.M.C. Functional Consequences of Wnt-Induced Dishevelled 2 Phosphorylation in Canonical and Noncanonical Wnt Signaling. J. Biol. Chem. 2013, 288, 9428–9437. [Google Scholar] [CrossRef] [PubMed]
- Esaki, N.; Enomoto, A.; Takagishi, M.; Mizutani, Y.; Iida, T.; Ushida, K.; Shiraki, Y.; Mii, S.; Takahashi, M. The Daple-CK1ε Complex Regulates Dvl2 Phosphorylation and Canonical Wnt Signaling. Biochem. Biophys. Res. Commun. 2020, 532, 406–413. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Rakus, D.; Gizak, A.; Steelman, L.S.; Abrams, S.L.; Lertpiriyapong, K.; Fitzgerald, T.L.; Yang, L.V.; Montalto, G.; Cervello, M.; et al. Effects of Mutations in Wnt/β-Catenin, Hedgehog, Notch and PI3K Pathways on GSK-3 Activity-Diverse Effects on Cell Growth, Metabolism and Cancer. Biochim. Biophys. Acta 2016, 1863, 2942–2976. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Abrams, S.L.; Montalto, G.; D’Assoro, A.B.; Libra, M.; Nicoletti, F.; Maestro, R.; et al. Multifaceted Roles of GSK-3 and Wnt/β-Catenin in Hematopoiesis and Leukemogenesis: Opportunities for Therapeutic Intervention. Leukemia 2014, 28, 15–33. [Google Scholar] [CrossRef]
- Wang, L.; Li, J.; Di, L. Glycogen Synthesis and beyond, a Comprehensive Review of GSK3 as a Key Regulator of Metabolic Pathways and a Therapeutic Target for Treating Metabolic Diseases. Med. Res. Rev. 2022, 42, 946–982. [Google Scholar] [CrossRef]
- Cormier, K.W.; Woodgett, J.R. Recent Advances in Understanding the Cellular Roles of GSK-3. F1000Res 2017, 6, F1000 Faculty Rev-167. [Google Scholar] [CrossRef]
- Doble, B.W.; Woodgett, J.R. GSK-3: Tricks of the Trade for a Multi-Tasking Kinase. J. Cell Sci. 2003, 116, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Grimes, C.A.; Jope, R.S. The Multifaceted Roles of Glycogen Synthase Kinase 3beta in Cellular Signaling. Prog. Neurobiol. 2001, 65, 391–426. [Google Scholar] [CrossRef] [PubMed]
- Frame, S.; Cohen, P. GSK3 Takes Centre Stage More than 20 Years after Its Discovery. Biochem. J. 2001, 359, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Eldar-Finkelman, H. Glycogen Synthase Kinase 3: An Emerging Therapeutic Target. Trends Mol. Med. 2002, 8, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Jope, R.S.; Cheng, Y.; Lowell, J.A.; Worthen, R.J.; Sitbon, Y.H.; Beurel, E. Stressed and Inflamed, Can GSK3 Be Blamed? Trends Biochem. Sci. 2017, 42, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, C.; Bienz, M. Inhibition of GSK3 by Wnt Signalling--Two Contrasting Models. J. Cell Sci. 2011, 124, 3537–3544. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Huang, H.; Garcia Abreu, J.; He, X. Inhibition of GSK3 Phosphorylation of Beta-Catenin via Phosphorylated PPPSPXS Motifs of Wnt Coreceptor LRP6. PLoS One 2009, 4, e4926. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Yu, S.X.; Lu, Y.; Bast, R.C.; Woodgett, J.R.; Mills, G.B. Phosphorylation and Inactivation of Glycogen Synthase Kinase 3 by Protein Kinase A. Proc. Natl. Acad. Sci. USA 2000, 97, 11960–11965. [Google Scholar] [CrossRef]
- Cohen, P.; Frame, S. The Renaissance of GSK3. Nat. Rev. Mol. Cell Biol. 2001, 2, 769–776. [Google Scholar] [CrossRef]
- Wu, D.; Pan, W. GSK3: A Multifaceted Kinase in Wnt Signaling. Trends Biochem. Sci. 2010, 35, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.J.; Williams, C.S. Protein Phosphatase 2A in the Regulation of Wnt Signaling, Stem Cells, and Cancer. Genes 2018, 9, 121. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Wu, C.; Liu, Y.; Zheng, P. Dimerization of Laforin Is Required for Its Optimal Phosphatase Activity, Regulation of GSK3beta Phosphorylation, and Wnt Signaling. J. Biol. Chem. 2006, 281, 34768–34774. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Menezes, M.E.; Pannell, L.K.; Mulekar, M.S.; Honkanen, R.E.; Shevde, L.A.; Samant, R.S. DNAJB6 Chaperones PP2A Mediated Dephosphorylation of GSK3β to Downregulate β-Catenin Transcription Target, Osteopontin. Oncogene 2012, 31, 4472–4483. [Google Scholar] [CrossRef] [PubMed]
- Jope, R.S. Lithium and GSK-3: One Inhibitor, Two Inhibitory Actions, Multiple Outcomes. Trends Pharmacol. Sci. 2003, 24, 441–443. [Google Scholar] [CrossRef] [PubMed]
- Alda, M. Lithium in the Treatment of Bipolar Disorder: Pharmacology and Pharmacogenetics. Mol. Psychiatry 2015, 20, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhu, B.; Zhan, M.; Hua, Z.-C. Lithium in Cancer Therapy: Friend or Foe? Cancers 2023, 15, 1095. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.A.; Mayo, J.A.; Flatow, L.; Cuthbertson, B.; O’Brien, B.E. Outcome of Bipolar Disorder on Long-Term Treatment with Lithium. Br. J. Psychiatry 1991, 159, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Valenta, T.; Hausmann, G.; Basler, K. The Many Faces and Functions of β-Catenin. EMBO J. 2012, 31, 2714–2736. [Google Scholar] [CrossRef]
- Huber, A.H.; Weis, W.I. The Structure of the β-Catenin/E-Cadherin Complex and the Molecular Basis of Diverse Ligand Recognition by β-Catenin. Cell 2001, 105, 391–402. [Google Scholar] [CrossRef]
- Daugherty, R.L.; Gottardi, C.J. Phospho-Regulation of β-Catenin Adhesion and Signaling Functions. Physiology 2007, 22, 303–309. [Google Scholar] [CrossRef]
- Tacchelly-Benites, O.; Wang, Z.; Yang, E.; Benchabane, H.; Tian, A.; Randall, M.P.; Ahmed, Y. Axin Phosphorylation in Both Wnt-off and Wnt-on States Requires the Tumor Suppressor APC. PLOS Genet. 2018, 14, e1007178. [Google Scholar] [CrossRef]
- Yamamoto, H.; Kishida, S.; Kishida, M.; Ikeda, S.; Takada, S.; Kikuchi, A. Phosphorylation of Axin, a Wnt Signal Negative Regulator, by Glycogen Synthase Kinase-3β Regulates Its Stability. J. Biol. Chem. 1999, 274, 10681–10684. [Google Scholar] [CrossRef] [PubMed]
- Jho, E.; Lomvardas, S.; Costantini, F. A GSK3β Phosphorylation Site in Axin Modulates Interaction with β-Catenin and Tcf-Mediated Gene Expression. Biochem. Biophys. Res. Commun. 1999, 266, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Peterson, A.; Garcia, B.A.; Coombs, G.; Kofahl, B.; Heinrich, R.; Shabanowitz, J.; Hunt, D.F.; Yost, H.J.; Virshup, D.M. Protein Phosphatase 1 Regulates Assembly and Function of the β-Catenin Degradation Complex. EMBO J. 2007, 26, 1511–1521. [Google Scholar] [CrossRef] [PubMed]
- Ishitani, T.; Ninomiya-Tsuji, J.; Nagai, S.; Nishita, M.; Meneghini, M.; Barker, N.; Waterman, M.; Bowerman, B.; Clevers, H.; Shibuya, H.; et al. The TAK1–NLK–MAPK-Related Pathway Antagonizes Signalling between β-Catenin and Transcription Factor TCF. Nature 1999, 399, 798–802. [Google Scholar] [CrossRef]
- Meneghini, M.D.; Ishitani, T.; Carter, J.C.; Hisamoto, N.; Ninomiya-Tsuji, J.; Thorpe, C.J.; Hamill, D.R.; Matsumoto, K.; Bowerman, B. MAP Kinase and Wnt Pathways Converge to Downregulate an HMG-Domain Repressor in Caenorhabditis Elegans. Nature 1999, 399, 793–797. [Google Scholar] [CrossRef]
- Behrens, J. Cross-Regulation of the Wnt Signalling Pathway: A Role of MAP Kinases. J. Cell Sci. 2000, 113 ( Pt. 6), 911–919. [Google Scholar] [CrossRef]
- Ota, S.; Ishitani, S.; Shimizu, N.; Matsumoto, K.; Itoh, M.; Ishitani, T. NLK Positively Regulates Wnt/β-Catenin Signalling by Phosphorylating LEF1 in Neural Progenitor Cells. EMBO J. 2012, 31, 1904–1915. [Google Scholar] [CrossRef]
- Lv, L.; Wan, C.; Chen, B.; Li, M.; Liu, Y.; Ni, T.; Yang, Y.; Liu, Y.; Cong, X.; Mao, G.; et al. Nemo-Like Kinase (NLK) Inhibits the Progression of NSCLC via Negatively Modulating WNT Signaling Pathway. J. Cell. Biochem. 2014, 115, 81–92. [Google Scholar] [CrossRef]
- Lee, E.; Salic, A.; Kirschner, M.W. Physiological Regulation of β-Catenin Stability by Tcf3 and CK1ϵ. J. Cell Biol. 2001, 154, 983–994. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Clevers, H. Wnt Signalling in Stem Cells and Cancer. Nature 2005, 434, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Klaus, A.; Birchmeier, W. Wnt Signalling and Its Impact on Development and Cancer. Nat. Rev. Cancer 2008, 8, 387–398. [Google Scholar] [CrossRef]
- Polakis, P. The Many Ways of Wnt in Cancer. Curr. Opin. Genet. Dev. 2007, 17, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Luga, V.; Zhang, L.; Viloria-Petit, A.M.; Ogunjimi, A.A.; Inanlou, M.R.; Chiu, E.; Buchanan, M.; Hosein, A.N.; Basik, M.; Wrana, J.L. Exosomes Mediate Stromal Mobilization of Autocrine Wnt-PCP Signaling in Breast Cancer Cell Migration. Cell 2012, 151, 1542–1556. [Google Scholar] [CrossRef] [PubMed]
- VanderVorst, K.; Dreyer, C.A.; Konopelski, S.E.; Lee, H.; Ho, H.-Y.H.; Carraway, K.L. Wnt/PCP Signaling Contribution to Carcinoma Collective Cell Migration and Metastasis. Cancer Res. 2019, 79, 1719–1729. [Google Scholar] [CrossRef]
- Wald, J.H.; Hatakeyama, J.; Printsev, I.; Cuevas, A.; Fry, W.H.D.; Saldana, M.J.; VanderVorst, K.; Rowson-Hodel, A.; Angelastro, J.M.; Sweeney, C.; et al. Suppression of Planar Cell Polarity Signaling and Migration in Glioblastoma by Nrdp1-Mediated Dvl Polyubiquitination. Oncogene 2017, 36, 5158–5167. [Google Scholar] [CrossRef]
- Pukrop, T.; Klemm, F.; Hagemann, T.; Gradl, D.; Schulz, M.; Siemes, S.; Trümper, L.; Binder, C. Wnt 5a Signaling Is Critical for Macrophage-Induced Invasion of Breast Cancer Cell Lines. Proc. Natl. Acad. Sci. USA 2006, 103, 5454–5459. [Google Scholar] [CrossRef]
- MacMillan, C.D.; Leong, H.S.; Dales, D.W.; Robertson, A.E.; Lewis, J.D.; Chambers, A.F.; Tuck, A.B. Stage of Breast Cancer Progression Influences Cellular Response to Activation of the WNT/Planar Cell Polarity Pathway. Sci. Rep. 2014, 4, 6315. [Google Scholar] [CrossRef]
- Uysal-Onganer, P.; Kawano, Y.; Caro, M.; Walker, M.M.; Diez, S.; Darrington, R.S.; Waxman, J.; Kypta, R.M. Wnt-11 Promotes Neuroendocrine-like Differentiation, Survival and Migration of Prostate Cancer Cells. Mol. Cancer 2010, 9, 55. [Google Scholar] [CrossRef]
- Kurayoshi, M.; Oue, N.; Yamamoto, H.; Kishida, M.; Inoue, A.; Asahara, T.; Yasui, W.; Kikuchi, A. Expression of Wnt-5a Is Correlated with Aggressiveness of Gastric Cancer by Stimulating Cell Migration and Invasion. Cancer Res. 2006, 66, 10439–10448. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.; Zhang, C.; Anagnostidis, V.; Liddle, C.; Fishel, M.L.; Gielen, F.; Scholpp, S. Cancer-Associated Fibroblasts Influence Wnt/PCP Signaling in Gastric Cancer Cells by Cytoneme-Based Dissemination of ROR2. Proc. Natl. Acad. Sci. USA 2023, 120, e2217612120. [Google Scholar] [CrossRef] [PubMed]
- Ueno, K.; Hiura, M.; Suehiro, Y.; Hazama, S.; Hirata, H.; Oka, M.; Imai, K.; Dahiya, R.; Hinoda, Y. Frizzled-7 as a Potential Therapeutic Target in Colorectal Cancer. Neoplasia 2008, 10, 697–705. [Google Scholar] [CrossRef]
- Nishioka, M.; Ueno, K.; Hazama, S.; Okada, T.; Sakai, K.; Suehiro, Y.; Okayama, N.; Hirata, H.; Oka, M.; Imai, K.; et al. Possible Involvement of Wnt11 in Colorectal Cancer Progression. Mol. Carcinog. 2013, 52, 207–217. [Google Scholar] [CrossRef]
- Feng, D.; Wang, J.; Yang, W.; Li, J.; Lin, X.; Zha, F.; Wang, X.; Ma, L.; Choi, N.T.; Mii, Y.; et al. Regulation of Wnt/PCP Signaling through P97/VCP-KBTBD7-Mediated Vangl Ubiquitination and Endoplasmic Reticulum-Associated Degradation. Sci. Adv. 2021, 7, eabg2099. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, J.; Wald, J.H.; Printsev, I.; Ho, H.-Y.H.; Carraway, K.L. Vangl1 and Vangl2: Planar Cell Polarity Components with a Developing Role in Cancer. Endocr. Relat. Cancer 2014, 21, R345–R356. [Google Scholar] [CrossRef]
- Papakrivopoulou, E.; Dean, C.H.; Copp, A.J.; Long, D.A. Planar Cell Polarity and the Kidney. Nephrol. Dial. Transplant. 2014, 29, 1320–1326. [Google Scholar] [CrossRef]
- Puvirajesinghe, T.M.; Bertucci, F.; Jain, A.; Scerbo, P.; Belotti, E.; Audebert, S.; Sebbagh, M.; Lopez, M.; Brech, A.; Finetti, P.; et al. Identification of P62/SQSTM1 as a Component of Non-Canonical Wnt VANGL2–JNK Signalling in Breast Cancer. Nat. Commun. 2016, 7, 10318. [Google Scholar] [CrossRef] [PubMed]
- Daulat, A.M.; Bertucci, F.; Audebert, S.; Sergé, A.; Finetti, P.; Josselin, E.; Castellano, R.; Birnbaum, D.; Angers, S.; Borg, J.-P. PRICKLE1 Contributes to Cancer Cell Dissemination through Its Interaction with mTORC2. Dev. Cell 2016, 37, 311–325. [Google Scholar] [CrossRef]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From Growth Signal Integration to Cancer, Diabetes and Ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef]
- Agarwal, N.K.; Chen, C.-H.; Cho, H.; Boulbès, D.R.; Spooner, E.; Sarbassov, D.D. Rictor Regulates Cell Migration by Suppressing RhoGDI2. Oncogene 2013, 32, 2521–2526. [Google Scholar] [CrossRef] [PubMed]
- Blanc, E.; Roux, G.L.; Bénard, J.; Raguénez, G. Low Expression of Wnt-5a Gene Is Associated with High-Risk Neuroblastoma. Oncogene 2005, 24, 1277–1283. [Google Scholar] [CrossRef] [PubMed]
- Blanc, E.; Goldschneider, D.; Douc-Rasy, S.; Bénard, J.; Raguénez, G. Wnt-5a Gene Expression in Malignant Human Neuroblasts. Cancer Lett. 2005, 228, 117–123. [Google Scholar] [CrossRef]
- Li, J.; Ying, J.; Fan, Y.; Wu, L.; Ying, Y.; Chan, A.T.; Srivastava, G.; Tao, Q. WNT5A Antagonizes WNT/β-Catenin Signaling and Is Frequently Silenced by Promoter CpG Methylation in Esophageal Squamous Cell Carcinoma. Cancer Biol. Ther. 2010, 10, 617–624. [Google Scholar] [CrossRef]
- Liang, H.; Chen, Q.; Coles, A.H.; Anderson, S.J.; Pihan, G.; Bradley, A.; Gerstein, R.; Jurecic, R.; Jones, S.N. Wnt5a Inhibits B Cell Proliferation and Functions as a Tumor Suppressor in Hematopoietic Tissue. Cancer Cell 2003, 4, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Roman-Gomez, J.; Jimenez-Velasco, A.; Cordeu, L.; Vilas-Zornoza, A.; San Jose-Eneriz, E.; Garate, L.; Castillejo, J.A.; Martin, V.; Prosper, F.; Heiniger, A.; et al. WNT5A, a Putative Tumour Suppressor of Lymphoid Malignancies, Is Inactivated by Aberrant Methylation in Acute Lymphoblastic Leukaemia. Eur. J. Cancer 2007, 43, 2736–2746. [Google Scholar] [CrossRef]
- Martín, V.; Valencia, A.; Agirre, X.; Cervera, J.; Jose-Eneriz, E.S.; Vilas-Zornoza, A.; Rodriguez-Otero, P.; Sanz, M.A.; Herrera, C.; Torres, A.; et al. Epigenetic Regulation of the Non-Canonical Wnt Pathway in Acute Myeloid Leukemia. Cancer Sci. 2010, 101, 425–432. [Google Scholar] [CrossRef]
- Prasad, C.P.; Manchanda, M.; Mohapatra, P.; Andersson, T. WNT5A as a Therapeutic Target in Breast Cancer. Cancer Metastasis Rev. 2018, 37, 767–778. [Google Scholar] [CrossRef]
- Prasad, C.P.; Chaurasiya, S.K.; Guilmain, W.; Andersson, T. WNT5A Signaling Impairs Breast Cancer Cell Migration and Invasion via Mechanisms Independent of the Epithelial-Mesenchymal Transition. J. Exp. Clin. Cancer Res. 2016, 35, 144. [Google Scholar] [CrossRef]
- Leris, A.C.A.; Roberts, T.R.; Jiang, W.G.; Newbold, R.F.; Mokbel, K. WNT5A Expression in Human Breast Cancer. Anticancer. Research 2005, 25, 731–734. [Google Scholar]
- Kremenevskaja, N.; von Wasielewski, R.; Rao, A.S.; Schöfl, C.; Andersson, T.; Brabant, G. Wnt-5a Has Tumor Suppressor Activity in Thyroid Carcinoma. Oncogene 2005, 24, 2144–2154. [Google Scholar] [CrossRef] [PubMed]
- Ying, J.; Li, H.; Yu, J.; Ng, K.M.; Poon, F.F.; Wong, S.C.C.; Chan, A.T.C.; Sung, J.J.Y.; Tao, Q. WNT5A Exhibits Tumor-Suppressive Activity through Antagonizing the Wnt/β-Catenin Signaling, and Is Frequently Methylated in Colorectal Cancer. Clin. Cancer Res. 2008, 14, 55–61. [Google Scholar] [CrossRef]
- MacLeod, R.J.; Hayes, M.; Pacheco, I. Wnt5a Secretion Stimulated by the Extracellular Calcium-Sensing Receptor Inhibits Defective Wnt Signaling in Colon Cancer Cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G403–G411. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Oue, N.; Sato, A.; Hasegawa, Y.; Yamamoto, H.; Matsubara, A.; Yasui, W.; Kikuchi, A. Wnt5a Signaling Is Involved in the Aggressiveness of Prostate Cancer and Expression of Metalloproteinase. Oncogene 2010, 29, 2036–2046. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chen, Q.; Xu, H. Wnt/β-Catenin Signal Transduction Pathway in Prostate Cancer and Associated Drug Resistance. Discov. Onc 2021, 12, 40. [Google Scholar] [CrossRef]
- Weeraratna, A.T.; Jiang, Y.; Hostetter, G.; Rosenblatt, K.; Duray, P.; Bittner, M.; Trent, J.M. Wnt5a Signaling Directly Affects Cell Motility and Invasion of Metastatic Melanoma. Cancer Cell 2002, 1, 279–288. [Google Scholar] [CrossRef]
- Anastas, J.N.; Kulikauskas, R.M.; Tamir, T.; Rizos, H.; Long, G.V.; von Euw, E.M.; Yang, P.-T.; Chen, H.-W.; Haydu, L.; Toroni, R.A.; et al. WNT5A Enhances Resistance of Melanoma Cells to Targeted BRAF Inhibitors. J. Clin. Invest. 2014, 124, 2877–2890. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Kadoya, T.; Amioka, A.; Hanaki, H.; Sasada, S.; Masumoto, N.; Yamamoto, H.; Arihiro, K.; Kikuchi, A.; Okada, M. Wnt5a-Induced Cell Migration Is Associated with the Aggressiveness of Estrogen Receptor-Positive Breast Cancer. Oncotarget 2018, 9, 20979–20992. [Google Scholar] [CrossRef]
- Ripka, S.; König, A.; Buchholz, M.; Wagner, M.; Sipos, B.; Klöppel, G.; Downward, J.; Gress, T.; Michl, P. WNT5A—Target of CUTL1 and Potent Modulator of Tumor Cell Migration and Invasion in Pancreatic Cancer. Carcinogenesis 2007, 28, 1178–1187. [Google Scholar] [CrossRef]
- Pilarsky, C.; Ammerpohl, O.; Sipos, B.; Dahl, E.; Hartmann, A.; Wellmann, A.; Braunschweig, T.; Löhr, M.; Jesnowski, R.; Friess, H.; et al. Activation of Wnt Signalling in Stroma from Pancreatic Cancer Identified by Gene Expression Profiling. J. Cell. Mol. Med. 2008, 12, 2823–2835. [Google Scholar] [CrossRef]
- Bo, H.; Zhang, S.; Gao, L.; Chen, Y.; Zhang, J.; Chang, X.; Zhu, M. Upregulation of Wnt5a Promotes Epithelial-to-Mesenchymal Transition and Metastasis of Pancreatic Cancer Cells. BMC Cancer 2013, 13, 496. [Google Scholar] [CrossRef]
- Gwak, J.; Cho, M.; Gong, S.-J.; Won, J.; Kim, D.-E.; Kim, E.-Y.; Lee, S.S.; Kim, M.; Kim, T.K.; Shin, J.-G.; et al. Protein-Kinase-C-Mediated β-Catenin Phosphorylation Negatively Regulates the Wnt/β-Catenin Pathway. J. Cell Sci. 2006, 119, 4702–4709. [Google Scholar] [CrossRef]
- Torii, K.; Nishizawa, K.; Kawasaki, A.; Yamashita, Y.; Katada, M.; Ito, M.; Nishimoto, I.; Terashita, K.; Aiso, S.; Matsuoka, M. Anti-Apoptotic Action of Wnt5a in Dermal Fibroblasts Is Mediated by the PKA Signaling Pathways. Cell. Signal. 2008, 20, 1256–1266. [Google Scholar] [CrossRef] [PubMed]
- Dissanayake, S.K.; Wade, M.; Johnson, C.E.; O’Connell, M.P.; Leotlela, P.D.; French, A.D.; Shah, K.V.; Hewitt, K.J.; Rosenthal, D.T.; Indig, F.E.; et al. The Wnt5A/Protein Kinase C Pathway Mediates Motility in Melanoma Cells via the Inhibition of Metastasis Suppressors and Initiation of an Epithelial to Mesenchymal Transition. J. Biol. Chem. 2007, 282, 17259–17271. [Google Scholar] [CrossRef] [PubMed]
- Dissanayake, S.K.; Weeraratna, A.T. Detecting PKC Phosphorylation as Part of the Wnt/Calcium Pathway in Cutaneous Melanoma. In Wnt Signaling: Pathway Methods and Mammalian Models; Methods in Molecular BiologyTM; Vincan, E., Ed.; Humana Press: Totowa, NJ, USA, 2008; pp. 157–172. ISBN 978-1-59745-249-6. [Google Scholar]
- Luna-Ulloa, L.B.; Hernández-Maqueda, J.G.; Castañeda-Patlán, M.C.; Robles-Flores, M. Protein Kinase C in Wnt Signaling: Implications in Cancer Initiation and Progression. IUBMB Life 2011, 63, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Williamson, M.; Bott, S.; Brookman-Amissah, N.; Freeman, A.; Nariculam, J.; Hubank, M.J.F.; Ahmed, A.; Masters, J.R. Hypomethylation of WNT5A, CRIP1 and S100P in Prostate Cancer. Oncogene 2007, 26, 6560–6565. [Google Scholar] [CrossRef]
- Chen, M.; Wang, J.; Lu, J.; Bond, M.C.; Ren, X.-R.; Lyerly, H.K.; Barak, L.S.; Chen, W. The Anti-Helminthic Niclosamide Inhibits Wnt/Frizzled1 Signaling. Biochemistry 2009, 48, 10267–10274. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Li, Y. Salinomycin Suppresses LRP6 Expression and Inhibits Both Wnt/β-Catenin and mTORC1 Signaling in Breast and Prostate Cancer Cells. J. Cell Biochem. 2014, 115, 1799–1807. [Google Scholar] [CrossRef]
- Osada, T.; Chen, M.; Yang, X.Y.; Spasojevic, I.; Vandeusen, J.B.; Hsu, D.; Clary, B.M.; Clay, T.M.; Chen, W.; Morse, M.A.; et al. Antihelminth Compound Niclosamide Downregulates Wnt Signaling and Elicits Antitumor Responses in Tumors with Activating APC Mutations. Cancer Res. 2011, 71, 4172–4182. [Google Scholar] [CrossRef]
- Kang, H.E.; Seo, Y.; Yun, J.S.; Song, S.H.; Han, D.; Cho, E.S.; Cho, S.B.; Jeon, Y.; Lee, H.; Kim, H.S.; et al. Metformin and Niclosamide Synergistically Suppress Wnt and YAP in APC-Mutated Colorectal Cancer. Cancers 2021, 13, 3437. [Google Scholar] [CrossRef]
- Yang, W.; Li, Y.; Ai, Y.; Obianom, O.N.; Guo, D.; Yang, H.; Sakamuru, S.; Xia, M.; Shu, Y.; Xue, F. Pyrazole-4-Carboxamide (YW2065): A Therapeutic Candidate for Colorectal Cancer via Dual Activities of Wnt/β-Catenin Signaling Inhibition and AMP-Activated Protein Kinase (AMPK) Activation. J. Med. Chem. 2019, 62, 11151–11164. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Yu, C.; Li, F.; Zuo, Y.; Wang, Y.; Yao, L.; Wu, C.; Wang, C.; Ye, L. Wnt/β-Catenin Signaling in Cancers and Targeted Therapies. Sig Transduct. Target. Ther. 2021, 6, 307. [Google Scholar] [CrossRef] [PubMed]
- Yuandani; Jantan, I.; Rohani, A.S.; Sumantri, I.B. Immunomodulatory Effects and Mechanisms of Curcuma Species and Their Bioactive Compounds: A Review. Front. Pharmacol. 2021, 12, 643119. [Google Scholar] [CrossRef] [PubMed]
- Mimeault, M.; Batra, S.K. Potential Applications of Curcumin and Its Novel Synthetic Analogs and Nanotechnology-Based Formulations in Cancer Prevention and Therapy. Chin. Med. 2011, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Ojo, O.A.; Adeyemo, T.R.; Rotimi, D.; Batiha, G.E.-S.; Mostafa-Hedeab, G.; Iyobhebhe, M.E.; Elebiyo, T.C.; Atunwa, B.; Ojo, A.B.; Lima, C.M.G.; et al. Anticancer Properties of Curcumin Against Colorectal Cancer: A Review. Front. Oncol. 2022, 12, 881641. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Simmen, R.C.M. Soy Isoflavone Genistein Upregulates Epithelial Adhesion Molecule E-Cadherin Expression and Attenuates Beta-Catenin Signaling in Mammary Epithelial Cells. Carcinogenesis 2009, 30, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Kleszcz, R.; Szymańska, A.; Krajka-Kuźniak, V.; Baer-Dubowska, W.; Paluszczak, J. Inhibition of CBP/β-Catenin and Porcupine Attenuates Wnt Signaling and Induces Apoptosis in Head and Neck Carcinoma Cells. Cell. Oncol. 2019, 42, 505–520. [Google Scholar] [CrossRef] [PubMed]
- Gabata, R.; Harada, K.; Mizutani, Y.; Ouchi, H.; Yoshimura, K.; Sato, Y.; Kitao, A.; Kimura, K.; Kouji, H.; Miyashita, T.; et al. Anti-Tumor Activity of the Small Molecule Inhibitor PRI-724 Against β-Catenin-Activated Hepatocellular Carcinoma. Anticancer. Res. 2020, 40, 5211–5219. [Google Scholar] [CrossRef]
- Jin, X.-F.; Spöttl, G.; Maurer, J.; Nölting, S.; Auernhammer, C.J. Inhibition of Wnt/β-Catenin Signaling in Neuroendocrine Tumors In Vitro: Antitumoral Effects. Cancers 2020, 12, 345. [Google Scholar] [CrossRef]
- Goldstein, S.D.; Trucco, M.; Guzman, W.B.; Hayashi, M.; Loeb, D.M. A Monoclonal Antibody against the Wnt Signaling Inhibitor Dickkopf-1 Inhibits Osteosarcoma Metastasis in a Preclinical Model. Oncotarget 2016, 7, 21114–21123. [Google Scholar] [CrossRef]
- Niida, A.; Hiroko, T.; Kasai, M.; Furukawa, Y.; Nakamura, Y.; Suzuki, Y.; Sugano, S.; Akiyama, T. DKK1, a Negative Regulator of Wnt Signaling, Is a Target of the β-Catenin/TCF Pathway. Oncogene 2004, 23, 8520–8526. [Google Scholar] [CrossRef]
- Rachner, T.D.; Thiele, S.; Göbel, A.; Browne, A.; Fuessel, S.; Erdmann, K.; Wirth, M.P.; Fröhner, M.; Todenhöfer, T.; Muders, M.H.; et al. High Serum Levels of Dickkopf-1 Are Associated with a Poor Prognosis in Prostate Cancer Patients. BMC Cancer 2014, 14, 649. [Google Scholar] [CrossRef] [PubMed]
- Säfholm, A.; Tuomela, J.; Rosenkvist, J.; Dejmek, J.; Härkönen, P.; Andersson, T. The Wnt-5a-Derived Hexapeptide Foxy-5 Inhibits Breast Cancer Metastasis in Vivo by Targeting Cell Motility. Clin. Cancer Res. 2008, 14, 6556–6563. [Google Scholar] [CrossRef] [PubMed]
- Canesin, G.; Evans-Axelsson, S.; Hellsten, R.; Krzyzanowska, A.; Prasad, C.P.; Bjartell, A.; Andersson, T. Treatment with the WNT5A-Mimicking Peptide Foxy-5 Effectively Reduces the Metastatic Spread of WNT5A-Low Prostate Cancer Cells in an Orthotopic Mouse Model. PLoS One 2017, 12, e0184418. [Google Scholar] [CrossRef]
- Futai, M.; Sun-Wada, G.-H.; Wada, Y.; Matsumoto, N.; Nakanishi-Matsui, M. Vacuolar-Type ATPase: A Proton Pump to Lysosomal Trafficking. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2019, 95, 261–277. [Google Scholar] [CrossRef]
- Sun-Wada, G.-H.; Wada, Y. Role of Vacuolar-Type Proton ATPase in Signal Transduction. Biochim. Et Biophys. Acta (BBA) Bioenerg. 2015, 1847, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Pamarthy, S.; Kulshrestha, A.; Katara, G.K.; Beaman, K.D. The Curious Case of Vacuolar ATPase: Regulation of Signaling Pathways. Mol. Cancer 2018, 17, 41. [Google Scholar] [CrossRef]
- Cruciat, C.-M.; Ohkawara, B.; Acebron, S.P.; Karaulanov, E.; Reinhard, C.; Ingelfinger, D.; Boutros, M.; Niehrs, C. Requirement of Prorenin Receptor and Vacuolar H+-ATPase-Mediated Acidification for Wnt Signaling. Science 2010, 327, 459–463. [Google Scholar] [CrossRef]
- Hermle, T.; Saltukoglu, D.; Grünewald, J.; Walz, G.; Simons, M. Regulation of Frizzled-Dependent Planar Polarity Signaling by a V-ATPase Subunit. Curr. Biol. 2010, 20, 1269–1276. [Google Scholar] [CrossRef]
- Cotter, K.; Liberman, R.; Sun-Wada, G.; Wada, Y.; Sgroi, D.; Naber, S.; Brown, D.; Breton, S.; Forgac, M. The A3 Isoform of Subunit a of the Vacuolar ATPase Localizes to the Plasma Membrane of Invasive Breast Tumor Cells and Is Overexpressed in Human Breast Cancer. Oncotarget 2016, 7, 46142–46157. [Google Scholar] [CrossRef]
- Liu, P.; Chen, H.; Han, L.; Zou, X.; Shen, W. Expression and Role of V1A Subunit of V-ATPases in Gastric Cancer Cells. Int. J. Clin. Oncol. 2015, 20, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T.; Numata, M.; Yagishita, H.; Futagami, F.; Tsukioka, Y.; Kitagawa, H.; Kayahara, M.; Nagakawa, T.; Miyazaki, I.; Yamamoto, M.; et al. Expression of 16 kDa Proteolipid of Vacuolar-Type H(+)-ATPase in Human Pancreatic Cancer. Br. J. Cancer 1996, 73, 1511–1517. [Google Scholar] [CrossRef]
- Son, S.W.; Kim, S.-H.; Moon, E.-Y.; Kim, D.-H.; Pyo, S.; Um, S.H. Prognostic Significance and Function of the Vacuolar H+-ATPase Subunit V1E1 in Esophageal Squamous Cell Carcinoma. Oncotarget 2016, 7, 49334–49348. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.-S.; Stratton, S.A.; Lee, S.H.; Kim, M.-J.; Jun, S.; Zhang, J.; Zheng, B.; Cervantes, C.L.; Cha, J.-H.; Barton, M.C.; et al. TMEM9-v-ATPase Activates Wnt/β-Catenin Signaling Via APC Lysosomal Degradation for Liver Regeneration and Tumorigenesis. Hepatology 2021, 73, 776–794. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wang, J.; Hassan, A.; Lee, C.-H.; Xie, X.-S.; Li, X. Molecular Basis of V-ATPase Inhibition by Bafilomycin A1. Nat. Commun. 2021, 12, 1782. [Google Scholar] [CrossRef]
- Kim, M.K. Novel Insight into the Function of Tankyrase (Review). Oncol. Lett. 2018, 16, 6895–6902. [Google Scholar] [CrossRef]
- Gunaydin, H.; Gu, Y.; Huang, X. Novel Binding Mode of a Potent and Selective Tankyrase Inhibitor. PLoS One 2012, 7, e33740. [Google Scholar] [CrossRef]

| Wnt Signaling Pathway | Receptors | Wnt-Ligands |
|---|---|---|
| Canonical Wnt/β-catenin Signaling pathway | LRP5/6 and FZD | Wnt1 |
| Wnt2 | ||
| Wnt2b | ||
| Wnt3 | ||
| Wnt3a | ||
| Wnt4 | ||
| Wnt5a | ||
| Wnt6 | ||
| Wnt7a | ||
| Wnt9a | ||
| Wnt10a | ||
| Wnt10b | ||
| Non-canonical Wnt/PCP Signaling pathway | ROR1/2, RYP, PTK7 and FZD | Wnt1 |
| Wnt2 | ||
| Wnt5a | ||
| Wnt5b | ||
| Wnt11 | ||
| Non-canonical Wnt/Ca2+ Signaling pathway | ROR1/2, RYP, PTK7 and FZD | Wnt1 |
| Wnt2 | ||
| Wnt5a | ||
| Wnt5b | ||
| Wnt11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tümen, D.; Heumann, P.; Huber, J.; Hahn, N.; Macek, C.; Ernst, M.; Kandulski, A.; Kunst, C.; Gülow, K. Unraveling Cancer’s Wnt Signaling: Dynamic Control through Protein Kinase Regulation. Cancers 2024, 16, 2686. https://doi.org/10.3390/cancers16152686
Tümen D, Heumann P, Huber J, Hahn N, Macek C, Ernst M, Kandulski A, Kunst C, Gülow K. Unraveling Cancer’s Wnt Signaling: Dynamic Control through Protein Kinase Regulation. Cancers. 2024; 16(15):2686. https://doi.org/10.3390/cancers16152686
Chicago/Turabian StyleTümen, Deniz, Philipp Heumann, Julia Huber, Nele Hahn, Celina Macek, Martha Ernst, Arne Kandulski, Claudia Kunst, and Karsten Gülow. 2024. "Unraveling Cancer’s Wnt Signaling: Dynamic Control through Protein Kinase Regulation" Cancers 16, no. 15: 2686. https://doi.org/10.3390/cancers16152686